
Hox Genes in Cardiovascular Development and Diseases


Abstract
:1. Introduction
2. Hox Genes and Lineage Commitment
3. Hox Genes and Patterning of the Second Heart Field
4. Role of Hox Genes in Pharyngeal Arch Development and Patterning
5. Hox Gene Specificity: Cofactors in Heart Development
6. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Poelmann, R.E.; Gittenberger-de Groot, A.C.; Hierck, B.P. The development of the heart and microcirculation: Role of shear stress. Med. Biol. Eng. Comput. 2008, 46, 479–484. [Google Scholar] [CrossRef] [PubMed]
- Hoffman, J.I.; Kaplan, S. The incidence of congenital heart disease. J. Am. Coll. Cardiol. 2002, 39, 1890–1900. [Google Scholar] [CrossRef]
- Vincent, S.D.; Buckingham, M.E. How to make a heart: The origin and regulation of cardiac progenitor cells. Curr. Top. Dev. Biol. 2010, 90, 1–41. [Google Scholar] [PubMed]
- Srivastava, D. Making or breaking the heart: From lineage determination to morphogenesis. Cell 2006, 126, 1037–1048. [Google Scholar] [CrossRef] [PubMed]
- Zaffran, S.; Kelly, R.G. New developments in the second heart field. Differentiation 2012, 84, 17–24. [Google Scholar] [CrossRef] [PubMed]
- Buckingham, M.; Meilhac, S.; Zaffran, S. Building the mammalian heart from two sources of myocardial cells. Nat. Rev. Genet. 2005, 6, 826–835. [Google Scholar] [CrossRef] [PubMed]
- Rochais, F.; Mesbah, K.; Kelly, R.G. Signaling pathways controlling second heart field development. Circ. Res. 2009, 104, 933–942. [Google Scholar] [CrossRef] [PubMed]
- Kirby, M.L.; Gale, T.F.; Stewart, D.E. Neural crest cells contribute to normal aorticopulmonary septation. Science 1983, 220, 1059–1061. [Google Scholar] [CrossRef] [PubMed]
- Etchevers, H.C.; Vincent, C.; le Douarin, N.M.; Couly, G.F. The cephalic neural crest provides pericytes and smooth muscle cells to all blood vessels of the face and forebrain. Development 2001, 128, 1059–1068. [Google Scholar] [PubMed]
- Deschamps, J.; van Nes, J. Developmental regulation of the hox genes during axial morphogenesis in the mouse. Development 2005, 132, 2931–2942. [Google Scholar] [CrossRef] [PubMed]
- Duboule, D.; Dolle, P. The structural and functional organization of the murine hox gene family resembles that of drosophila homeotic genes. EMBO J. 1989, 8, 1497–1505. [Google Scholar] [PubMed]
- Alexander, T.; Nolte, C.; Krumlauf, R. Hox genes and segmentation of the hindbrain and axial skeleton. Annu. Rev. Cell Dev. Biol. 2009, 25, 431–456. [Google Scholar] [CrossRef] [PubMed]
