
Role of Chondrocytes in Cartilage Formation, Progression of Osteoarthritis and Cartilage Regeneration


Abstract
1. Introduction
2. Chondrocyte Function and Regulation
3. ECM Production and Its Regulation by Chondrocytes
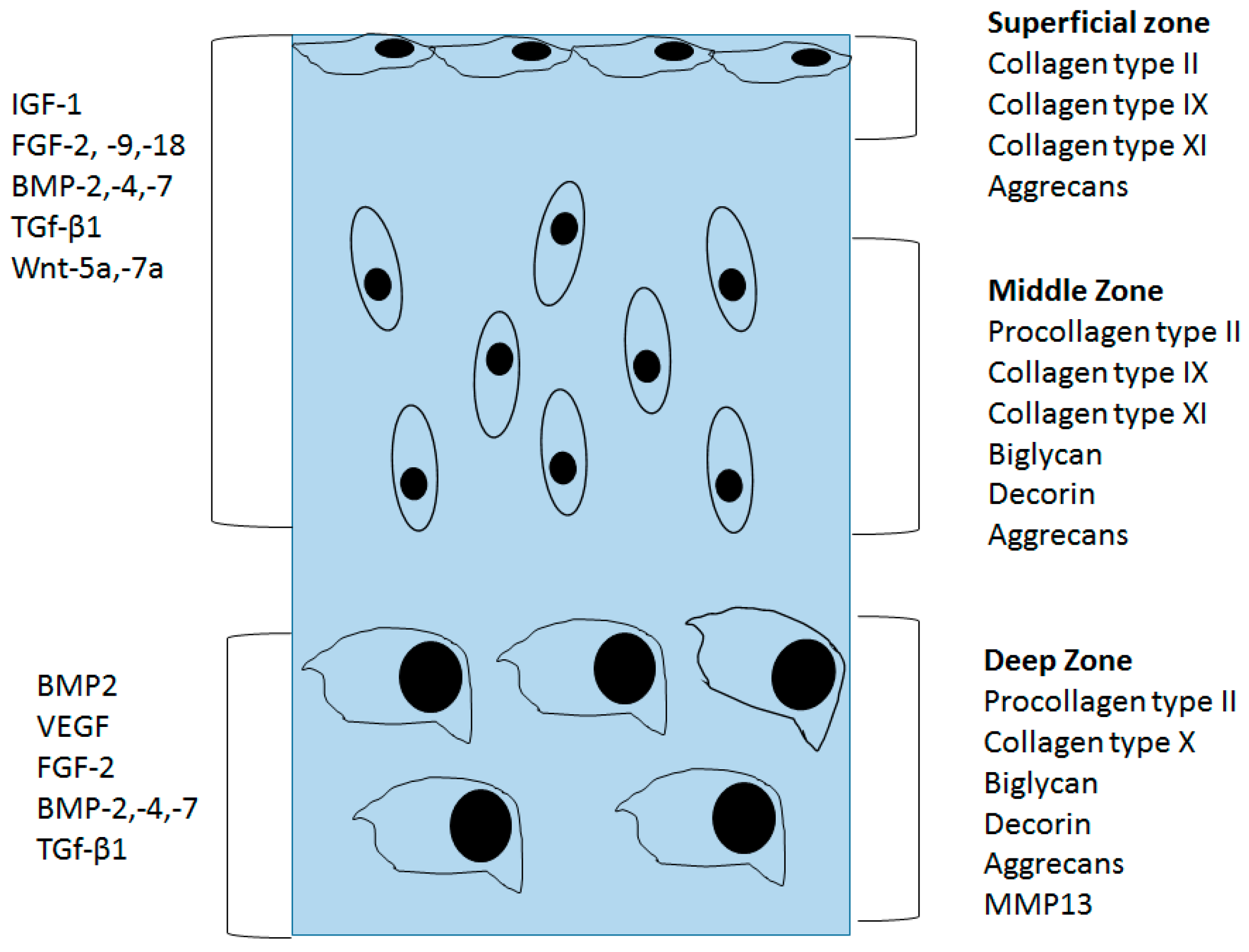
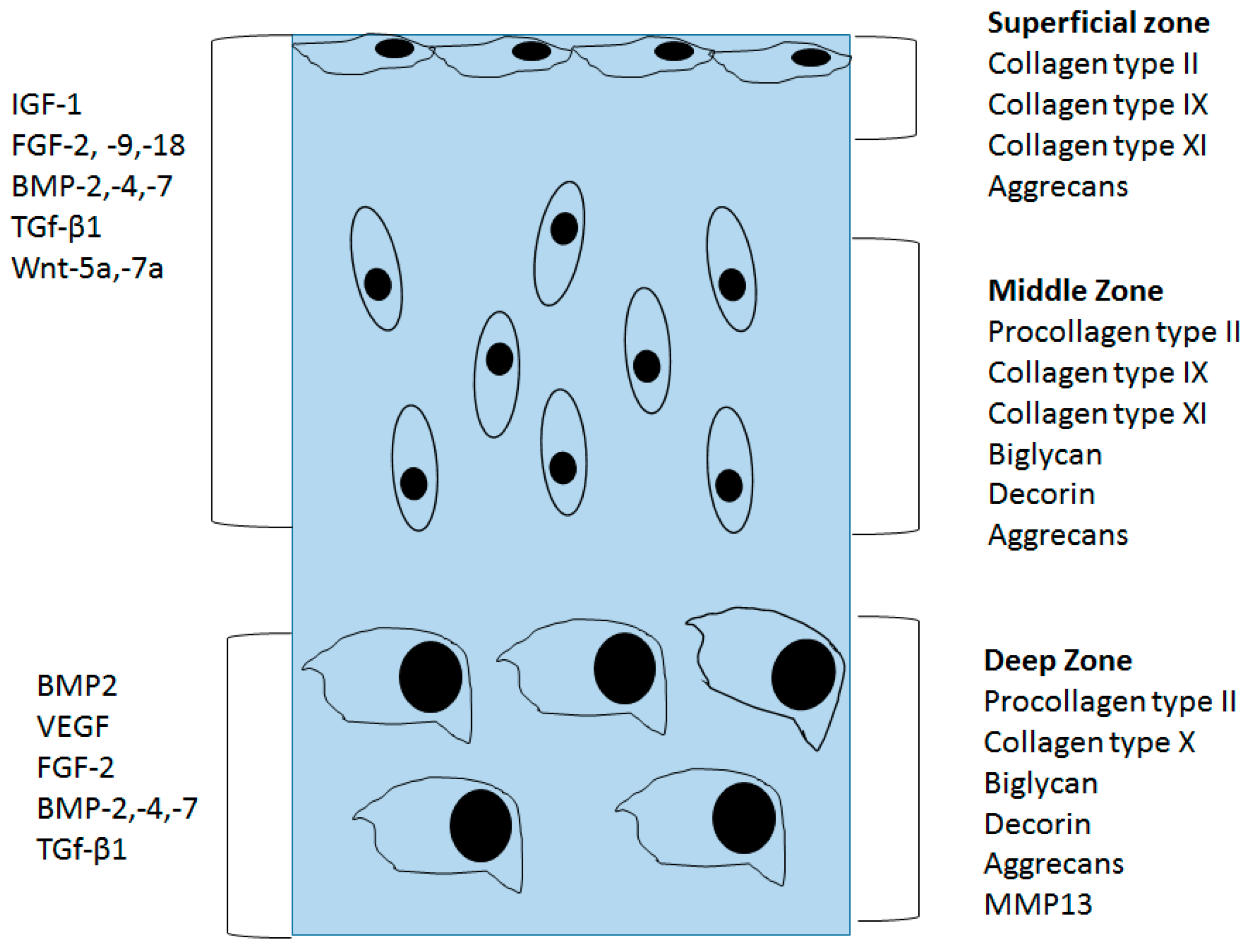
4. Structural Changes in OA Cartilage
5. OA Induced Osteophyte Formation and Fibrosis
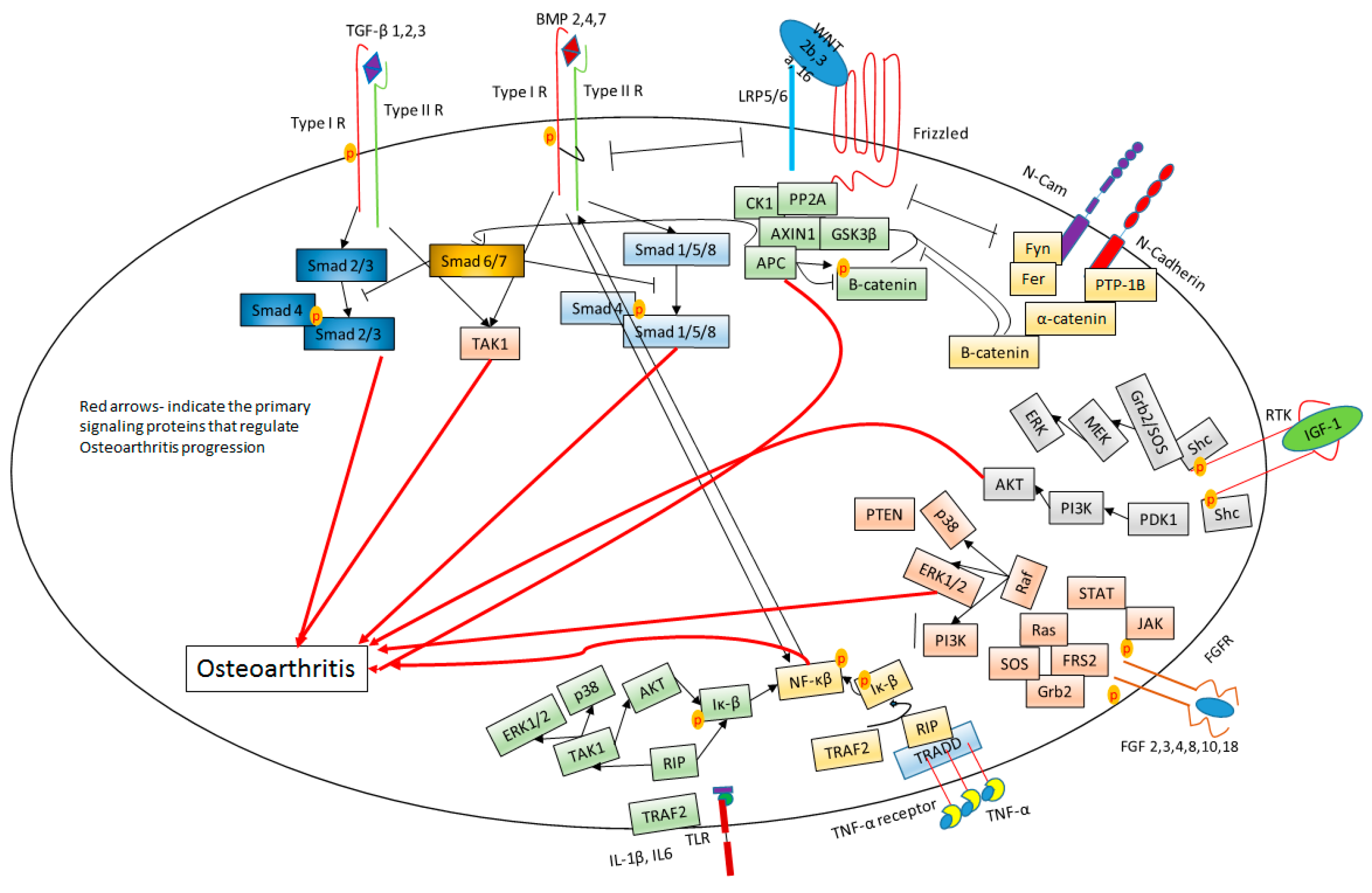
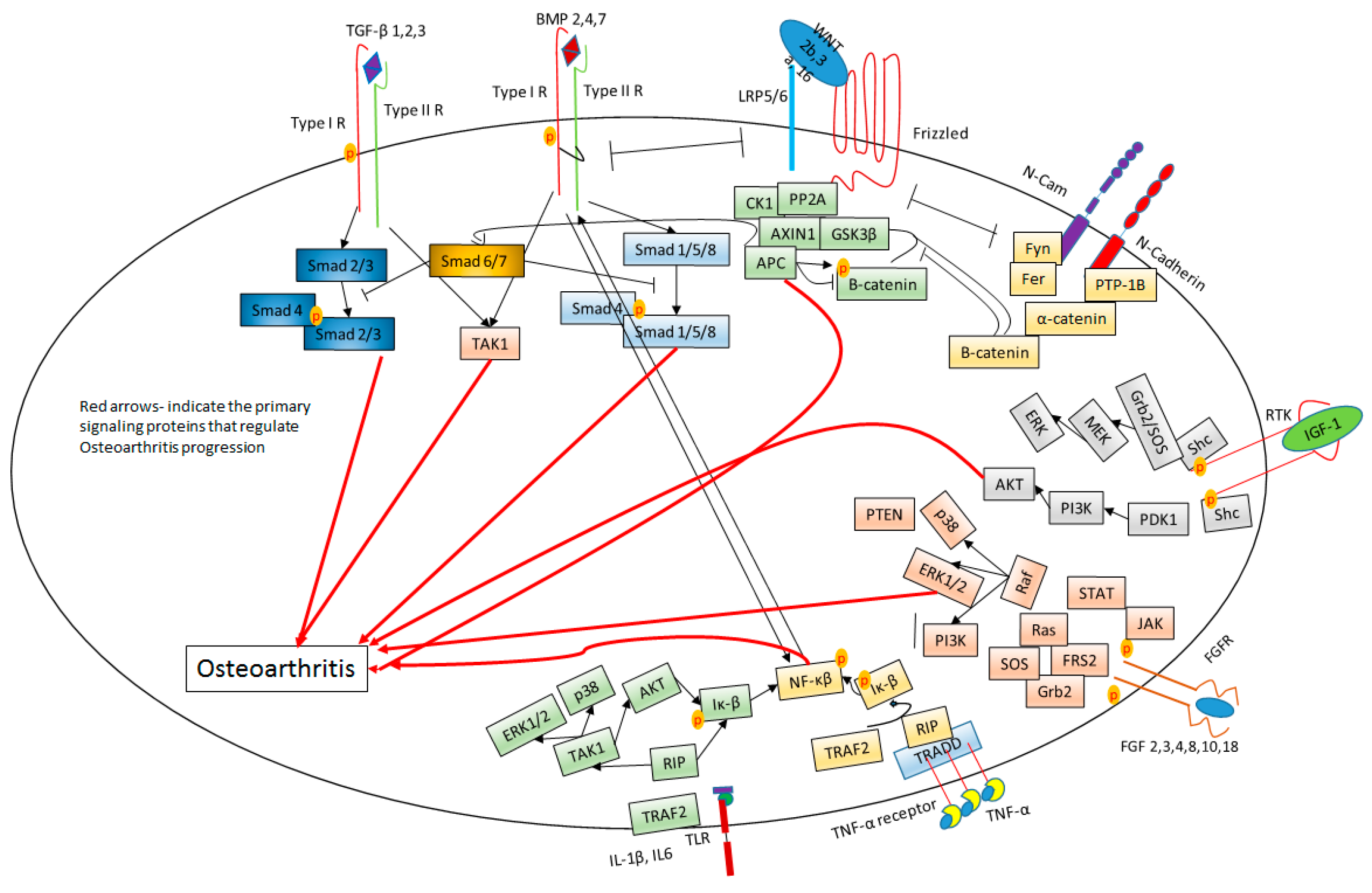
6. Signaling in OA Cartilage
7. Mesenchymal Stem Cell Niche for Cartilage Repair
8. Mesenchymal Stem Cell Therapy for OA Cartilage Repair

{kind=link}
{kind=link}
| Tissue Specific Progenitor Cells | ||||
|---|---|---|---|---|
| Tissue Source | Pros | Cons | Tissue Specific Cell Surface Markers | References |
| Bone Marrow | Can differentiate in to adipocytes, osteoblasts, and chondrocytes. High chondroprogenitor populations. High expansion potential | Heterogeneous population of cells. Invasive procedure to harvest | CD105, CD73, CD45, CD109, CD44, CD90, CD271, CD14, CD19, CD11b, CD13, CD166, CD146, CD34, SSEA-4, and HLADR | [3,86,87,94] |
| Synovium/synovial Fluid | Superior chondrogenic potential., regardless of donor age. High expansion potential | Heterogeneous populations of cells | CD105, CD73, CD44, CD90, CD271, CD13, CD166 | [3,95,96,97] |
| Infrapatellar Fat Pad | Superior source of progenitor cells for chondrogenic differentiation potential | Early senescence, differentiation capacity to other tissues | CD105, CD73, CD44, CD166, D271, CD13, CD90, CD34, CD45, CD31, VEGF-2 | [3,95,98,99] |
9. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Pearle, A.D.; Warren, R.F.; Rodeo, S.A. Basic science of articular cartilage and osteoarthritis. Clin. Sports Med. 2005, 24, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Archer, C.W.; Francis-West, P. The chondrocyte. Int. J. Biochem. Cell Biol. 2003, 35, 401–404. [Google Scholar] [CrossRef]
- O’Sullivan, J.; D’Arcy, S.; Barry, F.P.; Murphy, J.M.; Coleman, C.M. Mesenchymal chondroprogenitor cell origin and therapeutic potential. Stem Cell Res. Ther. 2011, 2, 8. [Google Scholar] [CrossRef] [PubMed]
- Kapoor, M.; Martel-Pelletier, J.; Lajeunesse, D.; Pelletier, J.P.; Fahmi, H. Role of proinflammatory cytokines in the pathophysiology of osteoarthritis. Nat. Rev. Rheumatol. 2011, 7, 33–42. [Google Scholar] [CrossRef] [PubMed]
- Fortier, L.A.; Barker, J.U.; Strauss, E.J.; McCarrel, T.M.; Cole, B.J. The role of growth factors in cartilage repair. Clin. Orthop. Relat. Res. 2011, 469, 2706–2715. [Google Scholar] [CrossRef] [PubMed]
- Goldring, M.B.; Otero, M.; Tsuchimochi, K.; Ijiri, K.; Li, Y. Defining the roles of inflammatory and anabolic cytokines in cartilage metabolism. Ann. Rheum. Dis. 2008, 67 (Suppl. 3), 75–82. [Google Scholar] [CrossRef] [PubMed]
- Bhosale, A.M.; Richardson, J.B. Articular cartilage: Structure, injuries and review of management. Br. Med. Bull. 2008, 87, 77–95. [Google Scholar] [CrossRef] [PubMed]
- Poole, C.A. Articular cartilage chondrons: Form, function and failure. J. Anat. 1997, 191, Pt 1. 1–13. [Google Scholar] [CrossRef] [PubMed]
- Sophia Fox, A.J.; Bedi, A.; Rodeo, S.A. The basic science of articular cartilage: Structure, composition, and function. Sports Health 2009, 1, 461–468. [Google Scholar] [CrossRef] [PubMed]
