Insights into the Etiology of Mammalian Neural Tube Closure Defects from Developmental, Genetic and Evolutionary Studies

Abstract
1. Introduction
2. The Basics of Neural Tube Formation
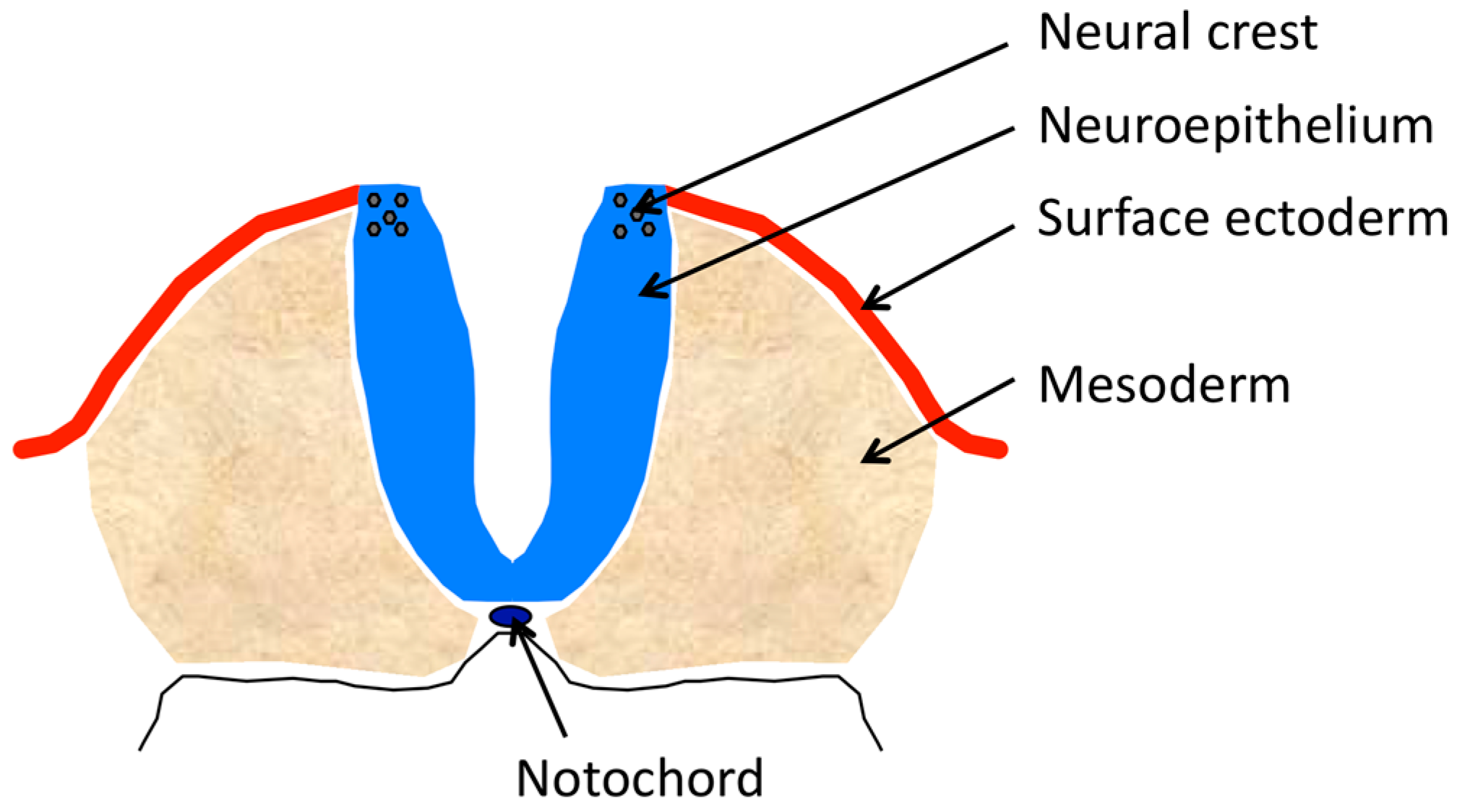
2.1. Components
2.2. Convergent Extension and Other Functions of the Planar Cell Polarity Pathway
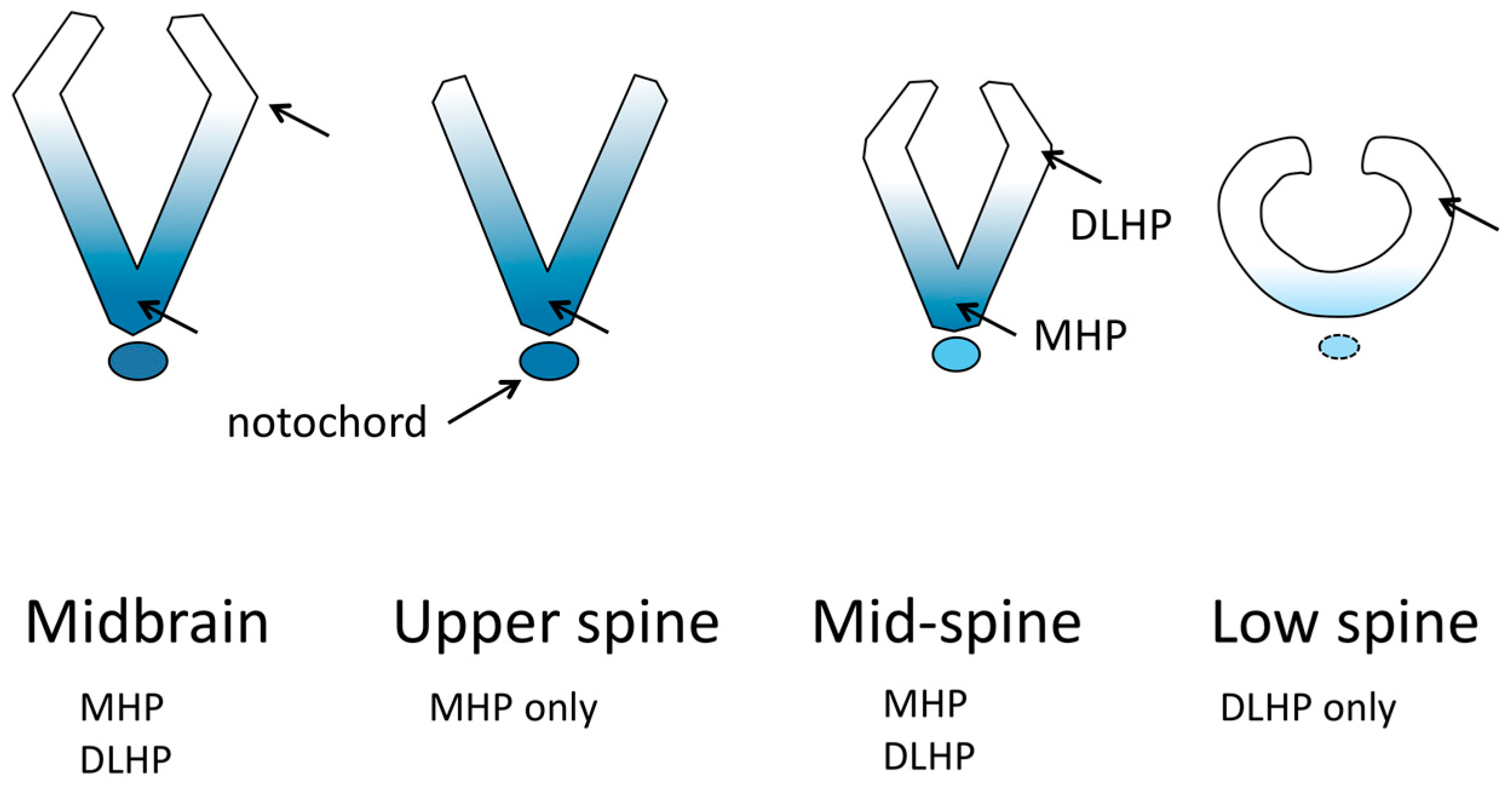
2.3. Bending and Hinge Points
2.4. Cilia
2.5. The Sonic Hedgehog Pathway Signaling in NTD
2.6. Adhesion and Fusion
3. Regional Differences in Closure Mechanisms
3.1. Planar Cell Polarity, MHP and DLHP
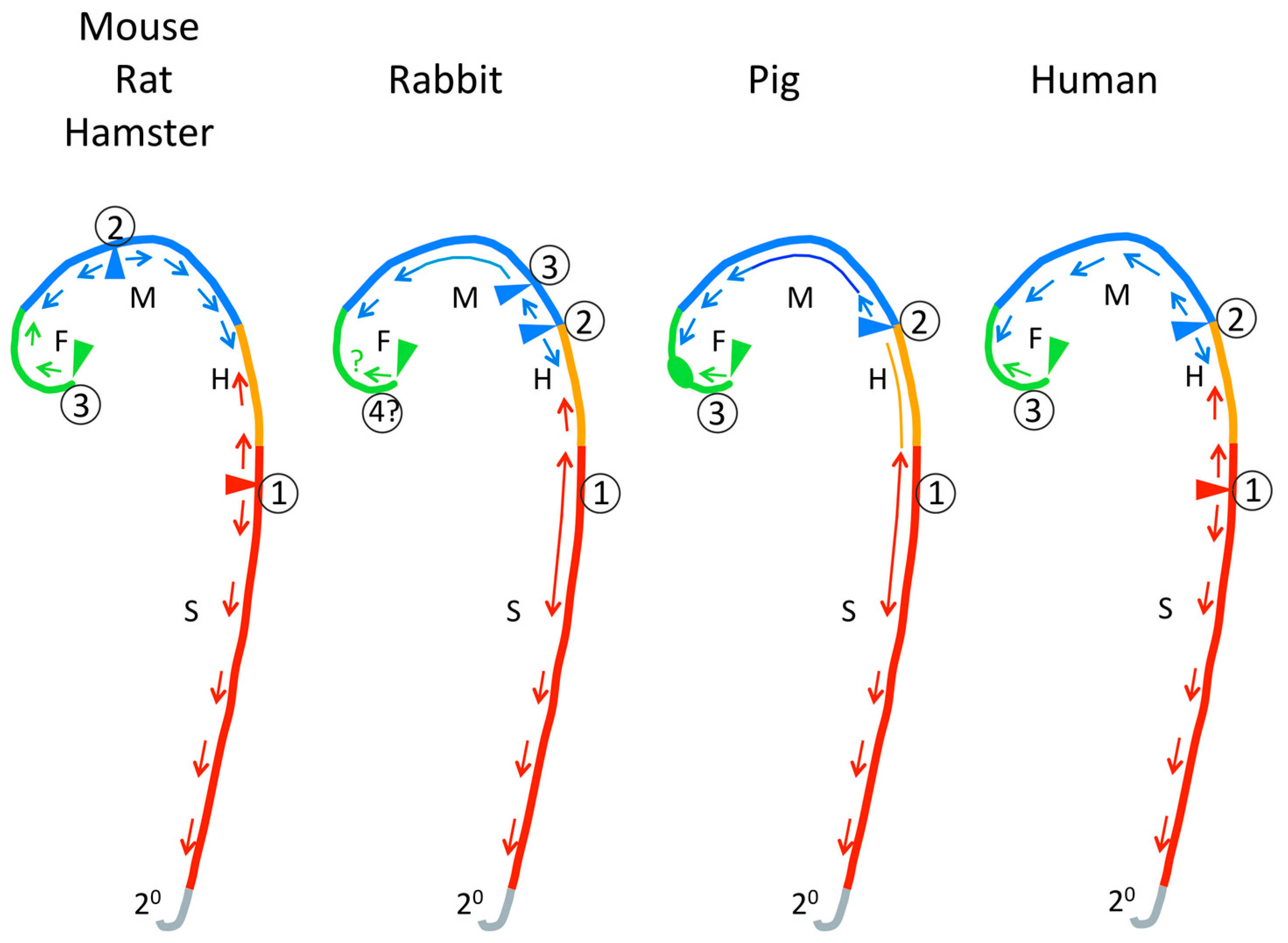
3.2. Closure Initiation Sites
3.3. Cell Type of Initial Contact
3.4. Cell Projection Types
3.5. Regional Differences in Ephrins and Ephrin Receptors
3.6. Requirement for Grhl Gene Family Expression
3.7. Dynamics of the Actomyosin Cytoskeleton of the Neuroepithelium
3.8. Other Examples: Hox, Neural Crest and Apoptosis
4. Evolutionary Aspects
4.1. Modern Vertebrates
4.2. Proxies for the Ancestral Pre-Vertebrate
5. The Cranial Neural Tube
5.1. The New Head and the Cranial Neural Crest
5.2. Absence of Hox Gene Expression
5.3. The Prechordal Plate and Cranial Flexure
5.4. Optic Sulci
5.5. Sonic Hedgehog and Cranial DLHP
5.6. The Actomyosin Cytoskeleton of the Cranial Neuroepithelium
5.7. Apoptosis in Cranial Neural Folds
5.8. Mutations in Genes Expressed in the Cranial Neural Crest That Are Associated with NTD
5.9. Role of Cranial Mesoderm in Neural Fold Elevation
5.10. Neuroepithelium-Expressed Genes and Mechanisms That Cause Exencephaly
5.11. Cranial Ephrins and Ephrin Receptors
5.12. Cranial Neural Fold Projections
5.13. The Phenomenon of Sex Ratio Distortions in Cranial NTD
6. The Spinal Region, PCP and Craniorachischisis
7. Studies of Digenic Mechanisms of NTD Involving PCP Gene Mutations
8. Folate and NTD
8.1. Human NTD Prevention by Folate
8.2. Mouse NTD Prevention by Folate
8.3. A Possible Effect of Folate on Cilia
8.4. Heterogeneity of Effects of Folate on Neural Tube Closure?
8.5. Mouse NTD Prevention by Formate
9. An Overview of the Genetics of NTD
9.1. Genetic Architecture of Human NTD
9.2. The Role of Environmental Effects
9.3. The Role of Mouse Mutants in NTD Genetics
10. Concluding Remarks
Funding
Conflicts of Interest
References
- Au, K.S.; Ashley-Koch, A.; Northrup, H. Epidemiologic and Genetic Aspects of Spina Bifida and Other Neural Tube Defects. Dev. Disabil. Res. Rev. 2010, 16, 6–15. [Google Scholar] [CrossRef] [PubMed]
- McBride, M.L. Sib Risks of Anencephaly and Spina Bifida in British Columbia. Am. J. Med. Genet. 1979, 3, 377–387. [Google Scholar] [CrossRef] [PubMed]
- Harris, M.J.; Juriloff, D.M. An Update to the List of Mouse Mutants with Neural Tube Closure Defects and Advances toward a Complete Genetic Perspective of Neural Tube Closure. Birth Defects Res. A Clin. Mol. Teratol. 2010, 88, 653–669. [Google Scholar] [CrossRef] [PubMed]
- Wilde, J.J.; Petersen, J.R.; Niswander, L. Genetic, Epigenetic, and Environmental Contributions to Neural Tube Closure. Annu. Rev. Genet. 2014, 48, 583–611. [Google Scholar] [CrossRef] [PubMed]
- Nikolopoulou, E.; Galea, G.L.; Rolo, A.; Greene, N.D.; Copp, A.J. Neural Tube Closure: Cellular, Molecular and Biomechanical Mechanisms. Development 2017, 144, 552–566. [Google Scholar] [CrossRef] [PubMed]
- Greene, N.D.; Copp, A.J. Neural Tube Defects. Annu. Rev. Neurosci. 2014, 37, 221–242. [Google Scholar] [CrossRef] [PubMed]
- Copp, A.J.; Stanier, P.; Greene, N.D. Neural Tube Defects: Recent Advances, Unsolved Questions, and Controversies. Lancet Neurol. 2013, 12, 799–810. [Google Scholar] [CrossRef]
- Wallingford, J.B.; Niswander, L.A.; Shaw, G.M.; Finnell, R.H. The Continuing Challenge of Understanding, Preventing, and Treating Neural Tube Defects. Science 2013, 339, 1222002. [Google Scholar] [CrossRef] [PubMed]
- Yamaguchi, Y.; Miura, M. How to Form and Close the Brain: Insight into the Mechanism of Cranial Neural Tube Closure in Mammals. Cell Mol. Life Sci. 2013, 70, 3171–3186. [Google Scholar] [CrossRef] [PubMed]
- Juriloff, D.M.; Harris, M.J. A Consideration of the Evidence that Genetic Defects in Planar Cell Polarity Contribute to the Etiology of Human Neural Tube Defects. Birth Defects Res. A Clin. Mol. Teratol. 2012, 94, 824–840. [Google Scholar] [CrossRef] [PubMed]
- Au, K.S.; Findley, T.O.; Northrup, H. Finding the Genetic Mechanisms of Folate Deficiency and Neural Tube Defects-Leaving no Stone Unturned. Am. J. Med. Genet. A 2017, 173, 3042–3057. [Google Scholar] [CrossRef] [PubMed]
- Harris, M.J.; Juriloff, D.M. Mouse Mutants with Neural Tube Closure Defects and their Role in Understanding Human Neural Tube Defects. Birth Defects Res. A Clin. Mol. Teratol. 2007, 79, 187–210. [Google Scholar] [CrossRef] [PubMed]
- Murdoch, J.N.; Copp, A.J. The Relationship between Sonic Hedgehog Signaling, Cilia, and Neural Tube Defects. Birth Defects Res. A Clin. Mol. Teratol. 2010, 88, 633–652. [Google Scholar] [CrossRef] [PubMed]
- Carvajal-Gonzalez, J.M.; Mulero-Navarro, S.; Mlodzik, M. Centriole Positioning in Epithelial Cells and its Intimate Relationship with Planar Cell Polarity. Bioessays 2016, 38, 1234–1245. [Google Scholar] [CrossRef] [PubMed]
- Montcouquiol, M.; Sans, N.; Huss, D.; Kach, J.; Dickman, J.D.; Forge, A.; Rachel, R.A.; Copeland, N.G.; Jenkins, N.A.; Bogani, D.; et al. Asymmetric Localization of Vangl2 and Fz3 Indicate Novel Mechanisms for Planar Cell Polarity in Mammals. J. Neurosci. 2006, 26, 5265–5275. [Google Scholar] [CrossRef] [PubMed]
- Williams, M.; Yen, W.; Lu, X.; Sutherland, A. Distinct Apical and Basolateral Mechanisms Drive Planar Cell Polarity-Dependent Convergent Extension of the Mouse Neural Plate. Dev. Cell. 2014, 29, 34–46. [Google Scholar] [CrossRef] [PubMed]
- Tissir, F.; Qu, Y.; Montcouquiol, M.; Zhou, L.; Komatsu, K.; Shi, D.; Fujimori, T.; Labeau, J.; Tyteca, D.; Courtoy, P.; et al. Lack of Cadherins Celsr2 and Celsr3 Impairs Ependymal Ciliogenesis, Leading to Fatal Hydrocephalus. Nat. Neurosci. 2010, 13, 700–707. [Google Scholar] [CrossRef] [PubMed]
- Zeng, H.; Hoover, A.N.; Liu, A. PCP Effector Gene Inturned is an Important Regulator of Cilia Formation and Embryonic Development in Mammals. Dev. Biol. 2010, 339, 418–428. [Google Scholar] [CrossRef] [PubMed]
- Escuin, S.; Vernay, B.; Savery, D.; Gurniak, C.B.; Witke, W.; Greene, N.D.; Copp, A.J. Rho-Kinase-Dependent Actin Turnover and Actomyosin Disassembly are Necessary for Mouse Spinal Neural Tube Closure. J. Cell Sci. 2015, 128, 2468–2481. [Google Scholar] [CrossRef] [PubMed]
- Rolo, A.; Escuin, S.; Greene, N.D.E.; Copp, A.J. Rho GTPases in Mammalian Spinal Neural Tube Closure. Small GTPases 2018, 9, 283–289. [Google Scholar] [CrossRef] [PubMed]
- Shum, A.S.; Copp, A.J. Regional Differences in Morphogenesis of the Neuroepithelium Suggest Multiple Mechanisms of Spinal Neurulation in the Mouse. Anat. Embryol. 1996, 194, 65–73. [Google Scholar] [CrossRef] [PubMed]
- Ybot-Gonzalez, P.; Cogram, P.; Gerrelli, D.; Copp, A.J. Sonic Hedgehog and the Molecular Regulation of Mouse Neural Tube Closure. Development 2002, 129, 2507–2517. [Google Scholar] [PubMed]
