
Asxl2−/− Mice Exhibit De Novo Cardiomyocyte Production during Adulthood

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Animals
2.2. Assessment of Heart Growth
2.3. Morphometric Analysis of Isolated Cardiomyocytes
2.4. Thymidine Analog Pulse and Pulse-Chase Assays
2.5. Preparation of Histological Sections
2.6. Immunofluorescence
3. Results
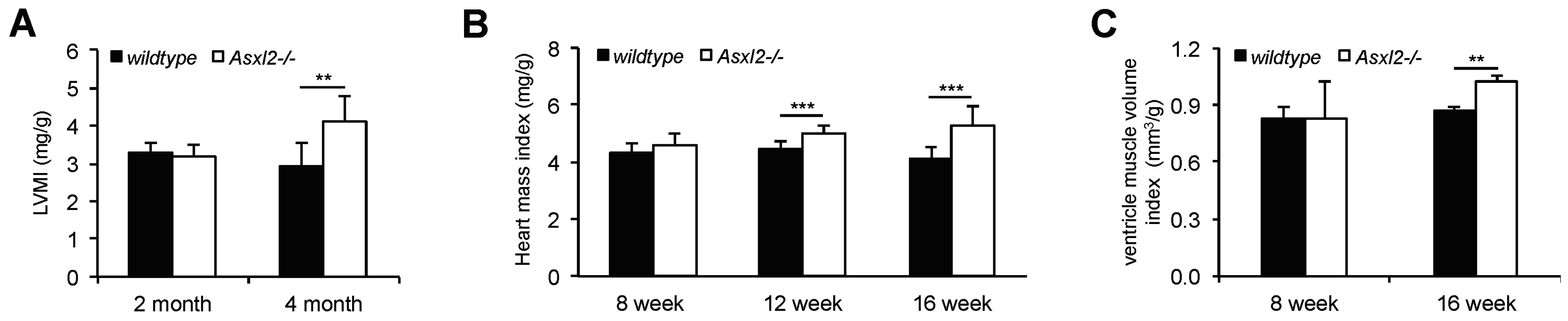
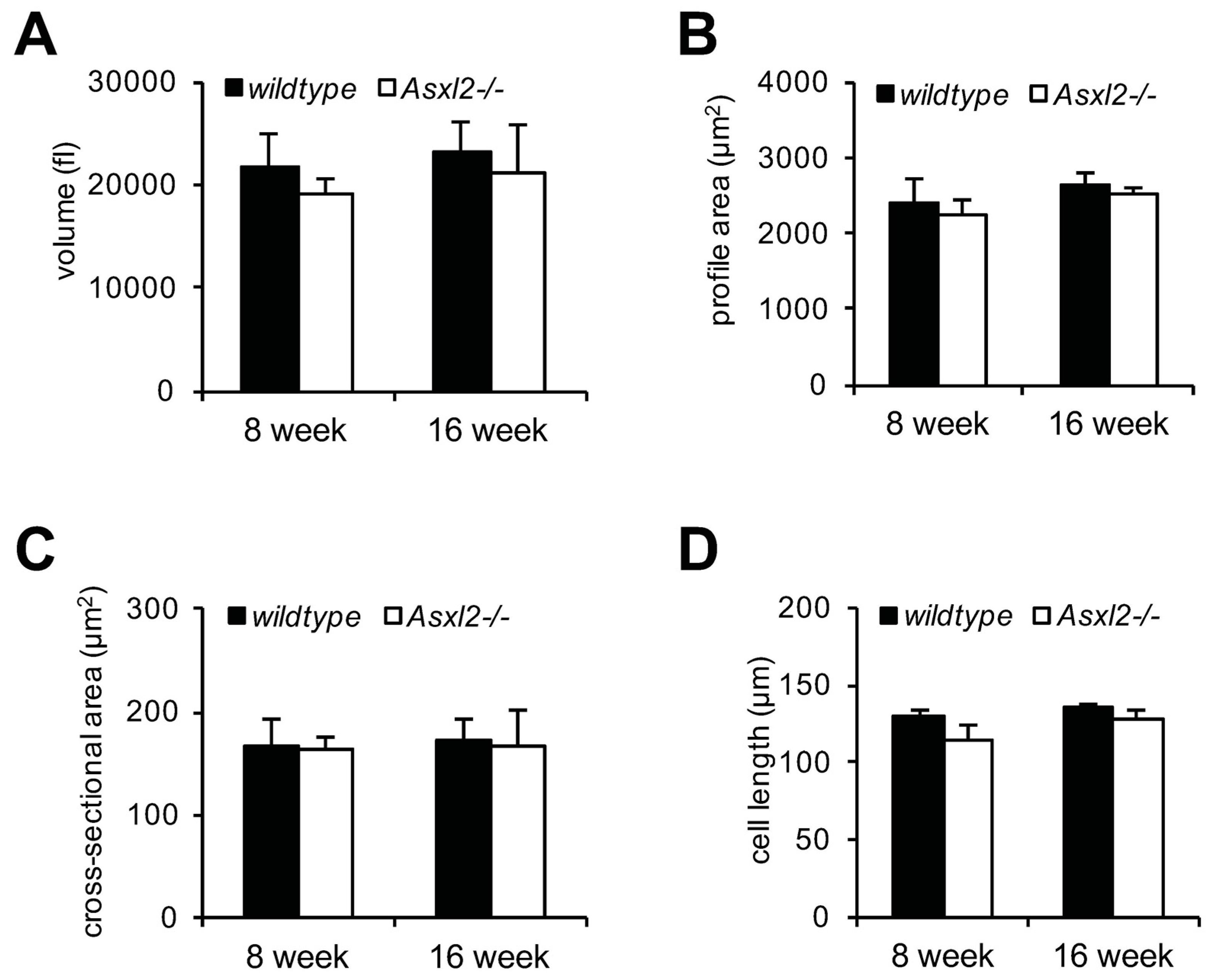
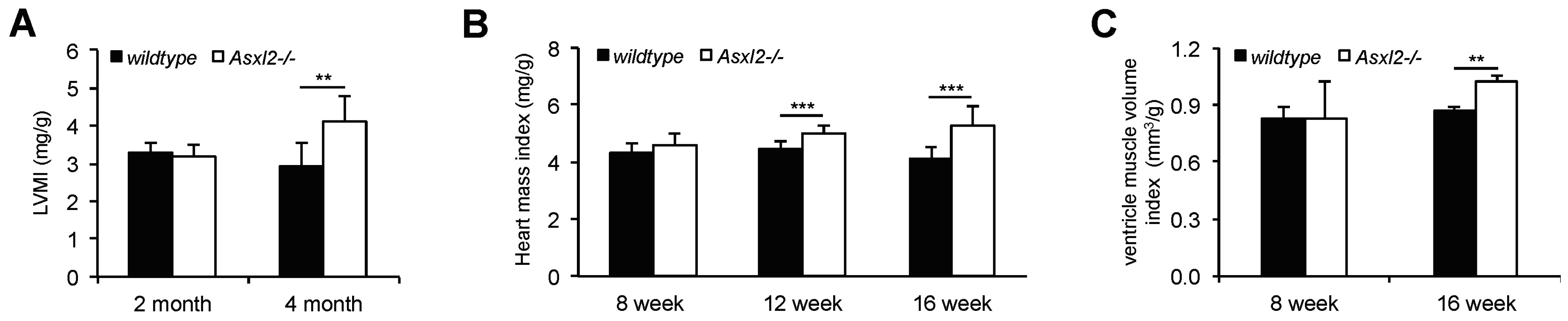
3.1. Overgrowth of the Adult Asxl2−/− Heart without Cardiomyocyte Hypertrophy
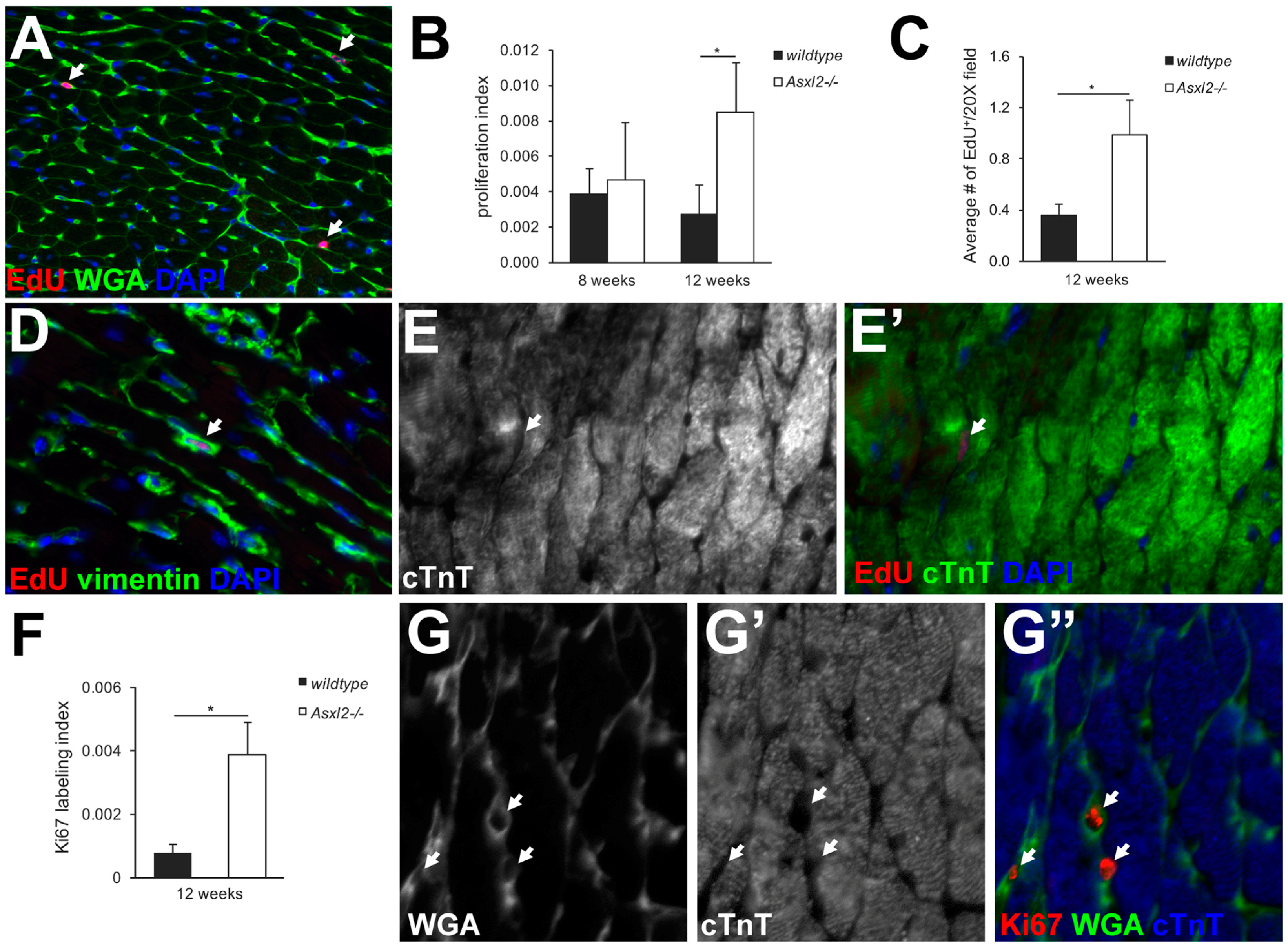
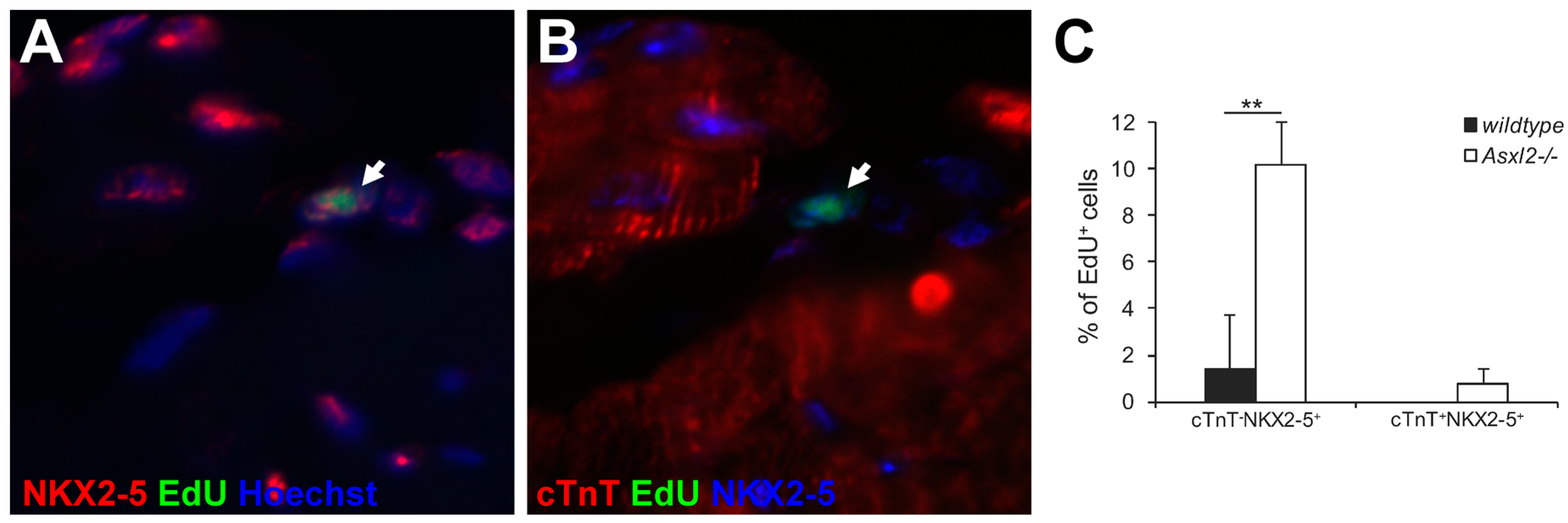
3.2. Asxl2−/− Hearts Exhibit Elevated Proliferative Activity, but Not in Cardiomyocytes
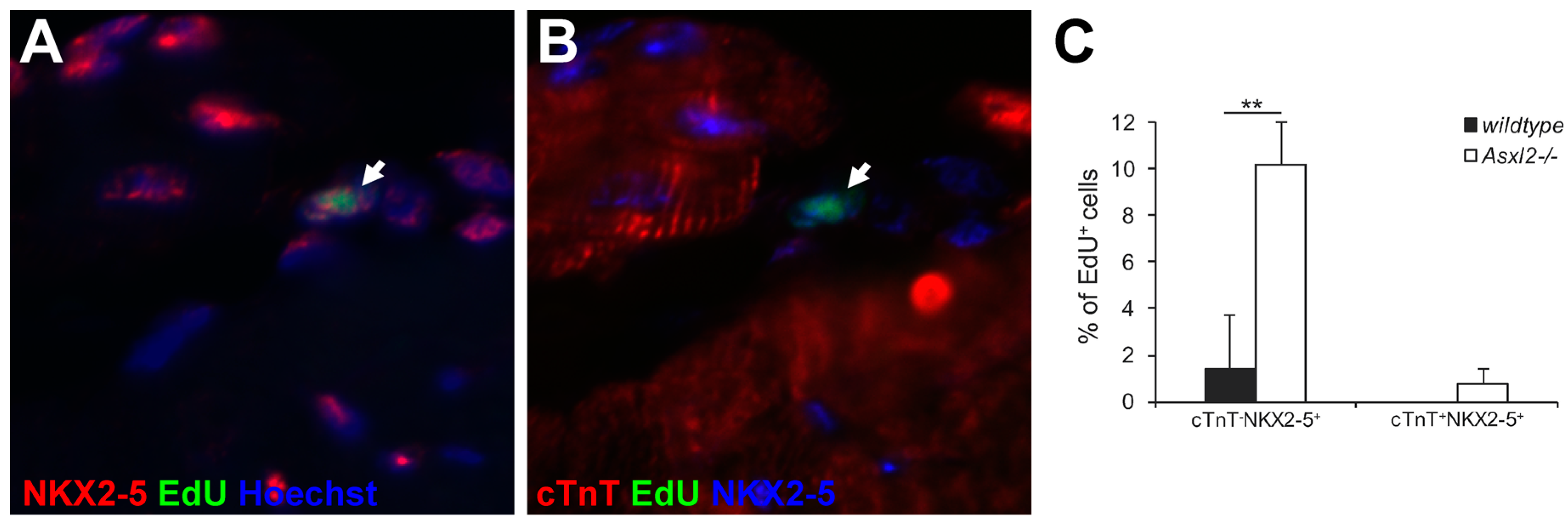
3.3. Expression of Cardiogenic Markers by Proliferative Cells in Asxl2−/− Hearts
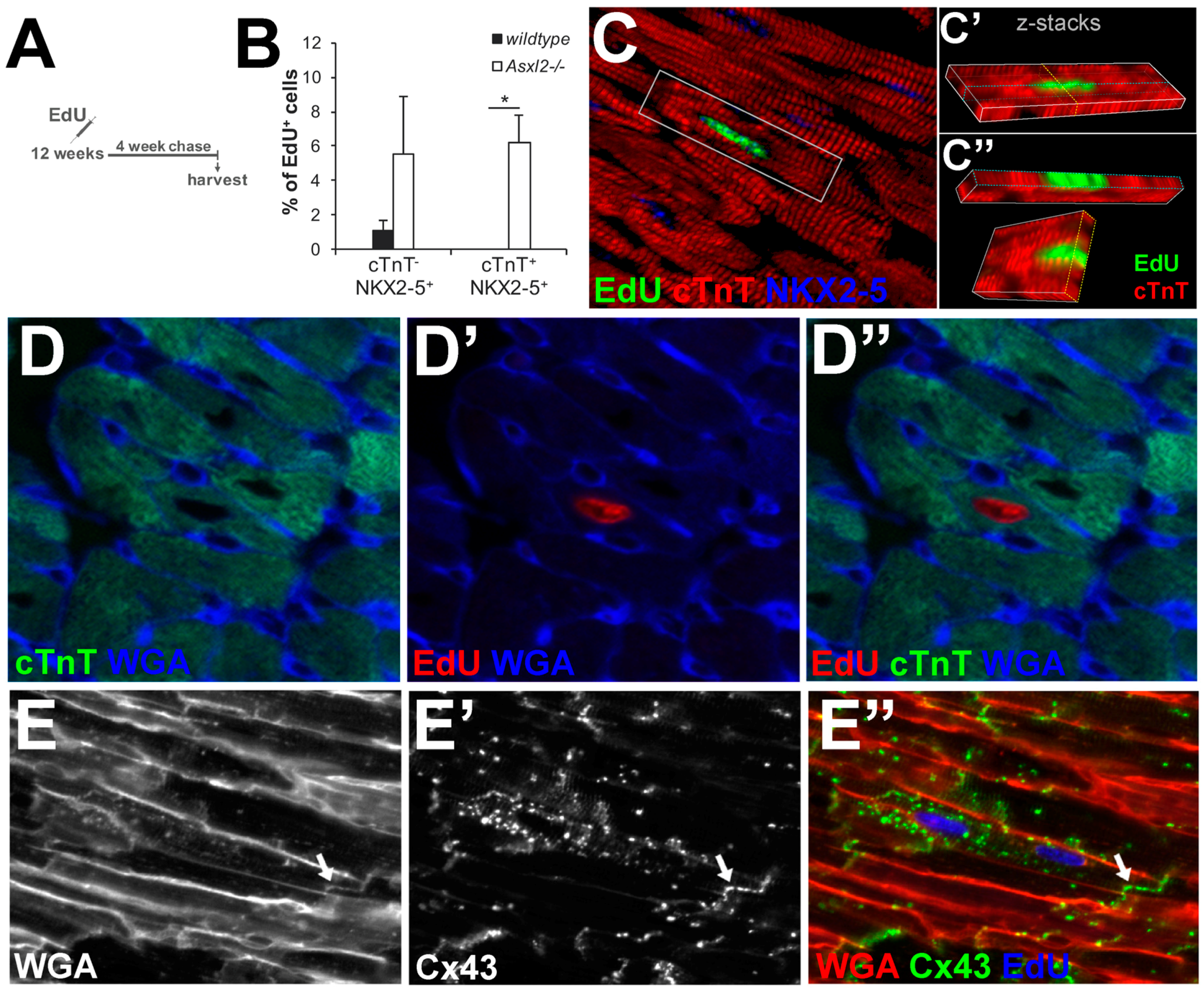
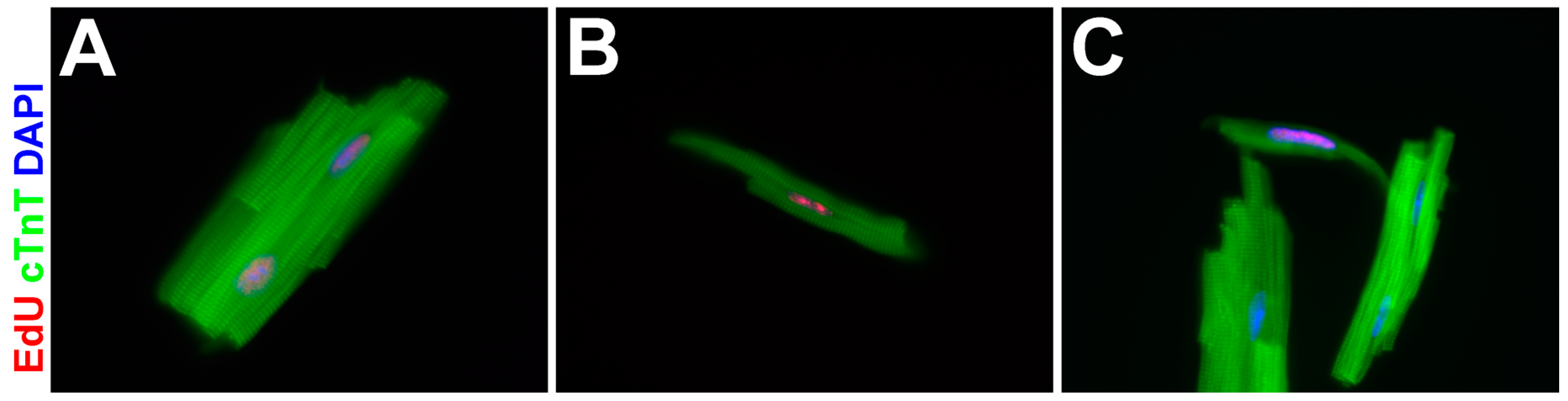
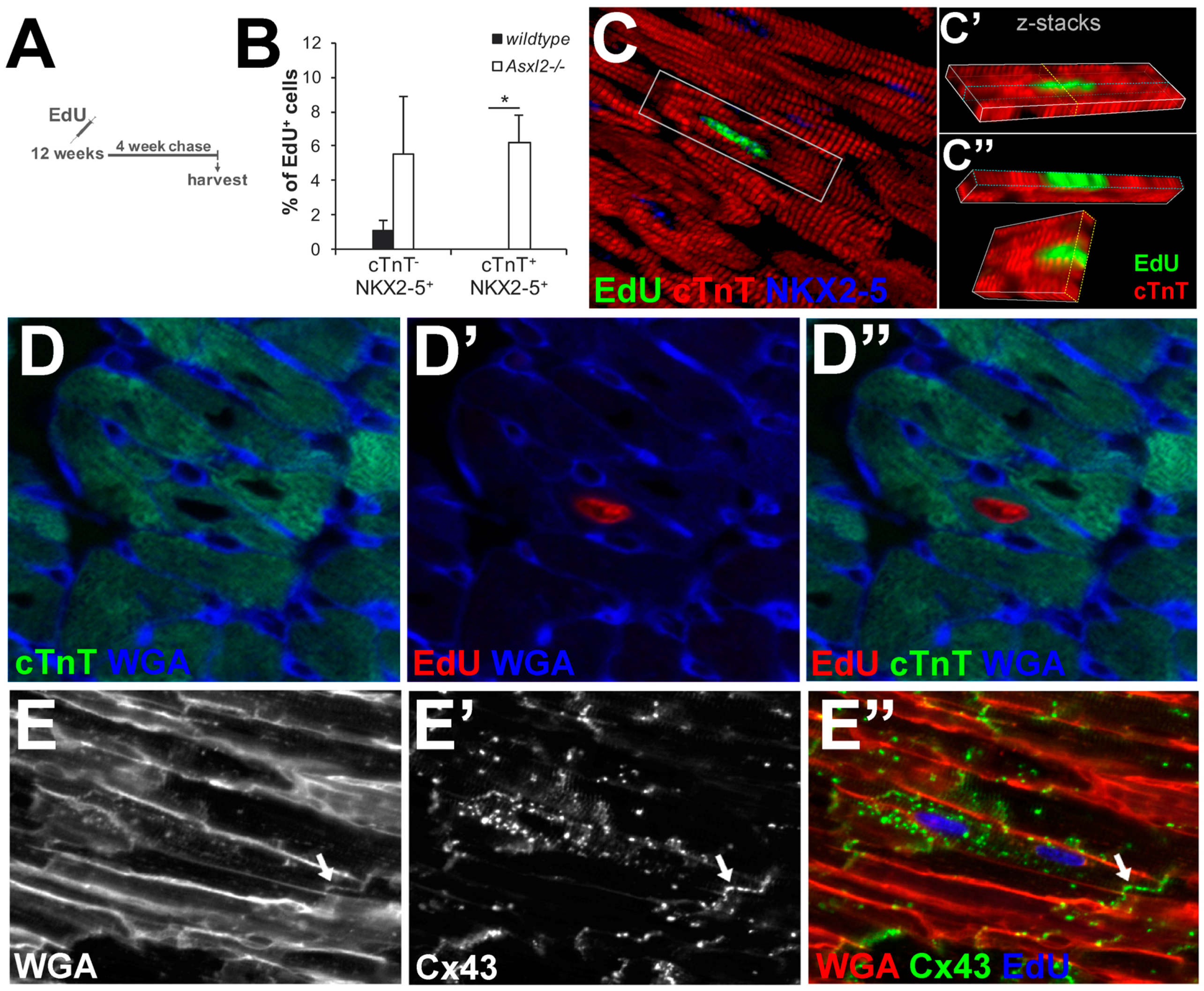
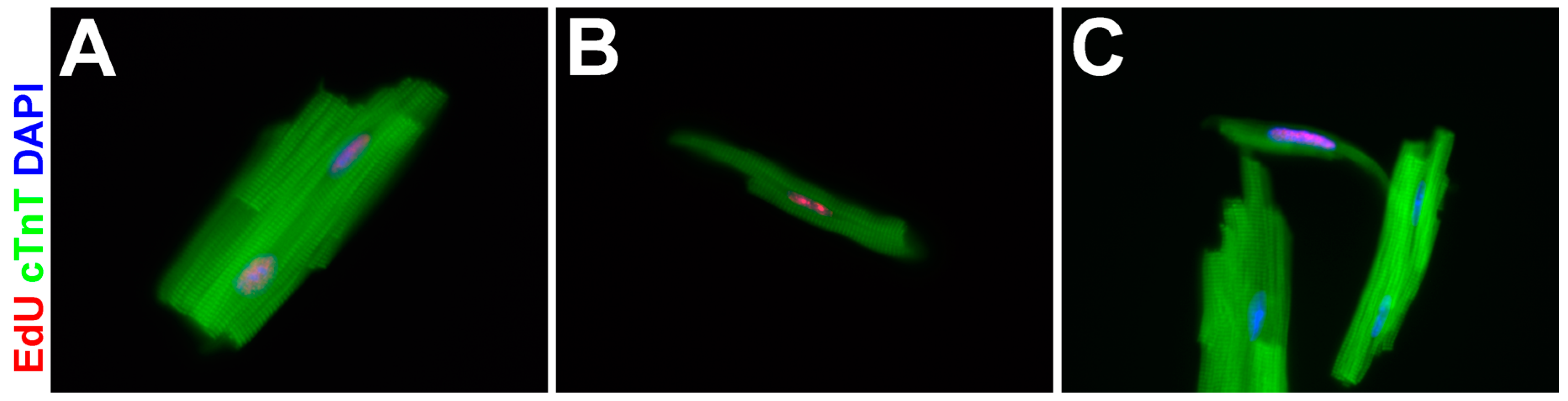
3.4. EdU-Labeled Cells Give Rise to Cardiomyocytes in Asxl2−/− Hearts after 4-Week Chase
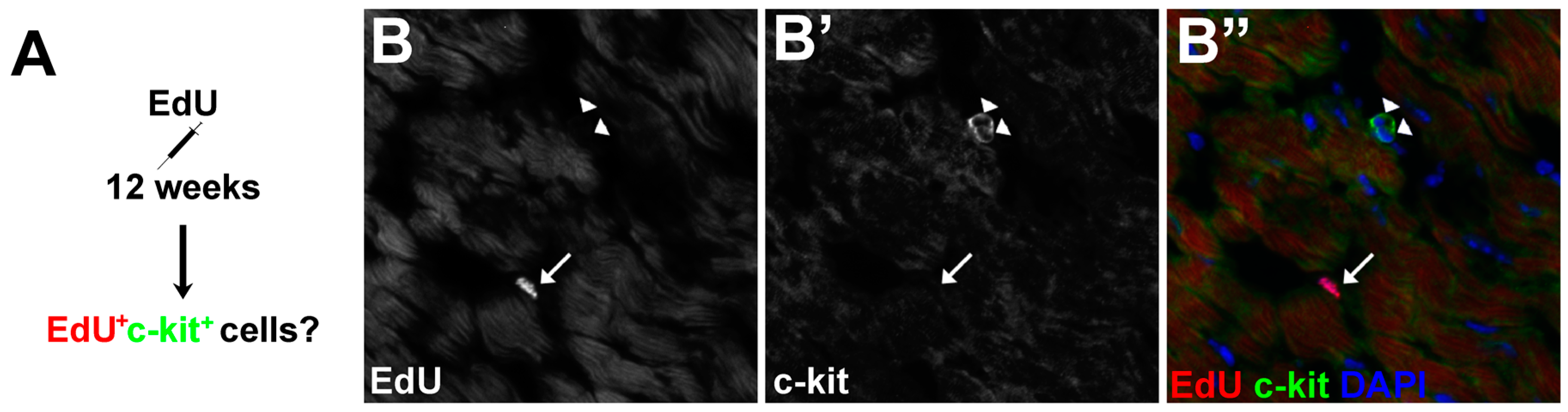
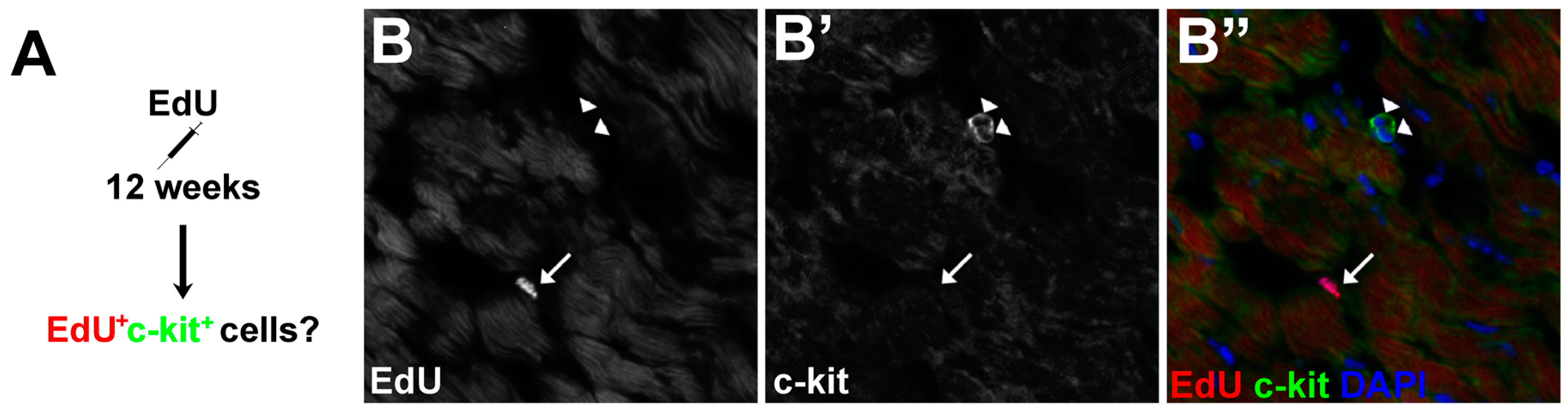
3.5. The Proliferative Cardiogenic Cells in Asxl2−/− Hearts are Distinct from c-kit+ Cardiac Stem Cells and cCFU-Fs
4. Discussion
4.1. Adult Asxl2−/− Hearts Exhibit De Novo Cardiomyocyte Production
4.2. What Are PCN Cells?
4.3. Epigenetic Regulation of Cardiogenicity?
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Quaini, F.; Cigola, E.; Lagrasta, C.; Saccani, G.; Quaini, E.; Rossi, C.; Olivetti, G.; Anversa, P. End-stage cardiac failure in humans is coupled with the induction of proliferating cell nuclear antigen and nuclear mitotic division in ventricular myocytes. Circ. Res. 1994, 75, 1050–1063. [Google Scholar] [CrossRef] [PubMed]
- Soonpaa, M.H.; Field, L.J. Assessment of cardiomyocyte DNA synthesis in normal and injured adult mouse hearts. Am. J. Physiol. 1997, 272, H220–H226. [Google Scholar] [PubMed]
- Kajstura, J.; Leri, A.; Finato, N.; di Loreto, C.; Beltrami, C.A.; Anversa, P. Myocyte proliferation in end-stage cardiac failure in humans. Proc. Natl. Acad. Sci. USA 1998, 95, 8801–8805. [Google Scholar] [CrossRef] [PubMed]
- Bergmann, O.; Bhardwaj, R.D.; Bernard, S.; Zdunek, S.; Walsh, S.; Zupicich, J.; Alkass, K.; Buchholz, B.A.; Jovinge, S.; Frisén, J.; et al. Evidence for Cardiomyocyte Renewal in Humans. Science 2009, 324, 98–102. [Google Scholar] [CrossRef] [PubMed]
- Malliaras, K.; Zhang, Y.; Seinfeld, J.; Galang, G.; Tseliou, E.; Cheng, K.; Sun, B.; Aminzadeh, M.; Marban, E. Cardiomyocyte proliferation and progenitor cell recruitment underlie therapeutic regeneration after myocardial infarction in the adult mouse heart. EMBO Mol. Med. 2013, 5, 191–209. [Google Scholar] [CrossRef] [PubMed]
