

Zebrafish Model of Stickler Syndrome Suggests a Role for Col2a1a in the Neural Crest during Early Eye Development


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Animal Husbandry and Animal Strains
2.2. Imaging
2.3. Pharmacological Treatments
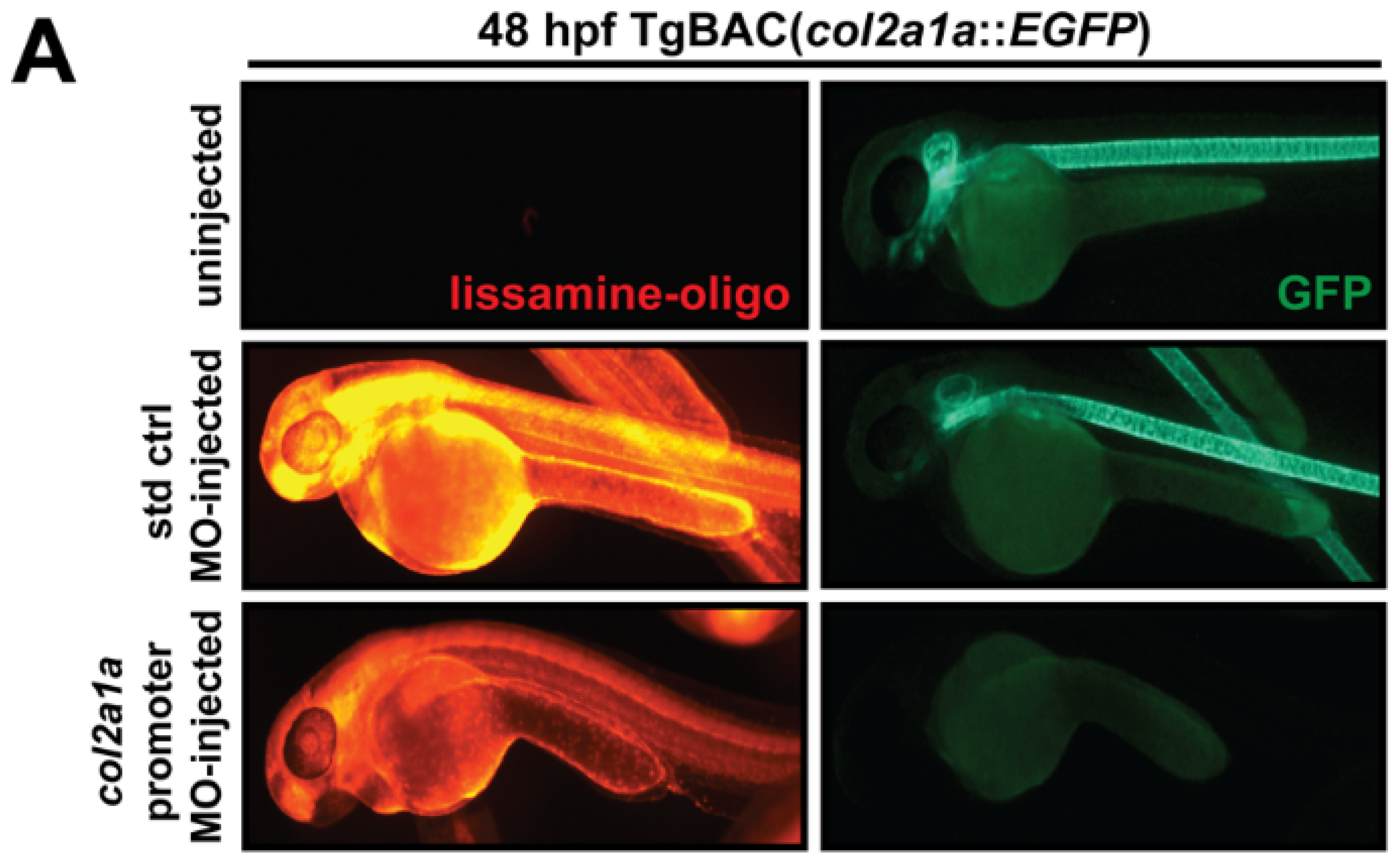
2.4. Morpholino Oligonucleotide Injections
2.5. Wholemount In Situ Hybridization and Immunostaining
3. Results
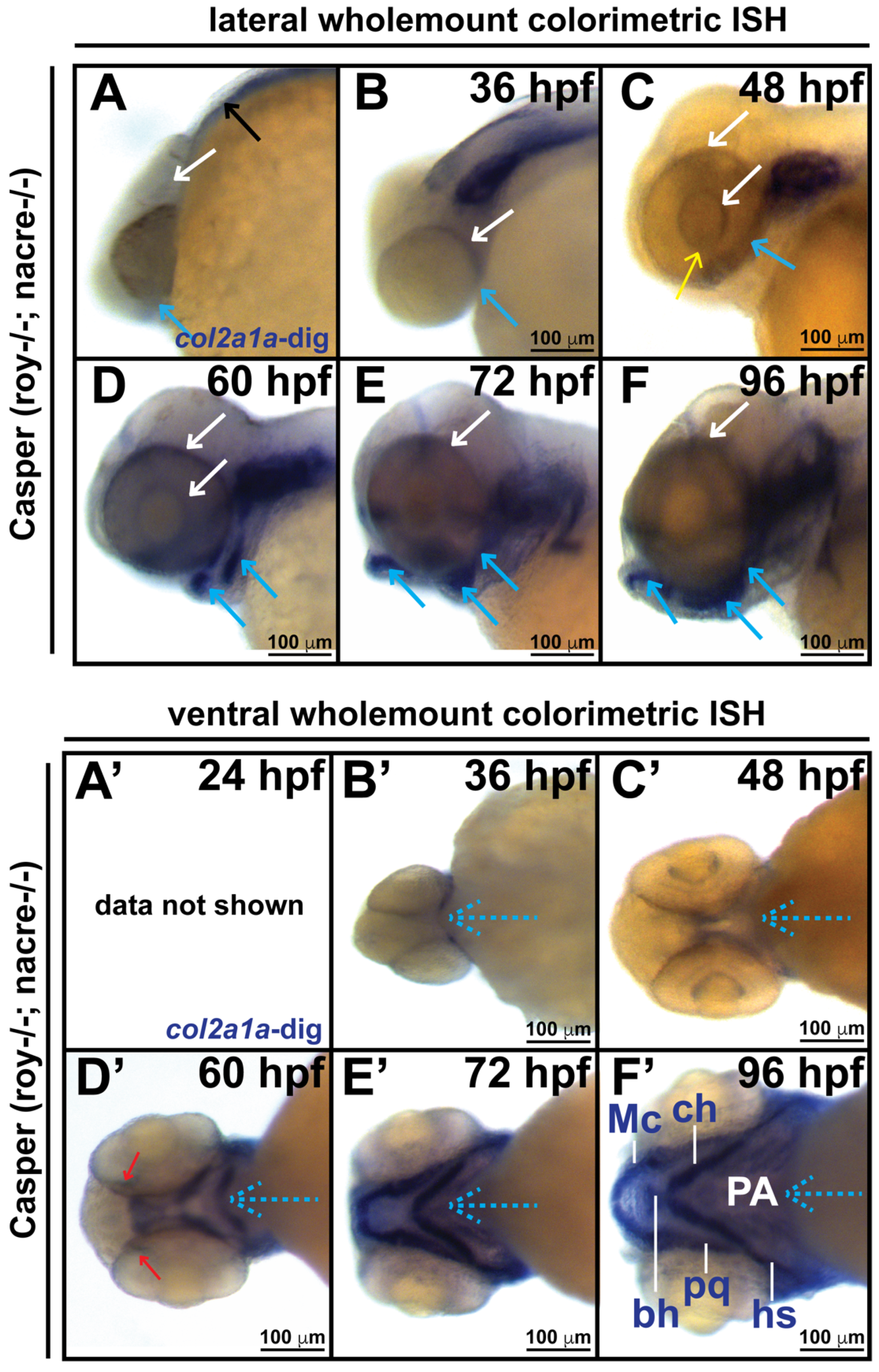
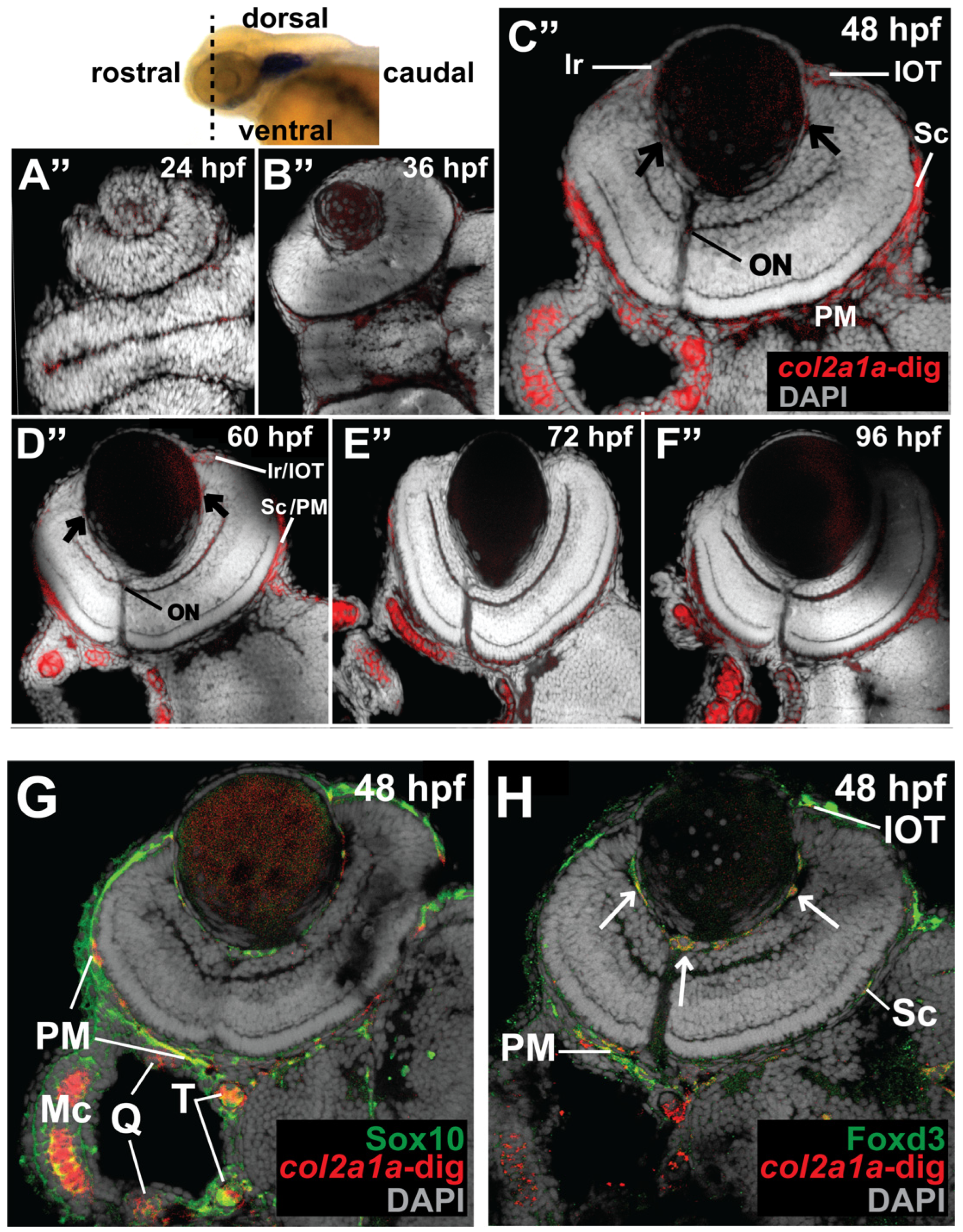
3.1. Col2a1a Is Expressed in the Ocular Neural Crest during Early Development
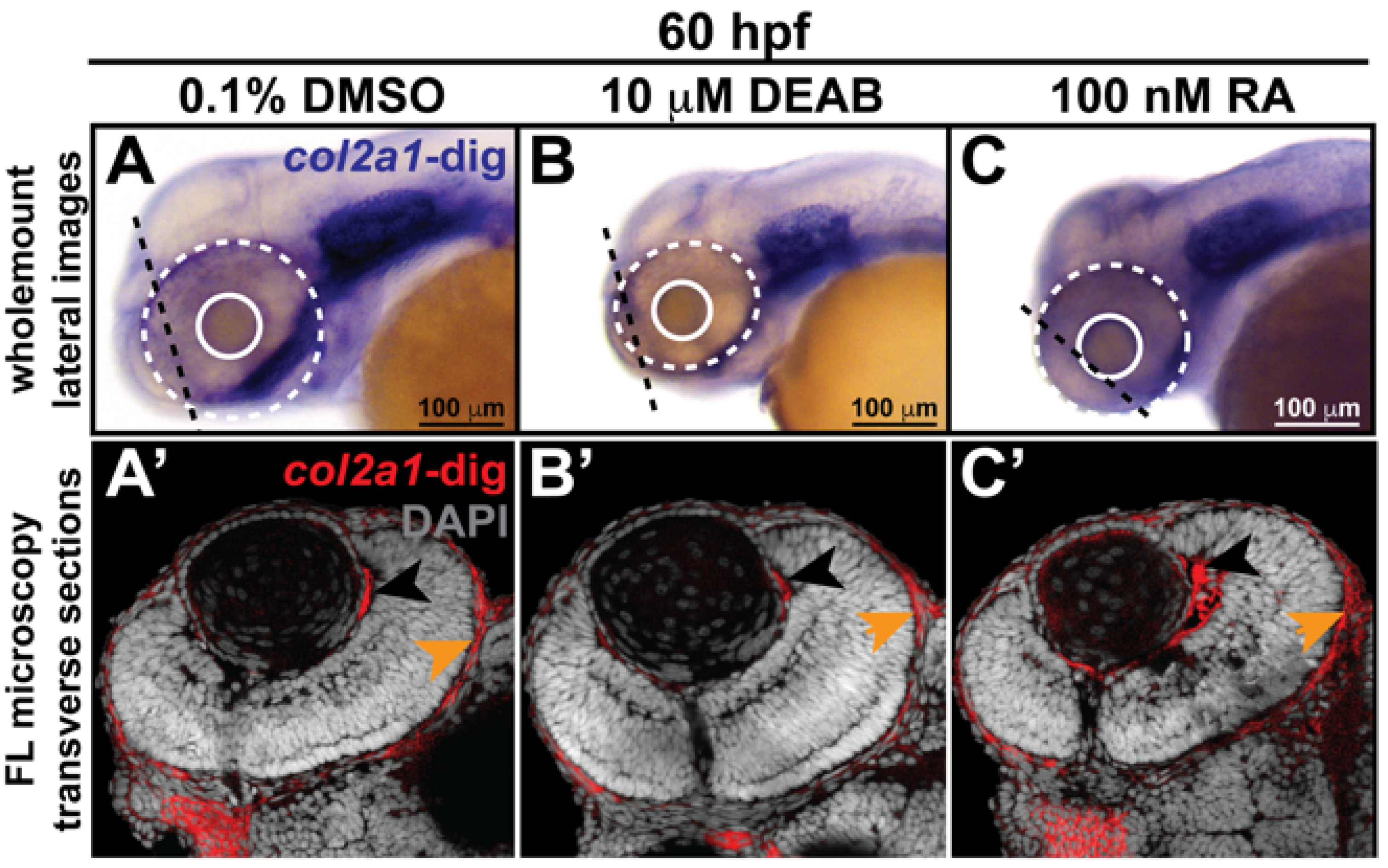
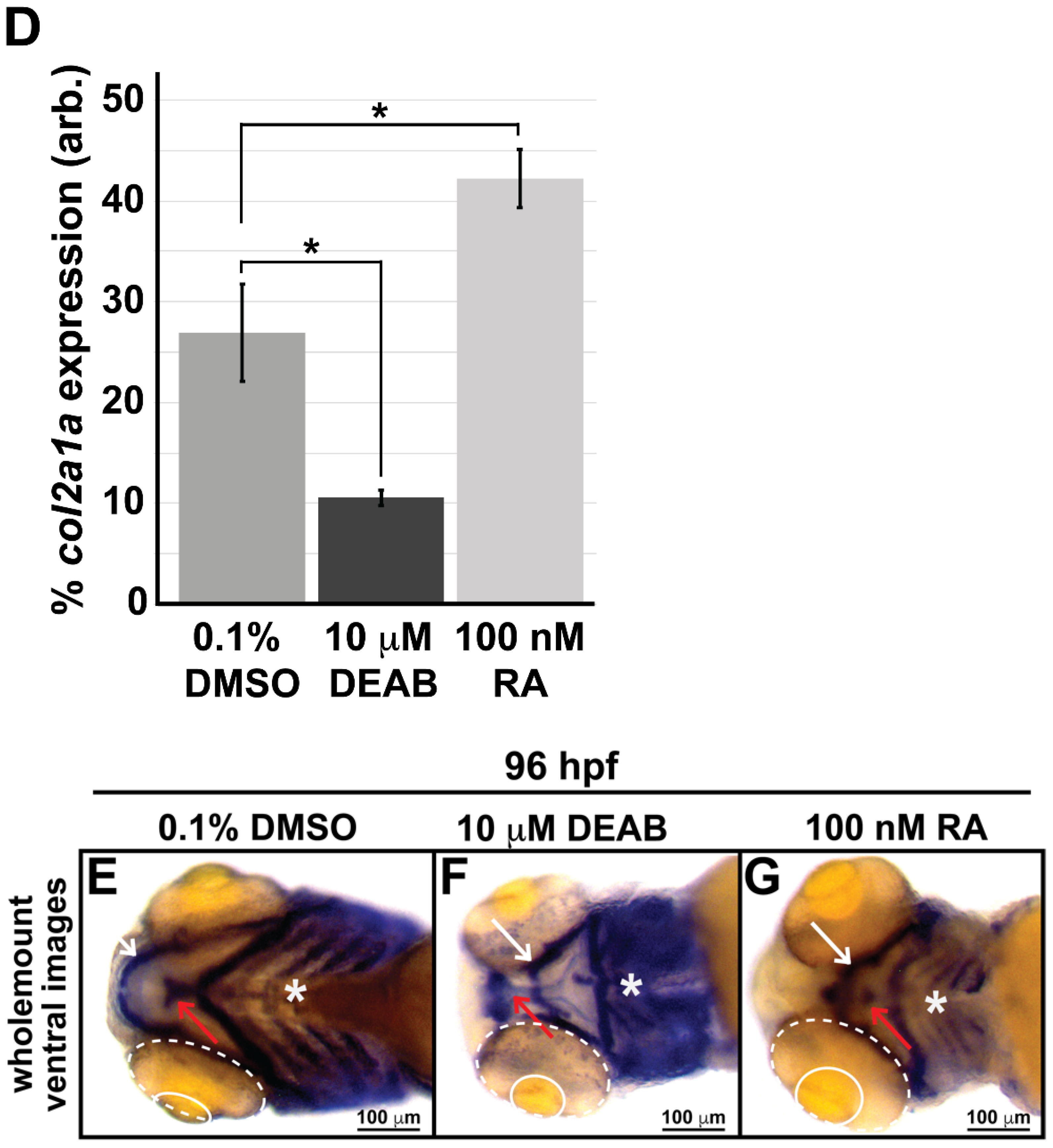
3.2. Retinoic Acid Regulates Col2a1a Expression in the Ocular Neural Crest
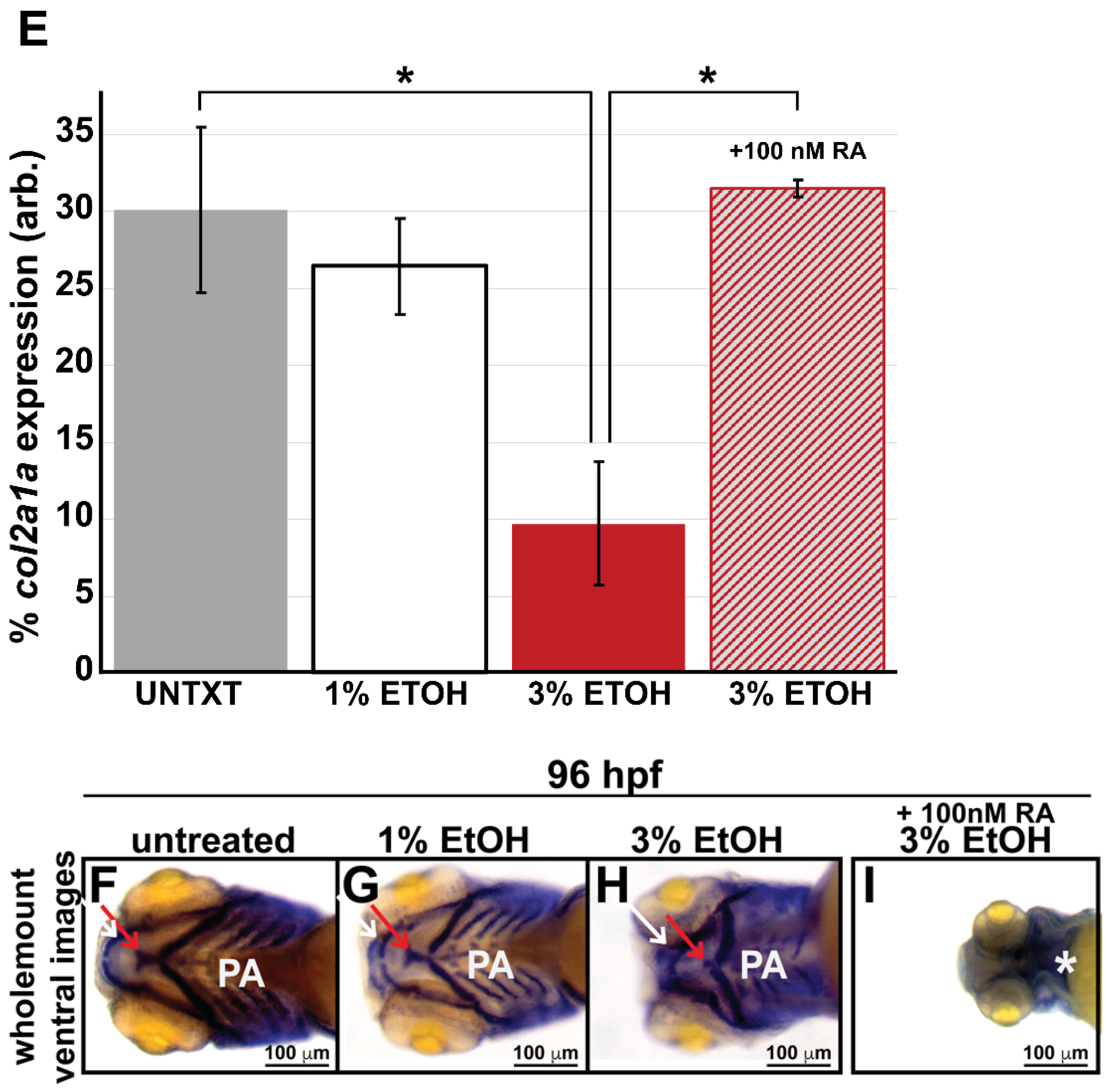
3.3. Ethanol Decreased Col2a1a Expression in the Ocular Neural Crest and Developing Jaw
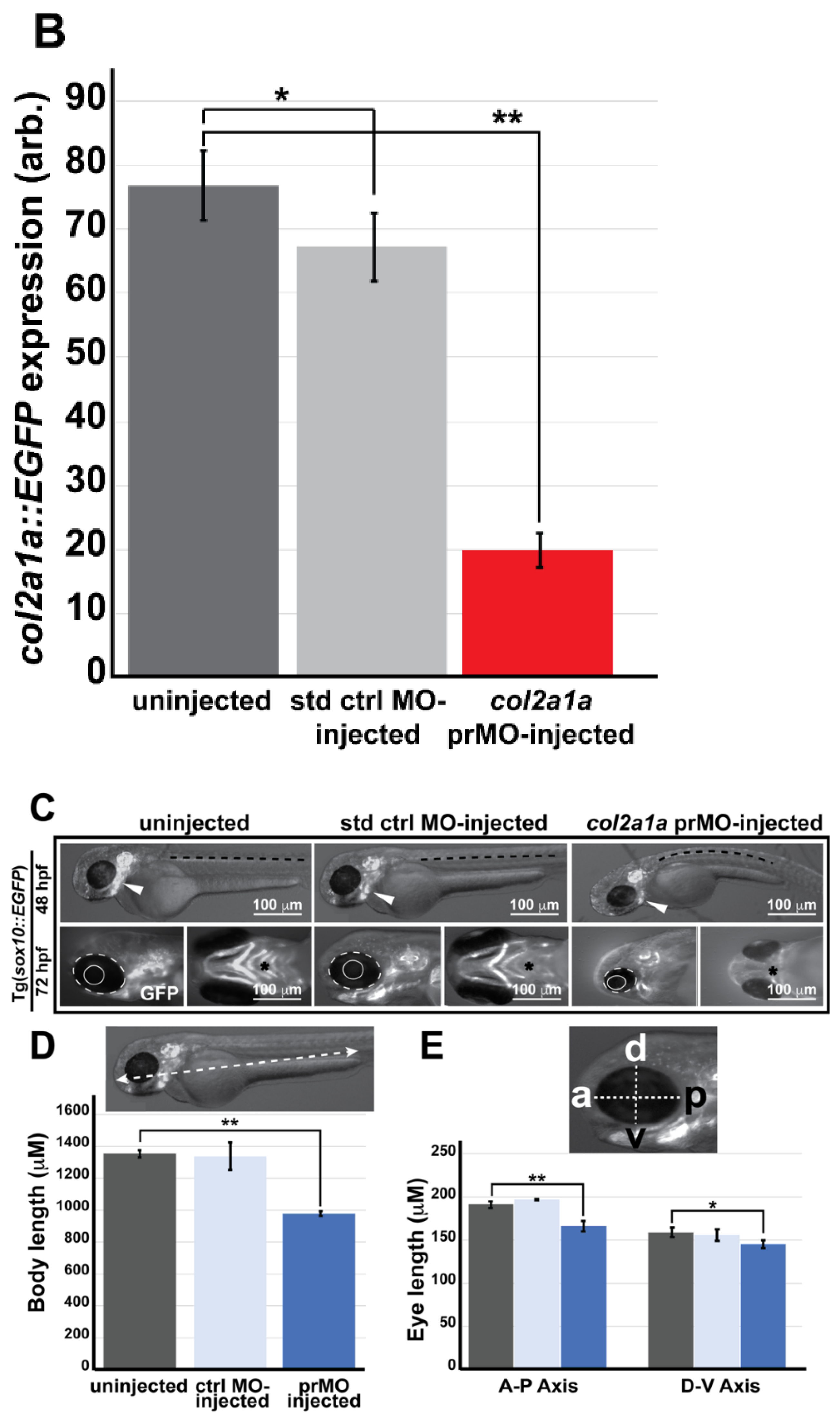
3.4. Col2a1a Knockdown in Zebrafish Mimics the Craniofacial and Ocular Defects Observed in Human Stickler Syndrome Patients
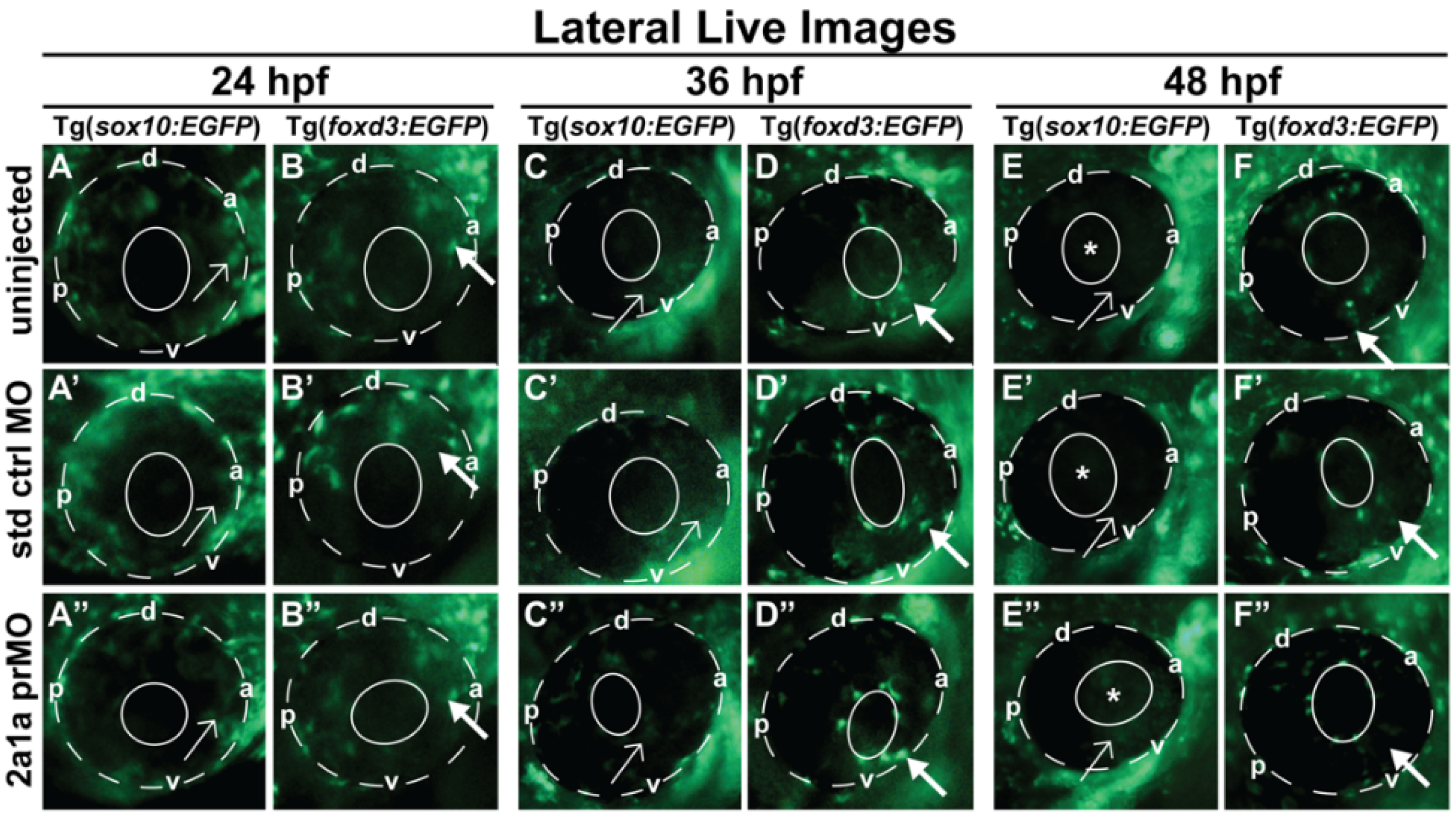
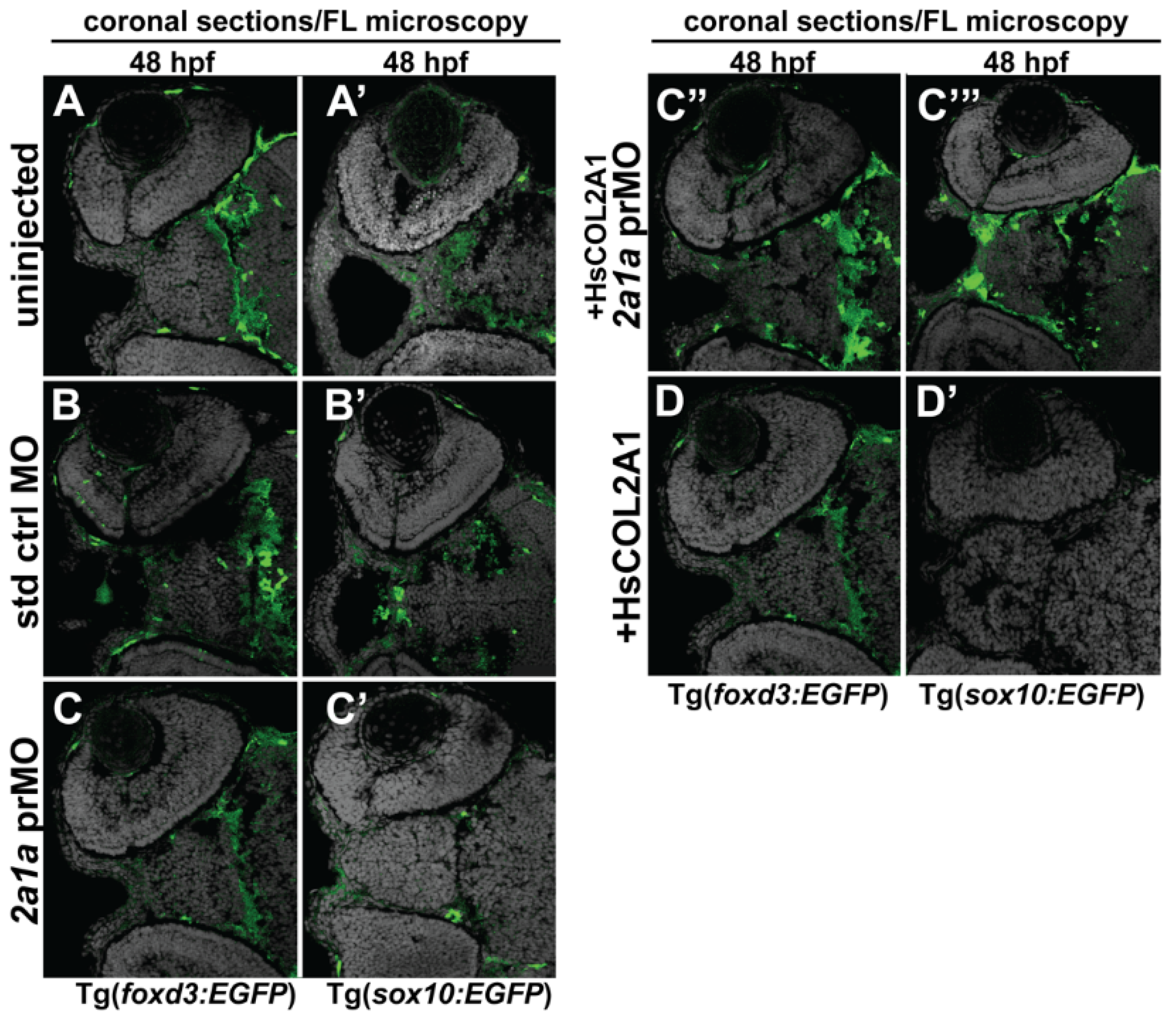
3.5. Col2a1a MO Knockdown Differentially Affected Sox10 and Foxd3 Expression and Ocular Neural Crest Cell Migration during Early Eye Development
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Boothe, M.; Morris, R.; Robin, N. Stickler Syndrome: A Review of Clinical Manifestations and the Genetics Evaluation. J. Personal. Med. 2020, 10, 105. [Google Scholar] [CrossRef] [PubMed]
- Wubben, T.J.; Branham, K.H.; Besirli, C.G.; Bohnsack, B.L. Retinal detachment and infantile-onset glaucoma in Stickler syndrome associated with known and novel COL2A1 mutations. Ophthalmic Genet. 2018, 39, 615–618. [Google Scholar] [CrossRef] [PubMed]
- Alshahrani, S.T.; Ghazi, N.G.; Al-Rashaed, S. Rhegmatogenous retinal detachments associated to Stickler syndrome in a tertiary eye care center in Saudi Arabia. Clin. Ophthalmol. 2016, 10, 1–6. [Google Scholar] [PubMed]
- Fincham, G.S.; Pasea, L.; Carroll, C.; McNinch, A.M.; Poulson, A.V.; Richards, A.J.; Scott, J.D.; Snead, M.P. Prevention of retinal detachment in Stickler syndrome: The Cambridge prophylactic cryotherapy protocol. Ophthalmology 2014, 121, 1588–1597. [Google Scholar] [CrossRef] [PubMed]
- Shenoy, B.H.; Mandal, A.K. Stickler syndrome associated with congenital glaucoma. Lancet 2013, 381, 422. [Google Scholar] [CrossRef]