- Eyre, D. Collagen of articular cartilage. Arthritis Res. 2002, 4, 30–35. [Google Scholar] [CrossRef] [PubMed]
- Roughley, P.J.; Lee, E.R. Cartilage proteoglycans: Structure and potential functions. Microsc Res. Tech. 1994, 28, 385–397. [Google Scholar] [CrossRef] [PubMed]
- Poole, A.R.; Kobayashi, M.; Yasuda, T.; Laverty, S.; Mwale, F.; Kojima, T.; Sakai, T.; Wahl, C.; El-Maadawy, S.; Webb, G.; et al. Type II collagen degradation and its regulation in articular cartilage in osteoarthritis. Ann. Rheum. Dis. 2002, 61 (Suppl. 2), 78–81. [Google Scholar] [CrossRef]
- Verzijl, N.; DeGroot, J.; Thorpe, S.R.; Bank, R.A.; Shaw, J.N.; Lyons, T.J.; Bijlsma, J.W.; Lafeber, F.P.; Baynes, J.W.; TeKoppele, J.M. Effect of collagen turnover on the accumulation of advanced glycation end products. J. Biol. Chem. 2000, 275, 39027–39031. [Google Scholar] [CrossRef] [PubMed]
- Hardingham, T.; Bayliss, M. Proteoglycans of articular cartilage: Changes in aging and in joint disease. Semin. Arthritis Rheum. 1990, 20, 12–33. [Google Scholar] [CrossRef]
- Martin, J.A.; Buckwalter, J.A. Roles of articular cartilage aging and chondrocyte senescence in the pathogenesis of osteoarthritis. Iowa Orthop. J. 2001, 21, 1–7. [Google Scholar] [PubMed]
- Martin, J.A.; Buckwalter, J.A. Telomere erosion and senescence in human articular cartilage chondrocytes. J. Gerontol. A Biol. Sci. Med. Sci. 2001, 56, B172–B179. [Google Scholar] [CrossRef] [PubMed]
- Martin, J.A.; Ellerbroek, S.M.; Buckwalter, J.A. Age-related decline in chondrocyte response to insulin-like growth factor-I: The role of growth factor binding proteins. J. Orthop. Res. 1997, 15, 491–498. [Google Scholar] [CrossRef] [PubMed]
- Sharma, A.R.; Jagga, S.; Lee, S.S.; Nam, J.S. Interplay between cartilage and subchondral bone contributing to pathogenesis of osteoarthritis. Int. J. Mol. Sci. 2013, 14, 19805–19830. [Google Scholar] [CrossRef] [PubMed]
- Hwang, J.; Bae, W.C.; Shieu, W.; Lewis, C.W.; Bugbee, W.D.; Sah, R.L. Increased hydraulic conductance of human articular cartilage and subchondral bone plate with progression of osteoarthritis. Arthritis Rheum. 2008, 58, 3831–3842. [Google Scholar] [CrossRef] [PubMed]
- Sandell, L.J.; Aigner, T. Articular cartilage and changes in arthritis. An introduction: Cell biology of osteoarthritis. Arthritis Res. 2001, 3, 107–113. [Google Scholar] [CrossRef] [PubMed]
- Falah, M.; Nierenberg, G.; Soudry, M.; Hayden, M.; Volpin, G. Treatment of articular cartilage lesions of the knee. Int. Orthop. 2010, 34, 621–630. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Manner, P.A.; Horner, A.; Shum, L.; Tuan, R.S.; Nuckolls, G.H. Regulation of MMP-13 expression by RUNX2 and FGF2 in osteoarthritic cartilage. Osteoarthr. Cartil. 2004, 12, 963–973. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Q.; Zhou, G.; Morello, R.; Chen, Y.; Garcia-Rojas, X.; Lee, B. Type X collagen gene regulation by RUNX2 contributes directly to its hypertrophic chondrocyte-specific expression in vivo. J. Cell. Biol. 2003, 162, 833–842. [Google Scholar] [CrossRef] [PubMed]