- Lo, P.C.; Frasch, M. Establishing A-P polarity in the embryonic heart tube. A conserved function of Hox genes in Drosophila and vertebrates? Trends Cardiovasc. Med. 2003, 13, 182–187. [Google Scholar] [CrossRef]
- Monier, B.; Tevy, M.F.; Perrin, L.; Capovilla, M.; Semeriva, M. Downstream of homeotic genes: In the heart of hox function. Fly 2007, 1, 59–67. [Google Scholar] [CrossRef] [PubMed]
- Zaffran, S.; Niederreither, K. Retinoic acid signaling and heart development. In The Retinoids; Dolle, P., Niederreither, K., Eds.; Wiley-Blackwell: Hoboken, NJ, USA, 2015. [Google Scholar]
- Searcy, R.D.; Yutzey, K.E. Analysis of Hox gene expression during early avian heart development. Dev. Dyn 1998, 213, 82–91. [Google Scholar] [CrossRef]
- Waxman, J.S.; Yelon, D. Increased Hox activity mimics the teratogenic effects of excess retinoic acid signaling. Dev. Dyn. 2009, 238, 1207–1213. [Google Scholar] [CrossRef] [PubMed]
- Van Vliet, P.; Wu, S.M.; Zaffran, S.; Puceat, M. Early cardiac development: A view from stem cells to embryos. Cardiovasc. Res. 2012, 96, 352–362. [Google Scholar] [CrossRef] [PubMed]
- Zaffran, S.; Frasch, M. Early signals in cardiac development. Circ. Res. 2002, 91, 457–469. [Google Scholar] [CrossRef] [PubMed]
- Lengerke, C.; Schmitt, S.; Bowman, T.V.; Jang, I.H.; Maouche-Chretien, L.; McKinney-Freeman, S.; Davidson, A.J.; Hammerschmidt, M.; Rentzsch, F.; Green, J.B.; et al. Bmp and Wnt specify hematopoietic fate by activation of the Cdx-Hox pathway. Cell Stem Cell 2008, 2, 72–82. [Google Scholar] [CrossRef] [PubMed]
- Yue, Q.; Wagstaff, L.; Yang, X.; Weijer, C.; Munsterberg, A. Wnt3a-mediated chemorepulsion controls movement patterns of cardiac progenitors and requires RhoA function. Development 2008, 135, 1029–1037. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Soshnikova, N. Hox genes regulation in vertebrates. Dev. Dyn. 2014, 243, 49–58. [Google Scholar] [CrossRef] [PubMed]
- Deng, C.; Li, Y.; Zhou, L.; Cho, J.; Patel, B.; Terada, N.; Li, Y.; Bungert, J.; Qiu, Y.; Huang, S. HoxBlinc RNA recruits Set1/MLL complexes to activate Hox gene expression patterns and mesoderm lineage development. Cell Rep. 2016, 14, 103–114. [Google Scholar] [CrossRef] [PubMed]
- Cai, C.L.; Liang, X.; Shi, Y.; Chu, P.H.; Pfaff, S.L.; Chen, J.; Evans, S. Isl1 identifies a cardiac progenitor population that proliferates prior to differentiation and contributes a majority of cells to the heart. Dev. Cell 2003, 5, 877–889. [Google Scholar] [CrossRef]
