Acute vs. Chronic vs. Cyclic Hypoxia: Their Differential Dynamics, Molecular Mechanisms, and Effects on Tumor Progression


Abstract
1. Introduction
2. Tumor Hypoxia
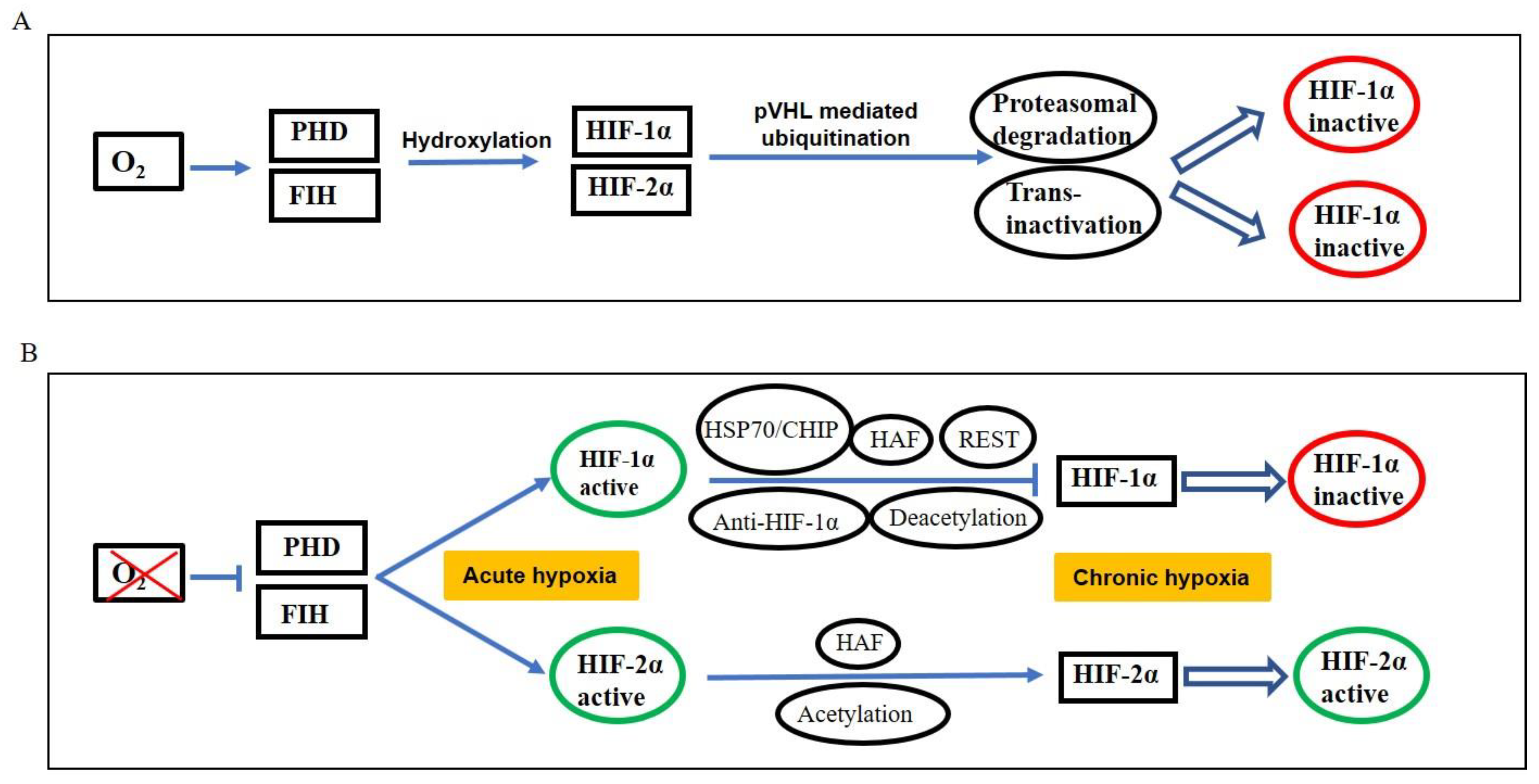
3. HIF Signaling: Key Regulators of Hypoxic Response
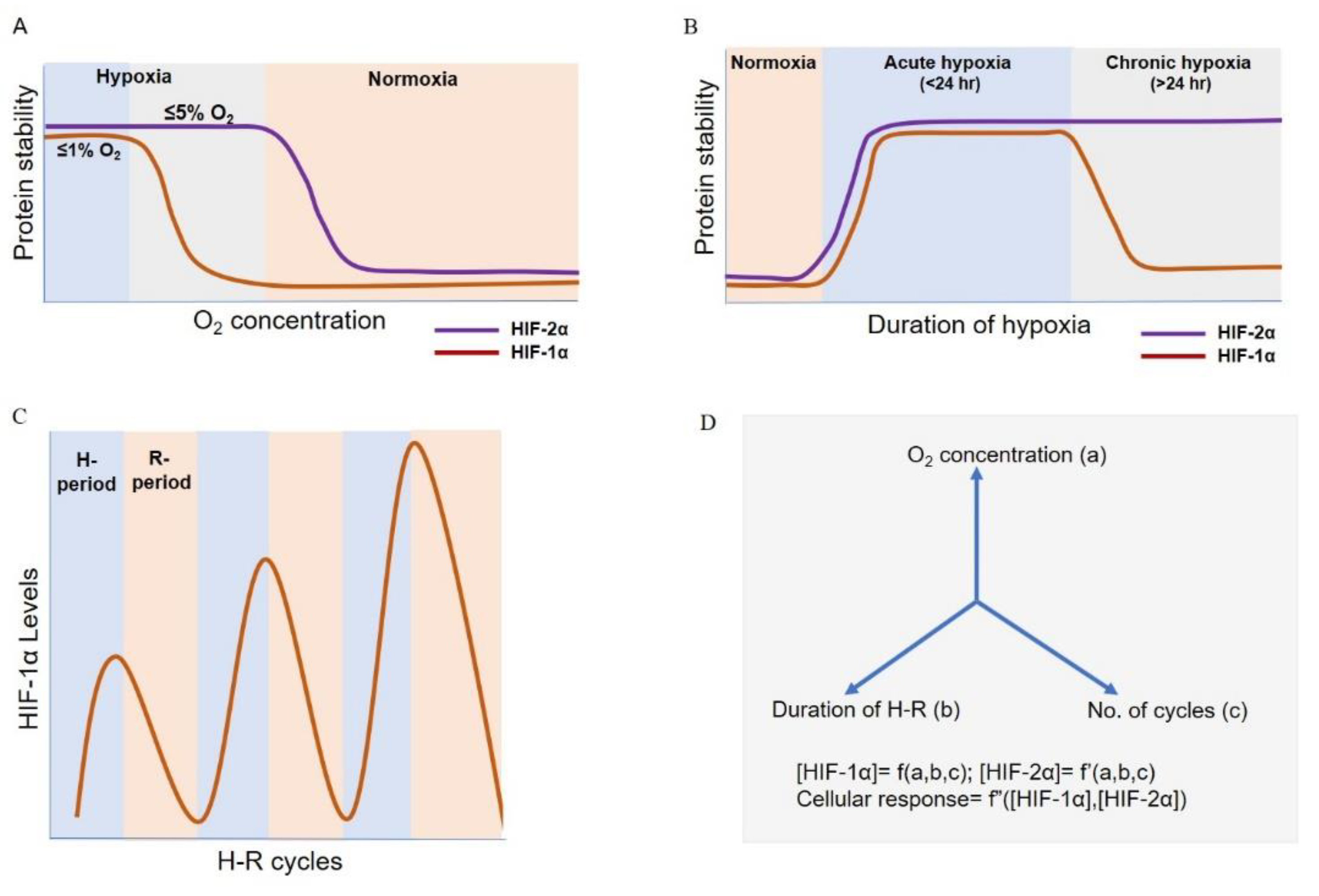
4. HIF Switch during Acute and Chronic Hypoxia
5. HIF Dynamics during Intermittent Hypoxia
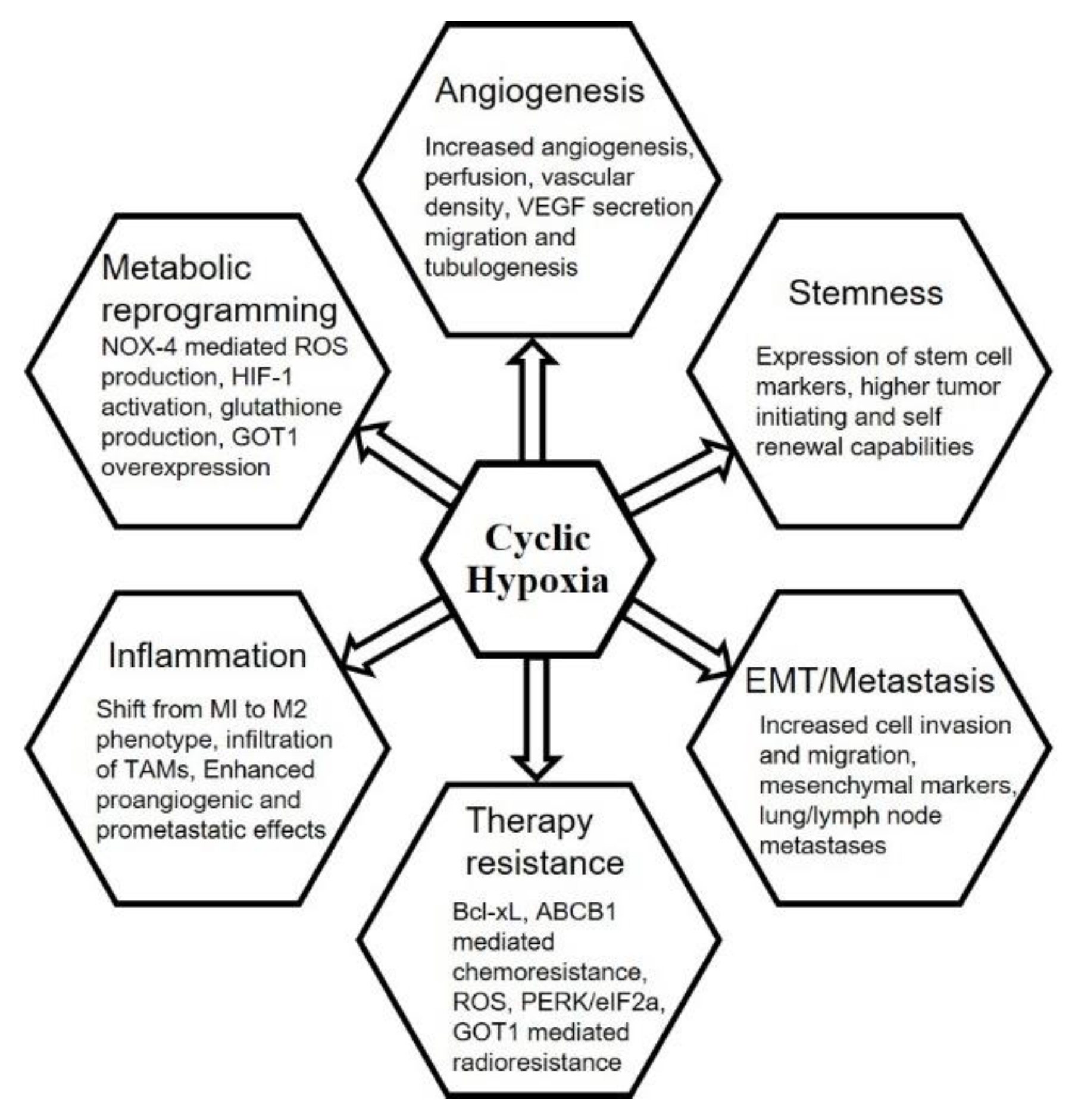
6. Chronic vs. Cyclic Hypoxia and Hallmarks of Cancer
6.1. Tumor Hypoxia and Angiogenesis
6.2. Stemness
6.3. Epithelial–Mesenchymal Transition, Invasion, Migration and Metastasis
6.4. Anti-Cancer Therapies
6.5. Inflammation
7. Intermittent Hypoxia Dynamics in Obstructive Sleep Apnea (OSA)
8. Mathematical Modelling As a Tool to Understand the Hypoxia Response Dynamics
9. Conclusions
Funding
Conflicts of Interest
References
- Gupta, G.P.; Massagué, J. Cancer metastasis: Building a framework. Cell 2006, 127, 679–695. [Google Scholar] [CrossRef] [PubMed]
- Rankin, E.B.; Giaccia, A.J. Hypoxic control of metastasis. Science 2016, 352, 175–180. [Google Scholar] [CrossRef] [PubMed]
- Dunwoodie, S.L. The Role of Hypoxia in Development of the Mammalian Embryo. Dev. Cell 2009, 17, 755–773. [Google Scholar] [CrossRef] [PubMed]
- Fisher, S.A.; Burggren, W.W. Role of Hypoxia in the Evolution and Development of the Cardiovascular System. Antioxid. Redox Signal. 2007, 9, 1339–1352. [Google Scholar] [CrossRef] [PubMed]
- Sendoel, A.; Hengartner, M.O. Apoptotic Cell Death Under Hypoxia. Physiology 2014, 29, 168–176. [Google Scholar] [CrossRef] [PubMed]
- Greijer, A.E.; Van Der Wall, E. The role of hypoxia inducible factor 1 (HIF-1) in hypoxia induced apoptosis. J. Clin. Pathol. 2004, 57, 1009–1014. [Google Scholar] [CrossRef] [PubMed]
- Harris, A.L. Hypoxia — a key regulatory factor in tumour growth. Nat. Rev. Cancer 2002, 2, 38–47. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef]
- Thomlinson, R.H.; Gray, L.H. The Histological Structure of Some Human Lung Cancers and the Possible Implications for Radiotherapy. Br. J. Cancer 1955, 9, 539–549. [Google Scholar] [CrossRef]
- Gray, L.H.; Conger, A.D.; Ebert, M.; Hornsey, S.; Scott, O.C.A. The Concentration of Oxygen Dissolved in Tissues at the Time of Irradiation as a Factor in Radiotherapy. Br. J. Radiol. 1953, 26, 638–648. [Google Scholar] [CrossRef]
- Hockel, M.; Vaupel, P. Tumor Hypoxia: Definitions and Current Clinical, Biologic, and Molecular Aspects. JNCI J. Natl. Cancer Inst. 2001, 93, 266–276. [Google Scholar] [CrossRef]
- Muz, B.; de la Puente, P.; Azab, F.; Azab, A.K. The role of hypoxia in cancer progression, angiogenesis, metastasis, and resistance to therapy. Hypoxia 2015, 11, 83–92. [Google Scholar] [CrossRef] [PubMed]
- Nobre, A.R.; Entenberg, D.; Wang, Y.; Condeelis, J.; Aguirre-ghiso, J.A. The Different Routes to Metastasis via Hypoxia-Regulated Programs. Trends Cell Biol. 2018, 28, 941–956. [Google Scholar] [CrossRef] [PubMed]
- Bristow, R.G.; Hill, R.P. Hypoxia, DNA repair and genetic instability. Nat. Rev. Cancer 2008, 8, 180–192. [Google Scholar] [CrossRef] [PubMed]
- Koh, M.Y.; Powis, G. Passing the baton: The HIF switch. Trends Biochem. Sci. 2012, 37, 364–372. [Google Scholar] [CrossRef] [PubMed]
- Kimura, H.; Braun, R.D.; Ong, E.T.; Hsu, R.; Secomb, T.W.; Papahadjopoulos, D.; Hong, K.; Dewhirst, M.W. Fluctuations in Red Cell Flux in Tumor Microvessels Can Lead to Transient Hypoxia and Reoxygenation in Tumor Parenchyma. Cancer Res. 1996, 56, 5522–5528. [Google Scholar] [PubMed]
- Almendros, I.; Gozal, D. Intermittent hypoxia and cancer: Undesirable bed partners? Respir. Physiol. Neurobiol. 2018, 256, 79–86. [Google Scholar] [CrossRef]
- Brown, J.M. Evidence for acutely hypoxic cells in mouse tumours, and a possible mechanism of reoxygenation. Br. J. Radiol. 1979, 52, 650–656. [Google Scholar] [CrossRef]
- Matsumoto, S.; Yasui, H.; Mitchell, J.B.; Krishna, M.C. Imaging cycling tumor hypoxia. Cancer Res. 2010, 70, 10019–10023. [Google Scholar] [CrossRef]
- Dewhirst, M.W. Intermittent Hypoxia Furthers the Rationale for Hypoxia-Inducible Factor-1 Targeting. Cancer Res. 2007, 854–856. [Google Scholar] [CrossRef]
- Michiels, C.; Tellier, C.; Feron, O. Cycling hypoxia: A key feature of the tumor microenvironment. Biochim. Biophys. Acta - Rev. Cancer 2016, 1866, 76–86. [Google Scholar] [CrossRef] [PubMed]
- Semenza, G.L. Targeting HIF-1 for cancer therapy. Nat. Rev. Cancer 2003, 3, 721–732. [Google Scholar] [CrossRef] [PubMed]
- Boareto, M.; Jolly, M.K.; Ben-Jacob, E.; Onuchic, J.N. Jagged mediates differences in normal and tumor angiogenesis by affecting tip-stalk fate decision. Proc. Natl. Acad. Sci. 2015, 112, E3836–E3844. [Google Scholar] [CrossRef] [PubMed]
