Altered Cogs of the Clock: Insights into the Embryonic Etiology of Spondylocostal Dysostosis



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
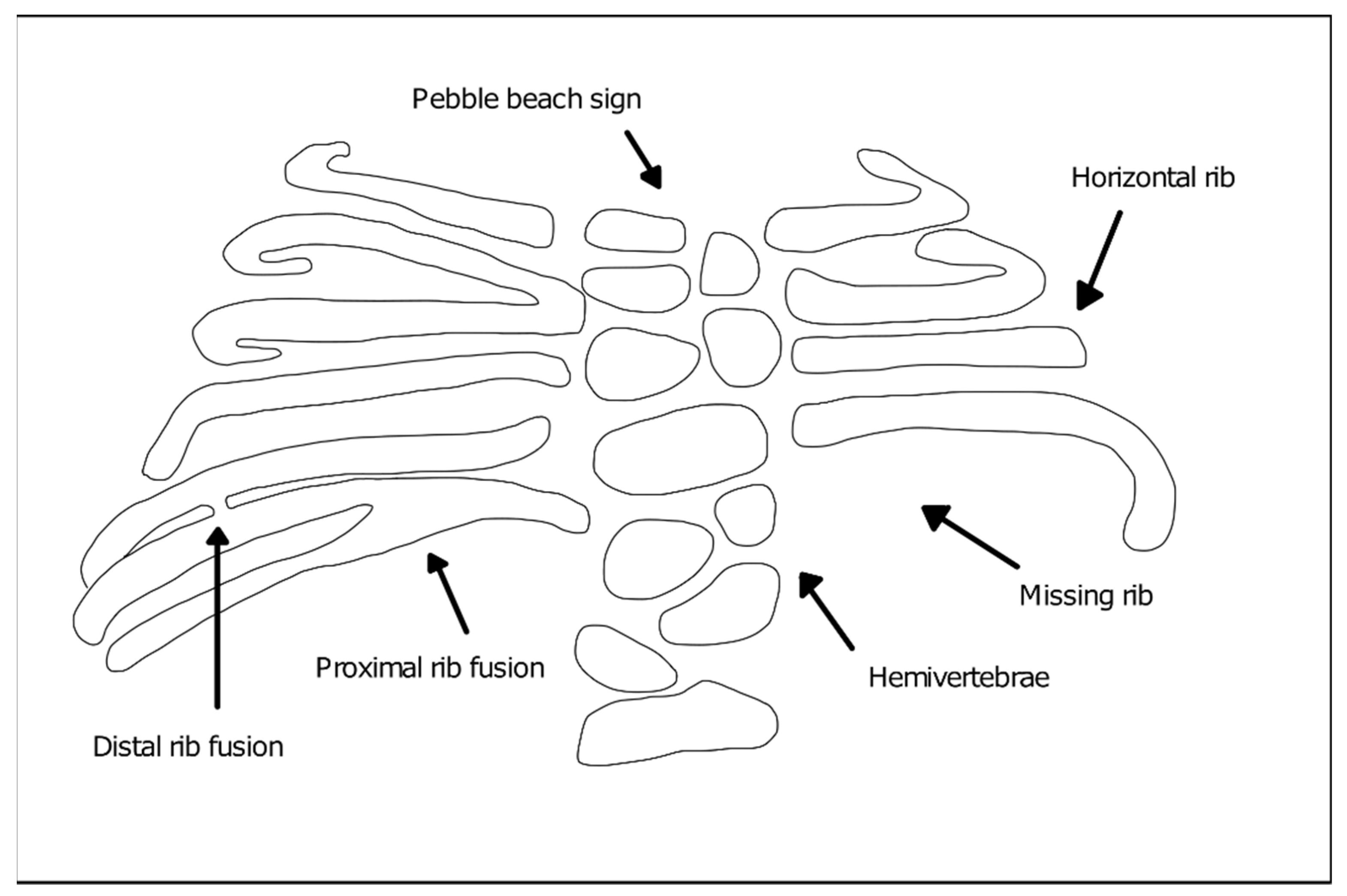
2. Spondylocostal Dysostosis
Genetics and Main Phenotypic Characteristics of SCDO Subtypes
- DLL3 (Delta-like protein 3)—Type 1 SCDO (OMIM #277300). Type 1 SCDO is the most common form found in clinical practice and the majority of the affected individuals result from inbreeding unions [6]. DLL3 mutations identified in patients include insertions, frameshift, splicing, and nonsense mutations leading to premature truncation or protein function impairment [11,12]. Phenotypically, the mutation of this gene leads to moderate, non-progressive scoliosis and rarely requires surgical intervention to stabilize the spine. The affected individuals consistently show an irregular ossification pattern, with the vertebral bodies assuming a rounded or oval shape during childhood (“pebble beach sign”), evolving into irregular vertebral bodies and hemivertebrae as ossification is completed [6];
- MESP2 (Mesoderm posterior protein 2)—Type 2 SCDO (OMIM #608681). The pathogenic variant of this gene results in straight ribs with fewer fusion points and therefore, more regularly aligned when compared to other types. SCDO type 2 has been described in three families, one of them with consanguineous parents [6]. Reported MESP2 missense mutations introduce premature stop codons leading to protein truncation [13]. Other mutations thought to severely reduce protein levels due to nonsense-mediated mRNA decay, are found in cases of STD and are associated with more severe phenotypes [6,13];
- LFNG (Lunatic Fringe)—Type 3 SCDO (OMIM #609813). There are two reports of LFNG mutations associated with SCDO. The first documented individual presented a more severe shortening of the spine than that found in the other SCDO subtypes, with all vertebral bodies exhibiting severe segmentation defects. Rib anomalies were similar to those in SCDO type 1 and 2 [14,15]. An additional report was made of an individual carrying two distinct mutations in LFNG, with multiple vertebral defects along the entire spine [16,17]. In both reported cases, the identified missense mutations were found to impair Lfng enzymatic activity and/or subcellular localization [17]. Comparable vertebral malformations were described in individuals with mutations in the SLC35A3 gene encoding the Golgi UDP-GlcNAc transporter [18] (required for Lfng substrate availability), which further supports the importance of Lfng activity in axial skeleton formation;
- HES7 (Hairy enhancer of split 7)—Type 4 SCDO (OMIM #613686). HES7 belongs to the family of hairy-enhancer-of-split transcription factors and is specifically expressed in the embryonic paraxial presomitic mesoderm (PSM) [19]. Mutations in HES7 were described in infants presenting a shortened spine, with segmentation defects predominantly in the thoracic region and irregularly aligned, fused ribs [20,21]. The identified missense mutations resulted in significant reduction of HES7 transcriptional inhibitory activity and alterations to its heterodimerization potential [20,21]. In some cases, neural tube closure defects were also present, although there is no evidence of a direct association of the two conditions. Type 4 SCDO was described in three families, one of which also had inbreeding [6];
- TBX6 (T-box transcription factor 6)—Type 5 SCDO (OMIM #122600). SCDO-associated mutations in TBX6 were described in three generations of the same family, following an autosomal dominant inheritance. The affected individuals, all male, had a mixture of hemivertebrae and blocks of fused vertebral segments, moderate scoliosis affecting the middle thoracic region, with little involvement of the ribs [22,23]. Multiple other cases have also been reported and the underlying TBX6 mutations include 16p11.2 genomic deletions, as well as nonsense and frameshift mutations, some of which were found to alter TBX6 subcellular localization and/or transcriptional activity [16,24,25];
- RIPPLY2 (Protein ripply 2)—Type 6 SCDO (OMIM #616566). The first report of mutations in this gene described two brothers who had vertebral segmentation defects in cervical and thoracic regions, including hemivertebrae and butterfly vertebrae but overall normal ribs, with marked cervical kyphosis and moderate thoracic scoliosis [6,26]. One of the reported mutations introduces a premature stop codon, with consequent loss of transcriptional repressor activity; the other is a missense mutation localized at a mRNA splice site, but its functional consequences have not yet been elucidated [26]. Since then, RIPPLY2 mutations were described in several other individuals with vertebral defects, many times associated with additional congenital malformations [27,28];
- DMRT2 (Doublesex And Mab-3 Related Transcription Factor 2; OMIM *604935). An homozygous DMRT2 variant, predicted to lead to the absence of full length DMRT2 protein product due to loss of the start codon, was recently associated with a severe form of a SCDO-like phenotype [29]. The newborn presented severe costovertebral defects, with all ribs affected either in size or shape (missing, fused, bifid, and hypoplastic), particularly in the most distal part. The vertebrae were also malformed (laminae intervertebral fusions and irregular ossification), despite the absence of clear segmentation defects of the vertebral bodies.