- Van der Kraan, P.M.; van den Berg, W.B. Chondrocyte hypertrophy and osteoarthritis: Role in initiation and progression of cartilage degeneration? Osteoarthr. Cartil. 2012, 20, 223–232. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.J.; Lark, M.W.; Chun, L.E.; Eyre, D.R. Sites of stromelysin cleavage in collagen types II, IX, X, and XI of cartilage. J. Biol. Chem. 1991, 266, 5625–5628. [Google Scholar] [PubMed]
- Troeberg, L.; Nagase, H. Proteases involved in cartilage matrix degradation in osteoarthritis. Biochim. Biophys. Acta 2012, 1824, 133–145. [Google Scholar] [CrossRef] [PubMed]
- Kostoulas, G.; Lang, A.; Nagase, H.; Baici, A. Stimulation of angiogenesis through cathepsin B inactivation of the tissue inhibitors of matrix metalloproteinases. FEBS Lett. 1999, 455, 286–290. [Google Scholar] [CrossRef]
- Buckwalter, J.A.; Anderson, D.D.; Brown, T.D.; Tochigi, Y.; Martin, J.A. The roles of mechanical stresses in the pathogenesis of osteoarthritis: Implications for treatment of joint injuries. Cartilage 2013, 4, 286–294. [Google Scholar] [CrossRef] [PubMed]
- Blain, E.J. Mechanical regulation of matrix metalloproteinases. Front. Biosci. 2007, 12, 507–527. [Google Scholar] [CrossRef] [PubMed]
- Lin, P.M.; Chen, C.T.; Torzilli, P.A. Increased stromelysin-1 (MMP-3), proteoglycan degradation (3B3- and 7D4) and collagen damage in cyclically load-injured articular cartilage. Osteoarthr. Cartil. 2004, 12, 485–496. [Google Scholar] [CrossRef] [PubMed]
- Choy, E.H.; Isenberg, D.A.; Garrood, T.; Farrow, S.; Ioannou, Y.; Bird, H.; Cheung, N.; Williams, B.; Hazleman, B.; Price, R.; et al. Therapeutic benefit of blocking interleukin-6 activity with an anti-interleukin-6 receptor monoclonal antibody in rheumatoid arthritis: A randomized, double-blind, placebo-controlled, dose-escalation trial. Arthritis Rheum. 2002, 46, 3143–3150. [Google Scholar] [CrossRef] [PubMed]
- Caron, J.P.; Fernandes, J.C.; Martel-Pelletier, J.; Tardif, G.; Mineau, F.; Geng, C.; Pelletier, J.P. Chondroprotective effect of intraarticular injections of interleukin-1 receptor antagonist in experimental osteoarthritis. Suppression of collagenase-1 expression. Arthritis Rheum. 1996, 39, 1535–1544. [Google Scholar] [CrossRef] [PubMed]
- Van de Loo, F.A.; Joosten, L.A.; van Lent, P.L.; Arntz, O.J.; van den Berg, W.B. Role of interleukin-1, tumor necrosis factor alpha, and interleukin-6 in cartilage proteoglycan metabolism and destruction. Effect of in situ blocking in murine antigen- and zymosan-induced arthritis. Arthritis Rheum. 1995, 38, 164–172. [Google Scholar] [PubMed]
- Saito, T.; Fukai, A.; Mabuchi, A.; Ikeda, T.; Yano, F.; Ohba, S.; Nishida, N.; Akune, T.; Yoshimura, N.; Nakagawa, T.; et al. Transcriptional regulation of endochondral ossification by HIF-2alpha during skeletal growth and osteoarthritis development. Nat. Med. 2010, 16, 678–686. [Google Scholar] [CrossRef] [PubMed]
- Stannus, O.; Jones, G.; Cicuttini, F.; Parameswaran, V.; Quinn, S.; Burgess, J.; Ding, C. Circulating levels of IL-6 and TNF-α are associated with knee radiographic osteoarthritis and knee cartilage loss in older adults. Osteoarthr. Cartil. 2010, 18, 1441–1447. [Google Scholar] [CrossRef] [PubMed]
- Rigoglou, S.; Papavassiliou, A.G. The NF-κB signalling pathway in osteoarthritis. Int. J. Biochem. Cell Biol. 2013, 45, 2580–2584. [Google Scholar] [CrossRef] [PubMed]