- Prall, O.W.; Menon, M.K.; Solloway, M.J.; Watanabe, Y.; Zaffran, S.; Bajolle, F.; Biben, C.; McBride, J.J.; Robertson, B.R.; Chaulet, H.; et al. An Nkx2–5/Bmp2/Smad1 negative feedback loop controls heart progenitor specification and proliferation. Cell 2007, 128, 947–959. [Google Scholar] [CrossRef] [PubMed]
- Baldini, A. Dissecting contiguous gene defects: Tbx1. Curr. Opin. Genet. Dev. 2005, 15, 279–284. [Google Scholar] [CrossRef] [PubMed]
- Galli, D.; Dominguez, J.N.; Zaffran, S.; Munk, A.; Brown, N.A.; Buckingham, M.E. Atrial myocardium derives from the posterior region of the second heart field, which acquires left-right identity as Pitx2c is expressed. Development 2008, 135, 1157–1167. [Google Scholar] [CrossRef] [PubMed]
- Zaffran, S.; Kelly, R.G.; Meilhac, S.M.; Buckingham, M.E.; Brown, N.A. Right ventricular myocardium derives from the anterior heart field. Circ. Res. 2004, 95, 261–268. [Google Scholar] [CrossRef] [PubMed]
- Bertrand, N.; Roux, M.; Ryckebusch, L.; Niederreither, K.; Dolle, P.; Moon, A.; Capecchi, M.; Zaffran, S. Hox genes define distinct progenitor sub-domains within the second heart field. Dev. Biol. 2011, 353, 266–274. [Google Scholar] [CrossRef] [PubMed]
- Makki, N.; Capecchi, M.R. Hoxa1 lineage tracing indicates a direct role for Hoxa1 in the development of the inner ear, the heart, and the third rhombomere. Dev. Biol. 2010, 341, 499–509. [Google Scholar] [CrossRef] [PubMed]
- Lescroart, F.; Mohun, T.; Meilhac, S.M.; Bennett, M.; Buckingham, M. Lineage tree for the venous pole of the heart: Clonal analysis clarifies controversial genealogy based on genetic tracing. Circ. Res. 2012, 111, 1313–1322. [Google Scholar] [CrossRef] [PubMed]
- Niederreither, K.; Subbarayan, V.; Dolle, P.; Chambon, P. Embryonic retinoic acid synthesis is essential for early mouse post-implantation development. Nat. Genet. 1999, 21, 444–448. [Google Scholar] [CrossRef] [PubMed]
- Ryckebusch, L.; Wang, Z.; Bertrand, N.; Lin, S.C.; Chi, X.; Schwartz, R.; Zaffran, S.; Niederreither, K. Retinoic acid deficiency alters second heart field formation. Proc. Natl. Acad. Sci. USA 2008, 105, 2913–2918. [Google Scholar] [CrossRef] [PubMed]
- Sirbu, I.O.; Zhao, X.; Duester, G. Retinoic acid controls heart anteroposterior patterning by down-regulating Isl1 through the Fgf8 pathway. Dev. Dyn. 2008, 237, 1627–1635. [Google Scholar] [CrossRef] [PubMed]