- Ziakas, N.G.; Ramsay, A.S.; Lynch, S.A.; Clarke, M.P. Stickler’s syndrome associated with congenital glaucoma. Ophthalmic Genet. 1998, 19, 55–58. [Google Scholar] [CrossRef] [PubMed]
- Liberfarb, R.M.; Levy, H.P.; Rose, P.S.; Wilkin, D.J.; Davis, J.; Balog, J.Z.; Griffith, A.J.; Szymko-Bennett, Y.M.; Johnston, J.J.; Francomano, C.A. The Stickler syndrome: Genotype/phenotype correlation in 10 families with Stickler syndrome resulting from seven mutations in the type II collagen gene locus COL2A1. Genet. Med. 2003, 5, 21–27. [Google Scholar] [CrossRef]
- Annunen, S.; Körkkö, J.; Czarny, M.; Warman, M.L.; Brunner, H.G.; Kääriäinen, H.; Mulliken, J.B.; Tranebjærg, L.; Brooks, D.G.; Cox, G.F.; et al. Splicing mutations of 54-bp exons in the COL11A1 gene cause Marshall syndrome, but other mutations cause overlapping Marshall/Stickler phenotypes. Am. J. Hum. Genet. 1999, 65, 974–983. [Google Scholar] [CrossRef]
- Baker, S.; Booth, C.; Fillman, C.; Shapiro, M.; Blair, M.P.; Hyland, J.C.; Ala-Kokko, L. A loss of function mutation in the COL9A2 gene causes autosomal recessive Stickler syndrome. Am. J. Med. Genet. A 2011, 155, 1668–1672. [Google Scholar] [CrossRef]
- Faletra, F.; D’Adamo, A.P.; Bruno, I.; Athanasakis, E.; Biskup, S.; Esposito, L.; Gasparini, P. Autosomal recessive Stickler syndrome due to a loss of function mutation in the COL9A3 gene. Am. J. Med. Genet. A 2014, 164, 42–47. [Google Scholar] [CrossRef]
- Khalifa, O.; Imtiaz, F.; Ramzan, K.; Allam, R.; Al Hemidan, A.; Faqeih, E.; Abuharb, G.; Balobaid, A.; Sakati, N.; Al Owain, M. Marshall syndrome: Further evidence of a distinct phenotypic entity and report of new findings. Am. J. Med. Genet. A 2014, 164, 2601–2606. [Google Scholar] [CrossRef] [PubMed]
- Van Camp, G.; Snoeckx, R.L.; Hilgert, N.; Ende, J.V.D.; Fukuoka, H.; Wagatsuma, M.; Suzuki, H.; Smets, R.E.; Vanhoenacker, F.; Declau, F.; et al. A new autosomal recessive form of Stickler syndrome is caused by a mutation in the COL9A1 gene. Am. J. Hum. Genet. 2006, 79, 449–457. [Google Scholar] [CrossRef] [PubMed]
- Line, S.R.; Torloni, H.; Junqueira, L.C. Diversity of collagen expression in the pleomorphic adenoma of the parotid gland. Virchows Arch. A Pathol. Anat. Histopathol. 1989, 414, 477–483. [Google Scholar] [CrossRef] [PubMed]