- Cárdenas-Navia, L.I.; Mace, D.; Richardson, R.A.; Wilson, D.F.; Shan, S.; Dewhirst, M.W. The pervasive presence of fluctuating oxygenation in tumors. Cancer Res. 2008, 68, 5812–5819. [Google Scholar] [CrossRef] [PubMed]
- Chaplin, D.J.; Olive, P.L.; Durand, R.E. Intermittent Blood Flow in a Murine Tumor: Radiobiological Effects. Cancer Res. 1987, 47, 597–601. [Google Scholar] [PubMed]
- Keith, B.; Johnson, R.S.; Simon, M.C. HIF1α and HIF2α: Sibling rivalry in hypoxic tumour growth and progression. Nat. Rev. Cancer 2012, 12, 9–22. [Google Scholar] [CrossRef] [PubMed]
- Loboda, A.; Jozkowicz, A.; Dulak, J. HIF-1 and HIF-2 transcription factors- similar but not identical. Mol. Cells 2010, 29, 435–442. [Google Scholar] [CrossRef] [PubMed]
- Mahon, P.C. FIH-1: A novel protein that interacts with HIF-1alpha and VHL to mediate repression of HIF-1 transcriptional activity. Genes Dev. 2001, 15, 2675–2686. [Google Scholar] [CrossRef] [PubMed]
- Koivunen, P.; Hirsilä, M.; Günzler, V.; Kivirikko, K.I.; Myllyharju, J. Catalytic Properties of the Asparaginyl Hydroxylase (FIH) in the Oxygen Sensing Pathway Are Distinct from Those of Its Prolyl 4-Hydroxylases. J. Biol. Chem. 2004, 279, 9899–9904. [Google Scholar] [CrossRef]
- Kaelin, W.G.; Ratcliffe, P.J. Oxygen Sensing by Metazoans: The Central Role of the HIF Hydroxylase Pathway. Mol. Cell 2008, 30, 393–402. [Google Scholar] [CrossRef]
- Bartoszewski, R.; Moszyńska, A.; Serocki, M.; Cabaj, A.; Polten, A.; Ochocka, R.; Dell’Italia, L.; Bartoszewska, S.; Króliczewski, J.; Dąbrowski, M.; et al. Primary endothelial cell–specific regulation of hypoxia-inducible factor (HIF)-1 and HIF-2 and their target gene expression profiles during hypoxia. FASEB J. 2019, 33, 7929–7941. [Google Scholar] [CrossRef] [PubMed]
- Ivanova, I.G.; Park, C.V.; Kenneth, N.S. Translating the Hypoxic Response—the Role of HIF Protein Translation in the Cellular Response to Low Oxygen. Cells 2019, 8, 114. [Google Scholar] [CrossRef] [PubMed]
- Gu, Y.Z.; Moran, S.M.; Hogenesch, J.B.; Wartman, L.; Bradfield, C.A. Molecular characterization and chromosomal localization of a third α- class hypoxia inducible factor subunit, HIF3α. Gene Expr. 1998, 7, 205–213. [Google Scholar] [PubMed]
- Makino, Y.; Kanopka, A.; Wilson, W.J.; Tanaka, H.; Poellinger, L. Inhibitory PAS domain protein (IPAS) is a hypoxia-inducible splicing variant of the hypoxia-inducible factor-3α locus. J. Biol. Chem. 2002, 277, 32405–32408. [Google Scholar] [CrossRef] [PubMed]
- Duan, C. Hypoxia-inducible factor 3 biology: Complexities and emerging themes. Am J Physiol Cell Physiol 2016, 310, C260–C269. [Google Scholar] [CrossRef] [PubMed]
- Jolly, M.K.; Preca, B.-T.; Tripathi, S.C.; Jia, D.; Hanash, S.M.; Brabletz, T.; Stemmler, M.P.; Maurer, J.; Levine, H. Interconnected feedback loops among ESRP1, HAS2, and CD44 regulate epithelial–mesenchymal plasticity in cancer. APL Bioeng. 2018, 2, 031908. [Google Scholar] [CrossRef]
- Tanaka, T.; Wiesener, M.; Bernhardt, W.; Eckardt, K.; Warnecke, C. The human HIF (hypoxia-inducible factor)-3alpha gene is a HIF-1 target gene and may modulate hypoxic gene induction. Biochem. J. 2009, 424, 143–151. [Google Scholar] [CrossRef]
- Holmquist-Mengelbier, L.; Fredlund, E.; Löfstedt, T.; Noguera, R.; Navarro, S.; Nilsson, H.; Pietras, A.; Vallon-Christersson, J.; Borg, Å.; Gradin, K.; et al. Recruitment of HIF-1α and HIF-2α to common target genes is differentially regulated in neuroblastoma: HIF-2α promotes an aggressive phenotype. Cancer Cell 2006, 10, 413–423. [Google Scholar] [CrossRef]
- Holmquist, L.; Jogi, A.; Påhlman, S. Phenotypic persistence after reoxygenation of hypoxic neuroblastoma cells. Int. J. Cancer 2005, 116, 218–225. [Google Scholar] [CrossRef]
- Koh, M.Y.; Lemos, R.; Liu, X.; Powis, G. The hypoxia-associated factor switches cells from HIF-1α- to HIF-2α-dependent signaling promoting stem cell characteristics, aggressive tumor growth and invasion. Cancer Res. 2011, 71, 4015–4027. [Google Scholar] [CrossRef]
- Uchida, T.; Rossignol, F.; Matthay, M.A.; Mounier, R.; Couette, S.; Clottes, E.; Clerici, C. Prolonged Hypoxia Differentially Regulates Hypoxia-inducible Factor (HIF) -1alpha and HIF-2alpha Expression in Lung Epithelial Cells: Implications of natural antisense HIF-1alpha. J Biol Chem 2004, 279, 14871–14878. [Google Scholar] [CrossRef] [PubMed]
- Nilsson, H.; Jögi, A.; Beckman, S.; Harris, A.L.; Poellinger, L.; Påhlman, S. HIF-2α expression in human fetal paraganglia and neuroblastoma: Relation to sympathetic differentiation, glucose deficiency, and hypoxia. Exp. Cell Res. 2005, 303, 447–456. [Google Scholar] [CrossRef] [PubMed]
- Lin, Q.; Cong, X.; Yun, Z. Differential hypoxic regulation of hypoxia-inducible factors 1alpha and 2alpha. Mol. Cancer Res. 2011, 9, 757–765. [Google Scholar] [CrossRef] [PubMed]