- Waxman, J.S.; Keegan, B.R.; Roberts, R.W.; Poss, K.D.; Yelon, D. Hoxb5b acts downstream of retinoic acid signaling in the forelimb field to restrict heart field potential in zebrafish. Dev. Cell 2008, 15, 923–934. [Google Scholar] [CrossRef] [PubMed]
- Duester, G. Retinoic acid synthesis and signaling during early organogenesis. Cell 2008, 134, 921–931. [Google Scholar] [CrossRef] [PubMed]
- Niederreither, K.; Dolle, P. Retinoids and heart development. In Heart Development; Rosenthal, N., Harvey, R.P., Eds.; Academic Press: San Diego, CA, USA, 2008. [Google Scholar]
- Diman, N.Y.; Remacle, S.; Bertrand, N.; Picard, J.J.; Zaffran, S.; Rezsohazy, R. A retinoic acid responsive Hoxa3 transgene expressed in embryonic pharyngeal endoderm, cardiac neural crest and a subdomain of the second heart field. PLoS One 2011, 6, e27624. [Google Scholar] [CrossRef] [PubMed]
- Nolte, C.; Jinks, T.; Wang, X.; Martinez Pastor, M.T.; Krumlauf, R. Shadow enhancers flanking the HoxB cluster direct dynamic hox expression in early heart and endoderm development. Dev. Biol. 2013, 383, 158–173. [Google Scholar] [CrossRef] [PubMed]
- Ward, C.; Stadt, H.; Hutson, M.; Kirby, M.L. Ablation of the secondary heart field leads to tetralogy of fallot and pulmonary atresia. Dev. Biol. 2005, 284, 72–83. [Google Scholar] [CrossRef] [PubMed]
- Holve, S.; Friedman, B.; Hoyme, H.E.; Tarby, T.J.; Johnstone, S.J.; Erickson, R.P.; Clericuzio, C.L.; Cunniff, C. Athabascan brainstem dysgenesis syndrome. Am. J. Med. Genet. Part A 2003, 120A, 169–173. [Google Scholar] [CrossRef] [PubMed]
- Tischfield, M.A.; Bosley, T.M.; Salih, M.A.; Alorainy, I.A.; Sener, E.C.; Nester, M.J.; Oystreck, D.T.; Chan, W.M.; Andrews, C.; Erickson, R.P.; et al. Homozygous hoxa1 mutations disrupt human brainstem, inner ear, cardiovascular and cognitive development. Nat. Genet. 2005, 37, 1035–1037. [Google Scholar] [CrossRef] [PubMed]
- Makki, N.; Capecchi, M.R. Cardiovascular defects in a mouse model of hoxa1 syndrome. Hum. Mol. Genet. 2012, 21, 26–31. [Google Scholar] [CrossRef] [PubMed]
- Roux, M.; Laforest, B.; Capecchi, M.; Bertrand, N.; Zaffran, S. Hoxb1 regulates proliferation and differentiation of second heart field progenitors in pharyngeal mesoderm and genetically interacts with Hoxa1 during cardiac outflow tract development. Dev. Biol. 2015, 406, 247–258. [Google Scholar] [CrossRef] [PubMed]