- McShane, S.G.; Mole, M.A.; Savery, D.; Greene, N.D.; Tam, P.P.; Copp, A.J. Cellular Basis of Neuroepithelial Bending during Mouse Spinal Neural Tube Closure. Dev. Biol. 2015, 404, 113–124. [Google Scholar] [CrossRef] [PubMed]
- Amarnath, S.; Agarwala, S. Cell-Cycle-Dependent TGFbeta-BMP Antagonism Regulates Neural Tube Closure by Modulating Tight Junctions. J. Cell Sci. 2017, 130, 119–131. [Google Scholar] [CrossRef] [PubMed]
- Cui, C.; Chatterjee, B.; Francis, D.; Yu, Q.; SanAgustin, J.T.; Francis, R.; Tansey, T.; Henry, C.; Wang, B.; Lemley, B.; et al. Disruption of Mks1 Localization to the Mother Centriole Causes Cilia Defects and Developmental Malformations in Meckel-Gruber Syndrome. Dis. Model. Mech. 2011, 4, 43–56. [Google Scholar] [CrossRef] [PubMed]
- Wallingford, J.B. Planar Cell Polarity and the Developmental Control of Cell Behavior in Vertebrate Embryos. Annu. Rev. Cell Dev. Biol. 2012, 28, 627–653. [Google Scholar] [CrossRef] [PubMed]
- Pala, R.; Alomari, N.; Nauli, S.M. Primary Cilium-Dependent Signaling Mechanisms. Int. J. Mol. Sci. 2017, 18, 18. [Google Scholar] [CrossRef] [PubMed]
- May-Simera, H.; Kelley, M.W. Planar Cell Polarity in the Inner Ear. Curr. Top. Dev. Biol. 2012, 101, 111–140. [Google Scholar] [PubMed]
- Adler, P.N.; Wallingford, J.B. From Planar Cell Polarity to Ciliogenesis and Back: The Curious Tale of the PPE and CPLANE Proteins. Trends Cell Biol. 2017, 27, 379–390. [Google Scholar] [CrossRef] [PubMed]
- Hui, C.C.; Angers, S. Gli Proteins in Development and Disease. Annu. Rev. Cell Dev. Biol. 2011, 27, 513–537. [Google Scholar] [CrossRef] [PubMed]
- Ybot-Gonzalez, P.; Gaston-Massuet, C.; Girdler, G.; Klingensmith, J.; Arkell, R.; Greene, N.D.; Copp, A.J. Neural Plate Morphogenesis during Mouse Neurulation is Regulated by Antagonism of Bmp Signalling. Development 2007, 134, 3203–3211. [Google Scholar] [CrossRef] [PubMed]
- Xue, Y.; Wang, X.; Li, Z.; Gotoh, N.; Chapman, D.; Skolnik, E.Y. Mesodermal Patterning Defect in Mice Lacking the Ste20 NCK Interacting Kinase (NIK). Development 2001, 128, 1559–1572. [Google Scholar] [PubMed]
- Chiang, C.; Litingtung, Y.; Lee, E.; Young, K.E.; Corden, J.L.; Westphal, H.; Beachy, P.A. Cyclopia and Defective Axial Patterning in Mice Lacking Sonic Hedgehog Gene Function. Nature 1996, 383, 407–413. [Google Scholar] [CrossRef] [PubMed]
- Aoto, K.; Shikata, Y.; Imai, H.; Matsumaru, D.; Tokunaga, T.; Shioda, S.; Yamada, G.; Motoyama, J. Mouse Shh is Required for Prechordal Plate Maintenance during Brain and Craniofacial Morphogenesis. Dev. Biol. 2009, 327, 106–120. [Google Scholar] [CrossRef] [PubMed]
- Patterson, V.L.; Damrau, C.; Paudyal, A.; Reeve, B.; Grimes, D.T.; Stewart, M.E.; Williams, D.J.; Siggers, P.; Greenfield, A.; Murdoch, J.N. Mouse Hitchhiker Mutants have Spina Bifida, Dorso-Ventral Patterning Defects and Polydactyly: Identification of Tulp3 as a Novel Negative Regulator of the Sonic Hedgehog Pathway. Hum. Mol. Genet. 2009, 18, 1719–1739. [Google Scholar] [CrossRef] [PubMed]
- Robbins, D.J.; Fei, D.L.; Riobo, N.A. The Hedgehog Signal Transduction Network. Sci. Signal. 2012, 5, re6. [Google Scholar] [CrossRef] [PubMed]
- Pyrgaki, C.; Trainor, P.; Hadjantonakis, A.K.; Niswander, L. Dynamic Imaging of Mammalian Neural Tube Closure. Dev. Biol. 2010, 344, 941–947. [Google Scholar] [CrossRef] [PubMed]
- Rolo, A.; Savery, D.; Escuin, S.; de Castro, S.C.; Armer, H.E.; Munro, P.M.; Mole, M.A.; Greene, N.D.; Copp, A.J. Regulation of Cell Protrusions by Small GTPases during Fusion of the Neural Folds. Elife 2016, 5, e13273. [Google Scholar] [CrossRef] [PubMed]
- Ray, H.J.; Niswander, L.A. Dynamic Behaviors of the Non-Neural Ectoderm during Mammalian Cranial Neural Tube Closure. Dev. Biol. 2016, 416, 279–285. [Google Scholar] [CrossRef] [PubMed]
- Holmberg, J.; Clarke, D.L.; Frisen, J. Regulation of Repulsion versus Adhesion by Different Splice Forms of an Eph Receptor. Nature 2000, 408, 203–206. [Google Scholar] [CrossRef] [PubMed]
- Rifat, Y.; Parekh, V.; Wilanowski, T.; Hislop, N.R.; Auden, A.; Ting, S.B.; Cunningham, J.M.; Jane, S.M. Regional Neural Tube Closure Defined by the Grainy Head-Like Transcription Factors. Dev. Biol. 2010, 345, 237–245. [Google Scholar] [CrossRef] [PubMed]
- Copp, A.J.; Greene, N.D.; Murdoch, J.N. The Genetic Basis of Mammalian Neurulation. Nat. Rev. Genet. 2003, 4, 784–793. [Google Scholar] [CrossRef] [PubMed]
- Abdul-Aziz, N.M.; Turmaine, M.; Greene, N.D.; Copp, A.J. EphrinA-EphA Receptor Interactions in Mouse Spinal Neurulation: Implications for Neural Fold Fusion. Int. J. Dev. Biol. 2009, 53, 559–568. [Google Scholar] [CrossRef] [PubMed]
- Lopez-Escobar, B.; Caro-Vega, J.M.; Vijayraghavan, D.S.; Plageman, T.F.; Sanchez-Alcazar, J.A.; Moreno, R.C.; Savery, D.; Marquez-Rivas, J.; Davidson, L.A.; Ybot-Gonzalez, P. The Non-Canonical Wnt-PCP Pathway Shapes the Mouse Caudal Neural Plate. Development 2018, 145, 145. [Google Scholar] [CrossRef] [PubMed]
- Saitsu, H.; Shiota, K. Involvement of the Axially Condensed Tail Bud Mesenchyme in Normal and Abnormal Human Posterior Neural Tube Development. Congenit. Anom. 2008, 48, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Dady, A.; Havis, E.; Escriou, V.; Catala, M.; Duband, J.L. Junctional Neurulation: A Unique Developmental Program Shaping a Discrete Region of the Spinal Cord Highly Susceptible to Neural Tube Defects. J. Neurosci. 2014, 34, 13208–13221. [Google Scholar] [CrossRef] [PubMed]
- Detrait, E.R.; George, T.M.; Etchevers, H.C.; Gilbert, J.R.; Vekemans, M.; Speer, M.C. Human Neural Tube Defects: Developmental Biology, Epidemiology, and Genetics. Neurotoxicol. Teratol. 2005, 27, 515–524. [Google Scholar] [CrossRef] [PubMed]
- Juriloff, D.M.; Harris, M.J.; Tom, C.; MacDonald, K.B. Normal Mouse Strains Differ in the Site of Initiation of Closure of the Cranial Neural Tube. Teratology 1991, 44, 225–233. [Google Scholar] [CrossRef] [PubMed]
- Kaufman, M. Cephalic Neurulation and Optic Vesicle Formation in the Early Mouse Embryo. Am. J. Anat. 1979, 155, 425–443. [Google Scholar] [CrossRef] [PubMed]
- Sakai, Y. Neurulation in the Mouse: Manner and Timing of Neural Tube Closure. Anat. Rec. 1989, 223, 194–203. [Google Scholar] [CrossRef] [PubMed]
- Golden, J.A.; Chernoff, G.F. Intermittent Pattern of Neural Tube Closure in Two Strains of Mice. Teratology 1993, 47, 73–80. [Google Scholar] [CrossRef] [PubMed]
- Morriss, G.M.; Solursh, M. Regional Differences in Mesenchymal Cell Morphology and Glycosaminoglycans in Early Neural-Fold Stage Rat Embryos. J. Embryol. Exp. Morphol. 1978, 46, 37–52. [Google Scholar] [PubMed]
- Christie, G.A. Developmental Stages in Somite and Post-Somite Rat Embryos, Based on External Appearance, and Including some Features of the Macroscopic Development of the Oral Cavity. J. Morphol. 1964, 114, 263–283. [Google Scholar] [CrossRef] [PubMed]
- Keyser, A. The Development of the Diencephalon of the Chinese Hamster. An Investigation of the Validity of the Criteria of Subdivision of the Brain. Acta Anat. Suppl. 1972, 59, 1–178. [Google Scholar] [PubMed]
- Waterman, R.E. Topographical Changes Along the Neural Fold Associated with Neurulation in the Hamster and Mouse. Am. J. Anat. 1976, 146, 151–171. [Google Scholar] [CrossRef] [PubMed]