- Mollova, M.; Bersell, K.; Walsh, S.; Savla, J.; Das, L.T.; Park, S.-Y.; Silberstein, L.E.; dos Remedios, C.G.; Graham, D.; Colan, S.; et al. Cardiomyocyte proliferation contributes to heart growth in young humans. Proc. Natl. Acad. Sci. USA 2013, 110, 1446–51. [Google Scholar] [CrossRef] [PubMed]
- Senyo, S.E.; Steinhauser, M.L.; Pizzimenti, C.L.; Yang, V.K.; Cai, L.; Wang, M.; Wu, T.-D.; Guerquin-Kern, J.-L.; Lechene, C.P.; Lee, R.T. Mammalian heart renewal by pre-existing cardiomyocytes. Nature 2013, 493, 433–436. [Google Scholar] [CrossRef] [PubMed]
- Ali, S.R.; Hippenmeyer, S.; Saadat, L.V.; Luo, L.; Weissman, I.L.; Ardehali, R. Existing cardiomyocytes generate cardiomyocytes at a low rate after birth in mice. Proc. Natl. Acad. Sci. USA 2014, 111, 8850–8855. [Google Scholar] [CrossRef] [PubMed]
- Bergmann, O.; Zdunek, S.; Felker, A.; Salehpour, M.; Alkass, K.; Bernard, S.; Sjostrom, S.L.; Szewczykowska, M.; Jackowska, T.; dos Remedios, C.; et al. Dynamics of Cell Generation and Turnover in the Human Heart. Cell 2015, 1566–1575. [Google Scholar] [CrossRef] [PubMed]
- Kimura, W.; Xiao, F.; Canseco, D.C.; Muralidhar, S.; Thet, S.; Zhang, H.M.; Abdulrahman, Y.; Chen, R.; Garcia, J.; Shelton, J.M.; et al. Hypoxia fate mapping identifies cycling cardiomyocytes in the adult heart. Nature 2015, 523, 226–230. [Google Scholar] [CrossRef] [PubMed]
- Chaudhry, H.W.; Dashoush, N.H.; Tang, H.; Zhang, L.; Wang, X.; Wu, E.X.; Wolgemuth, D.J. Cyclin A2 mediates cardiomyocyte mitosis in the postmitotic myocardium. J. Biol. Chem. 2004, 279, 35858–35866. [Google Scholar] [CrossRef] [PubMed]
- Sdek, P.; Zhao, P.; Wang, Y.; Huang, C.-J.; Ko, C.Y.; Butler, P.C.; Weiss, J.N.; Maclellan, W.R. Rb and p130 control cell cycle gene silencing to maintain the postmitotic phenotype in cardiac myocytes. J. Cell Biol. 2011, 194, 407–423. [Google Scholar] [CrossRef] [PubMed]
- Eulalio, A.; Mano, M.; Ferro, M.D.; Zentilin, L.; Sinagra, G.; Zacchigna, S.; Giacca, M. Functional screening identifies miRNAs inducing cardiac regeneration. Nature 2012, 492, 376–381. [Google Scholar] [CrossRef] [PubMed]
- Von Gise, A.; Lin, Z.; Schlegelmilch, K.; Honor, L.B.; Pan, G.M.; Buck, J.N. YAP1, the nuclear target of Hippo signaling, stimulates heart growth through cardiomyocyte proliferation but not hypertrophy. Proc. Natl. Acad. Sci. USA 2012, 109, 2–7. [Google Scholar] [CrossRef] [PubMed]