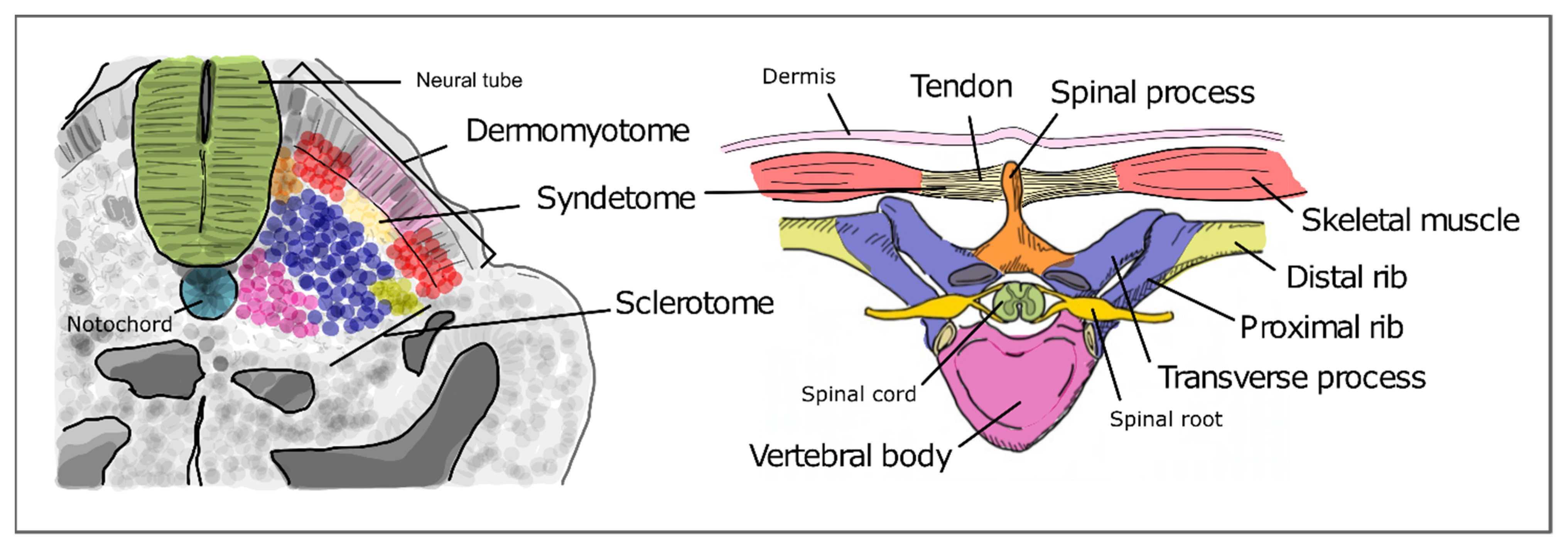
3. Formation of the Spine during Embryogenesis
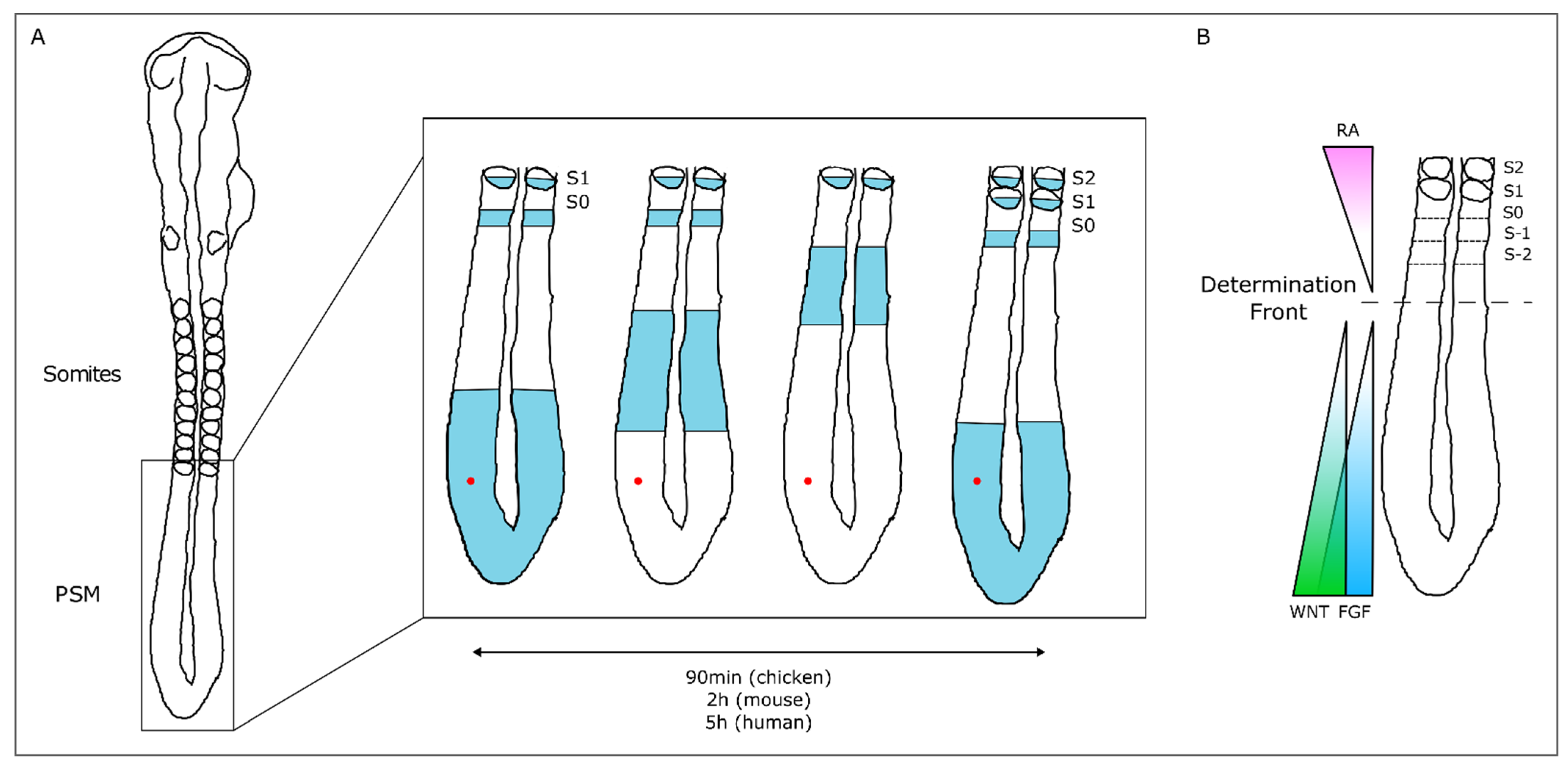
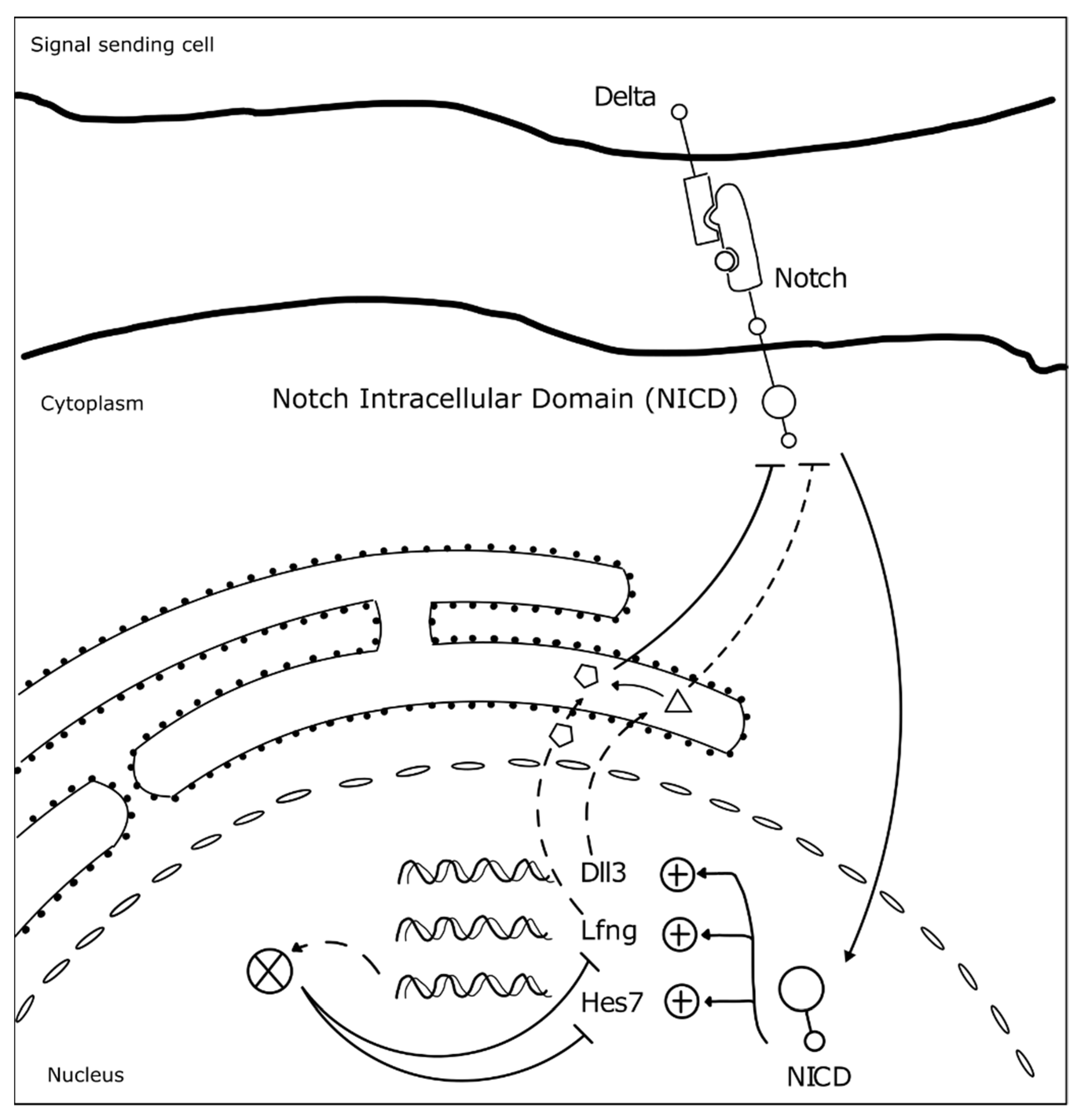
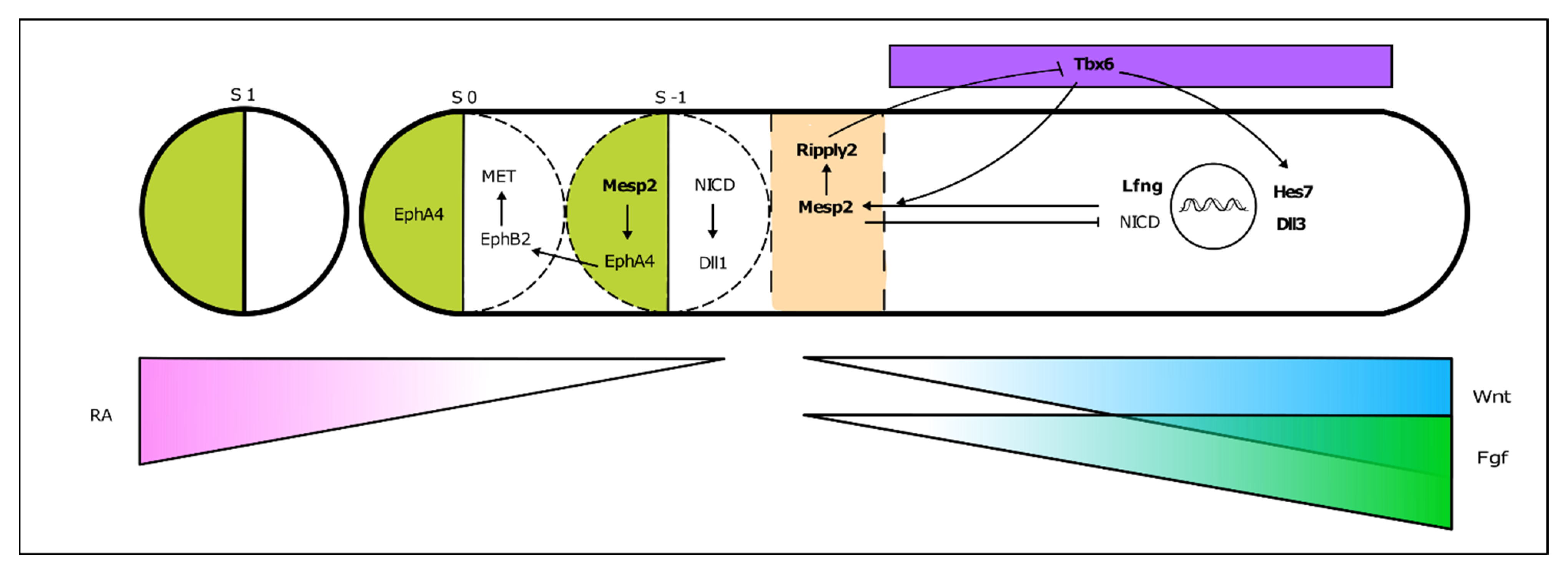
3.1. The Somitogenesis Molecular Clock
3.2. Somite Segmentation and Vertebrae Formation
4. Evidence for the Embryonic Origin of SCDO from Animal Models
- The first Dll3 mouse mutants were generated in 1961, in a series of experiments using X-rays to induce gene mutations [67,68]. These mice, known as pudgy mice, presented shortened tails, trunks, and extensive axial skeleton malformations, similarly to what can be observed in patients with SCDO type 1. The skeleton defects were traced back to somitogenesis, where these mutants presented irregularly shaped and missing posterior somites, and somite formation was delayed [69]. The somitogenesis clock oscillations were perturbed in these mutants, as well the Mesp2 expression domains, which most probably underlies the phenotypes observed [69,70];
- Studies using Mesp2 mutant mice showed that it is essential for the formation of boundaries between adjacent somites in the anterior PSM. These mutants present defects in somite segmentation and rostral-caudal polarity, which ultimately lead to severe skeletal malformations across the axis extension, including rib fusions and abnormally shaped vertebrae and ribs [71]. The similarity between the skeletal defects in Human SCDO type 2 caused by MESP2 homozygous mutation and in the mouse embryo was further confirmed by three-dimensional computed tomography [72];
- In Lfng homozygous mutant mice, somitic boundaries are unclear, generating somites that are irregular both in size and shape. Hes7 is overexpressed along the PSM instead of presenting the typical dynamic patterns, although Notch signaling remained dynamic [73]. Dorsal-ventral somite patterning is also affected [74,75]. Consequently, the axial skeleton of these animals presents severe malformations, including incompletely formed vertebrae and vertebral and rib fusions. This homozygous mutation is usually deadly in the neonatal period due to respiratory problems driven by rib cage abnormalities;