- Wehling, N.; Palmer, G.D.; Pilapil, C.; Liu, F.; Wells, J.W.; Müller, P.E.; Evans, C.H.; Porter, R.M. Interleukin-1beta and tumor necrosis factor alpha inhibit chondrogenesis by human mesenchymal stem cells through NF-kappaB-dependent pathways. Arthritis Rheum. 2009, 60, 801–812. [Google Scholar] [CrossRef] [PubMed]
- Hashimoto, S.; Creighton-Achermann, L.; Takahashi, K.; Amiel, D.; Coutts, R.D.; Lotz, M. Development and regulation of osteophyte formation during experimental osteoarthritis. Osteoarthr. Cartil. 2002, 10, 180–187. [Google Scholar] [CrossRef] [PubMed]
- Findlay, D.M.; Atkins, G.J. Osteoblast-chondrocyte interactions in osteoarthritis. Curr. Osteoporos. Rep. 2014, 12, 127–134. [Google Scholar] [CrossRef] [PubMed]
- Remst, D.F.; Blaney Davidson, E.N.; van der Kraan, P.M. Unravelling osteoarthritis-related synovial fibrosis: A step closer to solving joint stiffness. Rheumatology 2015, 54, 1954–1963. [Google Scholar] [CrossRef] [PubMed]
- Matyas, J.R.; Sandell, L.J.; Adams, M.E. Gene expression of type II collagens in chondro-osteophytes in experimental osteoarthritis. Osteoarthr. Cartil. 1997, 5, 99–105. [Google Scholar] [CrossRef]
- Blom, A.B.; van Lent, P.L.; Holthuysen, A.E.; van der Kraan, P.M.; Roth, J.; van Rooijen, N.; van den Berg, W.B. Synovial lining macrophages mediate osteophyte formation during experimental osteoarthritis. Osteoarthr. Cartil. 2004, 12, 627–635. [Google Scholar] [CrossRef] [PubMed]
- Gelse, K.; Söder, S.; Eger, W.; Diemtar, T.; Aigner, T. Osteophyte development—Molecular characterization of differentiation stages. Osteoarthr. Cartil. 2003, 11, 141–148. [Google Scholar] [CrossRef] [PubMed]
- Wong, S.P.; Rowley, J.E.; Redpath, A.N.; Tilman, J.D.; Fellous, T.G.; Johnson, J.R. Pericytes, mesenchymal stem cells and their contributions to tissue repair. Pharmacol. Ther. 2015, 151, 107–120. [Google Scholar] [CrossRef] [PubMed]
- Van Beuningen, H.M.; Glansbeek, H.L.; van der Kraan, P.M.; van den Berg, W.B. Differential effects of local application of BMP-2 or TGF-beta 1 on both articular cartilage composition and osteophyte formation. Osteoarthr. Cartil. 1998, 6, 306–317. [Google Scholar] [CrossRef] [PubMed]
- Uchino, M.; Izumi, T.; Tominaga, T.; Wakita, R.; Minehara, H.; Sekiguchi, M.; Itoman, M. Growth factor expression in the osteophytes of the human femoral head in osteoarthritis. Clin. Orthop. Relat. Res. 2000, 377, 119–125. [Google Scholar] [CrossRef] [PubMed]
- Cutolo, M.; Sulli, A.; Barone, A.; Seriolo, B.; Accardo, S. Macrophages, synovial tissue and rheumatoid arthritis. Clin. Exp. Rheumatol. 1993, 11, 331–339. [Google Scholar] [PubMed]
- Champagne, C.M.; Takebe, J.; Offenbacher, S.; Cooper, L.F. Macrophage cell lines produce osteoinductive signals that include bone morphogenetic protein-2. Bone 2002, 30, 26–31. [Google Scholar] [CrossRef]
- Aigner, T.; Dietz, U.; Stöss, H.; von der Mark, K. Differential expression of collagen types I, II, III, and X in human osteophytes. Lab. Invest. 1995, 73, 236–243. [Google Scholar] [PubMed]
- Leask, A.; Denton, C.P.; Abraham, D.J. Insights into the molecular mechanism of chronic fibrosis: The role of connective tissue growth factor in scleroderma. J. Invest. Dermatol. 2004, 122, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Leask, A.; Abraham, D.J. TGF-beta signaling and the fibrotic response. FASEB J. 2004, 18, 816–827. [Google Scholar] [CrossRef] [PubMed]