- Baas, D.; Malbouyres, M.; Haftek-Terreau, Z.; Le Guellec, D.; Ruggiero, F. Craniofacial cartilage morphogenesis requires zebrafish col11a1 activity. Matrix Biol. 2009, 28, 490–502. [Google Scholar] [CrossRef]
- Yan, Y.L.; Miller, C.T.; Nissen, R.; Singer, A.; Liu, D.; Kirn, A.; Draper, B.; Willoughby, J.; Morcos, P.A.; Amsterdam, A.; et al. A zebrafish sox9 gene required for cartilage morphogenesis. Development 2002, 129, 5065–5079. [Google Scholar] [CrossRef]
- Bishop, P.N. Structural macromolecules and supramolecular organisation of the vitreous gel. Progress Retin. Eye Res. 2000, 19, 323–344. [Google Scholar] [CrossRef]
- van Deemter, M.; Kuijer, R.; Pas, H.H.; Van Der Worp, R.J.; Hooymans, J.M.M.; Los, L.I. Trypsin-mediated enzymatic degradation of type II collagen in the human vitreous. Mol. Vis. 2013, 19, 1591–1599. [Google Scholar]
- van Deemter, M.; Pas, H.H.; Kuijer, R.; Van Der Worp, R.J.; Hooymans, J.M.M.; Los, L.I. Enzymatic breakdown of type II collagen in the human vitreous. Investig. Ophthalmol. Vis. Sci. 2009, 50, 4552–4560. [Google Scholar] [CrossRef]
- Kimmel, C.B.; Ballard, W.W.; Kimmel, S.R.; Ullmann, B.; Schilling, T.F. Stages of embryonic development of the zebrafish. Dev. Dyn. 1995, 203, 253–310. [Google Scholar] [CrossRef]
- Kucenas, S.; Snell, H.; Appel, B. nkx2.2a promotes specification and differentiation of a myelinating subset of oligodendrocyte lineage cells in zebrafish. Neuron. Glia Biol. 2008, 4, 71–81. [Google Scholar] [CrossRef]
- Kucenas, S.; Takada, N.; Park, H.-C.; Woodruff, E.; Broadie, K.; Appel, B. CNS-derived glia ensheath peripheral nerves and mediate motor root development. Nat. Neurosci. 2008, 11, 143–151. [Google Scholar] [CrossRef]
- Bohnsack, B.L.; Kasprick, D.S.; Kish, P.E.; Goldman, D.; Kahana, A. A zebrafish model of axenfeld-rieger syndrome reveals that pitx2 regulation by retinoic acid is essential for ocular and craniofacial development. Investig. Ophthalmol. Vis. Sci. 2012, 53, 7–22. [Google Scholar] [CrossRef] [PubMed]
- Barthel, L.K.; Raymond, P.A. In situ hybridization studies of retinal neurons. Methods Enzymol. 2000, 316, 579–590. [Google Scholar] [PubMed]
- Bohnsack, B.L.; Gallina, D.; Thompson, H.; Kasprick, D.S.; Lucarelli, M.J.; Dootz, G.; Nelson, C.; McGonnell, I.M.; Kahana, A. Development of extraocular muscles requires early signals from periocular neural crest and the developing eye. Arch. Ophthalmol. 2011, 129, 1030–1041. [Google Scholar] [CrossRef] [PubMed]