- Toffoli, S.; Roegiers, A.; Feron, O.; Van Steenbrugge, M.; Ninane, N.; Raes, M.; Michiels, C. Intermittent hypoxia is an angiogenic inducer for endothelial cells: Role of HIF-1. Angiogenesis 2009, 12, 47–67. [Google Scholar] [CrossRef] [PubMed]
- Dong, G.; Lin, X.; Liu, H.; Gao, D.; Cui, J.; Ren, Z.; Chen, R. Intermittent hypoxia alleviates increased VEGF and pro-angiogenic potential in liver cancer cells. Oncol. Lett. 2019, 18, 1831–1839. [Google Scholar] [CrossRef]
- Luo, W.; Zhong, J.; Chang, R.; Hu, H.; Pandey, A.; Semenza, G.L. Hsp70 and CHIP selectively mediate ubiquitination and degradation of hypoxia-inducible factor (HIF)-1α but not HIF-2α. J. Biol. Chem. 2010, 285, 3651–3663. [Google Scholar] [CrossRef]
- Bento, C.F.; Fernandes, R.; Ramalho, J.; Marques, C.; Shang, F.; Taylor, A.; Pereira, P. The chaperone-dependent ubiquitin ligase CHIP targets HIF-1α for degradation in the presence of methylglyoxal. PLoS One 2010, 5, e15062. [Google Scholar] [CrossRef]
- Koh, M.Y.; Darnay, B.G.; Powis, G. Hypoxia-Associated Factor, a Novel E3-Ubiquitin Ligase, Binds and Ubiquitinates Hypoxia-Inducible Factor 1alpha, Leading to Its oxyden-indepedent degradation. Mol Cell Biol 2008, 28, 7081–7095. [Google Scholar] [CrossRef]
- Cavadas, M.A.S.; Mesnieres, M.; Crifo, B.; Manresa, M.C.; Selfridge, A.C.; Scholz, C.C.; Cummins, E.P.; Cheong, A.; Taylor, C.T. REST mediates resolution of HIF-dependent gene expression in prolonged hypoxia. Sci. Rep. 2015, 5, 17851. [Google Scholar] [CrossRef]
- Serocki, M.; Bartoszewska, S.; Janaszak-Jasiecka, A.; Ochocka, R.J.; Collawn, J.F.; Bartoszewski, R. miRNAs regulate the HIF switch during hypoxia: A novel therapeutic target. Angiogenesis 2018, 21, 183–202. [Google Scholar] [CrossRef]
- Janaszak-Jasiecka, A.; Bartoszewska, S.; Kochan, K.; Piotrowski, A.; Kalinowski, L.; Kamysz, W.; Ochocka, R.J.; Bartoszewski, R.; Collawn, J.F. miR-429 regulates the transition between Hypoxia-Inducible Factor (HIF)1A and HIF3A expression in human endothelial cells. Sci. Rep. 2016, 6, 22775. [Google Scholar] [CrossRef] [PubMed]
- Guan, Z.; Ding, C.; Du, Y.; Zhang, K.; Zhu, J.N.; Zhang, T.; He, D.; Xu, S.; Wang, X.; Fan, J. HAF drives the switch of HIF-1α to HIF-2α by activating the NF-κB pathway, leading to malignant behavior of T24 bladder cancer cells. Int. J. Oncol. 2014, 44, 393–402. [Google Scholar] [CrossRef] [PubMed]
- Dai, Y.; Bae, K.; Siemann, D.W. Impact of Hypoxia on the Metastatic Potential of Human Prostate Cancer Cells. Int J Radiat Oncol Biol Phys 2011, 81, 521–528. [Google Scholar] [CrossRef] [PubMed]
- Stiehl, D.P.; Bordoli, M.R.; Abreu-Rodríguez, I.; Wollenick, K.; Schraml, P.; Gradin, K.; Poellinger, L.; Kristiansen, G.; Wenger, R.H. Non-canonical HIF-2α function drives autonomous breast cancer cell growth via an AREG-EGFR/ErbB4 autocrine loop. Oncogene 2012, 31, 2283–2297. [Google Scholar] [CrossRef] [PubMed]
- Martinive, P.; Defresne, F.; Quaghebeur, E.; Daneau, G.; Crokart, N.; Grégoire, V.; Gallez, B.; Dessy, C.; Feron, O. Impact of cyclic hypoxia on HIF-1α regulation in endothelial cells - New insights for anti-tumor treatments. FEBS J. 2009, 276, 509–518. [Google Scholar] [CrossRef] [PubMed]
- Malec, V.; Gottschald, O.R.; Li, S.; Rose, F.; Seeger, W.; Hänze, J. HIF-1α signaling is augmented during intermittent hypoxia by induction of the Nrf2 pathway in NOX1-expressing adenocarcinoma A549 cells. Free Radic. Biol. Med. 2010, 48, 1626–1635. [Google Scholar] [CrossRef] [PubMed]
- Martinive, P.; Defresne, F.; Bouzin, C.; Saliez, J.; Lair, F.; Grégoire, V.; Michiels, C.; Dessy, C.; Feron, O. Preconditioning of the tumor vasculature and tumor cells by intermittent hypoxia: Implications for anticancer therapies. Cancer Res. 2006, 66, 11736–11744. [Google Scholar] [CrossRef] [PubMed]
- Hsieh, C.-H.; Lee, C.-H.; Liang, J.-A.; Yu, C.-Y.; Shyu, W.-C. Cycling hypoxia increases U87 glioma cell radioresistance via ROS induced higher and long-term HIF-1 signal transduction activity. Oncol. Rep. 2010, 24, 1629–1636. [Google Scholar] [CrossRef]
- Yuan, G.; Nanduri, J.; Bhasker, C.R.; Semenza, G.L.; Prabhakar, N.R. Ca2+/calmodulin kinase-dependent activation of hypoxia inducible factor 1 transcriptional activity in cells subjected to intermittent hypoxia. J. Biol. Chem. 2005, 280, 4321–4328. [Google Scholar] [CrossRef]
- Chen, W.-L.; Wang, C.-C.; Lin, Y.-J.; Wu, C.-P.; Hsieh, C.-H. Cycling hypoxia induces chemoresistance through the activation of reactive oxygen species-mediated B-cell lymphoma extra-long pathway in glioblastoma multiforme. J. Transl. Med. 2015, 13, 389. [Google Scholar] [CrossRef]
- Cairns, R.A.; Kalliomaki, T.; Hill, R.P. Acute (cyclic) hypoxia enhances spontaneous metastasis of KHT murine tumors. Cancer Res. 2001, 61, 8903–8908. [Google Scholar] [PubMed]