- Shi, S.; Mercer, S.; Eckert, G.J.; Trippel, S.B. Growth factor regulation of growth factors in articular chondrocytes. J. Biol. Chem. 2009, 284, 6697–6704. [Google Scholar] [CrossRef] [PubMed]
- Mariani, E.; Pulsatelli, L.; Facchini, A. Signaling pathways in cartilage repair. Int. J. Mol. Sci. 2014, 15, 8667–8698. [Google Scholar] [CrossRef] [PubMed]
- Yan, D.; Chen, D.; Cool, S.M.; van Wijnen, A.J.; Mikecz, K.; Murphy, G.; Im, H.J. Fibroblast growth factor receptor 1 is principally responsible for fibroblast growth factor 2-induced catabolic activities in human articular chondrocytes. Arthritis Res. Ther. 2011, 13, R130. [Google Scholar] [CrossRef] [PubMed]
- Shintani, N.; Siebenrock, K.A.; Hunziker, E.B. Tgf-β1 enhances the BMP-2-induced chondrogenesis of bovine synovial explants and arrests downstream differentiation at an early stage of hypertrophy. PLoS ONE 2013, 8, e53086. [Google Scholar] [CrossRef] [PubMed]
- Blaney Davidson, E.N.; Vitters, E.L.; van Lent, P.L.; van de Loo, F.A.; van den Berg, W.B.; van der Kraan, P.M. Elevated extracellular matrix production and degradation upon bone morphogenetic protein-2 (BMP-2) stimulation point toward a role for BMP-2 in cartilage repair and remodeling. Arthritis Res. Ther. 2007, 9, R102. [Google Scholar] [CrossRef] [PubMed]
- Papathanasiou, I.; Malizos, K.N.; Tsezou, A. Bone morphogenetic protein-2-induced Wnt/β-catenin signaling pathway activation through enhanced low-density-lipoprotein receptor-related protein 5 catabolic activity contributes to hypertrophy in osteoarthritic chondrocytes. Arthritis Res. Ther. 2012, 14, R82. [Google Scholar] [CrossRef] [PubMed]
- Goldring, M.B.; Tsuchimochi, K.; Ijiri, K. The control of chondrogenesis. J. Cell. Biochem. 2006, 97, 33–44. [Google Scholar] [CrossRef] [PubMed]
- Olivotto, E.; Otero, M.; Marcu, K.B.; Goldring, M.B. Pathophysiology of osteoarthritis: Canonical NF-kappaB/IKKbeta-dependent and kinase-independent effects of IKKalpha in cartilage degradation and chondrocyte differentiation. RMD Open 2015, 1, e000061. [Google Scholar] [CrossRef] [PubMed]
- Van der Kraan, P.M.; Blaney Davidson, E.N.; van den Berg, W.B. Bone morphogenetic proteins and articular cartilage: To serve and protect or a wolf in sheep clothing’s? Osteoarthr. Cartil. 2010, 18, 735–741. [Google Scholar] [CrossRef] [PubMed]
- Retting, K.N.; Song, B.; Yoon, B.S.; Lyons, K.M. BMP canonical smad signaling through Smad1 and Smad5 is required for endochondral bone formation. Development 2009, 136, 1093–1104. [Google Scholar] [CrossRef] [PubMed]
- Nakase, T.; Miyaji, T.; Tomita, T.; Kaneko, M.; Kuriyama, K.; Myoui, A.; Sugamoto, K.; Ochi, T.; Yoshikawa, H. Localization of bone morphogenetic protein-2 in human osteoarthritic cartilage and osteophyte. Osteoarthr. Cartil. 2003, 11, 278–284. [Google Scholar] [CrossRef]
- Zoricic, S.; Maric, I.; Bobinac, D.; Vukicevic, S. Expression of bone morphogenetic proteins and cartilage-derived morphogenetic proteins during osteophyte formation in humans. J. Anat. 2003, 202, 269–277. [Google Scholar] [CrossRef] [PubMed]