- Peeters, M.C.; Viebahn, C.; Hekking, J.W.; van Straaten, H.W. Neurulation in the Rabbit Embryo. Anat. Embryol. 1998, 197, 167–175. [Google Scholar] [CrossRef] [PubMed]
- Van Straaten, H.W.; Peeters, M.C.; Hekking, J.W.; van der Lende, T. Neurulation in the Pig Embryo. Anat. Embryol. 2000, 202, 75–84. [Google Scholar] [CrossRef] [PubMed]
- Hamilton, W.J.; Mossman, H.W. Human Embryology: Prenatal Development of Form and Function, 4th ed.; Williams and Wilkins: Baltimore, MD, USA, 1972; pp. 1–646. [Google Scholar]
- Jirasek, J.E. Atlas of Human Prenatal Morphogenesis, 1st ed.; Springer: Dordrecht, The Netherlands, 1983; p. 168. [Google Scholar]
- Muller, F.; O’Rahilly, R. The First Appearance of the Neural Tube and Optic Primordium in the Human Embryo at Stage 10. Anat. Embryol. 1985, 172, 157–169. [Google Scholar] [CrossRef] [PubMed]
- Muller, F.; O’Rahilly, R. The Development of the Human Brain and the Closure of the Rostral Neuropore at Stage 11. Anat. Embryol. 1986, 175, 205–222. [Google Scholar] [CrossRef] [PubMed]
- Nakatsu, T.; Uwabe, C.; Shiota, K. Neural Tube Closure in Humans Initiates at Multiple Sites; Evidence from Human Embryos and Implications for the Pathogenesis of Neural Tube Defects. Anat. Embryol. 2000, 201, 455–466. [Google Scholar] [CrossRef] [PubMed]
- Peeters, M.C.; Hekking, J.W.; Shiota, K.; Drukker, J.; Van Straaten, H.W. Differences in Axial Curvature Correlate with Species-Specific Rate of Neural Tube Closure in Embryos of Chick, Rabbit, Mouse, Rat and Human. Anat. Embryol. 1998, 198, 185–194. [Google Scholar] [CrossRef] [PubMed]
- Juriloff, D.M.; Harris, M.J. Mouse Models for Neural Tube Closure Defects. Hum. Mol. Genet. 2000, 9, 993–1000. [Google Scholar] [CrossRef] [PubMed]
- Harris, M.J.; Juriloff, D.M.; Gunn, T.M.; Miller, J.E. Development of the Cerebellar Defect in Ataxic SELH/Bc Mice. Teratology 1994, 50, 63–73. [Google Scholar] [CrossRef] [PubMed]
- Golden, J.A.; Chernoff, G.F. Multiple Sites of Anterior Neural Tube Closure in Humans: Evidence from Anterior Neural Tube Defects (Anencephaly). Pediatrics 1995, 95, 506–510. [Google Scholar] [PubMed]
- Geelen, J.A.; Langman, J. Closure of the Neural Tube in the Cephalic Region of the Mouse Embryo. Anat. Rec. 1977, 189, 625–640. [Google Scholar] [CrossRef] [PubMed]
- Geelen, J.A.; Langman, J. Ultrastructural Observations on Closure of the Neural Tube in the Mouse. Anat. Embryol. 1979, 156, 73–88. [Google Scholar] [CrossRef] [PubMed]
- Nieto, M.A.; Gilardi-Hebenstreit, P.; Charnay, P.; Wilkinson, D.G. A Receptor Protein Tyrosine Kinase Implicated in the Segmental Patterning of the Hindbrain and Mesoderm. Development 1992, 116, 1137–1150. [Google Scholar] [PubMed]
- Niederreither, K.; Vermot, J.; Schuhbaur, B.; Chambon, P.; Dolle, P. Retinoic Acid Synthesis and Hindbrain Patterning in the Mouse Embryo. Development 2000, 127, 75–85. [Google Scholar] [PubMed]
- Ting, S.B.; Wilanowski, T.; Auden, A.; Hall, M.; Voss, A.K.; Thomas, T.; Parekh, V.; Cunningham, J.M.; Jane, S.M. Inositol- and Folate-Resistant Neural Tube Defects in Mice Lacking the Epithelial-Specific Factor Grhl-3. Nat. Med. 2003, 9, 1513–1519. [Google Scholar] [CrossRef] [PubMed]
- Ray, H.J.; Niswander, L.A. Grainyhead-Like 2 Downstream Targets Act to Suppress Epithelial-to-Mesenchymal Transition during Neural Tube Closure. Development 2016, 143, 1192–1204. [Google Scholar] [CrossRef] [PubMed]
- De Castro, S.C.P.; Hirst, C.S.; Savery, D.; Rolo, A.; Lickert, H.; Andersen, B.; Copp, A.J.; Greene, N.D.E. Neural Tube Closure Depends on Expression of Grainyhead-Like 3 in Multiple Tissues. Dev. Biol. 2018, 435, 130–137. [Google Scholar] [CrossRef] [PubMed]
- Deschamps, J.; van den Akker, E.; Forlani, S.; De Graaff, W.; Oosterveen, T.; Roelen, B.; Roelfsema, J. Initiation, Establishment and Maintenance of Hox Gene Expression Patterns in the Mouse. Int. J. Dev. Biol. 1999, 43, 635–650. [Google Scholar] [PubMed]
- Deschamps, J.; van Nes, J. Developmental Regulation of the Hox Genes during Axial Morphogenesis in the Mouse. Development 2005, 132, 2931–2942. [Google Scholar] [CrossRef] [PubMed]
- Morriss-Kay, G.; Tan, S. Mapping Cranial Neural Crest Cell Migration Pathways in Mammalian Embryos. TIG 1987, 3, 257–261. [Google Scholar] [CrossRef]
- Yamaguchi, Y.; Shinotsuka, N.; Nonomura, K.; Takemoto, K.; Kuida, K.; Yosida, H.; Miura, M. Live Imaging of Apoptosis in a Novel Transgenic Mouse Highlights its Role in Neural Tube Closure. J. Cell Biol. 2011, 195, 1047–1060. [Google Scholar] [CrossRef] [PubMed]
- Lowery, L.A.; Sive, H. Strategies of Vertebrate Neurulation and a Re-Evaluation of Teleost Neural Tube Formation. Mech. Dev. 2004, 121, 1189–1197. [Google Scholar] [CrossRef] [PubMed]
- Schroeder, T.E. Neurulation in Xenopus Laevis. An Analysis and Model Based upon Light and Electron Microscopy. J. Embryol. Exp. Morphol. 1970, 23, 427–462. [Google Scholar] [PubMed]
- Eagleson, G.W. Developmental Neurobiology of the Anterior Areas in Amphibians: Urodele Perspectives. Int. J. Dev. Biol. 1996, 40, 735–743. [Google Scholar] [PubMed]
- Holland, L.Z. The Origin and Evolution of Chordate Nervous Systems. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2015, 370, 370. [Google Scholar] [CrossRef] [PubMed]
- Shimeld, S.M.; Donoghue, P.C. Evolutionary Crossroads in Developmental Biology: Cyclostomes (Lamprey and Hagfish). Development 2012, 139, 2091–2099. [Google Scholar] [CrossRef] [PubMed]
- Holland, N.D.; Holland, L.Z. The Ups and Downs of Amphioxus Biology: A History. Int. J. Dev. Biol. 2017, 61, 575–583. [Google Scholar] [CrossRef] [PubMed]
- Putnam, N.H.; Butts, T.; Ferrier, D.E.; Furlong, R.F.; Hellsten, U.; Kawashima, T.; Robinson-Rechavi, M.; Shoguchi, E.; Terry, A.; Yu, J.K.; et al. The Amphioxus Genome and the Evolution of the Chordate Karyotype. Nature 2008, 453, 1064–1071. [Google Scholar] [CrossRef] [PubMed]
- Satoh, N. Cellular Morphology and Architecture during Early Morphogenesis of the Ascidian Egg: An SEM Study. Biol. Bull. 1978, 155, 608–614. [Google Scholar] [CrossRef] [PubMed]
- Jiang, D.; Munro, E.M.; Smith, W.C. Ascidian Prickle Regulates both Mediolateral and Anterior-Posterior Cell Polarity of Notochord Cells. Curr. Biol. 2005, 15, 79–85. [Google Scholar] [CrossRef] [PubMed]
- Sasakura, Y.; Mita, K.; Ogura, Y.; Horie, T. Ascidians as Excellent Chordate Models for Studying the Development of the Nervous System during Embryogenesis and Metamorphosis. Dev. Growth Differ. 2012, 54, 420–437. [Google Scholar] [CrossRef] [PubMed]
- Northcutt, R.G. The New Head Hypothesis Revisited. J. Exp. Zool. B Mol. Dev. Evol. 2005, 304, 274–297. [Google Scholar] [CrossRef] [PubMed]
- Bronner-Fraser, M. On the Trail of the ‘New Head’ in Les Treilles. Development 2008, 135, 2995–2999. [Google Scholar] [CrossRef] [PubMed]
- Holland, L.Z. Chordate Roots of the Vertebrate Nervous System: Expanding the Molecular Toolkit. Nat. Rev. Neurosci. 2009, 10, 736–746. [Google Scholar] [CrossRef] [PubMed]
- Bronner, M.E.; LeDouarin, N.M. Development and Evolution of the Neural Crest: An Overview. Dev. Biol. 2012, 366, 2–9. [Google Scholar] [CrossRef] [PubMed]