- Chakraborty, S.; Sengupta, A.; Yutzey, K.E. Tbx20 promotes cardiomyocyte proliferation and persistence of fetal characteristics in adult mouse hearts. J. Mol. Cell. Cardiol. 2013, 62, 203–213. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Huang, Z.P.; Seok, H.Y.; Ding, J.; Kataoka, M.; Zhang, Z.; Hu, X.; Wang, G.; Lin, Z.; Wang, S.; et al. Mir-17-92 cluster is required for and sufficient to induce cardiomyocyte proliferation in postnatal and adult hearts. Circ. Res. 2013, 112, 1557–1566. [Google Scholar] [CrossRef] [PubMed]
- Heallen, T.; Morikawa, Y.; Leach, J.; Tao, G.; Willerson, J.T.; Johnson, R.L.; Martin, J.F. Hippo signaling impedes adult heart regeneration. Development 2013, 140, 4683–4690. [Google Scholar] [CrossRef] [PubMed]
- Mahmoud, A.I.; Kocabas, F.; Muralidhar, S.A.; Kimura, W.; Koura, A.S.; Thet, S.; Porrello, E.R.; Sadek, H.A. Meis1 regulates postnatal cardiomyocyte cell cycle arrest. Nature 2013, 497, 249–53. [Google Scholar] [CrossRef] [PubMed]
- Xin, M.; Kim, Y.; Sutherland, L.B.; Murakami, M.; Qi, X.; McAnally, J.; Porrello, E.R.; Mahmoud, A.I.; Tan, W.; Shelton, J.M.; et al. Hippo pathway effector Yap promotes cardiac regeneration. Proc. Natl. Acad. Sci. USA 2013, 110, 13839–13844. [Google Scholar] [CrossRef] [PubMed]
- Rochais, F.; Sturny, R.; Chao, C.M.; Mesbah, K.; Bennett, M.; Mohun, T.J.; Bellusci, S.; Kelly, R.G. FGF10 promotes regional foetal cardiomyocyte proliferation and adult cardiomyocyte cell-cycle re-entry. Cardiovasc. Res. 2014, 104, 432–442. [Google Scholar] [CrossRef] [PubMed]
- Lin, Z.; von Gise, A.; Zhou, P.; Gu, F.; Ma, Q.; Jiang, J.; Yau, A.L.; Buck, J.N.; Gouin, K.A.; van Gorp, P.R.R.; et al. Cardiac-specific YAP activation improves cardiac function and survival in an experimental murine MI model. Circ. Res. 2014, 115, 354–363. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Gao, E.; Vite, A.; Yi, R.; Gomez, L.; Goossens, S.; van Roy, F.; Radice, G.L. Alpha-Catenins Control Cardiomyocyte Proliferation by Regulating Yap Activity. Circ. Res. 2015, 116, 70–79. [Google Scholar] [CrossRef] [PubMed]
- Tian, Y.; Liu, Y.; Wang, T.; Zhou, N.; Kong, J.; Chen, L.; Snitow, M.; Morley, M.; Li, D.; Petrenko, N.; et al. A microRNA-Hippo pathway that promotes cardiomyocyte proliferation and cardiac regeneration in mice. Sci. Transl. Med. 2015, 7, 279ra38. [Google Scholar] [CrossRef] [PubMed]
- Xiang, F.; Guo, M.; Yutzey, K.E. Overexpression of Tbx20 in Adult Cardiomyocytes Promotes Proliferation and Improves Cardiac Function after Myocardial Infarction. Circulation 2016, 133, 1081–1092. [Google Scholar] [CrossRef] [PubMed]
- Hsieh, P.C.H.; Segers, V.F.M.; Davis, M.E.; MacGillivray, C.; Gannon, J.; Molkentin, J.D.; Robbins, J.; Lee, R.T. Evidence from a genetic fate-mapping study that stem cells refresh adult mammalian cardiomyocytes after injury. Nat. Med. 2007, 13, 970–974. [Google Scholar] [CrossRef] [PubMed]