- Heterozygous Hes7 mutant mice show kinked tails in 43% of the animals. The homozygous embryos presented severe defects of the axial skeleton, as found in type 4 SCDO. They had shorter trunks and tails and the majority died shortly after birth, apparently due to respiratory problems. Vertebrae and ribs were abnormally formed and vertebral bodies and neural arches were fused across the vertebral column [19]. Further analysis showed that Hes7 homozygous mutations lead to loss of Notch-dependent oscillatory expression of Lfng, NCID, and Nrarp. Interestingly, dynamic expression patterns of genes belonging to Fgf and Wnt signaling were maintained [73];
- Homozygous Tbx6 mouse mutants lack somites [76]. This is essentially because Tbx6 is required for paraxial mesoderm specification. In these mutants, there is no PSM and three neural tubes are formed instead. A careful analysis of the heterozygous mutants however, unveiled mild defects in the axial skeleton at E14,5 [76]. Additionally, heterozygous mutations of Tbx6 in rats can lead to skeletal malformations, including lumbar vertebral distortion and abnormal number of vertebrae, which resembles the autosomal dominant form of human SCDO type 5 [77];
- Ripply2 mutant embryos fail to form clear boundaries between somites, which also present polarity abnormalities and the homozygous mice die shortly after birth [78]. These mutants present severe axial skeleton malformations, including fused arches and pedicles. This phenotype is very similar to that found in Mesp2 mutants, possibly because both genes are involved in the same process of somite boundary formation;