- Santagati, F.; Rijli, F.M. Cranial Neural Crest and the Building of the Vertebrate Head. Nat. Rev. Neurosci. 2003, 4, 806–818. [Google Scholar] [CrossRef] [PubMed]
- Jandzik, D.; Garnett, A.T.; Square, T.A.; Cattell, M.V.; Yu, J.K.; Medeiros, D.M. Evolution of the New Vertebrate Head by Co-Option of an Ancient Chordate Skeletal Tissue. Nature 2015, 518, 534–537. [Google Scholar] [CrossRef] [PubMed]
- Tan, S.S.; Morriss-Kay, G. The Development and Distribution of the Cranial Neural Crest in the Rat Embryo. Cell Tissue Res. 1985, 240, 403–416. [Google Scholar] [CrossRef] [PubMed]
- Osumi-Yamashita, N.; Ninomiya, Y.; Doi, H.; Eto, K. The Contribution of both Forebrain and Midbrain Crest Cells to the Mesenchyme in the Frontonasal Mass of Mouse Embryos. Dev. Biol. 1994, 164, 409–419. [Google Scholar] [CrossRef] [PubMed]
- Ezin, M.; Barembaum, M.; Bronner, M.E. Stage-Dependent Plasticity of the Anterior Neural Folds to Form Neural Crest. Differentiation 2014, 88, 42–50. [Google Scholar] [CrossRef] [PubMed]
- Masek, J.; Machon, O.; Korinek, V.; Taketo, M.M.; Kozmik, Z. Tcf7l1 Protects the Anterior Neural Fold from Adopting the Neural Crest Fate. Development 2016, 143, 2206–2216. [Google Scholar] [CrossRef] [PubMed]
- Nolte, C.; Krumlauf, R. Expression of Hox genes in the nervous system of vertebrates. In Hox Gene Expression; Papageorgiou, S., Ed.; Springer: New York, NY, USA, 2007; pp. 14–41. [Google Scholar]
- Muller, F.; O’Rahilly, R. The Prechordal Plate, the Rostral End of the Notochord and Nearby Median Features in Staged Human Embryos. Cells Tissues Organs 2003, 173, 1–20. [Google Scholar] [CrossRef] [PubMed]
- Ulmer, B.; Tingler, M.; Kurz, S.; Maerker, M.; Andre, P.; Monch, D.; Campione, M.; Deissler, K.; Lewandoski, M.; Thumberger, T.; et al. A Novel Role of the Organizer Gene Goosecoid as an Inhibitor of Wnt/PCP-Mediated Convergent Extension in Xenopus and Mouse. Sci. Rep. 2017, 7, 43010. [Google Scholar] [CrossRef] [PubMed]
- Shimamura, K.; Rubenstein, J.L. Inductive Interactions Direct Early Regionalization of the Mouse Forebrain. Development 1997, 124, 2709–2718. [Google Scholar] [PubMed]
- Belo, J.A.; Leyns, L.; Yamada, G.; De Robertis, E.M. The Prechordal Midline of the Chondrocranium is Defective in Goosecoid-1 Mouse Mutants. Mech. Dev. 1998, 72, 15–25. [Google Scholar] [CrossRef]
- Muller, F.; O’Rahilly, R. The First Appearance of the Major Divisions of the Human Brain at Stage 9. Anat. Embryol. 1983, 168, 419–432. [Google Scholar] [CrossRef] [PubMed]
- Jacobson, A.G.; Tam, P.P. Cephalic Neurulation in the Mouse Embryo Analyzed by SEM and Morphometry. Anat. Rec. 1982, 203, 375–396. [Google Scholar] [CrossRef] [PubMed]
- Takeuchi, T.; Kojima, M.; Nakajima, K.; Kondo, S. Jumonji Gene is Essential for the Neurulation and Cardiac Development of Mouse Embryos with a C3H/He Background. Mech. Dev. 1999, 86, 29–38. [Google Scholar] [CrossRef]
- Kaufman, M.H. The Role of Embryology in Teratological Research, with Particular Reference to the Development of the Neural Tube and Heart. J. Reprod. Fertil. 1981, 62, 607–623. [Google Scholar] [CrossRef] [PubMed]
- Massa, V.; Savery, D.; Ybot-Gonzalez, P.; Ferraro, E.; Rongvaux, A.; Cecconi, F.; Flavell, R.; Greene, N.D.; Copp, A.J. Apoptosis is Not Required for Mammalian Neural Tube Closure. Proc. Natl. Acad. Sci. USA 2009, 106, 8233–8238. [Google Scholar] [CrossRef] [PubMed]
- Brewer, S.; Feng, W.; Huang, J.; Sullivan, S.; Williams, T. Wnt1-Cre-Mediated Deletion of AP-2alpha Causes Multiple Neural Crest-Related Defects. Dev. Biol. 2004, 267, 135–152. [Google Scholar] [CrossRef] [PubMed]
- Luo, Y.; High, F.A.; Epstein, J.A.; Radice, G.L. N-Cadherin is Required for Neural Crest Remodeling of the Cardiac Outflow Tract. Dev. Biol. 2006, 299, 517–528. [Google Scholar] [CrossRef] [PubMed]
- Breau, M.A.; Pietri, T.; Stemmler, M.P.; Thiery, J.P.; Weston, J.A. A Nonneural Epithelial Domain of Embryonic Cranial Neural Folds Gives Rise to Ectomesenchyme. Proc. Natl. Acad. Sci. USA 2008, 105, 7750–7755. [Google Scholar] [CrossRef] [PubMed]
- Schorle, H.; Meier, P.; Buchert, M.; Jaenisch, R.; Mitchell, P.J. Transcription Factor AP-2 Essential for Cranial Closure and Craniofacial Development. Nature 1996, 381, 235–238. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Hagopian-Donaldson, S.; Serbedzija, G.; Elsemore, J.; Plehn-Dujowich, D.; McMahon, A.P.; Flavell, R.A.; Williams, T. Neural Tube, Skeletal and Body Wall Defects in Mice Lacking Transcription Factor AP-2. Nature 1996, 381, 238–241. [Google Scholar] [CrossRef] [PubMed]
- Radice, G.L.; Rayburn, H.; Matsunami, H.; Knudsen, K.A.; Takeichi, M.; Hynes, R.O. Developmental Defects in Mouse Embryos Lacking N-Cadherin. Dev. Biol. 1997, 181, 64–78. [Google Scholar] [CrossRef] [PubMed]
- Luo, Y.; Ferreira-Cornwell, M.; Baldwin, H.; Kostetskii, I.; Lenox, J.; Lieberman, M.; Radice, G. Rescuing the N-Cadherin Knockout by Cardiac-Specific Expression of N- Or E-Cadherin. Development 2001, 128, 459–469. [Google Scholar] [PubMed]
- Kinder, S.J.; Tsang, T.E.; Quinlan, G.A.; Hadjantonakis, A.K.; Nagy, A.; Tam, P.P. The Orderly Allocation of Mesodermal Cells to the Extraembryonic Structures and the Anteroposterior Axis during Gastrulation of the Mouse Embryo. Development 1999, 126, 4691–4701. [Google Scholar] [PubMed]
- Onai, T.; Aramaki, T.; Inomata, H.; Hirai, T.; Kuratani, S. Ancestral Mesodermal Reorganization and Evolution of the Vertebrate Head. Zool. Lett. 2015, 1, 29. [Google Scholar] [CrossRef] [PubMed]
- Zohn, I.E.; Sarkar, A.A. Does the Cranial Mesenchyme Contribute to Neural Fold Elevation during Neurulation? Birth Defects Res. A Clin. Mol. Teratol. 2012, 94, 841–848. [Google Scholar] [CrossRef] [PubMed]
- Copp, A.J. Neurulation in the Cranial Region—Normal and Abnormal. J. Anat. 2005, 207, 623–635. [Google Scholar] [CrossRef] [PubMed]
- Morris-Wiman, J.; Brinkley, L.L. Changes in Mesenchymal Cell and Hyaluronate Distribution Correlate with in vivo Elevation of the Mouse Mesencephalic Neural Folds. Anat. Rec. 1990, 226, 383–395. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.F.; Behringer, R.R. Twist is Required in Head Mesenchyme for Cranial Neural Tube Morphogenesis. Genes Dev. 1995, 9, 686–699. [Google Scholar] [CrossRef] [PubMed]
- Bildsoe, H.; Loebel, D.A.; Jones, V.J.; Hor, A.C.; Braithwaite, A.W.; Chen, Y.T.; Behringer, R.R.; Tam, P.P. The Mesenchymal Architecture of the Cranial Mesoderm of Mouse Embryos is Disrupted by the Loss of Twist1 Function. Dev. Biol. 2013, 374, 295–307. [Google Scholar] [CrossRef] [PubMed]
- Wolf, C.; Thisse, C.; Stoetzel, C.; Thisse, B.; Gerlinger, P.; Perrin-Schmitt, F. The M-Twist Gene of Mus is Expressed in Subsets of Mesodermal Cells and is Closely Related to the Xenopus X-Twi and the Drosophila Twist Genes. Dev. Biol. 1991, 143, 363–373. [Google Scholar] [CrossRef]
- Soo, K.; O’Rourke, M.P.; Khoo, P.L.; Steiner, K.A.; Wong, N.; Behringer, R.R.; Tam, P.P. Twist Function is Required for the Morphogenesis of the Cephalic Neural Tube and the Differentiation of the Cranial Neural Crest Cells in the Mouse Embryo. Dev. Biol. 2002, 247, 251–270. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Q.; Behringer, R.R.; de Crombrugghe, B. Prenatal Folic Acid Treatment Suppresses Acrania and Meroanencephaly in Mice Mutant for the Cart1 Homeobox Gene. Nat. Genet. 1996, 13, 275–283. [Google Scholar] [CrossRef] [PubMed]