- Beltrami, A.P.; Barlucchi, L.; Torella, D.; Baker, M.; Limana, F.; Chimenti, S.; Kasahara, H.; Rota, M.; Musso, E.; Urbanek, K.; et al. Adult Cardiac Stem Cells Are Multipotent and Support Myocardial Regeneration. Cell 2003, 114, 763–776. [Google Scholar] [CrossRef]
- Ellison, G.M.; Vicinanza, C.; Smith, A.J.; Aquila, I.; Leone, A.; Waring, C.D.; Henning, B.J.; Stirparo, G.G.; Papait, R.; Scarfò, M.; et al. Adult c-kitpos cardiac stem cells are necessary and sufficient for functional cardiac regeneration and repair. Cell 2013, 154, 827–842. [Google Scholar] [CrossRef] [PubMed]
- Hesse, M.; Fleischmann, B.K.; Kotlikoff, M.I. Concise review: The role of C-kit expressing cells in heart repair at the neonatal and adult stage. Stem Cells 2014, 32, 1701–1712. [Google Scholar] [CrossRef] [PubMed]
- Van Berlo, J.H.; Kanisicak, O.; Maillet, M.; Vagnozzi, R.J.; Karch, J.; Lin, S.-C.J.; Middleton, R.C.; Marbán, E.; Molkentin, J.D. C-Kit+ Cells Minimally Contribute Cardiomyocytes to the Heart. Nature 2014, 509, 337–341. [Google Scholar] [CrossRef] [PubMed]
- Sultana, N.; Zhang, L.; Yan, J.; Chen, J.; Cai, W.; Razzaque, S.; Jeong, D.; Sheng, W.; Bu, L.; Xu, M.; et al. Resident c-kit(+) cells in the heart are not cardiac stem cells. Nat. Commun. 2015, 6, 8701. [Google Scholar] [CrossRef] [PubMed]
- Smart, N.; Bollini, S.; Dubé, K.N.; Vieira, J.M.; Zhou, B.; Davidson, S.; Yellon, D.; Riegler, J.; Price, A.N.; Lythgoe, M.F.; et al. De novo cardiomyocytes from within the activated adult heart after injury. Nature 2011, 474, 640–644. [Google Scholar] [CrossRef] [PubMed]
- Smart, N.; Bollini, S.; Dube, K.N.; Vieira, J.M.; Zhou, B.; Riegler, J.; Price, A.N.; Lythgoe, M.F.; Davidson, S.; Yellon, D.; et al. Myocardial regeneration: Expanding the repertoire of thymosin beta 4 in the ischaemic heart. October 2012, 1269, 92–101. [Google Scholar]
- Chen, S.; Shimoda, M.; Chen, J.; Grayburn, P.A. Stimulation of adult resident cardiac progenitor cells by durable myocardial expression of thymosin beta 4 with ultrasound-targeted microbubble delivery. Gene Ther. 2013, 20, 225–233. [Google Scholar] [CrossRef] [PubMed]
- Zhou, B.; Honor, L.B.; Ma, Q.; Oh, J.H.; Lin, R.Z.; Melero-Martin, J.M.; von Gise, A.; Zhou, P.; Hu, T.; He, L.; et al. Thymosin beta 4 treatment after myocardial infarction does not reprogram epicardial cells into cardiomyocytes. J. Mol. Cell. Cardiol. 2012, 52, 43–47. [Google Scholar] [CrossRef] [PubMed]
- Oh, H.; Bradfute, S.B.; Gallardo, T.D.; Nakamura, T.; Gaussin, V.; Mishina, Y.; Pocius, J.; Michael, L.H.; Behringer, R.R.; Garry, D.J.; et al. Cardiac progenitor cells from adult myocardium: Homing, differentiation, and fusion after infarction. Proc. Natl. Acad. Sci. USA 2003, 100, 12313–12318. [Google Scholar] [CrossRef] [PubMed]