- Dupays, L.; Shang, C.; Wilson, R.; Kotecha, S.; Wood, S.; Towers, N.; Mohun, T. Sequential binding of MEIS1 and NKX2–5 on the Popdc2 gene: A mechanism for spatiotemporal regulation of enhancers during cardiogenesis. Cell Rep. 2015, 13, 183–195. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez-Lazaro, M.; Rosello-Diez, A.; Delgado, I.; Carramolino, L.; Sanguino, M.A.; Giovinazzo, G.; Torres, M. Two new targeted alleles for the comprehensive analysis of Meis1 functions in the mouse. Genesis 2014, 52, 967–975. [Google Scholar] [CrossRef] [PubMed]
- Stankunas, K.; Shang, C.; Twu, K.Y.; Kao, S.C.; Jenkins, N.A.; Copeland, N.G.; Sanyal, M.; Selleri, L.; Cleary, M.L.; Chang, C.P. Pbx/Meis deficiencies demonstrate multigenetic origins of congenital heart disease. Circ. Res. 2008, 103, 702–709. [Google Scholar] [CrossRef] [PubMed]
- Lufkin, T.; Dierich, A.; LeMeur, M.; Mark, M.; Chambon, P. Disruption of the Hox-1.6 homeobox gene results in defects in a region corresponding to its rostral domain of expression. Cell 1991, 66, 1105–1119. [Google Scholar] [CrossRef]
- Godwin, A.R.; Stadler, H.S.; Nakamura, K.; Capecchi, M.R. Detection of targeted GFP-Hox gene fusions during mouse embryogenesis. Proc. Natl. Acad. Sci. USA 1998, 95, 13042–13047. [Google Scholar] [CrossRef] [PubMed]
- Chisaka, O.; Capecchi, M.R. Regionally restricted developmental defects resulting from targeted disruption of the mouse homeobox gene Hox-1.5. Nature 1991, 350, 473–479. [Google Scholar] [CrossRef] [PubMed]
- Chisaka, O.; Kameda, Y. Hoxa3 regulates the proliferation and differentiation of the third pharyngeal arch mesenchyme in mice. Cell Tissue Res. 2005, 320, 77–89. [Google Scholar] [CrossRef] [PubMed]
- Kameda, Y.; Watari-Goshima, N.; Nishimaki, T.; Chisaka, O. Disruption of the hoxa3 homeobox gene results in anomalies of the carotid artery system and the arterial baroreceptors. Cell Tissue Res. 2003, 311, 343–352. [Google Scholar] [PubMed]
- Soshnikova, N.; Dewaele, R.; Janvier, P.; Krumlauf, R.; Duboule, D. Duplications of Hox gene clusters and the emergence of vertebrates. Dev. Biol. 2013, 378, 194–199. [Google Scholar] [CrossRef] [PubMed]
- Kao, R.M.; Rurik, J.G.; Farr, G.H., 3rd; Dong, X.R.; Majesky, M.W.; Maves, L. Pbx4 is required for the temporal onset of zebrafish myocardial differentiation. J. Dev. Biol. 2015, 3, 93–111. [Google Scholar] [CrossRef] [PubMed]
- Mahmoud, A.I.; Kocabas, F.; Muralidhar, S.A.; Kimura, W.; Koura, A.S.; Thet, S.; Porrello, E.R.; Sadek, H.A. Meis1 regulates postnatal cardiomyocyte cell cycle arrest. Nature 2013, 497, 249–253. [Google Scholar] [CrossRef] [PubMed]