- ten Berge, D.; Brouwer, A.; el Bahi, S.; Guenet, J.L.; Robert, B.; Meijlink, F. Mouse Alx3: An Aristaless-Like Homeobox Gene Expressed during Embryogenesis in Ectomesenchyme and Lateral Plate Mesoderm. Dev. Biol. 1998, 199, 11–25. [Google Scholar] [CrossRef] [PubMed]
- Lakhwani, S.; Garcia-Sanz, P.; Vallejo, M. Alx3-Deficient Mice Exhibit Folic Acid-Resistant Craniofacial Midline and Neural Tube Closure Defects. Dev. Biol. 2010, 344, 869–880. [Google Scholar] [CrossRef] [PubMed]
- Reid, B.S.; Sargent, T.D.; Williams, T. Generation and Characterization of a Novel Neural Crest Marker Allele, Inka1-LacZ, Reveals a Role for Inka1 in Mouse Neural Tube Closure. Dev. Dyn. 2010, 239, 1188–1196. [Google Scholar] [CrossRef] [PubMed]
- Baskaran, Y.; Ang, K.C.; Anekal, P.V.; Chan, W.L.; Grimes, J.M.; Manser, E.; Robinson, R.C. An in Cellulo-Derived Structure of PAK4 in Complex with its Inhibitor Inka1. Nat. Commun. 2015, 6, 8681. [Google Scholar] [CrossRef] [PubMed]
- Baranek, C.; Atanasoski, S. Modulating Epigenetic Mechanisms: The Diverse Functions of Ski during Cortical Development. Epigenetics 2012, 7, 676–679. [Google Scholar] [CrossRef] [PubMed]
- Berk, M.; Desai, S.Y.; Heyman, H.C.; Colmenares, C. Mice Lacking the Ski Proto-Oncogene have Defects in Neurulation, Craniofacial, Patterning, and Skeletal Muscle Development. Genes Dev. 1997, 11, 2029–2039. [Google Scholar] [CrossRef] [PubMed]
- Lyons, G.E.; Micales, B.K.; Herr, M.J.; Horrigan, S.K.; Namciu, S.; Shardy, D.; Stavnezer, E. Protooncogene c-Ski is Expressed in both Proliferating and Postmitotic Neuronal Populations. Dev. Dyn. 1994, 201, 354–365. [Google Scholar] [CrossRef] [PubMed]
- Bultman, S.; Gebuhr, T.; Yee, D.; La Mantia, C.; Nicholson, J.; Gilliam, A.; Randazzo, F.; Metzger, D.; Chambon, P.; Crabtree, G.; et al. A Brg1 Null Mutation in the Mouse Reveals Functional Differences among Mammalian SWI/SNF Complexes. Mol. Cell 2000, 6, 1287–1295. [Google Scholar] [CrossRef]
- Randazzo, F.M.; Khavari, P.; Crabtree, G.; Tamkun, J.; Rossant, J. Brg1: A Putative Murine Homologue of the Drosophila Brahma Gene, a Homeotic Gene Regulator. Dev. Biol. 1994, 161, 229–242. [Google Scholar] [CrossRef] [PubMed]
- Zohn, I.E.; Anderson, K.V.; Niswander, L. The Hectd1 Ubiquitin Ligase is Required for Development of the Head Mesenchyme and Neural Tube Closure. Dev. Biol. 2007, 306, 208–221. [Google Scholar] [CrossRef] [PubMed]
- Sarkar, A.A.; Zohn, I.E. Hectd1 Regulates Intracellular Localization and Secretion of Hsp90 to Control Cellular Behavior of the Cranial Mesenchyme. J. Cell Biol. 2012, 196, 789–800. [Google Scholar] [CrossRef] [PubMed]
- Banting, G.S.; Barak, O.; Ames, T.M.; Burnham, A.C.; Kardel, M.D.; Cooch, N.S.; Davidson, C.E.; Godbout, R.; McDermid, H.E.; Shiekhattar, R. CECR2, a Protein Involved in Neurulation, Forms a Novel Chromatin Remodeling Complex with SNF2L. Hum. Mol. Genet. 2005, 14, 513–524. [Google Scholar] [CrossRef] [PubMed]
- Dawe, C.E.; Kooistra, M.K.; Fairbridge, N.A.; Pisio, A.C.; McDermid, H.E. Role of Chromatin Remodeling Gene Cecr2 in Neurulation and Inner Ear Development. Dev. Dyn. 2011, 240, 372–383. [Google Scholar] [CrossRef] [PubMed]
- Fairbridge, N.A.; Dawe, C.E.; Niri, F.H.; Kooistra, M.K.; King-Jones, K.; McDermid, H.E. Cecr2 Mutations Causing Exencephaly Trigger Misregulation of mesenchymal/ectodermal Transcription Factors. Birth Defects Res. A Clin. Mol. Teratol. 2010, 88, 619–625. [Google Scholar] [CrossRef] [PubMed]
- Nishi, T.; Nakano, R. First-Trimester Diagnosis of Exencephaly by Transvaginal Ultrasonography. J. Ultrasound Med. 1994, 13, 149–151. [Google Scholar] [CrossRef] [PubMed]
- Chatzipapas, I.K.; Whitlow, B.J.; Economides, D.L. The ‘Mickey Mouse’ Sign and the Diagnosis of Anencephaly in Early Pregnancy. Ultrasound Obstet. Gynecol. 1999, 13, 196–199. [Google Scholar] [CrossRef] [PubMed]
- Grego-Bessa, J.; Hildebrand, J.; Anderson, K.V. Morphogenesis of the Mouse Neural Plate Depends on Distinct Roles of Cofilin 1 in Apical and Basal Epithelial Domains. Development 2015, 142, 1305–1314. [Google Scholar] [CrossRef] [PubMed]
- Gurniak, C.B.; Perlas, E.; Witke, W. The Actin Depolymerizing Factor n-Cofilin is Essential for Neural Tube Morphogenesis and Neural Crest Cell Migration. Dev. Biol. 2005, 278, 231–241. [Google Scholar] [CrossRef] [PubMed]
- Cox, B.; Hadjantonakis, A.K.; Collins, J.E.; Magee, A.I. Cloning and Expression Throughout Mouse Development of mfat1, a Homologue of the Drosophila Tumour Suppressor Gene Fat. Dev. Dyn. 2000, 217, 233–240. [Google Scholar] [CrossRef]
- Tanoue, T.; Takeichi, M. Mammalian Fat1 Cadherin Regulates Actin Dynamics and Cell-Cell Contact. J. Cell Biol. 2004, 165, 517–528. [Google Scholar] [CrossRef] [PubMed]
- Badouel, C.; Zander, M.A.; Liscio, N.; Bagherie-Lachidan, M.; Sopko, R.; Coyaud, E.; Raught, B.; Miller, F.D.; McNeill, H. Fat1 Interacts with Fat4 to Regulate Neural Tube Closure, Neural Progenitor Proliferation and Apical Constriction during Mouse Brain Development. Development 2015, 142, 2781–2791. [Google Scholar] [CrossRef] [PubMed]
- Barbera, J.P.; Rodriguez, T.A.; Greene, N.D.; Weninger, W.J.; Simeone, A.; Copp, A.J.; Beddington, R.S.; Dunwoodie, S. Folic Acid Prevents Exencephaly in Cited2 Deficient Mice. Hum. Mol. Genet. 2002, 11, 283–293. [Google Scholar] [CrossRef] [PubMed]
- Bamforth, S.D.; Braganca, J.; Eloranta, J.J.; Murdoch, J.N.; Marques, F.I.; Kranc, K.R.; Farza, H.; Henderson, D.J.; Hurst, H.C.; Bhattacharya, S. Cardiac Malformations, Adrenal Agenesis, Neural Crest Defects and Exencephaly in Mice Lacking Cited2, a New Tfap2 Co-Activator. Nat. Genet. 2001, 29, 469–474. [Google Scholar] [CrossRef] [PubMed]
- Kranc, K.R.; Oliveira, D.V.; Armesilla-Diaz, A.; Pacheco-Leyva, I.; Catarina Matias, A.; Luisa Escapa, A.; Subramani, C.; Wheadon, H.; Trindade, M.; Nichols, J.; et al. Acute Loss of Cited2 Impairs Nanog Expression and Decreases Self-Renewal of Mouse Embryonic Stem Cells. Stem Cells 2015, 33, 699–712. [Google Scholar] [CrossRef] [PubMed]
- Dunwoodie, S.L.; Rodriguez, T.A.; Beddington, R.S. Msg1 and Mrg1, Founding Members of a Gene Family, show Distinct Patterns of Gene Expression during Mouse Embryogenesis. Mech. Dev. 1998, 72, 27–40. [Google Scholar] [CrossRef]
- Sakaki-Yumoto, M.; Kobayashi, C.; Sato, A.; Fujimura, S.; Matsumoto, Y.; Takasato, M.; Kodama, T.; Aburatani, H.; Asashima, M.; Yoshida, N.; et al. The Murine Homolog of SALL4, a Causative Gene in Okihiro Syndrome, is Essential for Embryonic Stem Cell Proliferation, and Cooperates with Sall1 in Anorectal, Heart, Brain and Kidney Development. Development 2006, 133, 3005–3013. [Google Scholar] [CrossRef] [PubMed]
- Buck, A.; Kispert, A.; Kohlhase, J. Embryonic Expression of the Murine Homologue of SALL1, the Gene Mutated in Townes–Brocks Syndrome. Mech. Dev. 2001, 104, 143–146. [Google Scholar] [CrossRef]