- Chan, B.Y.; Little, C.B. The interaction of canonical bone morphogenetic protein- and Wnt-signaling pathways may play an important role in regulating cartilage degradation in osteoarthritis. Arthritis Res. Ther. 2012, 14, 119. [Google Scholar] [CrossRef] [PubMed]
- Van der Kraan, P.M.; Blaney Davidson, E.N.; Blom, A.; van den Berg, W.B. TGF-beta signaling in chondrocyte terminal differentiation and osteoarthritis: Modulation and integration of signaling pathways through receptor-smads. Osteoarthr. Cartil. 2009, 17, 1539–1545. [Google Scholar] [CrossRef] [PubMed]
- Blom, A.B.; Brockbank, S.M.; van Lent, P.L.; van Beuningen, H.M.; Geurts, J.; Takahashi, N.; van der Kraan, P.M.; van de Loo, F.A.; Schreurs, B.W.; Clements, K.; et al. Involvement of the Wnt signaling pathway in experimental and human osteoarthritis: Prominent role of Wnt-induced signaling protein 1. Arthritis Rheum. 2009, 60, 501–512. [Google Scholar] [CrossRef] [PubMed]
- Hall, B.K.; Miyake, T. All for one and one for all: Condensations and the initiation of skeletal development. Bioessays 2000, 22, 138–147. [Google Scholar] [CrossRef]
- DeLise, A.M.; Fischer, L.; Tuan, R.S. Cellular interactions and signaling in cartilage development. Osteoarthr. Cartil. 2000, 8, 309–334. [Google Scholar] [CrossRef] [PubMed]
- Tuan, R.S. Cellular signaling in developmental chondrogenesis: N-cadherin, Wnts, and BMP-2. J. Bone Joint Surg. Am. 2003, 85-A (Suppl. 2), 137–141. [Google Scholar]
- Price, J.S.; Waters, J.G.; Darrah, C.; Pennington, C.; Edwards, D.R.; Donell, S.T.; Clark, I.M. The role of chondrocyte senescence in osteoarthritis. Aging Cell 2002, 1, 57–65. [Google Scholar] [CrossRef] [PubMed]
- Maroudas, A.; Bannon, C. Measurement of swelling pressure in cartilage and comparison with the osmotic pressure of constituent proteoglycans. Biorheology 1981, 18, 619–632. [Google Scholar] [PubMed]
- Urban, J.P.; Maroudas, A.; Bayliss, M.T.; Dillon, J. Swelling pressures of proteoglycans at the concentrations found in cartilaginous tissues. Biorheology 1979, 16, 447–464. [Google Scholar] [PubMed]
- Morales, T.I. Chondrocyte moves: Clever strategies? Osteoarthr. Cartil. 2007, 15, 861–871. [Google Scholar] [CrossRef] [PubMed]
- Kouri, J.B.; Jiménez, S.A.; Quintero, M.; Chico, A. Ultrastructural study of chondrocytes from fibrillated and non-fibrillated human osteoarthritic cartilage. Osteoarthr. Cartil. 1996, 4, 111–125. [Google Scholar] [CrossRef]
- Jones, E.A.; Crawford, A.; English, A.; Henshaw, K.; Mundy, J.; Corscadden, D.; Chapman, T.; Emery, P.; Hatton, P.; McGonagle, D. Synovial fluid mesenchymal stem cells in health and early osteoarthritis: Detection and functional evaluation at the single-cell level. Arthritis Rheum. 2008, 58, 1731–1740. [Google Scholar] [CrossRef] [PubMed]
- Harvanová, D.; Tóthová, T.; Sarišský, M.; Amrichová, J.; Rosocha, J. Isolation and characterization of synovial mesenchymal stem cells. Folia Biol. 2011, 57, 119–124. [Google Scholar]
- Iwata, H.; Ono, S.; Sato, K.; Sato, T.; Kawamura, M. Bone morphogenetic protein-induced muscle- and synovium-derived cartilage differentiation in vitro. Clin. Orthop. Relat. Res. 1993, 296, 295–300. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Muneta, T.; Morito, T.; Mochizuki, T.; Sekiya, I. Autologous synovial fluid enhances migration of mesenchymal stem cells from synovium of osteoarthritis patients in tissue culture system. J. Orthop. Res. 2008, 26, 1413–1418. [Google Scholar] [CrossRef] [PubMed]