- In 2006, Seo and collaborators engineered a Dmrt2 knock-out mouse in order to study the role of this gene during embryonic development. Homozygous mutants showed kinked tails and respiratory distress due to malformations of the thoracic cage [59]. Mutants had truncated ribs, rib bifurcations and fusions, along with other vertebral defects. Dmrt2 mutant mice die perinatally, similarly to the reported case of DMRT2-associated SCDO [59].
4.1. Environmental Contributions to SCDO
4.2. New Insights on SCDO from Emerging Experimental Models
5. Conclusions
Funding
Conflicts of Interest
References
- UNICEF; WHO; World Bank. UN DESA Levels & Trends in Child Mortality 2019. UN IGME Rep. 2019, 52. [Google Scholar]
- Krauss, R.S.; Hong, M. Gene–environment interactions and the etiology of birth defects. Curr. Top. Dev. Biol. 2016, 116, 569–580. [Google Scholar] [PubMed]
- Geister, K.A.; Camper, S.A. Advances in Skeletal Dysplasia Genetics. Annu. Rev. Genomics Hum. Genet. 2015, 16, 199–227. [Google Scholar] [CrossRef] [PubMed]
- Berdon, W.E.; Lampl, B.S.; Cornier, A.S.; Ramirez, N.; Turnpenny, P.D.; Vitale, M.G.; Seimon, L.P.; Cowles, R.A. Clinical and radiological distinction between spondylothoracic dysostosis (Lavy-Moseley syndrome) and spondylocostal dysostosis (Jarcho-Levin syndrome). Pediatr. Radiol. 2011, 41, 384–388. [Google Scholar] [CrossRef]
- Jarcho, S. Hereditary malformation of the vertebral bodies. Bull. Johns Hopkins Hosp. 1938, 62, 216–226. [Google Scholar]
- Turnpenny, P.D.; Sloman, M.; Dunwoodie, S.; ICVS (International Consortium for Vertebral Anomalies and Scoliosis). Spondylocostal Dysostosis, Autosomal Recessive. In GeneReviews [Internet]; Adam, M.P., Ardinger, H.H., Pagon, R.A., Wallace, S.E., Bean, L.J.H., Stephens, K., Amemiya, A., Eds.; University of Washington Press: Seattle, WA, USA, 2017. Available online: https://www.ncbi.nlm.nih.gov/books/NBK8828/ (accessed on 24 January 2021).