- Bohm, J.; Buck, A.; Borozdin, W.; Mannan, A.U.; Matysiak-Scholze, U.; Adham, I.; Schulz-Schaeffer, W.; Floss, T.; Wurst, W.; Kohlhase, J.; et al. Sall1, sall2, and sall4 are Required for Neural Tube Closure in Mice. Am. J. Pathol. 2008, 173, 1455–1463. [Google Scholar] [CrossRef] [PubMed]
- Kiefer, S.M.; Ohlemiller, K.K.; Yang, J.; McDill, B.W.; Kohlhase, J.; Rauchman, M. Expression of a Truncated Sall1 Transcriptional Repressor is Responsible for Townes-Brocks Syndrome Birth Defects. Hum. Mol. Genet. 2003, 12, 2221–2227. [Google Scholar] [CrossRef] [PubMed]
- Ohmura, T.; Shioi, G.; Hirano, M.; Aizawa, S. Neural Tube Defects by NUAK1 and NUAK2 Double Mutation. Dev. Dyn. 2012, 241, 1350–1364. [Google Scholar] [CrossRef] [PubMed]
- Hildebrand, J.D.; Soriano, P. Shroom, a PDZ Domain-Containing Actin-Binding Protein, is Required for Neural Tube Morphogenesis in Mice. Cell 1999, 99, 485–497. [Google Scholar] [CrossRef]
- Sousa-Nunes, R.; Rana, A.A.; Kettleborough, R.; Brickman, J.M.; Clements, M.; Forrest, A.; Grimmond, S.; Avner, P.; Smith, J.C.; Dunwoodie, S.L.; et al. Characterizing Embryonic Gene Expression Patterns in the Mouse using Nonredundant Sequence-Based Selection. Genome Res. 2003, 13, 2609–2620. [Google Scholar] [CrossRef] [PubMed]
- McGreevy, E.M.; Vijayraghavan, D.; Davidson, L.A.; Hildebrand, J.D. Shroom3 Functions Downstream of Planar Cell Polarity to Regulate Myosin II Distribution and Cellular Organization during Neural Tube Closure. Biol. Open 2015, 4, 186–196. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Zhang, H.; Hu, G.; Wang, H.; Abate-Shen, C.; Shen, M.M. An Early Phase of Embryonic Dlx5 Expression Defines the Rostral Boundary of the Neural Plate. J. Neurosci. 1998, 18, 8322–8330. [Google Scholar] [CrossRef] [PubMed]
- Depew, M.J.; Liu, J.K.; Long, J.E.; Presley, R.; Meneses, J.J.; Pedersen, R.A.; Rubenstein, J.L. Dlx5 Regulates Regional Development of the Branchial Arches and Sensory Capsules. Development 1999, 126, 3831–3846. [Google Scholar] [PubMed]
- Lee, J.; Corcoran, A.; Han, M.; Gardiner, D.M.; Muneoka, K. Dlx5 and Msx2 Regulate Mouse Anterior Neural Tube Closure through ephrinA5-EphA7. Dev. Growth Differ. 2013, 55, 341–349. [Google Scholar] [CrossRef] [PubMed]
- Ishii, M.; Han, J.; Yen, H.Y.; Sucov, H.M.; Chai, Y.; Maxson, R.E., Jr. Combined Deficiencies of Msx1 and Msx2 Cause Impaired Patterning and Survival of the Cranial Neural Crest. Development 2005, 132, 4937–4950. [Google Scholar] [CrossRef] [PubMed]
- Trainor, P.A.; Sobieszczuk, D.; Wilkinson, D.; Krumlauf, R. Signalling between the Hindbrain and Paraxial Tissues Dictates Neural Crest Migration Pathways. Development 2002, 129, 433–442. [Google Scholar] [PubMed]
- Han, J.; Ishii, M.; Bringas, P., Jr.; Maas, R.L.; Maxson, R.E., Jr.; Chai, Y. Concerted Action of Msx1 and Msx2 in Regulating Cranial Neural Crest Cell Differentiation during Frontal Bone Development. Mech. Dev. 2007, 124, 729–745. [Google Scholar] [CrossRef] [PubMed]
- Arvanitis, D.N.; Behar, A.; Tryoen-Toth, P.; Bush, J.O.; Jungas, T.; Vitale, N.; Davy, A. Ephrin B1 Maintains Apical Adhesion of Neural Progenitors. Development 2013, 140, 2082–2092. [Google Scholar] [CrossRef] [PubMed]
- Heimsath, E.G., Jr.; Yim, Y.I.; Mustapha, M.; Hammer, J.A.; Cheney, R.E. Myosin-X Knockout is Semi-Lethal and Demonstrates that Myosin-X Functions in Neural Tube Closure, Pigmentation, Hyaloid Vasculature Regression, and Filopodia Formation. Sci. Rep. 2017, 7, 17354. [Google Scholar] [CrossRef] [PubMed]
- Juriloff, D.M.; Harris, M.J. Hypothesis: The Female Excess in Cranial Neural Tube Defects Reflects an Epigenetic Drag of the Inactivating X Chromosome on the Molecular Mechanisms of Neural Fold Elevation. Birth Defects Res. A Clin. Mol. Teratol. 2012, 94, 849–855. [Google Scholar] [CrossRef] [PubMed]
- Poletta, F.A.; Rittler, M.; Saleme, C.; Campana, H.; Gili, J.A.; Pawluk, M.S.; Gimenez, L.G.; Cosentino, V.R.; Castilla, E.E.; Lopez-Camelo, J.S. Neural Tube Defects: Sex Ratio Changes After Fortification with Folic Acid. PLoS ONE 2018, 13, e0193127. [Google Scholar] [CrossRef] [PubMed]
- Davidson, C.E.; Li, Q.; Churchill, G.A.; Osborne, L.R.; McDermid, H.E. Modifier Locus for Exencephaly in Cecr2 Mutant Mice is Syntenic to the 10q25.3 Region Associated with Neural Tube Defects in Humans. Physiol. Genom. 2007, 31, 244–251. [Google Scholar] [CrossRef] [PubMed]
- Santander, N.G.; Contreras-Duarte, S.; Awad, M.F.; Lizama, C.; Passalacqua, I.; Rigotti, A.; Busso, D. Developmental Abnormalities in Mouse Embryos Lacking the HDL Receptor SR-BI. Hum. Mol. Genet. 2013, 22, 1086–1096. [Google Scholar] [CrossRef] [PubMed]
- Pai, Y.J.; Leung, K.Y.; Savery, D.; Hutchin, T.; Prunty, H.; Heales, S.; Brosnan, M.E.; Brosnan, J.T.; Copp, A.J.; Greene, N.D. Glycine Decarboxylase Deficiency Causes Neural Tube Defects and Features of Non-Ketotic Hyperglycinemia in Mice. Nat. Commun. 2015, 6, 6388. [Google Scholar] [CrossRef] [PubMed]
- Zvetkova, I.; Apedaile, A.; Ramsahoye, B.; Mermoud, J.E.; Crompton, L.A.; John, R.; Feil, R.; Brockdorff, N. Global Hypomethylation of the Genome in XX Embryonic Stem Cells. Nat. Genet. 2005, 37, 1274–1279. [Google Scholar] [CrossRef] [PubMed]
- Dobbs, K.B.; Rodriguez, M.; Sudano, M.J.; Ortega, M.S.; Hansen, P.J. Dynamics of DNA Methylation during Early Development of the Preimplantation Bovine Embryo. PLoS ONE 2013, 8, e66230. [Google Scholar] [CrossRef] [PubMed]
- Vallot, C.; Ouimette, J.F.; Rougeulle, C. Establishment of X Chromosome Inactivation and Epigenomic Features of the Inactive X Depend on Cellular Contexts. Bioessays 2016, 38, 869–880. [Google Scholar] [CrossRef] [PubMed]
- Carrel, L.; Brown, C.J. When the Lyon(Ized Chromosome) Roars: Ongoing Expression from an Inactive X Chromosome. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2017, 372, 372. [Google Scholar] [CrossRef] [PubMed]
- Wijchers, P.J.; Festenstein, R.J. Epigenetic Regulation of Autosomal Gene Expression by Sex Chromosomes. Trends Genet. 2011, 27, 132–140. [Google Scholar] [CrossRef] [PubMed]
- Blewitt, M.E.; Vickaryous, N.K.; Hemley, S.J.; Ashe, A.; Bruxner, T.J.; Preis, J.I.; Arkell, R.; Whitelaw, E. An N-Ethyl-N-Nitrosourea Screen for Genes Involved in Variegation in the Mouse. Proc. Natl. Acad. Sci. USA 2005, 102, 7629–7634. [Google Scholar] [CrossRef] [PubMed]
- Wijchers, P.J.; Yandim, C.; Panousopoulou, E.; Ahmad, M.; Harker, N.; Saveliev, A.; Burgoyne, P.S.; Festenstein, R. Sexual Dimorphism in Mammalian Autosomal Gene Regulation is Determined Not Only by Sry but by Sex Chromosome Complement as Well. Dev. Cell. 2010, 19, 477–484. [Google Scholar] [CrossRef] [PubMed]
- Evans, J.A. Comment on Changes in Sex Ratio in Neural Tube Defects since Food Fortification with Folic Acid: Re “Hypothesis: The Female Excess in Cranial Neural Tube Defects Reflects an Epigenetic Drag of the Inactivating X Chromosome on the Molecular Mechanisms of Neural Tube Fold Elevation”. Birth Defects Res. A Clin. Mol. Teratol. 2012, 94, 958. [Google Scholar] [CrossRef] [PubMed]
- Greene, N.D.; Gerrelli, D.; van Straaten, H.W.; Copp, A.J. Abnormalities of Floor Plate, Notochord and Somite Differentiation in the Loop-Tail (Lp) Mouse: A Model of Severe Neural Tube Defects. Mech. Dev. 1998, 73, 59–72. [Google Scholar] [CrossRef]