- Pfister, O.; Mouquet, F.; Jain, M.; Summer, R.; Helmes, M.; Fine, A.; Colucci, W.S.; Liao, R. CD31− but not CD31+ cardiac side population cells exhibit functional cardiomyogenic differentiation. Circ. Res. 2005, 97, 52–61. [Google Scholar] [CrossRef] [PubMed]
- Fuentes, T.I.; Appleby, N.; Tsay, E.; Martinez, J.J.; Bailey, L.; Hasaniya, N.; Kearns-Jonker, M. Human Neonatal Cardiovascular Progenitors: Unlocking the Secret to Regenerative Ability. PLoS ONE 2013, 8, e77464. [Google Scholar]
- Chong, J.J.H.; Chandrakanthan, V.; Xaymardan, M.; Asli, N.S.; Li, J.; Ahmed, I.; Heffernan, C.; Menon, M.K.; Scarlett, C.J.; Rashidianfar, A.; et al. Adult cardiac-resident MSC-like stem cells with a proepicardial origin. Cell Stem Cell 2011, 9, 527–540. [Google Scholar] [PubMed]
- Ieda, M.; Fu, J.D.; Delgado-Olguin, P.; Vedantham, V.; Hayashi, Y.; Bruneau, B.G.; Srivastava, D. Direct reprogramming of fibroblasts into functional cardiomyocytes by defined factors. Cell 2010, 142, 375–386. [Google Scholar] [CrossRef] [PubMed]
- Song, K.; Nam, Y.-J.; Luo, X.; Qi, X.; Tan, W.; Huang, G.N.; Acharya, A.; Smith, C.L.; Tallquist, M.D.; Neilson, E.G.; et al. Heart repair by reprogramming non-myocytes with cardiac transcription factors. Nature 2012, 485, 599–604. [Google Scholar] [CrossRef] [PubMed]
- Qian, L.; Huang, Y.; Spencer, C.I.; Foley, A.; Vedantham, V.; Liu, L.; Conway, S.J.; Fu, J.; Srivastava, D. In vivo reprogramming of murine cardiac fibroblasts into induced cardiomyocytes. Nature 2012, 485, 593–598. [Google Scholar] [CrossRef] [PubMed]
- Furtado, M.B.; Costa, M.W.; Pranoto, E.A.; Salimova, E.; Pinto, A.R.; Lam, N.T.; Park, A.; Snider, P.; Chandran, A.; Harvey, R.P.; et al. Cardiogenic genes expressed in cardiac fibroblasts contribute to heart development and repair. Circ. Res. 2014, 114, 1422–1434. [Google Scholar] [CrossRef] [PubMed]
- Baskind, H.A.; Na, L.; Ma, Q.; Patel, M.P.; Geenen, D.L.; Wang, Q.T. Functional conservation of Asxl2, a murine homolog for the Drosophila enhancer of trithorax and polycomb group gene Asx. PLoS ONE 2009, 4, e4750. [Google Scholar] [CrossRef] [PubMed]
- Lai, H.L.; Wang, Q.T. Additional Sex Combs-Like 2 Is Required for Polycomb Repressive Complex 2 Binding at Select Targets. PLoS ONE 2013, 8, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Lai, H.L.; Grachoff, M.; McGinley, A.L.; Khan, F.F.; Warren, C.M.; Chowdhury, S.A.K.; Wolska, B.M.; Solaro, R.J.; Geenen, D.L.; Wang, Q.T. Maintenance of adult cardiac function requires the chromatin factor Asxl2. J. Mol. Cell. Cardiol. 2012, 53, 734–741. [Google Scholar] [CrossRef] [PubMed]