- Karlsson, C.; Thornemo, M.; Henriksson, H.B.; Lindahl, A. Identification of a stem cell niche in the zone of ranvier within the knee joint. J. Anat. 2009, 215, 355–363. [Google Scholar] [CrossRef] [PubMed]
- Dragoo, J.L.; Samimi, B.; Zhu, M.; Hame, S.L.; Thomas, B.J.; Lieberman, J.R.; Hedrick, M.H.; Benhaim, P. Tissue-engineered cartilage and bone using stem cells from human infrapatellar fat pads. J. Bone Joint Surg. Br. 2003, 85, 740–747. [Google Scholar] [PubMed]
- De Bari, C.; Dell’Accio, F.; Tylzanowski, P.; Luyten, F.P. Multipotent mesenchymal stem cells from adult human synovial membrane. Arthritis Rheum. 2001, 44, 1928–1942. [Google Scholar] [CrossRef]
- Candela, M.E.; Yasuhara, R.; Iwamoto, M.; Enomoto-Iwamoto, M. Resident mesenchymal progenitors of articular cartilage. Matrix Biol. 2014, 39, 44–49. [Google Scholar] [CrossRef] [PubMed]
- Yoshimura, H.; Muneta, T.; Nimura, A.; Yokoyama, A.; Koga, H.; Sekiya, I. Comparison of rat mesenchymal stem cells derived from bone marrow, synovium, periosteum, adipose tissue, and muscle. Cell Tissue Res. 2007, 327, 449–462. [Google Scholar] [CrossRef] [PubMed]
- Grogan, S.P.; Miyaki, S.; Asahara, H.; D’Lima, D.D.; Lotz, M.K. Mesenchymal progenitor cell markers in human articular cartilage: Normal distribution and changes in osteoarthritis. Arthritis Res. Ther. 2009, 11, R85. [Google Scholar] [CrossRef] [PubMed]
- Hass, R.; Kasper, C.; Böhm, S.; Jacobs, R. Different populations and sources of human mesenchymal stem cells (MSC): A comparison of adult and neonatal tissue-derived MSC. Cell. Commun. Signal. 2011, 9, 12. [Google Scholar] [CrossRef] [PubMed]
- Dominici, M.; le Blanc, K.; Mueller, I.; Slaper-Cortenbach, I.; Marini, F.; Krause, D.; Deans, R.; Keating, A.; Prockop, D.; Horwitz, E. Minimal criteria for defining multipotent mesenchymal stromal cells. The international society for cellular therapy position statement. Cytotherapy 2006, 8, 315–317. [Google Scholar] [CrossRef] [PubMed]
- Gupta, P.K.; Das, A.K.; Chullikana, A.; Majumdar, A.S. Mesenchymal stem cells for cartilage repair in osteoarthritis. Stem Cell Res. Ther. 2012, 3, 25. [Google Scholar] [CrossRef] [PubMed]
- Prockop, D.J. Marrow stromal cells as stem cells for nonhematopoietic tissues. Science 1997, 276, 71–74. [Google Scholar] [CrossRef] [PubMed]
- Pittenger, M.F.; Mackay, A.M.; Beck, S.C.; Jaiswal, R.K.; Douglas, R.; Mosca, J.D.; Moorman, M.A.; Simonetti, D.W.; Craig, S.; Marshak, D.R. Multilineage potential of adult human mesenchymal stem cells. Science 1999, 284, 143–147. [Google Scholar] [CrossRef] [PubMed]
- Nejadnik, H.; Hui, J.H.; Feng Choong, E.P.; Tai, B.C.; Lee, E.H. Autologous bone marrow-derived mesenchymal stem cells versus autologous chondrocyte implantation: An observational cohort study. Am. J. Sports Med. 2010, 38, 1110–1116. [Google Scholar] [CrossRef] [PubMed]
- Richardson, S.M.; Curran, J.M.; Chen, R.; Vaughan-Thomas, A.; Hunt, J.A.; Freemont, A.J.; Hoyland, J.A. The differentiation of bone marrow mesenchymal stem cells into chondrocyte-like cells on poly-l-lactic acid (PLLA) scaffolds. Biomaterials 2006, 27, 4069–4078. [Google Scholar] [CrossRef] [PubMed]