- Southam, B.R.; Schumaier, A.P.; Crawford, A.H. Spondylocostal Dysostosis: A Literature Review and Case Report with Long-Term Follow-Up of a Conservatively Managed Patient. Case Rep. Orthop. 2018, 2018, 1–6. [Google Scholar] [CrossRef]
- Campbell, R.M.J. Spine Deformities in Rare Congenital Syndromes: Clinical Issues. Spine 2009, 34, 1815–1827. [Google Scholar] [CrossRef]
- Lavy, N.W.; Palmer, C.G.; Merritt, A.D. A syndrome of bizarre vertebral anomalies. J. Pediatrics 1966, 69, 1121–1125. [Google Scholar] [CrossRef]
- Beine, O.; Bolland, J.; Verloes, A.; Lebrun, F.; Khamis, J.; Muller, C. Dysostosis spondylo-costale: It unites maladie génétique rare. Rev. Medicale Liege 2004, 59, 513–516. [Google Scholar]
- Bulman, M.P.; Kusumi, K.; Frayling, T.M.; McKeown, C.; Garrett, C.; Lander, E.S.; Krumlauf, R.; Hattersley, A.T.; Ellard, S.; Turnpenny, P.D. Mutations in the human delta homologue, DLL3, cause axial skeletal defects in spondylocostal dysostosis. Nat. Genet. 2000, 24, 438–441. [Google Scholar] [CrossRef]
- Turnpenny, P.D.; Whittock, N.; Duncan, J.; Dunwoodie, S.; Kusumi, K.; Ellard, S. Novel mutations in DLL3, a somitogenesis gene encoding a ligand for the Notch signalling pathway, cause a consistent pattern of abnormal vertebral segmentation in spondylocostal dysostosis. J. Med. Genet. 2003, 40, 333–339. [Google Scholar] [CrossRef] [PubMed]
- Whittock, N.V.; Sparrow, D.B.; Wouters, M.A.; Sillence, D.; Ellard, S.; Dunwoodie, S.L.; Turnpenny, P.D. Mutated MESP2 causes spondylocostal dysostosis in humans. Am. J. Hum. Genet. 2004, 74, 1249–1254. [Google Scholar] [CrossRef] [PubMed]
- Sparrow, D.B.; Chapman, G.; Wouters, M.A.; Whittock, N.V.; Ellard, S.; Fatkin, D.; Turnpenny, P.D.; Kusumi, K.; Sillence, D.; Dunwoodie, S.L. Mutation of the LUNATIC FRINGE gene in humans causes spondylocostal dysostosis with a severe vertebral phenotype. Am. J. Hum. Genet. 2006, 78, 28–37. [Google Scholar] [CrossRef]
- Shifley, E.T.; VanHorn, K.M.; Perez-Balaguer, A.; Franklin, J.D.; Weinstein, M.; Cole, S.E. Oscillatory lunatic fringe activity is crucial for segmentation of the anterior but not posterior skeleton. Development 2008, 135, 899–908. [Google Scholar] [CrossRef]
- Otomo, N.; Takeda, K.; Kawai, S.; Kou, I.; Guo, L.; Osawa, M.; Alev, C.; Kawakami, N.; Miyake, N.; Matsumoto, N.; et al. Bi-allelic loss of function variants of TBX6 causes a spectrum of malformation of spine and rib including congenital scoliosis and spondylocostal dysostosis. J. Med. Genet. 2019, 56, 622–628. [Google Scholar] [CrossRef]
- Otomo, N.; Mizumoto, S.; Lu, H.F.; Takeda, K.; Campos-Xavier, B.; Mittaz-Crettol, L.; Guo, L.; Takikawa, K.; Nakamura, M.; Yamada, S.; et al. Identification of novel LFNG mutations in spondylocostal dysostosis. J. Hum. Genet. 2019, 64, 261–264. [Google Scholar] [CrossRef]
- Edmondson, A.C.; Bedoukian, E.C.; Deardorff, M.A.; McDonald-McGinn, D.M.; Li, X.; He, M.; Zackai, E.H. A human case of SLC35A3-related skeletal dysplasia. Am. J. Med. Genet. A 2017, 173, 2758–2762. [Google Scholar] [CrossRef]
- Bessho, Y.; Sakata, R.; Komatsu, S.; Shiota, K.; Yamada, S.; Kageyama, R. Dynamic expression and essential functions of Hes7 in somite segmentation. Genes Dev. 2001, 15, 2642–2647. [Google Scholar] [CrossRef]
- Sparrow, D.B.; Guillén-Navarro, E.; Fatkin, D.; Dunwoodie, S.L. Mutation of HAIRY-AND-ENHANCER-OF-SPLIT-7 in humans causes spondylocostal dysostosis. Hum. Mol. Genet. 2008, 17, 3761–3766. [Google Scholar] [CrossRef]
- Sparrow, D.B.; Sillence, D.; Wouters, M.A.; Turnpenny, P.D.; Dunwoodie, S.L. Two novel missense mutations in HAIRY-AND-ENHANCER-OF-SPLIT-7 in a family with spondylocostal dysostosis. Eur. J. Hum. Genet. 2010, 18, 674–679. [Google Scholar] [CrossRef]
- Sparrow, D.B.; McInerney-Leo, A.; Gucev, Z.S.; Gardiner, B.; Marshall, M.; Leo, P.J.; Chapman, D.L.; Tasic, V.; Shishko, A.; Brown, M.A.; et al. Autosomal dominant spondylocostal dysostosis is caused by mutation in TBX6. Hum. Mol. Genet. 2013, 22, 1625–1631. [Google Scholar] [CrossRef]
- Chen, W.; Liu, J.; Yuan, D.; Zuo, Y.; Liu, Z.; Liu, S.; Zhu, Q.; Qiu, G.; Huang, S.; Giampietro, P.F.; et al. Progress and perspective of TBX6 gene in congenital vertebral malformations. Oncotarget 2016, 7, 57430–57441. [Google Scholar] [CrossRef]
- Wu, N.; Ming, X.; Xiao, J.; Wu, Z.; Chen, X.; Shinawi, M.; Shen, Y.; Yu, G.; Liu, J.; Xie, H.; et al. TBX6 null variants and a common hypomorphic allele in congenital scoliosis. N. Engl. J. Med. 2015, 372, 341–350. [Google Scholar] [CrossRef]
- Takeda, K.; Kou, I.; Kawakami, N.; Iida, A.; Nakajima, M.; Ogura, Y.; Imagawa, E.; Miyake, N.; Matsumoto, N.; Yasuhiko, Y.; et al. Compound Heterozygosity for Null Mutations and a Common Hypomorphic Risk Haplotype in TBX6 Causes Congenital Scoliosis. Hum. Mutat. 2017, 38, 317–323. [Google Scholar] [CrossRef]
- McInerney-Leo, A.M.; Sparrow, D.B.; Harris, J.E.; Gardiner, B.B.; Marshall, M.S.; O’Reilly, V.C.; Shi, H.; Brown, M.A.; Leo, P.J.; Zankl, A.; et al. Compound heterozygous mutations in RIPPLY2 associated with vertebral segmentation defects. Hum. Mol. Genet. 2015, 24, 1234–1242. [Google Scholar] [CrossRef]