- Schiller, N.B.; Shah, P.M.; Crawford, M.; DeMaria, A.; Devereux, R.; Feigenbaum, H.; Gutgesell, H.; Reicheck, N.; Sahn, D.; Schnittger, I.; et al. Recommendations for quantification of the left ventricle by two-dimensional echocardiography. American Society of Echocardiography Committee on Standards, Subcommittee on Quantification of Two-dimensional Echocardiograms. J. Am. Soc. Echocardiogr. 1989, 2, 358–367. [Google Scholar] [PubMed]
- Louch, W.E.; Sheehan, K.A.; Wolska, B.M. Methods in cardiomyocyte isolation, culture, and gene transfer. J. Mol. Cell. Cardiol. 2011, 51, 288–298. [Google Scholar] [CrossRef] [PubMed]
- Lamprecht, M.R.; Sabatini, D.M.; Carpenter, A.E. Short Technical Reports CellProfiler™: Free, versatile software for automated biological image analysis Short Technical Reports. Biotechniques 2007, 42, 71–75. [Google Scholar] [CrossRef] [PubMed]
- Li, F.; Wang, X.; Capasso, J.M.; Gerdes, A.M. Rapid transition of cardiac myocytes from hyperplasia to hypertrophy during postnatal development. J. Mol. Cell. Cardiol. 1996, 28, 1737–1746. [Google Scholar] [CrossRef] [PubMed]
- Bai, S.L.; Campbell, S.E.; Moore, J.A.; Morales, M.C.; Gerdes, A.M. Influence of age, growth, and sex on cardiac myocyte size and number in rats. Anat. Rec. 1990, 226, 207–212. [Google Scholar] [CrossRef] [PubMed]
- Gerdes, A.M. A reliable, efficient, and comprehensive approach to assess myocyte remodeling in cardiac hypertrophy and failure. J. Card. Fail. 1997, 3, 63–68. [Google Scholar] [CrossRef]
- Adler, C.P.; Friedburg, H.; Herget, G.W.; Neuburger, M.; Schwalb, H. Variability of cardiomyocyte DNA content, ploidy level and nuclear number in mammalian hearts. Virchows Arch. 1996, 429, 159–164. [Google Scholar] [CrossRef] [PubMed]
- Matiasova, A.; Sevc, J.; Mikes, J.; Jendzelovsky, R.; Daxnerova, Z.; Fedorocko, P. Flow cytometric determination of 5-bromo-2’-deoxyuridine pharmokinetics in blood serum after intraperitoneal administration to rats and mice. Histochem. Cell Biol. 2014, 142, 703–12. [Google Scholar] [CrossRef] [PubMed]
- Packard, D.; Menzies, R.; Skalko, R. Incorporation of thymidine and its analogue, bromodeoxyuridine, into embryos and maternal tissues of the mouse. Differentiation 1973, 1, 397–405. [Google Scholar] [CrossRef] [PubMed]
- Soonpaa, M.H.; Rubart, M.; Field, L.J. Challenges measuring cardiomyocyte renewal. Biochim. Biophys. Acta Mol. Cell Res. 2013, 1833, 799–803. [Google Scholar] [CrossRef] [PubMed]
- Fioret, B.A.; Heimfeld, J.D.; Paik, D.T.; Hatzopoulos, A.K. Endothelial cells contribute to generation of adult ventricular myocytes during cardiac homeostasis. Cell Rep. 2014, 8, 229–241. [Google Scholar] [CrossRef] [PubMed]