- Ybot-Gonzalez, P.; Savery, D.; Gerrelli, D.; Signore, M.; Mitchell, C.E.; Faux, C.H.; Greene, N.D.; Copp, A.J. Convergent Extension, Planar-Cell-Polarity Signalling and Initiation of Mouse Neural Tube Closure. Development 2007, 134, 789–799. [Google Scholar] [CrossRef] [PubMed]
- Robinson, A.; Escuin, S.; Doudney, K.; Vekemans, M.; Stevenson, R.E.; Greene, N.D.; Copp, A.J.; Stanier, P. Mutations in the Planar Cell Polarity Genes CELSR1 and SCRIB are Associated with the Severe Neural Tube Defect Craniorachischisis. Hum. Mutat. 2012, 33, 440–447. [Google Scholar] [CrossRef] [PubMed]
- Kirillova, I.; Novikova, I.; Auge, J.; Audollent, S.; Esnault, D.; Encha-Razavi, F.; Lazjuk, G.; Attie-Bitach, T.; Vekemans, M. Expression of the Sonic Hedgehog Gene in Human Embryos with Neural Tube Defects. Teratology 2000, 61, 347–354. [Google Scholar] [CrossRef]
- Murdoch, J.N.; Doudney, K.; Paternotte, C.; Copp, A.J.; Stanier, P. Severe Neural Tube Defects in the Loop-Tail Mouse Result from Mutation of Lpp1, a Novel Gene Involved in Floor Plate Specification. Hum. Mol. Genet. 2001, 10, 2593–2601. [Google Scholar] [CrossRef] [PubMed]
- Mahaffey, J.P.; Grego-Bessa, J.; Liem, K.F., Jr.; Anderson, K.V. Cofilin and Vangl2 Cooperate in the Initiation of Planar Cell Polarity in the Mouse Embryo. Development 2013, 140, 1262–1271. [Google Scholar] [CrossRef] [PubMed]
- Murdoch, J.N.; Damrau, C.; Paudyal, A.; Bogani, D.; Wells, S.; Greene, N.D.; Stanier, P.; Copp, A.J. Genetic Interactions between Planar Cell Polarity Genes Cause Diverse Neural Tube Defects in Mice. Dis. Model. Mech. 2014, 7, 1153–1163. [Google Scholar] [CrossRef] [PubMed]
- Doudney, K.; Ybot-Gonzalez, P.; Paternotte, C.; Stevenson, R.E.; Greene, N.D.; Moore, G.E.; Copp, A.J.; Stanier, P. Analysis of the Planar Cell Polarity Gene Vangl2 and its Co-Expressed Paralogue Vangl1 in Neural Tube Defect Patients. Am. J. Med. Genet. A 2005, 136, 90–92. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y.; Ding, Y.; Lei, Y.P.; Yang, X.Y.; Xie, G.M.; Wen, J.; Cai, C.Q.; Li, H.; Chen, Y.; Zhang, T.; et al. Identification of Novel Rare Mutations of DACT1 in Human Neural Tube Defects. Hum. Mutat. 2012, 33, 1450–1455. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Xiao, Y.; Tian, T.; Jin, L.; Lei, Y.; Finnell, R.H.; Ren, A. Digenic Variants of Planar Cell Polarity Genes in Human Neural Tube Defect Patients. Mol. Genet. Metab. 2018, 124, 94–100. [Google Scholar] [CrossRef] [PubMed]
- Scott, J.M.; Kirke, P.N.; Weir, D.G. The Role of Nutrition in Neural Tube Defects. Annu. Rev. Nutr. 1990, 10, 277–295. [Google Scholar] [CrossRef] [PubMed]
- MRC Vitamin Study Research Group. Prevention of Neural Tube Defects: Results of the Medical Research Council Vitamin Study. MRC Vitamin Study Research Group. Lancet 1991, 338, 131–137. [Google Scholar] [CrossRef]
- Czeizel, A.E.; Dudas, I. Prevention of the First Occurrence of Neural-Tube Defects by Periconceptional Vitamin Supplementation. N. Engl. J. Med. 1992, 327, 1832–1835. [Google Scholar] [CrossRef] [PubMed]
- Garrett, G.S.; Bailey, L.B. A Public Health Approach for Preventing Neural Tube Defects: Folic Acid Fortification and Beyond. Ann. N. Y. Acad. Sci. 2018, 1414, 47–58. [Google Scholar] [CrossRef] [PubMed]
- Greene, N.D.; Leung, K.Y.; Copp, A.J. Inositol, Neural Tube Closure and the Prevention of Neural Tube Defects. Birth Defects Res. 2017, 109, 68–80. [Google Scholar] [CrossRef] [PubMed]
- Kirke, P.N.; Molloy, A.M.; Daly, L.E.; Burke, H.; Weir, D.G.; Scott, J.M. Maternal Plasma Folate and Vitamin B12 are Independent Risk Factors for Neural Tube Defects. Q. J. Med. 1993, 86, 703–708. [Google Scholar] [PubMed]
- Scott, J.M. Folate and Vitamin B12. Proc. Nutr. Soc. 1999, 58, 441–448. [Google Scholar] [CrossRef] [PubMed]
- Heid, M.K.; Bills, N.D.; Hinrichs, S.H.; Clifford, A.J. Folate Deficiency Alone does Not Produce Neural Tube Defects in Mice. J. Nutr. 1992, 122, 888–894. [Google Scholar] [CrossRef] [PubMed]
- Burgoon, J.M.; Selhub, J.; Nadeau, M.; Sadler, T.W. Investigation of the Effects of Folate Deficiency on Embryonic Development through the Establishment of a Folate Deficient Mouse Model. Teratology 2002, 65, 219–227. [Google Scholar] [CrossRef] [PubMed]
- Blom, H.J.; Shaw, G.M.; den Heijer, M.; Finnell, R.H. Neural Tube Defects and Folate: Case Far from Closed. Nat. Rev. Neurosci. 2006, 7, 724–731. [Google Scholar] [CrossRef] [PubMed]
- Doudney, K.; Grinham, J.; Whittaker, J.; Lynch, S.A.; Thompson, D.; Moore, G.E.; Copp, A.J.; Greene, N.D.; Stanier, P. Evaluation of Folate Metabolism Gene Polymorphisms as Risk Factors for Open and Closed Neural Tube Defects. Am. J. Med. Genet. A 2009, 149A, 1585–1589. [Google Scholar] [CrossRef] [PubMed]
- Balashova, O.A.; Visina, O.; Borodinsky, L.N. Folate Action in Nervous System Development and Disease. Dev. Neurobiol. 2018, 78, 391–402. [Google Scholar] [CrossRef] [PubMed]
- Harris, M.J. Insights into Prevention of Human Neural Tube Defects by Folic Acid Arising from Consideration of Mouse Mutants. Birth Defects Res. A Clin. Mol. Teratol. 2009, 85, 331–339. [Google Scholar] [CrossRef] [PubMed]
- Gray, J.D.; Nakouzi, G.; Slowinska-Castaldo, B.; Dazard, J.E.; Rao, J.S.; Nadeau, J.H.; Ross, M.E. Functional Interactions between the LRP6 WNT Co-Receptor and Folate Supplementation. Hum. Mol. Genet. 2010, 19, 4560–4572. [Google Scholar] [CrossRef] [PubMed]
- Fleming, A.; Copp, A.J. Embryonic Folate Metabolism and Mouse Neural Tube Defects. Science 1998, 280, 2107–2109. [Google Scholar] [CrossRef] [PubMed]
- Greene, N.D.; Massa, V.; Copp, A.J. Understanding the Causes and Prevention of Neural Tube Defects: Insights from the Splotch Mouse Model. Birth Defects Res. A Clin. Mol. Teratol. 2009, 85, 322–330. [Google Scholar] [CrossRef] [PubMed]
- Toriyama, M.; Toriyama, M.; Wallingford, J.B.; Finnell, R.H. Folate-Dependent Methylation of Septins Governs Ciliogenesis during Neural Tube Closure. FASEB J. 2017, 31, 3622–3635. [Google Scholar] [CrossRef] [PubMed]
- Saitsu, H.; Ishibashi, M.; Nakano, H.; Shiota, K. Spatial and Temporal Expression of Folate-Binding Protein 1 (Fbp1) is Closely Associated with Anterior Neural Tube Closure in Mice. Dev. Dyn. 2003, 226, 112–117. [Google Scholar] [CrossRef] [PubMed]
- Saitsu, H. Folate Receptors and Neural Tube Closure. Congenit. Anom. 2017, 57, 130–133. [Google Scholar] [CrossRef] [PubMed]
- Tang, L.S.; Finnell, R.H. Neural and Orofacial Defects in Folp1 Knockout Mice [Corrected]. Birth Defects Res. A Clin. Mol. Teratol. 2003, 67, 209–218. [Google Scholar] [CrossRef] [PubMed]
- Ishida, M.; Cullup, T.; Boustred, C.; James, C.; Docker, J.; English, C.; GOSgene; Lench, N.; Copp, A.J.; Moore, G.E.; et al. A Targeted Sequencing Panel Identifies Rare Damaging Variants in Multiple Genes in the Cranial Neural Tube Defect, Anencephaly. Clin. Genet. 2018, 93, 870–879. [Google Scholar] [CrossRef] [PubMed]
- O’Byrne, M.R.; Au, K.S.; Morrison, A.C.; Lin, J.I.; Fletcher, J.M.; Ostermaier, K.K.; Tyerman, G.H.; Doebel, S.; Northrup, H. Association of Folate Receptor (FOLR1, FOLR2, FOLR3) and Reduced Folate Carrier (SLC19A1) Genes with Meningomyelocele. Birth Defects Res. A Clin. Mol. Teratol. 2010, 88, 689–694. [Google Scholar] [CrossRef] [PubMed]