- Dale, R.M.; Topczewski, J. Identification of an evolutionarily conserved regulatory element of the zebrafish col2a1a gene. Dev. Biol. 2011, 357, 518–531. [Google Scholar] [CrossRef] [PubMed]
- Zhang, T.; Sun, X.; Li, M.; Huang, H. De novo mutation in COL2A1 leads to lethal foetal skeletal dysplasia. Bone 2021, 153, 116169. [Google Scholar] [CrossRef] [PubMed]
- Dutton, K.A.; Pauliny, A.; Lopes, S.S.; Elworthy, S.; Carney, T.J.; Rauch, J.; Geisler, R.; Haffter, P.; Kelsh, R.N. Zebrafish colourless encodes sox10 and specifies non-ectomesenchymal neural crest fates. Development 2001, 128, 4113–4125. [Google Scholar] [CrossRef]
- Eason, J.; Williams, A.L.; Chawla, B.; Apsey, C.; Bohnsack, B.L. Differences in neural crest sensitivity to ethanol account for the infrequency of anterior segment defects in the eye compared with craniofacial anomalies in a zebrafish model of fetal alcohol syndrome. Birth Defects Res. 2017, 109, 1212–1227. [Google Scholar] [CrossRef]
- Honore, S.M.; Aybar, M.J.; Mayor, R. Sox10 is required for the early development of the prospective neural crest in Xenopus embryos. Dev. Biol. 2003, 260, 79–96. [Google Scholar] [CrossRef]
- Montero-Balaguer, M.; Lang, M.R.; Sachdev, S.W.; Knappmeyer, C.; Stewart, R.A.; De La Guardia, A.; Hatzopoulos, A.K.; Knapik, E.W. The mother superior mutation ablates foxd3 activity in neural crest progenitor cells and depletes neural crest derivatives in zebrafish. Dev. Dyn. 2006, 235, 3199–3212. [Google Scholar] [CrossRef]
- Stewart, R.A.; Arduini, B.L.; Berghmans, S.; George, R.E.; Kanki, J.P.; Henion, P.D.; Look, A.T. Zebrafish foxd3 is selectively required for neural crest specification, migration and survival. Dev. Biol. 2006, 292, 174–188. [Google Scholar] [CrossRef] [PubMed]
- Berenguer, M.; Duester, G. Retinoic acid, RARs and early development. J. Mol. Endocrinol. 2022. [Google Scholar] [CrossRef] [PubMed]
- Chawla, B.; Schley, E.; Williams, A.L.; Bohnsack, B.L. Retinoic Acid and Pitx2 Regulate Early Neural Crest Survival and Migration in Craniofacial and Ocular Development. Birth Defects Res. B Dev. Reprod. Toxicol. 2016, 107, 126–135. [Google Scholar] [CrossRef] [PubMed]
- Cvekl, A.; Wang, W.L. Retinoic acid signaling in mammalian eye development. Exp. Eye Res. 2009, 89, 280–291. [Google Scholar] [CrossRef] [PubMed]