- Cai, C.L.; Liang, X.; Shi, Y.; Chu, P.H.; Pfaff, S.L.; Chen, J.; Evans, S. Isl1 identifies a cardiac progenitor population that proliferates prior to differentiation and contributes a majority of cells to the heart. Dev. Cell 2003, 5, 877–889. [Google Scholar] [CrossRef]
- Laugwitz, K.-L.; Moretti, A.; Lam, J.; Gruber, P.; Chen, Y.; Woodard, S.; Lin, L.-Z.; Cai, C.-L.; Lu, M.M.; Reth, M.; et al. Postnatal isl1+ cardioblasts enter fully differentiated cardiomyocyte lineages. Nature 2005, 433, 647–53. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Liu, Z.; Yin, C.; Asfour, H.; Chen, O.; Li, Y.; Bursac, N.; Liu, J.; Qian, L. Stoichiometry of Gata4, Mef2c, and Tbx5 influences the efficiency and quality of induced cardiac myocyte reprogramming. Circ. Res. 2015, 116, 237–244. [Google Scholar] [CrossRef] [PubMed]
- Dave, J.M.; Bayless, K.J. Vimentin as an integral regulator of cell adhesion and endothelial sprouting. Microcirculation 2014, 21, 333–344. [Google Scholar] [CrossRef] [PubMed]
- Chong, J.J.H.; Reinecke, H.; Iwata, M.; Torok-Storb, B.; Stempien-Otero, A.; Murry, C.E. Progenitor Cells Identified by PDGFR-Alpha Expression in the Developing and Diseased Human Heart. Stem Cells Dev. 2013, 22, 1932–1943. [Google Scholar] [CrossRef] [PubMed]
- Struhl, G.; Akam, M. Altered distributions of Ultrabithorax transcripts in extra sex combs mutant embryos of Drosophila. EMBO J. 1985, 4, 3259–3264. [Google Scholar] [PubMed]
- Bantignies, F.; Cavalli, G. Cellular memory and dynamic regulation of polycomb group proteins. Curr. Opin. Cell Biol. 2006, 18, 275–283. [Google Scholar] [CrossRef] [PubMed]
- Margueron, R.; Reinberg, D. Chromatin structure and the inheritance of epigenetic information. Nat. Rev. Genet. 2010, 11, 285–296. [Google Scholar] [CrossRef] [PubMed]
- Barrero, M.J.; Boué, S.; Izpisúa Belmonte, J.C. Epigenetic Mechanisms that Regulate Cell Identity. Cell Stem Cell 2010, 7, 565–570. [Google Scholar] [CrossRef] [PubMed]
- Griffiths, E.A.; Gore, S.D. Epigenetic Therapies in MDS and AML; Springer: New York, NY, USA, 2013; Volume 754. [Google Scholar]
- Nie, J.; Liu, L.; Li, X.; Han, W. Decitabine, a new star in epigenetic therapy: The clinical application and biological mechanism in solid tumors. Cancer Lett. 2014, 354, 12–20. [Google Scholar] [CrossRef] [PubMed]
- Nervi, C.; de Marinis, E.; Codacci-Pisanelli, G. Epigenetic treatment of solid tumours: A review of clinical trials. Clin. Epigenet. 2015, 7, 127. [Google Scholar] [CrossRef] [PubMed]
- Van Weerd, J.H.; Koshiba-Takeuchi, K.; Kwon, C.; Takeuchi, J.K. Epigenetic factors and cardiac development. Cardiovasc. Res. 2011, 91, 203–211. [Google Scholar] [CrossRef] [PubMed]