- Paige, S.L.; Thomas, S.; Stoick-Cooper, C.L.; Wang, H.; Maves, L.; Sandstrom, R.; Pabon, L.; Reinecke, H.; Pratt, G.; Keller, G.; et al. A temporal chromatin signature in human embryonic stem cells identifies regulators of cardiac development. Cell 2012, 151, 221–232. [Google Scholar] [CrossRef] [PubMed]
- Machon, O.; Masek, J.; Machonova, O.; Krauss, S.; Kozmik, Z. Meis2 is essential for cranial and cardiac neural crest development. BMC Dev. Biol. 2015, 15, 40. [Google Scholar] [CrossRef] [PubMed]
- Yashiro, K.; Shiratori, H.; Hamada, H. Haemodynamics determined by a genetic programme govern asymmetric development of the aortic arch. Nature 2007, 450, 285–288. [Google Scholar] [CrossRef] [PubMed]
- Le Lievre, C.S.; le Douarin, N.M. Mesenchymal derivatives of the neural crest: Analysis of chimaeric quail and chick embryos. J. Embryol Exp. Morphol. 1975, 34, 125–154. [Google Scholar] [PubMed]
- Kirby, M.L.; Waldo, K.L. Neural crest and cardiovascular patterning. Circ. Res. 1995, 77, 211–215. [Google Scholar] [CrossRef] [PubMed]
- Jiang, X.; Choudhary, B.; Merki, E.; Chien, K.R.; Maxson, R.E.; Sucov, H.M. Normal fate and altered function of the cardiac neural crest cell lineage in retinoic acid receptor mutant embryos. Mech. Dev. 2002, 117, 115–122. [Google Scholar] [CrossRef]
- Boot, M.J.; Gittenberger-De Groot, A.C.; van Iperen, L.; Hierck, B.P.; Poelmann, R.E. Spatiotemporally separated cardiac neural crest subpopulations that target the outflow tract septum and pharyngeal arch arteries. Anat. Rec. Part A Discov. Mol. Cell Evol. Biol. 2003, 275, 1009–1018. [Google Scholar] [CrossRef] [PubMed]
- Waldo, K.L.; Kumiski, D.; Kirby, M.L. Cardiac neural crest is essential for the persistence rather than the formation of an arch artery. Dev. Dyn 1996, 205, 281–292. [Google Scholar] [CrossRef]
- Hutson, M.R.; Kirby, M.L. Model systems for the study of heart development and disease cardiac neural crest and conotruncal malformations. Semin. Cell Dev. Biol. 2007, 18, 101–110. [Google Scholar] [CrossRef] [PubMed]
- Gouti, M.; Briscoe, J.; Gavalas, A. Anterior Hox genes interact with components of the neural crest specification network to induce neural crest fates. Stem Cells 2011, 29, 858–870. [Google Scholar] [CrossRef] [PubMed]
- Studer, M.; Gavalas, A.; Marshall, H.; Ariza-McNaughton, L.; Rijli, F.M.; Chambon, P.; Krumlauf, R. Genetic interactions between Hoxa1 and Hoxb1 reveal new roles in regulation of early hindbrain patterning. Development 1998, 125, 1025–1036. [Google Scholar] [PubMed]