- Serey-Gaut, M.; Scala, M.; Reversade, B.; Ruaud, L.; Cabrol, C.; Musacchia, F.; Torella, A.; Accogli, A.; Escande-Beillard, N.; Langlais, J.; et al. Congenital posterior cervical spine malformation due to biallelic c.240-4T>G RIPPLY2 variant: A discrete entity. Am. J. Med. Genet. Part A 2020, 182, 1466–1472. [Google Scholar] [CrossRef]
- Wegler, M.; Roth, C.; Schumann, E.; Kogan, J.; Totten, E.; Guillen Sacoto, M.J.; Abou Jamra, R.; Hornemann, F. Congenital cervical spine malformation due to bi-allelic RIPPLY2 variants in spondylocostal dysostosis type 6. Clin. Genet. 2021. [Google Scholar] [CrossRef]
- Bouman, A.; Waisfisz, Q.; Admiraal, J.; van de Loo, M.; van Rijn, R.R.; Dimitra, M.; Oostra, R.J.; Mathijssen, I.B. Homozygous DMRT2 variant associates with severe rib malformations in a newborn. Am. J. Med. Genet. Part A 2018, 176, 1216–1221. [Google Scholar] [CrossRef]
- Williams, S.; Alkhatib, B.; Serra, R. Development of the axial skeleton and intervertebral disc. Curr. Top. Dev. Biol. 2019, 133, 49–90. [Google Scholar] [CrossRef]
- Pourquié, O. Segmentation of the vertebrate spine: From clock to scoliosis. Cell 2012, 145, 650–663. [Google Scholar] [CrossRef]
- Gibb, S.; Maroto, M.; Dale, J.K. The segmentation clock mechanism moves up a notch. Trends Cell Biol. 2010, 20, 593–600. [Google Scholar] [CrossRef]
- Kageyama, R.; Niwa, Y.; Isomura, A.; González, A.; Harima, Y. Oscillatory gene expression and somitogenesis. Wiley Interdiscip. Rev. Dev. Biol. 2012, 1, 629–641. [Google Scholar] [CrossRef]
- Chu, L.-F.; Mamott, D.; Ni, Z.; Bacher, R.; Liu, C.; Swanson, S.; Kendziorski, C.; Stewart, R.; Thomson, J.A. An In Vitro Human Segmentation Clock Model Derived from Embryonic Stem Cells. Cell Rep. 2019, 28, 2247–2255. [Google Scholar] [CrossRef]
- Cooke, J.; Zeeman, E.C. A clock and wavefront model for control of the number of repeated structures during animal morphogenesis. J. Theor. Biol. 1976, 58, 455–476. [Google Scholar] [CrossRef]
- Palmeirim, I.; Henrique, D.; Ish-Horowicz, D.; Pourquié, O. Avian hairy Gene Expression Identifies a Molecular Clock Linked to Vertebrate Segmentation and Somitogenesis. Cell 1997, 91, 639–648. [Google Scholar] [CrossRef]
- Pourquie, O. The vertebrate segmentation clock. J. Anat. 2001, 199, 169–175. [Google Scholar] [CrossRef]
- Dubrulle, J.; McGrew, M.J.; Pourquié, O. FGF signaling controls somite boundary position and regulates segmentation clock control of spatiotemporal Hox gene activation. Cell 2001, 106, 219–232. [Google Scholar] [CrossRef]
- Aulehla, A.; Wehrle, C.; Brand-Saberi, B.; Kemler, R.; Gossler, A.; Kanzler, B.; Herrmann, B.G. Wnt3a Plays a Major Role in the Segmentation Clock Controlling Somitogenesis. Dev. Cell 2003, 4, 395–406. [Google Scholar] [CrossRef]
- Diez del Corral, R.; Storey, K.G. Opposing FGF and retinoid pathways: A signalling switch that controls differentiation and patterning onset in the extending vertebrate body axis. Bioessays 2004, 26, 857–869. [Google Scholar] [CrossRef]
- Boulet, A.M.; Capecchi, M.R. Signaling by FGF4 and FGF8 is required for axial elongation of the mouse embryo. Dev. Biol. 2012, 371, 235–245. [Google Scholar] [CrossRef]
- Nakaya, M.; Biris, K.; Tsukiyama, T.; Jaime, S.; Rawls, J.A.; Yamaguchi, T.P. Wnt3alinks left-right determination with segmentation and anteroposterior axis elongation. Development 2005, 132, 5425–5436. [Google Scholar] [CrossRef] [PubMed]
- Takashima, Y.; Ohtsuka, T.; González, A.; Miyachi, H.; Kageyama, R. Intronic delay is essential for oscillatory expression in the segmentation clock. Proc. Natl. Acad. Sci. USA 2011, 108, 3300–3305. [Google Scholar] [CrossRef] [PubMed]
- Harima, Y.; Takashima, Y.; Ueda, Y.; Ohtsuka, T.; Kageyama, R. Accelerating the tempo of the segmentation clock by reducing the number of introns in the Hes7 gene. Cell Rep. 2013, 3, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Nitanda, Y.; Matsui, T.; Matta, T.; Higami, A.; Kohno, K.; Nakahata, Y.; Bessho, Y. 3′–UTR-dependent regulation of mRNA turnover is critical for differential distribution patterns of cyclic gene mRNAs. FEBS J. 2014, 281, 146–156. [Google Scholar] [CrossRef] [PubMed]
- Bonev, B.; Stanley, P.; Papalopulu, N. MicroRNA-9 Modulates Hes1 ultradian oscillations by forming a double-negative feedback loop. Cell Rep. 2012, 2, 10–18. [Google Scholar] [CrossRef] [PubMed]
- Fujino, Y.; Yamada, K.; Sugaya, C.; Ooka, Y.; Ovara, H.; Ban, H.; Akama, K.; Otosaka, S.; Kinoshita, H.; Yamasu, K.; et al. Deadenylation by the CCR4-NOT complex contributes to the turnover of hairy-related mRNAs in the zebrafish segmentation clock. FEBS Lett. 2018, 592, 3388–3398. [Google Scholar] [CrossRef]
- Hirata, H.; Bessho, Y.; Kokubu, H.; Masamizu, Y.; Yamada, S.; Lewis, J.; Kageyama, R. Instability of Hes7 protein is crucial for the somite segmentation clock. Nat. Genet. 2004, 36, 750–754. [Google Scholar] [CrossRef]