- Vallaster, M.; Vallaster, C.D.; Wu, S.M. Epigenetic mechanisms in cardiac development and disease Overview of Epigenetic Mechanisms. Acta Biochim. Biophys. Hung. 2012, 92–102. [Google Scholar]
- Wang, Q.T. Epigenetic regulation of cardiac development and function by polycomb group and trithorax group proteins. Dev. Dyn. 2012, 241, 1021–1033. [Google Scholar] [CrossRef] [PubMed]
- Daou, S.; Hammond-Martel, I.; Mashtalir, N.; Barbour, H.; Gagnon, J.; Iannantuono, N.V.; Nkwe, N.S.; Motorina, A.; Pak, H.; Yu, H.; et al. The BAP1/ASXL2 histone H2A deubiquitinase complex regulates cell proliferation and is disrupted in cancer. J. Biol. Chem. 2015, 290, 28643–28663. [Google Scholar] [CrossRef] [PubMed]
- Park, U.; Kang, M.; Kim, E.; Kwon, Y.; Hur, W.; Yoon, S.; Song, B.; Park, J.; Hwang, J.; Jeong, J.; et al. ASXL2 promotes proliferation of breast cancer cells by linking ERalpha to histone methylation. Oncogene 2016, 14, 3742–3752. [Google Scholar] [CrossRef] [PubMed]
- Valiente-alandi, I.; Albo-castellanos, C.; Herrero, D.; Arza, E.; Garcia-gomez, M.; Segovia, J.C.; Capecchi, M.; Bernad, A. Cardiac Bmi1+ cells contribute to myocardial renewal in the murine adult heart. Stem Cell Res. Ther. 2015. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Wang, L.; Vaseghi, H.R.; Liu, Z.; Lu, R.; Alimohamadi, S.; Yin, C.; Fu, J.D.; Wang, G.G.; Liu, J.; et al. Bmi1 Is a Key Epigenetic Barrier to Direct Cardiac Reprogramming. Cell Stem Cell 2016, 18, 382–395. [Google Scholar] [CrossRef] [PubMed]
- Valiente-alandi, I.; Albo-castellanos, C.; Herrero, D.; Sanchez, I.; Bernad, A. Bmi1+ cardiac progenitor cells contribute to myocardial repair following acute injury. Stem Cell Res. Ther. 2016. [Google Scholar] [CrossRef] [PubMed]

© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Brunner, R.; Lai, H.-L.; Deliu, Z.; Melman, E.; Geenen, D.L.; Wang, Q.T. Asxl2−/− Mice Exhibit De Novo Cardiomyocyte Production during Adulthood. J. Dev. Biol. 2016, 4, 32. https://doi.org/10.3390/jdb4040032
Brunner R, Lai H-L, Deliu Z, Melman E, Geenen DL, Wang QT. Asxl2−/− Mice Exhibit De Novo Cardiomyocyte Production during Adulthood. Journal of Developmental Biology. 2016; 4(4):32. https://doi.org/10.3390/jdb4040032
Chicago/Turabian StyleBrunner, Rachel, Hsiao-Lei Lai, Zane Deliu, Elan Melman, David L. Geenen, and Q. Tian Wang. 2016. "Asxl2−/− Mice Exhibit De Novo Cardiomyocyte Production during Adulthood" Journal of Developmental Biology 4, no. 4: 32. https://doi.org/10.3390/jdb4040032
APA StyleBrunner, R., Lai, H.-L., Deliu, Z., Melman, E., Geenen, D. L., & Wang, Q. T. (2016). Asxl2−/− Mice Exhibit De Novo Cardiomyocyte Production during Adulthood. Journal of Developmental Biology, 4(4), 32. https://doi.org/10.3390/jdb4040032