- Liu, Y.; Song, X.; Wang, X.; Wei, L.; Liu, X.; Yuan, S.; Lv, L. Effect of chronic intermittent hypoxia on biological behavior and hypoxia-associated gene expression in lung cancer cells. J. Cell. Biochem. 2010, 111, 554–563. [Google Scholar] [CrossRef] [PubMed]
- Toffoli, S.; Feron, O.; Raes, M.; Michiels, C. Intermittent hypoxia changes HIF-1α phosphorylation pattern in endothelial cells: Unravelling of a new PKA-dependent regulation of HIF-1α. Biochim. Biophys. Acta - Mol. Cell Res. 2007, 1773, 1558–1571. [Google Scholar] [CrossRef] [PubMed]
- Moeller, B.J.; Cao, Y.; Li, C.Y.; Dewhirst, M.W. Radiation activates HIF-1 to regulate vascular radiosensitivity in tumors: Role of reoxygenation, free radicals, and stress granules. Cancer Cell 2004, 5, 429–441. [Google Scholar] [CrossRef]
- Schlaepfer, I.R.; Nambiar, D.K.; Ramteke, A.; Kumar, R.; Dhar, D.; Agarwal, C.; Bergman, B.; Graner, M.; Maroni, P.; Singh, R.P.; et al. Hypoxia induces triglycerides accumulation in prostate cancer cells and extracellular vesicles supporting growth and invasiveness following reoxygenation. Oncotarget 2015, 6, 22836–22856. [Google Scholar] [CrossRef] [PubMed]
- Galanis, A.; Pappa, A.; Giannakakis, A.; Lanitis, E.; Dangaj, D.; Sandaltzopoulos, R. Reactive oxygen species and HIF-1 signalling in cancer. Cancer Lett. 2008, 266, 12–20. [Google Scholar] [CrossRef] [PubMed]
- Granger, D.N.; Kvietys, P.R. Reperfusion injury and reactive oxygen species: The evolution of a concept. Redox Biol. 2015, 6, 524–551. [Google Scholar] [CrossRef] [PubMed]
- Glasauer, A.; Chandel, N.S. Targeting antioxidants for cancer therapy. Biochem. Pharmacol. 2014, 92, 90–101. [Google Scholar] [CrossRef]
- Huang, L.E.; Arany, Z.; Livingston, D.M.; Franklin Bunn, H. Activation of hypoxia-inducible transcription factor depends primarily upon redox-sensitive stabilization of its α subunit. J. Biol. Chem. 1996, 271, 32253–32259. [Google Scholar] [CrossRef]
- Nanduri, J.; Wang, N.; Yuan, G.; Khan, S.A.; Souvannakitti, D.; Peng, Y.-J.; Kumar, G.K.; Garcia, J.A.; Prabhakar, N.R. Intermittent hypoxia degrades HIF-2 via calpains resulting in oxidative stress: Implications for recurrent apnea-induced morbidities. Proc. Natl. Acad. Sci. 2009, 106, 1199–1204. [Google Scholar] [CrossRef]
- Bhaskara, V.K.; Mohanam, I.; Rao, J.S.; Mohanam, S. Intermittent Hypoxia Regulates Stem-like Characteristics and Differentiation of Neuroblastoma Cells. PLoS One 2012, 7, e30905. [Google Scholar] [CrossRef] [PubMed]
- Miao, Z.F.; Zhao, T.T.; Wang, Z.N.; Xu, Y.Y.; Mao, X.Y.; Wu, J.H.; Liu, X.Y.; Xu, H.; You, Y.; Xu, H.M. Influence of different hypoxia models on metastatic potential of SGC-7901 gastric cancer cells. Tumor Biol. 2014, 35, 6801–6808. [Google Scholar] [CrossRef] [PubMed]
- Zhu, H.; Wang, D.; Zhang, L.; Xie, X.; Wu, Y.; Liu, Y.; Shao, G.; Su, Z. Upregulation of autophagy by hypoxia-inducible factor-1α promotes EMT and metastatic ability of CD133+ pancreatic cancer stem-like cells during intermittent hypoxia. Oncol. Rep. 2014, 32, 935–942. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Liu, W.; Wang, L.; Zhu, T.; Zhong, J.; Xie, N. Hypoxia-inducible factor 1 mediates intermittent hypoxia-induced migration of human breast cancer MDA-MB-231 cells. Oncol. Lett. 2017, 14, 7715–7722. [Google Scholar] [CrossRef]
- Chou, C.; Wang, C.; Wu, C.; Lin, Y.; Lee, Y. Tumor cycling hypoxia induces chemoresistance in glioblastoma multiforme by upregulating the expression and function. Neuro-oncology 2012, 14, 1227–1238. [Google Scholar] [CrossRef] [PubMed]
- Martinez, C.A.; Kerr, B.; Jin, C.; Cistulli, P.A.; Cook, K.M. Obstructive sleep apnea activates HIF-1 in a hypoxia dose-dependent manner in HCT116 colorectal carcinoma cells. Int. J. Mol. Sci. 2019, 20, 445. [Google Scholar] [CrossRef] [PubMed]
- Conway, E.; Collen, D.; Carmeliet, P. Molecular mechanisms of blood vessel formation. Trends Biochem. Sci. 2001, 22, 251–256. [Google Scholar]
- Semenza, G.L.; Wang, G.L. A Nuclear Factor Induced by Hypoxia via De Novo Protein Synthesis Binds to the Human Erythropoietin Gene Enhancer at a Site Required for Transcriptional Activation. Mol. Cell. Biol. 1992, 12, 5447–5454. [Google Scholar] [CrossRef]
- Toffoli, S.; Michiels, C. Intermittent hypoxia is a key regulator of cancer cell and endothelial cell interplay in tumours. FEBS J. 2008, 275, 2991–3002. [Google Scholar] [CrossRef]
- Kochan-Jamrozy, K.; Króliczewski, J.; Moszyńska, A.; Collawn, J.F.; Bartoszewski, R. miRNA networks modulate human endothelial cell adaptation to cyclic hypoxia. Cell. Signal. 2019, 54, 150–160. [Google Scholar] [CrossRef]
- Gaustad, J.V.; Simonsen, T.G.; Roa, A.M.A.; Rofstad, E.K. Tumors exposed to acute cyclic hypoxia show increased vessel density and delayed blood supply. Microvasc. Res. 2013, 85, 10–15. [Google Scholar] [CrossRef] [PubMed]