- Kur, E.; Mecklenburg, N.; Cabrera, R.M.; Willnow, T.E.; Hammes, A. LRP2 Mediates Folate Uptake in the Developing Neural Tube. J. Cell Sci. 2014, 127, 2261–2268. [Google Scholar] [CrossRef] [PubMed]
- Sabatino, J.A.; Stokes, B.A.; Zohn, I.E. Prevention of Neural Tube Defects in Lrp2 Mutant Mouse Embryos by Folic Acid Supplementation. Birth Defects Res. 2017, 109, 16–26. [Google Scholar] [CrossRef] [PubMed]
- Boshnjaku, V.; Shim, K.W.; Tsurubuchi, T.; Ichi, S.; Szany, E.V.; Xi, G.; Mania-Farnell, B.; McLone, D.G.; Tomita, T.; Mayanil, C.S. Nuclear Localization of Folate Receptor Alpha: A New Role as a Transcription Factor. Sci. Rep. 2012, 2, 980. [Google Scholar] [CrossRef] [PubMed]
- Balashova, O.A.; Visina, O.; Borodinsky, L.N. Folate Receptor 1 is Necessary for Neural Plate Cell Apical Constriction during Xenopus Neural Tube Formation. Development 2017, 144, 1518–1530. [Google Scholar] [CrossRef] [PubMed]
- Narisawa, A.; Komatsuzaki, S.; Kikuchi, A.; Niihori, T.; Aoki, Y.; Fujiwara, K.; Tanemura, M.; Hata, A.; Suzuki, Y.; Relton, C.L.; et al. Mutations in Genes Encoding the Glycine Cleavage System Predispose to Neural Tube Defects in Mice and Humans. Hum. Mol. Genet. 2012, 21, 1496–1503. [Google Scholar] [CrossRef] [PubMed]
- Momb, J.; Lewandowski, J.P.; Bryant, J.D.; Fitch, R.; Surman, D.R.; Vokes, S.A.; Appling, D.R. Deletion of Mthfd1l Causes Embryonic Lethality and Neural Tube and Craniofacial Defects in Mice. Proc. Natl. Acad. Sci. USA 2013, 110, 549–554. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Lei, Y.; Guo, J.; Kim, S.E.; Wlodarczyk, B.J.; Cabrera, R.M.; Lin, Y.L.; Nilsson, T.K.; Zhang, T.; Ren, A.; et al. Formate Rescues Neural Tube Defects Caused by Mutations in Slc25a32. Proc. Natl. Acad. Sci. USA 2018, 115, 4690–4695. [Google Scholar] [PubMed]
- Di Pietro, E.; Sirois, J.; Tremblay, M.L.; MacKenzie, R.E. Mitochondrial NAD-Dependent Methylenetetrahydrofolate Dehydrogenase-Methenyltetrahydrofolate Cyclohydrolase is Essential for Embryonic Development. Mol. Cell. Biol. 2002, 22, 4158–4166. [Google Scholar] [CrossRef] [PubMed]
- Leung, K.Y.; De Castro, S.C.; Savery, D.; Copp, A.J.; Greene, N.D. Nucleotide Precursors Prevent Folic Acid-Resistant Neural Tube Defects in the Mouse. Brain 2013, 136, 2836–2841. [Google Scholar] [CrossRef] [PubMed]
- Sudiwala, S.; De Castro, S.C.; Leung, K.Y.; Brosnan, J.T.; Brosnan, M.E.; Mills, K.; Copp, A.J.; Greene, N.D. Formate Supplementation Enhances Folate-Dependent Nucleotide Biosynthesis and Prevents Spina Bifida in a Mouse Model of Folic Acid-Resistant Neural Tube Defects. Biochimie 2016, 126, 63–70. [Google Scholar] [CrossRef] [PubMed]
- Lalouel, J.M.; Rao, D.C.; Morton, N.E.; Elston, R.C. A Unified Model for Complex Segregation Analysis. Am. J. Hum. Genet. 1983, 35, 816–826. [Google Scholar] [PubMed]
- Morton, N.E.; MacLean, C.J. Analysis of Family Resemblance. III. Complex Segregation of Quantitative Traits. Am. J. Hum. Genet. 1974, 26, 489–503. [Google Scholar] [PubMed]
- Greene, N.D.E.; Stanier, P.; Copp, A.J. Genetics of Human Neural Tube Defects. Hum. Mol. Genet. 2009, 18, R113–R129. [Google Scholar] [CrossRef] [PubMed]
- Lu, W.; Guzman, A.R.; Yang, W.; Chapa, C.J.; Shaw, G.M.; Greene, R.M.; Pisano, M.M.; Lammer, E.J.; Finnell, R.H.; Zhu, H. Genes Encoding Critical Transcriptional Activators for Murine Neural Tube Development and Human Spina Bifida: A Case-Control Study. BMC Med. Genet. 2010, 11, 141. [Google Scholar] [CrossRef] [PubMed]
- Boyle, E.A.; Li, Y.; Pritchard, J.K. An Expanded View of Complex Traits: From Polygenic to Omnigenic. Cell 2017, 169, 1177–1186. [Google Scholar] [CrossRef] [PubMed]
- Hill, W.G. Understanding and Using Quantitative Genetic Variation. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2010, 365, 73–85. [Google Scholar] [CrossRef] [PubMed]
- Lynch, S.A. Non-multifactorial Neural Tube Defects. Am. J. Med. Genet. C Semin. Med. Genet. 2005, 135C, 69–76. [Google Scholar] [CrossRef] [PubMed]
- Juriloff, D.M.; Harris, M.J.; McMahon, A.P.; Carroll, T.J.; Lidral, A.C. Wnt9b is the Mutated Gene Involved in Multifactorial Nonsyndromic Cleft Lip with or without Cleft Palate in A/WySn Mice, as Confirmed by a Genetic Complementation Test. Birth Defects Res. A Clin. Mol. Teratol. 2006, 76, 574–579. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Lack of convergent extension |
| Lack of medial hinge point (MHP) 1 |
| Lack of dorsolateral hinge point (DLHP) 1 |
| Lack of neuroepithelial bending by apical constriction (cranial region) |
| Lack of structural integrity of the neuroepithelium |
| Lack of support from surrounding mesenchyme Lack of midline fusion of neuroepithelium and/or surface ectoderm Interference by excessive anterior-posterior curvature of the neural plate |
| Mechanism | Difference between Regions of Neural Tube |
|---|---|
| Convergent extension | Absent in forebrain |
| Notochord in head | Absent in forebrain |
| Notochord in spinal region | Absent at posterior neuropore during closure |
| Level of Shh expression | Gradient; lowest in posterior neuropore |
| Medial Hinge Point | Absent in posterior neuropore |
| Dorsolateral Hinge Point | Absent in upper spinal region |
| Convex mesenchymal expansion | Midbrain only |
| Grhl2, Grhl3 expression | Cranial, caudal and upper spine differ |
| Ephrins and Ephrin receptors | Different members of gene family regionally |
| Hox gene expression | Absent in midbrain and forebrain regions |
| Closure initiation site spacing | Caudal closure furthest from an initiation site |
| “Zipping” vs. simultaneous closure | Regional differences in axial bending |
| Meeting of neural folds in midline | Contact of neuroepithelium vs. surface ectoderm |
| Ruffles versus filopodia | Forebrain, midbrain, hindbrain, spine differ |
| Apical actomyosin contractility | Required for cranial closure, not spinal |
| Neural crest emigration | Spinal after closure; cranial before closure |
| Apoptosis | Required for cranial closure, not caudal |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Juriloff, D.M.; Harris, M.J. Insights into the Etiology of Mammalian Neural Tube Closure Defects from Developmental, Genetic and Evolutionary Studies. J. Dev. Biol. 2018, 6, 22. https://doi.org/10.3390/jdb6030022
Juriloff DM, Harris MJ. Insights into the Etiology of Mammalian Neural Tube Closure Defects from Developmental, Genetic and Evolutionary Studies. Journal of Developmental Biology. 2018; 6(3):22. https://doi.org/10.3390/jdb6030022
Chicago/Turabian StyleJuriloff, Diana M., and Muriel J. Harris. 2018. "Insights into the Etiology of Mammalian Neural Tube Closure Defects from Developmental, Genetic and Evolutionary Studies" Journal of Developmental Biology 6, no. 3: 22. https://doi.org/10.3390/jdb6030022
APA StyleJuriloff, D. M., & Harris, M. J. (2018). Insights into the Etiology of Mammalian Neural Tube Closure Defects from Developmental, Genetic and Evolutionary Studies. Journal of Developmental Biology, 6(3), 22. https://doi.org/10.3390/jdb6030022