- Duester, G. Towards a Better Vision of Retinoic Acid Signaling during Eye Development. Cells 2022, 11, 322. [Google Scholar] [CrossRef] [PubMed]
- Williams, A.L.; Bohnsack, B.L. Neural crest derivatives in ocular development: Discerning the eye of the storm. Birth Defects Res. C Embryo Today 2015, 105, 87–95. [Google Scholar] [CrossRef]
- Williams, A.L.; Bohnsack, B.L. What’s retinoic acid got to do with it? Retinoic acid regulation of the neural crest in craniofacial and ocular development. Genesis 2019, 57, e23308. [Google Scholar] [CrossRef]
- Chawla, B.; Swain, W.; Williams, A.L.; Bohnsack, B.L. Retinoic Acid Maintains Function of Neural Crest-Derived Ocular and Craniofacial Structures in Adult Zebrafish. Investig. Ophthalmol. Vis. Sci. 2018, 59, 1924–1935. [Google Scholar] [CrossRef]
- Chute, J.P.; Muramoto, G.G.; Whitesides, J.; Colvin, M.; Safi, R.; Chao, N.J.; McDonnell, D.P. Inhibition of aldehyde dehydrogenase and retinoid signaling induces the expansion of human hematopoietic stem cells. Proc. Natl. Acad. Sci. USA 2006, 103, 11707–11712. [Google Scholar] [CrossRef]
- Moreb, J.S.; Ucar, D.; Han, S.; Amory, J.; Goldstein, A.S.; Ostmark, B.; Chang, L.J. The enzymatic activity of human aldehyde dehydrogenases 1A2 and 2 (ALDH1A2 and ALDH2) is detected by Aldefluor, inhibited by diethylaminobenzaldehyde and has significant effects on cell proliferation and drug resistance. Chem. Biol. Interact. 2012, 195, 52–60. [Google Scholar] [CrossRef]
- Aliferis, K.; Marsal, C.; Pelletier, V.; Doray, B.; Weiss, M.; Tops, C.; Speeg-Schatz, C.; Lesnik, S.; Dollfus, H. A novel nonsense B3GALTL mutation confirms Peters plus syndrome in a patient with multiple malformations and Peters anomaly. Ophthalmic Genet. 2010, 31, 205–208. [Google Scholar] [CrossRef]
- Dressler, S.; Meyer-Marcotty, P.; Weisschuh, N.; Jablonski-Momeni, A.; Pieper, K.; Gramer, G.; Gramer, E. Dental and Craniofacial Anomalies Associated with Axenfeld-Rieger Syndrome with PITX2 Mutation. Case Rep. Med. 2010, 2010, 621984. [Google Scholar] [CrossRef] [PubMed]
- Ozeki, H.; Shirai, S.; Ikeda, K.; Ogura, Y. Anomalies associated with Axenfeld-Rieger syndrome. Graefe’s Arch. Clin. Exp. Ophthalmol. 1999, 237, 730–734. [Google Scholar] [CrossRef] [PubMed]
- Ozeki, H.; Shirai, S.; Nozaki, M.; Sakurai, E.; Mizuno, S.; Ashikari, M.; Matsunaga, N.; Ogura, Y. Ocular and systemic features of Peters’ anomaly. Graefe’s Arch. Clin. Exp. Ophthalmol. 2000, 238, 833–839. [Google Scholar] [CrossRef]