- Rofstad, E.K.; Gaustad, J.; Egeland, T.A.M.; Mathiesen, B.; Galappathi, K. Tumors exposed to acute cyclic hypoxic stress show enhanced angiogenesis, perfusion and metastatic dissemination. Int. J. Cancer 2010, 127, 1535–1546. [Google Scholar] [CrossRef] [PubMed]
- Viscor, G.; Torrella, J.R.; Corral, L.; Ricart, A.; Javierre, C.; Pages, T.; Ventura, J.L. Physiological and biological responses to short-term intermittent hypobaric hypoxia exposure: From sports and mountain medicine to new biomedical applications. Front. Physiol. 2018, 9, 814. [Google Scholar] [CrossRef] [PubMed]
- Jolly, M.K.; Kulkarni, P.; Weninger, K.; Orban, J.; Levine, H. Phenotypic Plasticity, Bet-Hedging, and Androgen Independence in Prostate Cancer: Role of Non-Genetic Heterogeneity. Front. Oncol. 2018, 8, 50. [Google Scholar] [CrossRef] [PubMed]
- Hinohara, K.; Polyak, K. Intratumoral Heterogeneity: More Than Just Mutations. Trends Cell Biol. 2019, 29, 569–579. [Google Scholar] [CrossRef]
- Al-Hajj, M.; Wicha, M.S.; Benito-Hernandez, A.; Morrison, S.J.; Clarke, M.F. Prospective identification of tumorigenic breast cancer cells. Proc. Natl. Acad. Sci. 2003, 100, 3983–3988. [Google Scholar] [CrossRef]
- Mani, S.A.; Guo, W.; Liao, M.-J.; Eaton, E.N.; Ayyanan, A.; Zhou, A.Y.; Brooks, M.; Reinhard, F.; Zhang, C.C.; Shipitsin, M.; et al. The epithelial–mesenchymal transition generates cells with properties of stem cells. Cell 2008, 133, 704–715. [Google Scholar] [CrossRef]
- Araos, J.; Sleeman, J.P.; Garvalov, B.K. The role of hypoxic signalling in metastasis: Towards translating knowledge of basic biology into novel anti-tumour strategies. Clin Exp Metastasis 2018, 35, 563–599. [Google Scholar] [CrossRef]
- Yang, G.; Quan, Y.; Wang, W.; Fu, Q.; Wu, J.; Mei, T.; Li, J.; Tang, Y.; Luo, C.; Ouyang, Q.; et al. Dynamic equilibrium between cancer stem cells and non-stem cancer cells in human SW620 and MCF-7 cancer cell populations. Br. J. Cancer 2012, 106, 1512–1519. [Google Scholar] [CrossRef]
- Jolly, M.K.; Jia, D.; Boareto, M.; Mani, S.A.; Pienta, K.J.; Ben-Jacob, E.; Levine, H. Coupling the modules of EMT and stemness: A tunable ‘ stemness window ’ model. Oncotarget 2015, 6, 25161–25174. [Google Scholar] [CrossRef]
- Li, Z.; Bao, S.; Wu, Q.; Wang, H.; Eyler, C.; Sathornsumetee, S.; Shi, Q.; Cao, Y.; Lathia, J.; Mclendon, R.E.; et al. Hypoxia-Inducible Factors Regulate Tumorigenic Capacity of Glioma Stem Cells cancer stem cell specific molecules involved in neoangiogenesis, including HIF2α and its regulated factors. Cancer Cell 2009, 15, 501–513. [Google Scholar] [CrossRef] [PubMed]
- Louie, E.; Nik, S.; Chen, J.; Schmidt, M.; Song, B.; Pacson, C.; Chen, X.F.; Park, S.; Ju, J.; Chen, E.I. Identification of a stem-like cell population by exposing metastatic breast cancer cell lines to repetitive cycles of hypoxia and reoxygenation. Breast Cancer Res. 2010, 12, R94. [Google Scholar] [CrossRef] [PubMed]
- Alhawarat, F.M.; Hammad, H.M.; Hijjawi, M.S.; Sharab, A.S.; Abuarqoub, D.A.; Al Shhab, M.A.; Zihlif, M.A. The effect of cycling hypoxia on MCF-7 cancer stem cells and the impact of their microenvironment on angiogenesis using human umbilical vein endothelial cells (HUVECs) as a model. PeerJ 2019, 7, e5990. [Google Scholar] [CrossRef] [PubMed]
- Gupta, R.; Chetty, C.; Bhoopathi, P.; Lakka, S.; Mohanam, S.; Rao, J.S.; Dinh, D.H. Downregulation of uPA/uPAR inhibits intermittent hypoxia-induced epithelial–mesenchymal transition (EMT) in DAOY and D283 medulloblastoma cells. Int. J. Oncol. 2011, 38, 733–744. [Google Scholar] [PubMed]
- Liu, H.; Jiang, F.; Jia, X.; Lan, J.; Guo, H.; Li, E.; Yan, A.; Wang, Y. Cycling hypoxia affects cell invasion and proliferation through direct regulation of claudin1/claudin7 expression, and indirect regulation of P18 through claudin7. Oncotarget 2017, 8, 10298–10311. [Google Scholar] [CrossRef] [PubMed]
- Cairns, R.A.; Hill, R.P. Acute Hypoxia Enhances Spontaneous Lymph Node Metastasis in an Orthotopic Murine Model of Human Cervical Carcinoma. Cancer Res. 2004, 64, 2054–2061. [Google Scholar] [CrossRef] [PubMed]
- Chen, A.; Sceneay, J.; Gödde, N.; Kinwel, T.; Ham, S.; Thompson, E.W.; Humbert, P.O.; Möller, A.; Humbert, P.O. Intermittent hypoxia induces a metastatic phenotype in breast cancer. Oncogene 2018, 4214–4225. [Google Scholar] [CrossRef]
- Celià-Terrassa, T.; Kang, Y. Distinctive properties of metastasis-initiating cells. Genes Dev. 2016, 30, 892–908. [Google Scholar] [CrossRef]
- Nieto, M.A. Epithelial plasticity: A common theme in embryonic and cancer cells. Science 2013, 342, 1234850. [Google Scholar] [CrossRef]