- Bosley, T.M.; Alorainy, I.A.; Salih, M.A.; Aldhalaan, H.M.; Abu-Amero, K.K.; Oystreck, D.T.; Tischfield, M.A.; Engle, E.C.; Erickson, R.P. The clinical spectrum of homozygous hoxa1 mutations. Am. J. Med. Genet. Part A 2008, 146A, 1235–1240. [Google Scholar] [CrossRef] [PubMed]
- Krasnow, M.A.; Saffman, E.E.; Kornfeld, K.; Hogness, D.S. Transcriptional activation and repression by ultrabithorax proteins in cultured drosophila cells. Cell 1989, 57, 1031–1043. [Google Scholar] [CrossRef]
- Moens, C.B.; Selleri, L. Hox cofactors in vertebrate development. Dev. Biol. 2006, 291, 193–206. [Google Scholar] [CrossRef] [PubMed]
- Ladam, F.; Sagerstrom, C.G. Hox regulation of transcription: More complex(es). Dev. Dyn. 2014, 243, 4–15. [Google Scholar] [CrossRef] [PubMed]
- Amin, S.; Donaldson, I.J.; Zannino, D.A.; Hensman, J.; Rattray, M.; Losa, M.; Spitz, F.; Ladam, F.; Sagerstrom, C.; Bobola, N. Hoxa2 selectively enhances meis binding to change a branchial arch ground state. Dev. Cell 2015, 32, 265–277. [Google Scholar] [CrossRef] [PubMed]
- Ferretti, E.; Marshall, H.; Popperl, H.; Maconochie, M.; Krumlauf, R.; Blasi, F. Segmental expression of Hoxb2 in r4 requires two separate sites that integrate cooperative interactions between Prep1, Pbx and Hox proteins. Development 2000, 127, 155–166. [Google Scholar] [PubMed]
- Choe, S.K.; Lu, P.; Nakamura, M.; Lee, J.; Sagerstrom, C.G. Meis cofactors control HDAC and CBP accessibility at Hox-regulated promoters during zebrafish embryogenesis. Dev. Cell 2009, 17, 561–567. [Google Scholar] [CrossRef] [PubMed]
- Choe, S.K.; Ladam, F.; Sagerstrom, C.G. Tale factors poise promoters for activation by Hox proteins. Dev. Cell 2014, 28, 203–211. [Google Scholar] [CrossRef] [PubMed]
- Chang, C.P.; Stankunas, K.; Shang, C.; Kao, S.C.; Twu, K.Y.; Cleary, M.L. Pbx1 functions in distinct regulatory networks to pattern the great arteries and cardiac outflow tract. Development 2008, 135, 3577–3586. [Google Scholar] [CrossRef] [PubMed]
- Maves, L.; Tyler, A.; Moens, C.B.; Tapscott, S.J. Pbx acts with Hand2 in early myocardial differentiation. Dev. Biol. 2009, 333, 409–418. [Google Scholar] [CrossRef] [PubMed]
- Crowley, M.A.; Conlin, L.K.; Zackai, E.H.; Deardorff, M.A.; Thiel, B.D.; Spinner, N.B. Further evidence for the possible role of MEIS2 in the development of cleft palate and cardiac septum. Am. J. Med. Genet. Part A 2010, 152A, 1326–1327. [Google Scholar] [CrossRef] [PubMed]