- Dougherty, M.; Kamel, G.; Grimaldi, M.; Gfrerer, L.; Shubinets, V.; Ethier, R.; Hickey, G.; Cornell, R.A.; Liao, E.C. Distinct requirements for wnt9a and irf6 in extension and integration mechanisms during zebrafish palate morphogenesis. Development 2013, 140, 76–81. [Google Scholar] [CrossRef] [PubMed]
- Dougherty, M.; Kamel, G.; Shubinets, V.; Hickey, G.; Grimaldi, M.; Liao, E.C. Embryonic fate map of first pharyngeal arch structures in the sox10: Kaede zebrafish transgenic model. J. Craniofac. Surg. 2012, 23, 1333–1337. [Google Scholar] [CrossRef]
- Trainor, P.A. Specification and patterning of neural crest cells during craniofacial development. Brain Behav. Evol. 2005, 66, 266–280. [Google Scholar] [CrossRef]
- Trainor, P.A. Specification of neural crest cell formation and migration in mouse embryos. Semin. Cell Dev. Biol. 2005, 16, 683–693. [Google Scholar] [CrossRef]
- Williams, A.L.; Bohnsack, B.L. The Ocular Neural Crest: Specification, Migration, and Then What? Front. Cell Dev. Biol. 2020, 8, 595896. [Google Scholar] [CrossRef]
- Bohnsack, B.L.; Kahana, A. Thyroid hormone and retinoic acid interact to regulate zebrafish craniofacial neural crest development. Dev. Biol. 2013, 373, 300–309. [Google Scholar] [CrossRef]
- Williams, A.L.; Bohnsack, B.L. Multi-Photon Time Lapse Imaging to Visualize Development in Real-time: Visualization of Migrating Neural Crest Cells in Zebrafish Embryos. J. Vis. Exp. 2017, 126, 56214. [Google Scholar] [CrossRef] [PubMed]
- Ghyselinck, N.B.; Duester, G. Retinoic acid signaling pathways. Development 2019, 146, dev167502. [Google Scholar] [CrossRef] [PubMed]
- Smith, J.N.; Walker, H.M.; Thompson, H.; Collinson, J.M.; Vargesson, N.; Erskine, L. Lens-regulated retinoic acid signalling controls expansion of the developing eye. Development 2018, 145, dev167171. [Google Scholar] [CrossRef] [PubMed]
- Hale, L.A.; Tallafuss, A.; Yan, Y.-L.; Dudley, L.; Eisen, J.S.; Postlethwait, J.H. Characterization of the retinoic acid receptor genes raraa, rarab and rarg during zebrafish development. Gene Exp. Patterns 2006, 6, 546–555. [Google Scholar] [CrossRef] [PubMed]
- Linville, A.; Radtke, K.; Waxman, J.S.; Yelon, D.; Schilling, T.F. Combinatorial roles for zebrafish retinoic acid receptors in the hindbrain, limbs and pharyngeal arches. Dev. Biol. 2009, 325, 60–70. [Google Scholar] [CrossRef]