- Yang, M.H.; Wu, M.Z.; Chiou, S.H.; Chen, P.M.; Chang, S.Y.; Liu, C.J.; Teng, S.C.; Wu, K.J. Direct regulation of TWIST by HIF-1α promotes metastasis. Nat. Cell Biol. 2008, 10, 295–305. [Google Scholar] [CrossRef]
- Jolly, M.K.; Ware, K.E.; Gilja, S.; Somarelli, J.A.; Levine, H. EMT and MET: Necessary or permissive for metastasis? Mol. Oncol. 2017, 11, 755–769. [Google Scholar] [CrossRef] [PubMed]
- Tripathi, S.; Levine, H.; Jolly, M.K. A mechanism for epithelial–mesenchymal heterogeneity in a population of cancer cells. bioRxiv 2019, 592691. [Google Scholar]
- Jia, W.; Deshmukh, A.; Mani, S.A.; Jolly, M.K.; Levine, H. A possible role for epigenetic feedback regulation in the dynamics of the Epithelial–mesenchymal Transition (EMT). Phys. Biol. 2019, in press. [Google Scholar] [CrossRef] [PubMed]
- Karacosta, L.G.; Anchang, B.; Ignatiadis, N.; Kimmey, S.C.; Benson, J.A.; Shrager, J.B.; Tibshirani, R.; Bendall, S.C.; Plevritis, S.K. Mapping lung cancer epithelial–mesenchymal transition states and trajectories with single-cell resolution. bioRxiv 2019, 570341. [Google Scholar]
- Celià-Terrassa, T.; Bastian, C.; Liu, D.D.; Ell, B.; Aiello, N.M.; Wei, Y.; Zamalloa, J.; Blanco, A.M.; Hang, X.; Kunisky, D.; et al. Hysteresis control of epithelial–mesenchymal transition dynamics conveys a distinct program with enhanced metastatic ability. Nat. Commun. 2018, 9, 5005. [Google Scholar] [CrossRef] [PubMed]
- Lehmann, S.; te Boekhorst, V.; Odenthal, J.; Bianchi, R.; van Helvert, S.; Ikenberg, K.; Ilina, O.; Stoma, S.; Xandry, J.; Jiang, L.; et al. Hypoxia Induces a HIF-1-Dependent Transition from Collective-to-Amoeboid Dissemination in Epithelial Cancer Cells. Curr. Biol. 2017, 27, 392–400. [Google Scholar] [CrossRef] [PubMed]
- Huang, B.; Jolly, M.K.; Lu, M.; Tsarfaty, I.; Ben-Jacob, E.; Onuchic, J.N. Modeling the Transitions between Collective and Solitary Migration Phenotypes in Cancer Metastasis. Sci. Rep. 2015, 5, 17379. [Google Scholar] [CrossRef] [PubMed]
- Rofstad, E.K.; Galappathi, K.; Mathiesen, B.; Ruud, E.B.M. Fluctuating and diffusion-limited hypoxia in hypoxia-induced metastasis. Clin. Cancer Res. 2007, 13, 1971–1978. [Google Scholar] [CrossRef] [PubMed]
- Rockwell, S.; Dobrucki, I.T.; Kim, E.Y.; Marrison, S.T.; Vu, V.T. Hypoxia and radiation therapy: Past history, ongoing research, and future promise. Curr. Mol. Med. 2009, 9, 442–458. [Google Scholar] [CrossRef]
- Reginato, M.; Karakashev, S. Progress toward overcoming hypoxia-induced resistance to solid tumor therapy. Cancer Manag. Res. 2015, 7, 253–264. [Google Scholar] [CrossRef]
- Gil, M.; Thomas, R.J. Apoptosis and Cancer; Humana Press: New Jersey, 2007; Volume 414, ISBN 1-59745-339-0. [Google Scholar]
- Plesca, D.; Mazumder, S.; Almasan, A. Chapter 6 DNA Damage Response and Apoptosis. Methods Enzymol. 2008; Volume 6879, 107–122ISBN 9780123744647. [Google Scholar]
- Williams, A.B.; Schumacher, B. p53 in the DNA-Damage-Repair Process. Cold Spring Harb. Perspect. Med. 2016, 6, a026070. [Google Scholar] [CrossRef] [PubMed]
- Shannon, A.M.; Bouchier-hayes, D.J.; Condron, C.M.; Toomey, D. Tumour hypoxia, chemotherapeutic resistance and hypoxia-related therapies. Cancer Treat. Rev. 2003, 7372, 297–307. [Google Scholar] [CrossRef]
- Graeber, T.G.; Osmanian, C.; Jacks, T.; Housman, D.E.; Koch, C.J.; Lowe, S.W.; Giaccia, A.J. Hypoxia-mediated selection of cells with diminished apoptotic potential in solid tumours. Nature 1996, 379, 88–91. [Google Scholar] [CrossRef] [PubMed]
- Verduzco, D.; Lloyd, M.; Xu, L.; Ibrahim-Hashim, A.; Balagurunathan, Y.; Gatenby, R.A.; Gillies, R.J. Intermittent Hypoxia Selects for Genotypes and Phenotypes That Increase Survival, Invasion, and Therapy Resistance. PLoS One 2015, 10, e0120958. [Google Scholar] [CrossRef] [PubMed]
- Jolly, M.K.; Somarelli, J.A.; Sheth, M.; Biddle, A.; Tripathi, S.C.; Armstrong, A.J.; Hanash, S.M.; Bapat, S.A.; Rangarajan, A.; Levine, H. Hybrid epithelial/mesenchymal phenotypes promote metastasis and therapy resistance across carcinomas. Pharmacol. Ther. 2019, 194, 161–184. [Google Scholar] [CrossRef] [PubMed]
- Hsieh, C.-H.; Wu, C.-P.; Lee, H.-T.; Liang, J.-A.; Yu, C.-Y.; Lin, Y.-J. NADPH oxidase subunit 4 mediates cycling hypoxia-promoted radiation resistance in glioblastoma multiforme. Free Radic. Biol. Med. 2012, 53, 649–658. [Google Scholar] [CrossRef] [PubMed]
- Rouschop, K.M.; Dubois, L.J.; Keulers, T.G.; van den Beucken, T.; Lambin, P.; Bussink, J.; van der Kogel, A.J.; Koritzinsky, M.; Wouters, B.G. PERK/eIF2 signaling protects therapy resistant hypoxic cells through induction of glutathione synthesis and protection against ROS. Proc. Natl. Acad. Sci. 2013, 110, 4622–4627. [Google Scholar] [CrossRef] [PubMed]