- Matsuda, M.; Hayashi, H.; Garcia-Ojalvo, J.; Yoshioka-Kobayashi, K.; Kageyama, R.; Yamanaka, Y.; Ikeya, M.; Toguchida, J.; Alev, C.; Ebisuya, M. Species-specific segmentation clock periods are due to differential biochemical reaction speeds. Science 2020, 369, 1450–1455. [Google Scholar] [CrossRef]
- Chapman, G.; Sparrow, D.B.; Kremmer, E.; Dunwoodie, S.L. Notch inhibition by the ligand DELTA-LIKE 3 defines the mechanism of abnormal vertebral segmentation in spondylocostal dysostosis. Hum. Mol. Genet. 2011, 20, 905–916. [Google Scholar] [CrossRef]
- Niwa, Y.; Masamizu, Y.; Liu, T.; Nakayama, R.; Deng, C.X.; Kageyama, R. The Initiation and Propagation of Hes7 Oscillation Are Cooperatively Regulated by Fgf and Notch Signaling in the Somite Segmentation Clock. Dev. Cell 2007, 13, 298–304. [Google Scholar] [CrossRef]
- Soza-Ried, C.; Öztürk, E.; Ish-Horowicz, D.; Lewis, J. Pulses of Notch activation synchronise oscillating somite cells and entrain the zebrafish segmentation clock. Development 2014, 141, 1780–1788. [Google Scholar] [CrossRef] [PubMed]
- Yoshioka-Kobayashi, K.; Matsumiya, M.; Niino, Y.; Isomura, A.; Kori, H.; Miyawaki, A.; Kageyama, R. Coupling delay controls synchronized oscillation in the segmentation clock. Nature 2020, 580, 119–123. [Google Scholar] [CrossRef]
- Dequéant, M.L.; Glynn, E.; Gaudenz, K.; Wahl, M.; Chen, J.; Mushegian, A.; Pourquié, O. A complex oscillating network of signaling genes underlies the mouse segmentation clock. Science 2006, 314, 1595–1598. [Google Scholar] [CrossRef]
- Krol, A.J.; Roellig, D.; Dequéant, M.L.; Tassy, O.; Glynn, E.; Hattem, G.; Mushegian, A.; Oates, A.C.; Pourquié, O. Evolutionary plasticity of segmentation clock networks. Development 2011, 138, 2783–2792. [Google Scholar] [CrossRef]
- Sonnen, K.F.; Lauschke, V.M.; Uraji, J.; Falk, H.J.; Petersen, Y.; Funk, M.C.; Beaupeux, M.; François, P.; Merten, C.A.; Aulehla, A. Modulation of Phase Shift between Wnt and Notch Signaling Oscillations Controls Mesoderm Segmentation. Cell 2018, 172, 1079–1090.e12. [Google Scholar] [CrossRef]
- Saúde, L.; Lourenço, R.; Gonçalves, A.; Palmeirim, I. Terra Is a Left-Right Asymmetry Gene Required for Left-Right Synchronization of the Segmentation Clock. Nat. Cell Biol. 2005, 7, 918–920. [Google Scholar] [CrossRef]
- Lourenço, R.; Lopes, S.S.; Saúde, L. Left-Right Function of dmrt2 Genes Is Not Conserved between Zebrafish and Mouse. PLoS ONE 2011, 5, 1–8. [Google Scholar] [CrossRef]
- Seo, K.W.; Wang, Y.; Kokubo, H.; Kettlewell, J.R.; Zarkower, D.A.; Johnson, R.L. Targeted disruption of the DM domain containing transcription factor Dmrt2 reveals an essential role in somite patterning. Dev. Biol. 2006, 290, 200–210. [Google Scholar] [CrossRef]
- Saga, Y. The mechanism of somite formation in mice. Curr. Opin. Genet. Dev. 2012, 22, 331–338. [Google Scholar] [CrossRef]
- Zhao, W.; Ajima, R.; Ninomiya, Y.; Saga, Y. Segmental border is defined by Ripply2-mediated Tbx6 repression independent of Mesp2. Dev. Biol. 2015, 400, 105–117. [Google Scholar] [CrossRef]
- Rifes, P.; Thorsteinsdóttir, S. Extracellular matrix assembly and 3D organization during paraxial mesoderm development in the chick embryo. Dev. Biol. 2012, 368, 370–381. [Google Scholar] [CrossRef]
- Maroto, M.; Bone, R.A.; Kim Dale, J. Somitogenesis. Development 2012, 139, 2453–2456. [Google Scholar] [CrossRef]
- Sato, T.; Rocancourt, D.; Marques, L.; Thorsteinsdóttir, S.; Buckingham, M. A Pax3/Dmrt2/Myf5 Regulatory Cascade Functions at the Onset of Myogenesis. PLoS Genet. 2010, 6, e1000897. [Google Scholar] [CrossRef]
- Fleming, A.; Kishida, M.G.; Kimmel, C.B.; Keynes, R.J. Building the backbone: The development and evolution of vertebral patterning. Development 2015, 142, 1733–1744. [Google Scholar] [CrossRef]
- Keynes, R. Patterning spinal nerves and vertebral bones. J. Anat. 2018, 232, 534–539. [Google Scholar] [CrossRef]
- Grüneberg, H. Genetical studies on the skeleton of the mouse. J. Genet. 1953, 51, 327–358. [Google Scholar] [CrossRef]
- Kusumi, K.; Sun, E.S.; Kerrebrock, A.W.; Bronson, R.T.; Chi, D.C.; Bulotsky, M.S.; Spencer, J.B.; Birren, B.W.; Frankel, W.N.; Lander, E.S. The mouse pudgy mutation disrupts Delta homologue DII3 and initiation of early somite boundaries. Nat. Genet. 1998, 19, 274–278. [Google Scholar] [CrossRef]
- Dunwoodie, S.L.; Clements, M.; Sparrow, D.B.; Sa, X.; Conlon, R.A.; Beddington, R.S.P. Axial skeletal defects caused by mutation in the spondylocostal dysplasia/pudgy gene Dll3 are associated with disruption of the segmentation clock within the presomitic mesoderm. Development 2002, 129, 1795–1806. [Google Scholar]
- Kusumi, K.; Mimoto, M.S.; Covello, K.L.; Beddington, R.S.P.; Krumlauf, R.; Dunwoodie, S.L. Dll3 pudgy mutation differentially disrupts dynamic expression of somite genes. Genesis 2004, 39, 115–121. [Google Scholar] [CrossRef]
- Saga, Y.; Hata, N.; Koseki, H.; Taketo, M.M. Mesp2: A novel mouse gene expressed in the presegmented mesoderm and essential for segmentation initiation. Genes Dev. 1997, 11, 1827–1839. [Google Scholar] [CrossRef]
- Makinoa, Y.; Kanekob, K.; Yamaguchia, A.; Iimura, T. Developmental biology and etiology of axial skeleton: Lessons from a mouse model of spondylocostal dysostosis and spondylothoracic dysostosis. J. Oral Biosci. 2013, 55, 175–179. [Google Scholar] [CrossRef]