- Thisse, B.; Heyer, V.; Lux, A.; Alunni, V.; Degrave, A.; Seiliez, I.; Kirchner, J.; Parkhill, J.-P.; Thisse, C. Spatial and temporal expression of the zebrafish genome by large-scale in situ hybridization screening. Methods Cell Biol. 2004, 77, 505–519. [Google Scholar]
- Duester, G, Keeping an eye on retinoic acid signaling during eye development. Chem. Biol. Interact. 2009, 178, 178–181. [CrossRef]
- Deltour, L.; Ang, H.L.; Duester, G. Ethanol inhibition of retinoic acid synthesis as a potential mechanism for fetal alcohol syndrome. FASEB J. 1996, 10, 1050–1057. [Google Scholar] [CrossRef]
- Muralidharan, P.; Sarmah, S.; Marrs, J.A. Zebrafish retinal defects induced by ethanol exposure are rescued by retinoic acid and folic acid supplement. Alcohol 2015, 49, 149–163. [Google Scholar] [CrossRef]
- Flentke, G.R.; Garic, A.; Amberger, E.; Hernandez, M.; Smith, S.M. Calcium-mediated repression of beta-catenin and its transcriptional signaling mediates neural crest cell death in an avian model of fetal alcohol syndrome. Birth Defects Res. A Clin. Mol. Teratol. 2011, 91, 591–602. [Google Scholar] [CrossRef]
- Flentke, G.R.; Garic, A.; Hernandez, M.; Smith, S.M. CaMKII represses transcriptionally active beta-catenin to mediate acute ethanol neurodegeneration and can phosphorylate beta-catenin. J. Neurochem. 2014, 128, 523–535. [Google Scholar] [CrossRef]
- Garic, A.; Flentke, G.R.; Amberger, E.; Hernandez, M.; Smith, S.M. CaMKII activation is a novel effector of alcohol’s neurotoxicity in neural crest stem/progenitor cells. J. Neurochem. 2011, 118, 646–657. [Google Scholar] [CrossRef]
- Smith, S.M.; Garic, A.; Berres, M.E.; Flentke, G.R. Genomic factors that shape craniofacial outcome and neural crest vulnerability in FASD. Front. Genet. 2014, 5, 224. [Google Scholar] [CrossRef]
- Smith, S.M.; Garic, A.; Flentke, G.R.; Berres, M.E. Neural crest development in fetal alcohol syndrome. Birth Defects Res. C Embryo Today 2014, 102, 210–220. [Google Scholar] [CrossRef]
- Chen, S.Y.; Sulik, K.K. Free radicals and ethanol-induced cytotoxicity in neural crest cells. Alcohol Clin. Exp. Res. 1996, 20, 1071–1076. [Google Scholar] [CrossRef]
- Davis, W.L.; Crawford, L.A.; Cooper, O.J.; Farmer, G.R.; Thomas, D.L.; Freeman, B.L. Ethanol induces the generation of reactive free radicals by neural crest cells in vitro. J. Craniofac Genet. Dev. Biol. 1990, 10, 277–293. [Google Scholar]