- Louw, J.J.; Corveleyn, A.; Jia, Y.; Hens, G.; Gewillig, M.; Devriendt, K. MEIS2 involvement in cardiac development, cleft palate, and intellectual disability. Am. J. Med. Genet. Part A 2015, 167A, 1142–1146. [Google Scholar] [CrossRef] [PubMed]
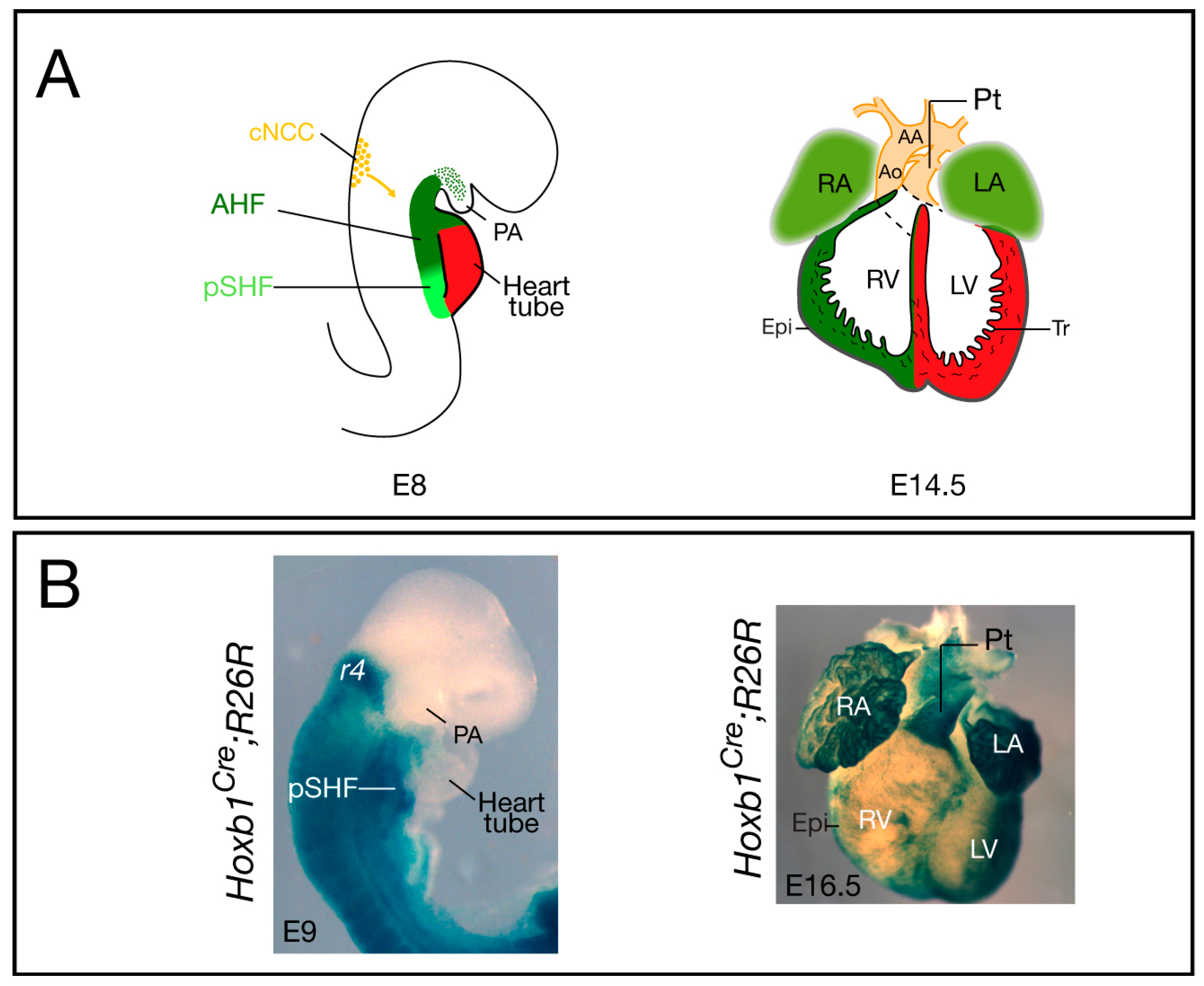

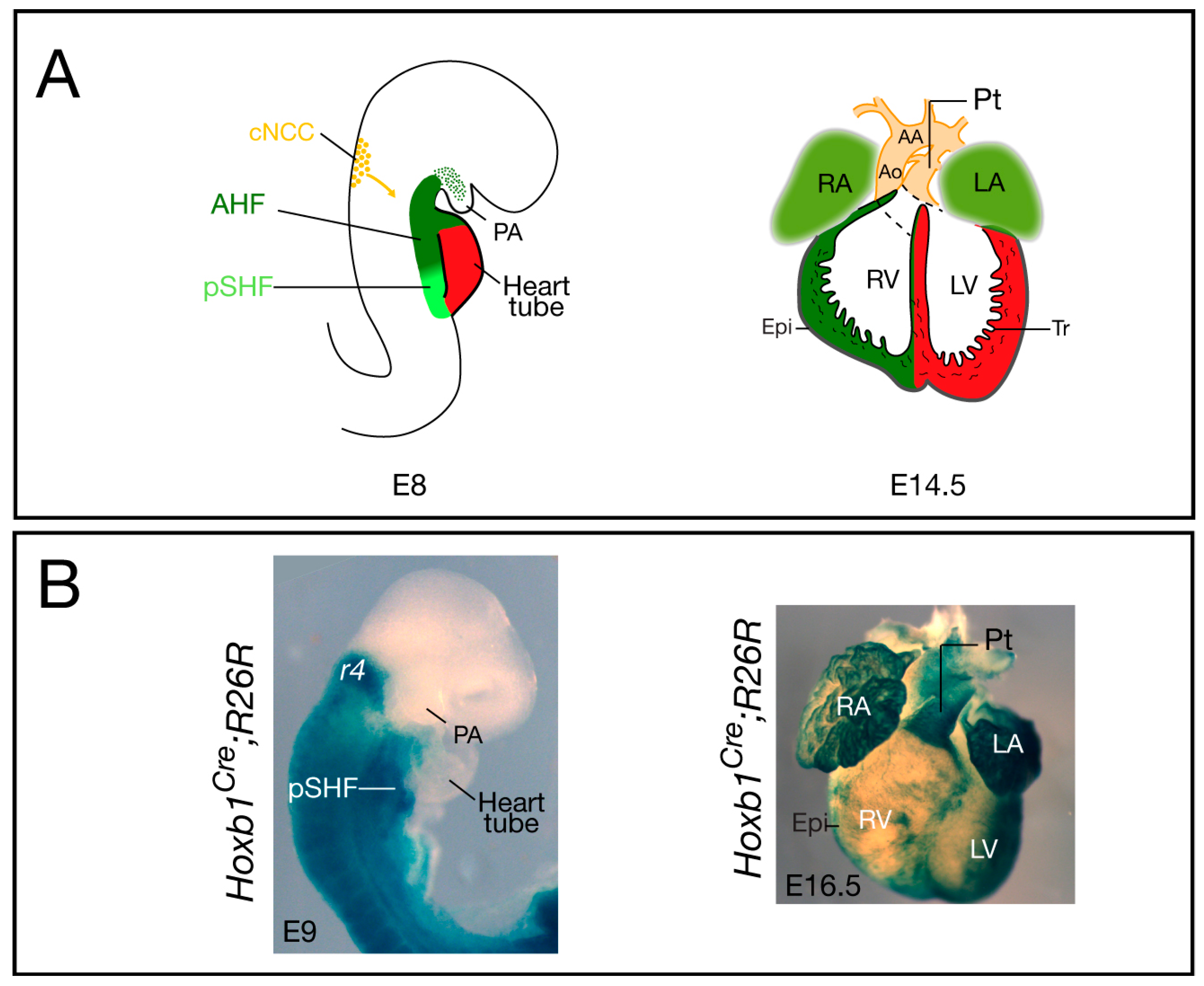{kind=link}
| Gene | Mutants | Anomalies | References |
|---|---|---|---|
| Hoxa1 | Hoxa-1.6−/− | No cardiac phenotype VSD | [48] |
| Hoxa-1.6−/−;Hoxb1GFP/+ | OFT defects, VSD | [44] | |
| Hoxa1GFPneo/GFPneo | No cardiac phenotype | [49] | |
| Hoxa1−/− | IAA-B, ASC, RAA, VSD, ToF | [43] | |
| Hoxa3 | Hox-1.5−/− | No cardiac phenotype | [50] |
| Hoxa3−/− | Degeneration of the 3rd arch artery Malformation of the carotid artery system | [51,52] | |
| Hoxb1 | Hoxb1GFP/GFP | OFT defects, VSD | [44] |
| HoxA/HoxB | Hoxa−/−;Hoxb−/− | Heart looping defects | [53] |
| Pbx1 | Pbx1−/− | Die around E15; PTA and VSD | [47] |
| Pbx1+/−;Pbx2+/−;Pbx3+/− | Bicuspid aortic valve (BAV) | [47] | |
| Pbx1+/−;Pbx2−/− | Overriding aorta, VSD, BAV, bicuspid pulmonary valve | [47] | |
| Pbx1+/−;Pbx2−/−;Pbx3+/− | ToF | [47] | |
| Pbx2 | Pbx2−/− | No cardiac phenotype | [47] |
| Pbx3 | Pbx3−/− | No cardiac phenotype | [47] |
| Pbx4 | pbx4b557−/− (zebrafish) | OFT defects | [54] |
| Meis1 | Meis1−/− | Overriding aorta, VSD | [47] |
| Meis1ECFP/ECFP | Die around E14; VSD | [46] | |
| α-MHC-Cre;Meis1f/f | Increased postnatal cardiomyocyte proliferation | [55] | |
| Meis2 | meis2b-MO (zebrafish) | Heart looping defects | [56] |
| Meis2−/− | Lethality by E13.5-E15; PTA and VSD | [57] | |
| AP2α-Cre;Meis2f/f | DORV, abnormal semilunar valves | [57] |
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Roux, M.; Zaffran, S. Hox Genes in Cardiovascular Development and Diseases. J. Dev. Biol. 2016, 4, 14. https://doi.org/10.3390/jdb4020014
Roux M, Zaffran S. Hox Genes in Cardiovascular Development and Diseases. Journal of Developmental Biology. 2016; 4(2):14. https://doi.org/10.3390/jdb4020014
Chicago/Turabian StyleRoux, Marine, and Stéphane Zaffran. 2016. "Hox Genes in Cardiovascular Development and Diseases" Journal of Developmental Biology 4, no. 2: 14. https://doi.org/10.3390/jdb4020014
APA StyleRoux, M., & Zaffran, S. (2016). Hox Genes in Cardiovascular Development and Diseases. Journal of Developmental Biology, 4(2), 14. https://doi.org/10.3390/jdb4020014