- Ferjentsik, Z.; Hayashi, S.; Dale, J.K.; Bessho, Y.; Herreman, A.; De Strooper, B.; del Monte, G.; de la Pompa, J.L.; Maroto, M. Notch Is a Critical Component of the Mouse Somitogenesis Oscillator and Is Essential for the Formation of the Somites. PLoS Genet. 2009, 5, e1000662. [Google Scholar] [CrossRef]
- Zhang, N.; Gridley, T. Defects in somite formation in lunatic fringe-deficient mice. Nature 1998, 394, 374–377. [Google Scholar] [CrossRef]
- Evrard, Y.A.; Lun, Y.; Aulehla, A.; Gan, L.; Johnson, R.L. Lunatic fringe is an essential mediator of somite segmentation and patterning. Nature 1998, 394, 377–381. [Google Scholar] [CrossRef]
- Chapman, D.L.; Papaioannou, V.E. Three neural tubes in mouse embryos with mutations in the T-box gene Tbx6. Nature 1998, 391, 695–697. [Google Scholar] [CrossRef]
- Abe, K.; Takamatsu, N.; Ishikawa, K.; Tsurumi, T.; Tanimoto, S.; Sakurai, Y.; Lisse, T.; Imai, K.; Serikawa, T.; Mashimo, T. Novel ENU-induced mutation in Tbx6 causes dominant spondylocostal dysostosis-like vertebral malformations in the rat. PLoS ONE 2015, 10, 134527. [Google Scholar] [CrossRef]
- Morimoto, M.; Sasaki, N.; Oginuma, M.; Kiso, M.; Igarashi, K.; Aizaki, K.I.; Kanno, J.; Saga, Y. The negative regulation of Mesp2 by mouse Ripply2 is required to establish the rostro-caudal patterning within a somite. Development 2007, 134, 1561–1569. [Google Scholar] [CrossRef]
- Sparrow, D.B.; Chapman, G.; Smith, A.J.; Mattar, M.Z.; Major, J.A.; O’Reilly, V.C.; Saga, Y.; Zackai, E.H.; Dormans, J.P.; Alman, B.A.; et al. A mechanism for gene-environment interaction in the etiology of congenital scoliosis. Cell 2012, 149, 295–306. [Google Scholar] [CrossRef]
- Hou, D.; Kang, N.; Yin, P.; Hai, Y. Abnormalities associated with congenital scoliosis in high-altitude geographic regions. Int. Orthop. 2018, 42, 575–581. [Google Scholar] [CrossRef]
- Mills, J.L. Malformations in infants of diabetic mothers. Teratology 25:385-94. 1982. Birth Defects Res. A Clin. Mol. Teratol. 2010, 88, 769–778. [Google Scholar] [CrossRef]
- Lemire, G.T.; Beauregard-Lacroix, É.; Campeau, P.M.; Parent, S.; Roy-Beaudry, M.; Soglio, D.D.; Grignon, A.; Rypens, F.; Wavrant, S.; Laberge, A.M.; et al. Retrospective analysis of fetal vertebral defects: Associated anomalies, etiologies, and outcome. Am. J. Med. Genet. Part A 2020, 182, 664–672. [Google Scholar] [CrossRef]
- Hakyemez Toptan, H.; Karadag, N.; Tuten, A.; Gokmen Yildirim, T.; Karatekin, G. Infant of a Diabetic Mother with Spondylocostal Dysostosis and Multiple Congenital Anomalies. Istanbul Med. J. 2017, 18, 40–43. [Google Scholar] [CrossRef]
- Oginuma, M.; Moncuquet, P.; Xiong, F.; Karoly, E.; Chal, J.; Guevorkian, K.; Pourquié, O. A Gradient of Glycolytic Activity Coordinates FGF and Wnt Signaling during Elongation of the Body Axis in Amniote Embryos. Dev. Cell 2017, 40, 342–353.e10. [Google Scholar] [CrossRef]
- Cho, W.; Shepard, N.; Arlet, V. The etiology of congenital scoliosis: Genetic vs. environmental-a report of three monozygotic twin cases. Eur. Spine J. 2018, 27, 533–537. [Google Scholar] [CrossRef]
- Kaspiris, A.; Grivas, T.B.; Weiss, H.R. Congenital scoliosis in monozygotic twins: Case report and review of possible factors contributing to its development. Scoliosis 2008, 3, 1–6. [Google Scholar] [CrossRef]
- Mckinley, L.M.; Leatherman, K.D. Idiopathic and Congenital Scoliosis in Twins. Spine 1978, 3, 227–229. [Google Scholar] [CrossRef]
- Matsuda, M.; Yamanaka, Y.; Uemura, M.; Osawa, M.; Saito, M.K.; Nagahashi, A.; Nishio, M.; Guo, L.; Ikegawa, S.; Sakurai, S.; et al. Recapitulating the human segmentation clock with pluripotent stem cells. Nature 2020, 580, 124–129. [Google Scholar] [CrossRef]
- Moris, N.; Anlas, K.; van den Brink, S.C.; Alemany, A.; Schröder, J.; Ghimire, S.; Balayo, T.; van Oudenaarden, A.; Martinez Arias, A. An in vitro model of early anteroposterior organization during human development. Nature 2020, 582, 410–415. [Google Scholar] [CrossRef]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nóbrega, A.; Maia-Fernandes, A.C.; Andrade, R.P. Altered Cogs of the Clock: Insights into the Embryonic Etiology of Spondylocostal Dysostosis. J. Dev. Biol. 2021, 9, 5. https://doi.org/10.3390/jdb9010005
Nóbrega A, Maia-Fernandes AC, Andrade RP. Altered Cogs of the Clock: Insights into the Embryonic Etiology of Spondylocostal Dysostosis. Journal of Developmental Biology. 2021; 9(1):5. https://doi.org/10.3390/jdb9010005
Chicago/Turabian StyleNóbrega, Ana, Ana C. Maia-Fernandes, and Raquel P. Andrade. 2021. "Altered Cogs of the Clock: Insights into the Embryonic Etiology of Spondylocostal Dysostosis" Journal of Developmental Biology 9, no. 1: 5. https://doi.org/10.3390/jdb9010005
APA StyleNóbrega, A., Maia-Fernandes, A. C., & Andrade, R. P. (2021). Altered Cogs of the Clock: Insights into the Embryonic Etiology of Spondylocostal Dysostosis. Journal of Developmental Biology, 9(1), 5. https://doi.org/10.3390/jdb9010005