- Hlouschek, J.; Hansel, C.; Jendrossek, V.; Matschke, J. The Mitochondrial Citrate Carrier (SLC25A1) Sustains Redox Homeostasis and Mitochondrial Metabolism Supporting Radioresistance of Cancer Cells With Tolerance to Cycling Severe Hypoxia. Front. Oncol. 2018, 8, 170. [Google Scholar] [CrossRef]
- Hlouschek, J.; Ritter, V.; Wirsdörfer, F.; Klein, D.; Jendrossek, V.; Matschke, J. Targeting SLC25A10 alleviates improved antioxidant capacity and associated radioresistance of cancer cells induced by chronic-cycling hypoxia. Cancer Lett. 2018, 439, 24–38. [Google Scholar] [CrossRef]
- Matschke, J.; Riffkin, H.; Klein, D.; Handrick, R.; Lüdemann, L.; Metzen, E.; Shlomi, T.; Stuschke, M.; Jendrossek, V. Targeted Inhibition of Glutamine-Dependent Glutathione Metabolism Overcomes Death Resistance Induced by Chronic Cycling Hypoxia. Antioxid. Redox Signal. 2016, 25, 89–107. [Google Scholar] [CrossRef]
- Barsoum, I.B.; Koti, M.; Siemens, D.R.; Graham, C.H. Mechanisms of hypoxia-mediated immune escape in cancer. Cancer Res. 2014, 74, 7185–7190. [Google Scholar] [CrossRef] [PubMed]
- Hao, N.-B.; Lü, M.-H.; Fan, Y.-H.; Cao, Y.-L.; Zhang, Z.-R.; Yang, S.-M. Macrophages in Tumor Microenvironments and the Progression of Tumors. Clin. Dev. Immunol. 2012, 2012, 948098. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.-J.; Wu, S.; Yan, R.-M.; Fan, L.-S.; Yu, L.; Zhang, Y.-M.; Wei, W.-F.; Zhou, C.-F.; Wu, X.-G.; Zhong, M.; et al. The role of the hypoxia-Nrp-1 axis in the activation of M2-like tumor-associated macrophages in the tumor microenvironment of cervical cancer. Mol. Carcinog. 2018, 58, 388–397. [Google Scholar] [CrossRef] [PubMed]
- Wrzesinski, S.H.; Wan, Y.Y.; Flavell, R.A. Transforming growth factor-β and the immune response: Implications for anticancer therapy. Clin. Cancer Res. 2007, 13, 5262–5270. [Google Scholar] [CrossRef] [PubMed]
- Tellier, C.; Desmet, D.; Petit, L.; Finet, L.; Graux, C.; Raes, M.; Feron, O.; Michiels, C. Cycling Hypoxia Induces a Specific Amplified Inflammatory Phenotype in Endothelial Cells and Enhances Tumor-Promoting Inflammation In Vivo. Neoplasia 2015, 17, 66–78. [Google Scholar] [CrossRef] [PubMed]
- Gutsche, K.; Randi, E.B.; Blank, V.; Fink, D.; Wenger, R.H.; Leo, C.; Scholz, C.C. Intermittent hypoxia confers pro-metastatic gene expression selectively through NF-κB in inflammatory breast cancer cells. Free Radic. Biol. Med. 2016, 101, 129–142. [Google Scholar] [CrossRef] [PubMed]
- Bocci, F.; Gearhart-Serna, L.; Boareto, M.; Ribeiro, M.; Ben-Jacob, E.; Devi, G.R.; Levine, H.; Onuchic, J.N.; Jolly, M.K. Toward understanding cancer stem cell heterogeneity in the tumor microenvironment. Proc. Natl. Acad. Sci. USA 2019, 116, 148–157. [Google Scholar] [CrossRef]
- Gozal, D.; Almendros, I.; Hakim, F. Sleep apnea awakens cancer. Oncoimmunology 2014, 3, e28326. [Google Scholar] [CrossRef]
- Dewan, N.A.; Nieto, F.J.; Somers, V.K. Intermittent hypoxemia and OSA: Implications for comorbidities. Chest 2015, 147, 266–274. [Google Scholar] [CrossRef]
- Cao, J.; Feng, J.; Li, L.; Chen, B. Obstructive sleep apnea promotes cancer development and progression: A concise review. Sleep Breath. 2015, 19, 453–457. [Google Scholar] [CrossRef]
- Gildeh, N.; Drakatos, P.; Higgins, S.; Rosenzweig, I.; Kent, B.D. Emerging co-morbidities of obstructive sleep apnea: Cognition, kidney disease, and cancer. J. Thorac. Dis. 2016, 8, E901–E917. [Google Scholar] [CrossRef]
- Lim, D.C.; Pack, A.I. Obstructive Sleep Apnea: Update and Future. Annu. Rev. Med. 2017, 68, 99–112. [Google Scholar] [CrossRef] [PubMed]
- Ali, M.; Kowkuntla, S.; Delloro, D.J.; Galambos, C.; Hathi, D.; Janz, S.; Shokeen, M.; Tripathi, C.; Xu, H.; Yuk, J.; et al. Chronic intermittent hypoxia enhances disease progression in myeloma-resistant mice. Am. J. Physiol. Integr. Comp. Physiol. 2019, 316, R678–R686. [Google Scholar] [CrossRef] [PubMed]
- Gallego-Martin, T.; Farré, R.; Almendros, I.; Gonzalez-Obeso, E.; Obeso, A. Chronic intermittent hypoxia mimicking sleep apnoea increases spontaneous tumorigenesis in mice. Eur. Respir. J. 2017, 49, 1602111. [Google Scholar] [CrossRef] [PubMed]
- Hunyor, I.; Cook, K.M. Models of intermittent hypoxia and obstructive sleep apnea: Molecular pathways and their contribution to cancer. Am. J. Physiol. Integr. Comp. Physiol. 2018, 315, R669–R687. [Google Scholar] [CrossRef] [PubMed]
- Almendros, I.; Montserrat, J.M.; Ramirez, J.; Torres, M.; Duran-Cantolla, J.; Navajas, D.; Farre, R. Intermittent hypoxia enhances cancer progression in a mouse model of sleep apnoea. Eur. Respir. J. 2012, 39, 215–217. [Google Scholar] [CrossRef] [PubMed]
- Yoon, D.W.; So, D.; Min, S.; Kim, J.; Lee, M.; Roza, K.; Park, J.-W.; Shin, H.-W. Accelerated tumor growth under intermittent hypoxia is associated with hypoxia-inducible factor-1-dependent adaptive responses to hypoxia. Sleep Med. 2017, 40, e356. [Google Scholar]