- Floyd, R.A.; Carney, J.M. Free radical damage to protein and DNA: Mechanisms involved and relevant observations on brain undergoing oxidative stress. Ann. Neurol. 1992, 32, S22–S27. [Google Scholar] [CrossRef]
- Henderson, G.I.; Devi, B.G.; Perez, A.; Schenker, S. In utero ethanol exposure elicits oxidative stress in the rat fetus. Alcohol Clin. Exp. Res. 1995, 19, 714–720. [Google Scholar] [CrossRef]
- Mork, L.; Crump, G. Zebrafish Craniofacial Development: A Window into Early Patterning. Curr. Top. Dev. Biol. 2015, 115, 235–269. [Google Scholar]
- Van Der Meulen, K.L.; Vöcking, O.; Weaver, M.L.; Meshram, N.N.; Famulski, J.K. Spatiotemporal Characterization of Anterior Segment Mesenchyme Heterogeneity During Zebrafish Ocular Anterior Segment Development. Front. Cell Dev. Biol. 2020, 8, 379. [Google Scholar] [CrossRef]
- Duband, J.L.; Dady, A.; Fleury, V. Resolving time and space constraints during neural crest formation and delamination. Curr. Top. Dev. Biol. 2015, 111, 27–67. [Google Scholar]
- Perris, R.; Krotoski, D.; Bronner-Fraser, M. Collagens in avian neural crest development: Distribution in vivo and migration-promoting ability in vitro. Development 1991, 113, 969–984. [Google Scholar] [CrossRef]










Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Williams, A.L.; Bohnsack, B.L. Zebrafish Model of Stickler Syndrome Suggests a Role for Col2a1a in the Neural Crest during Early Eye Development. J. Dev. Biol. 2022, 10, 42. https://doi.org/10.3390/jdb10040042
Williams AL, Bohnsack BL. Zebrafish Model of Stickler Syndrome Suggests a Role for Col2a1a in the Neural Crest during Early Eye Development. Journal of Developmental Biology. 2022; 10(4):42. https://doi.org/10.3390/jdb10040042
Chicago/Turabian StyleWilliams, Antionette L., and Brenda L. Bohnsack. 2022. "Zebrafish Model of Stickler Syndrome Suggests a Role for Col2a1a in the Neural Crest during Early Eye Development" Journal of Developmental Biology 10, no. 4: 42. https://doi.org/10.3390/jdb10040042
APA StyleWilliams, A. L., & Bohnsack, B. L. (2022). Zebrafish Model of Stickler Syndrome Suggests a Role for Col2a1a in the Neural Crest during Early Eye Development. Journal of Developmental Biology, 10(4), 42. https://doi.org/10.3390/jdb10040042