- Connelly, J.T.; Wilson, C.G.; Levenston, M.E. Characterization of proteoglycan production and processing by chondrocytes and bmscs in tissue engineered constructs. Osteoarthr. Cartil. 2008, 16, 1092–1100. [Google Scholar] [CrossRef] [PubMed]
- Farrar, C.A.; Kupiec-Weglinski, J.W.; Sacks, S.H. The innate immune system and transplantation. Cold Spring Harb. Perspect. Med. 2013, 3, a015479. [Google Scholar] [CrossRef] [PubMed]
- Boxall, S.A.; Jones, E. Markers for characterization of bone marrow multipotential stromal cells. Stem Cells Int. 2012, 2012, 975871. [Google Scholar] [CrossRef] [PubMed]
- Peng, L.; Jia, Z.; Yin, X.; Zhang, X.; Liu, Y.; Chen, P.; Ma, K.; Zhou, C. Comparative analysis of mesenchymal stem cells from bone marrow, cartilage, and adipose tissue. Stem Cells Dev. 2008, 17, 761–773. [Google Scholar] [CrossRef] [PubMed]
- De Sousa, E.B.; Casado, P.L.; Moura Neto, V.; Duarte, M.E.; Aguiar, D.P. Synovial fluid and synovial membrane mesenchymal stem cells: Latest discoveries and therapeutic perspectives. Stem Cell Res. Ther. 2014, 5, 112. [Google Scholar] [CrossRef] [PubMed]
- Sakaguchi, Y.; Sekiya, I.; Yagishita, K.; Muneta, T. Comparison of human stem cells derived from various mesenchymal tissues: Superiority of synovium as a cell source. Arthritis Rheum. 2005, 52, 2521–2529. [Google Scholar] [CrossRef] [PubMed]
- Jones, E.A.; Kinsey, S.E.; English, A.; Jones, R.A.; Straszynski, L.; Meredith, D.M.; Markham, A.F.; Jack, A.; Emery, P.; McGonagle, D. Isolation and characterization of bone marrow multipotential mesenchymal progenitor cells. Arthritis Rheum. 2002, 46, 3349–3360. [Google Scholar] [CrossRef] [PubMed]
- Khan, W.S.; Tew, S.R.; Adesida, A.B.; Hardingham, T.E. Human infrapatellar fat pad-derived stem cells express the pericyte marker 3G5 and show enhanced chondrogenesis after expansion in fibroblast growth factor-2. Arthritis Res. Ther. 2008, 10, R74. [Google Scholar] [CrossRef] [PubMed]
- Chevalier, X. Intraarticular treatments for osteoarthritis: New perspectives. Curr. Drug Targets 2010, 11, 546–560. [Google Scholar] [CrossRef] [PubMed]
- Buckwalter, J.A.; Saltzman, C.; Brown, T. The impact of osteoarthritis: Implications for research. Clin. Orthop. Relat. Res. 2004, 427, S6–S15. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Akkiraju, H.; Nohe, A. Role of Chondrocytes in Cartilage Formation, Progression of Osteoarthritis and Cartilage Regeneration. J. Dev. Biol. 2015, 3, 177-192. https://doi.org/10.3390/jdb3040177
Akkiraju H, Nohe A. Role of Chondrocytes in Cartilage Formation, Progression of Osteoarthritis and Cartilage Regeneration. Journal of Developmental Biology. 2015; 3(4):177-192. https://doi.org/10.3390/jdb3040177
Chicago/Turabian StyleAkkiraju, Hemanth, and Anja Nohe. 2015. "Role of Chondrocytes in Cartilage Formation, Progression of Osteoarthritis and Cartilage Regeneration" Journal of Developmental Biology 3, no. 4: 177-192. https://doi.org/10.3390/jdb3040177
APA StyleAkkiraju, H., & Nohe, A. (2015). Role of Chondrocytes in Cartilage Formation, Progression of Osteoarthritis and Cartilage Regeneration. Journal of Developmental Biology, 3(4), 177-192. https://doi.org/10.3390/jdb3040177