- Almendros, I.; Wang, Y.; Becker, L.; Lennon, F.E.; Zheng, J.; Coats, B.R.; Schoenfelt, K.S.; Carreras, A.; Hakim, F.; Zhang, S.X.; et al. Intermittent hypoxia-induced changes in tumor-associated macrophages and tumor malignancy in a mouse model of sleep apnea. Am. J. Respir. Crit. Care Med. 2014, 189, 593–601. [Google Scholar] [CrossRef]
- Li, X.; Jolly, M.K.; George, J.T.; Pienta, K.J.; Levine, H. Computational Modeling of the Crosstalk Between Macrophage Polarization and Tumor Cell Plasticity in the Tumor Microenvironment. Front. Oncol. 2019, 9, 10. [Google Scholar] [CrossRef]
- Almendros, I.; Gileles-Hillel, A.; Khalyfa, A.; Wang, Y.; Zhang, S.X.; Carreras, A.; Farré, R.; Gozal, D. Adipose tissue macrophage polarization by intermittent hypoxia in a mouse model of OSA: Effect of tumor microenvironment. Cancer Lett. 2015, 361, 233–239. [Google Scholar] [CrossRef]
- Vilaseca, A.; Campillo, N.; Torres, M.; Musquera, M.; Gozal, D.; Montserrat, J.M.; Alcaraz, A.; Touijer, K.A.; Farré, R.; Almendros, I. Intermittent hypoxia increases kidney tumor vascularization in a murine model of sleep apnea. PLoS One 2017, 12, e0179444. [Google Scholar] [CrossRef] [PubMed]
- Campillo, N.; Torres, M.; Vilaseca, A.; Nonaka, P.N.; Gozal, D.; Roca-Ferrer, J.; Picado, C.; Montserrat, J.M.; Farré, R.; Navajas, D.; et al. Role of cyclooxygenase-2 on intermittent hypoxia-induced lung tumor malignancy in a mouse model of sleep apnea. Sci. Rep. 2017, 7, 44693. [Google Scholar] [CrossRef] [PubMed]
- Cubillos-Zapata, C.; Hernández-Jiménez, E.; Avendaño-Ortiz, J.; Toledano, V.; Varela-Serrano, A.; Fernández-Navarro, I.; Casitas, R.; Carpio, C.; Aguirre, L.A.; García-Río, F.; et al. Obstructive Sleep Apnea Monocytes Exhibit High Levels of Vascular Endothelial Growth Factor Secretion, Augmenting Tumor Progression. Mediators Inflamm. 2018, 2018, 7373921. [Google Scholar] [CrossRef] [PubMed]
- Cubillos-Zapata, C.; Avendaño-Ortiz, J.; Hernandez-Jimenez, E.; Toledano, V.; Casas-Martin, J.; Varela-Serrano, A.; Torres, M.; Almendros, I.; Casitas, R.; Fernández-Navarro, I.; et al. Hypoxia-induced PD-L1/PD-1 crosstalk impairs T-cell function in sleep apnoea. Eur. Respir. J. 2017, 50, 1700833. [Google Scholar] [CrossRef] [PubMed]
- Huang, M.-H.; Zhang, X.-B.; Wang, H.-L.; Li, L.-X.; Zeng, Y.-M.; Wang, M.; Zeng, H.-Q. Intermittent hypoxia enhances the tumor programmed death ligand 1 expression in a mouse model of sleep apnea. Ann. Transl. Med. 2019, 7, 97. [Google Scholar] [CrossRef]
- Akbarpour, M.; Khalyfa, A.; Qiao, Z.; Gileles-Hillel, A.; Almendros, I.; Farré, R.; Gozal, D. Altered CD8+ t-cell lymphocyte function and tc1 cell stemness contribute to enhanced malignant tumor properties in murine models of sleep apnea. Sleep 2017, 40, zsw040. [Google Scholar] [CrossRef] [PubMed]
- Tripathi, S.C.; Peters, H.L.; Taguchi, A.; Katayama, H.; Wang, H.; Momin, A.; Jolly, M.K.; Celiktas, M.; Rodriguez-Canales, J.; Liu, H.; et al. Immunoproteasome deficiency is a feature of non-small cell lung cancer with a mesenchymal phenotype and is associated with a poor outcome. Proc. Natl. Acad. Sci. 2016, 113, E1555–E1564. [Google Scholar] [CrossRef] [PubMed]
- Noman, M.Z.; Van Moer, K.; Marani, V.; Gemmill, R.M.; Tranchevent, L.C.; Azuaje, F.; Muller, A.; Chouaib, S.; Thiery, J.P.; Berchem, G.; et al. CD47 is a direct target of SNAI1 and ZEB1 and its blockade activates the phagocytosis of breast cancer cells undergoing EMT. Oncoimmunology 2018, 7, e1345415. [Google Scholar] [CrossRef]
- Chae, Y.K.; Chang, S.; Ko, T.; Anker, J.; Agte, S.; Iams, W.; Choi, W.M.; Lee, K.; Cruz, M. Epithelial–mesenchymal transition (EMT) signature is inversely associated with T-cell infiltration in non-small cell lung cancer (NSCLC). Sci. Rep. 2018, 8, 2918. [Google Scholar] [CrossRef]
- Gaoatswe, G.; Kent, B.D.; Corrigan, M.A.; Nolan, G.; Hogan, A.E.; McNicholas, W.T.; O’Shea, D. Invariant Natural Killer T Cell Deficiency and Functional Impairment in Sleep Apnea: Links to Cancer Comorbidity. Sleep 2015, 38, 1629–1634. [Google Scholar] [CrossRef]
- Kohn, K.W.; Riss, J.; Aprelikova, O.; Weinstein, J.N.; Pommier, Y.; Barrett, J.C. Properties of Switch-like Bioregulatory Networks Studied by Simulation of the Hypoxia Response Control System. Mol. Biol. Cell 2004, 15, 3042–3052. [Google Scholar] [CrossRef] [PubMed]
- Cavadas, M.A.S.; Nguyen, L.K.; Cheong, A. Hypoxia-inducible factor (HIF) network: Insights from mathematical models. Cell Commun. Signal. 2013, 11, 42. [Google Scholar] [CrossRef] [PubMed]
- Dhawan, A.; Madani Tonekaboni, S.A.; Taube, J.H.; Hu, S.; Sphyris, N.; Mani, S.A.; Kohandel, M. Mathematical modelling of phenotypic plasticity and conversion to a stem-cell state under hypoxia. Sci. Rep. 2016, 6, 18074. [Google Scholar] [CrossRef] [PubMed]
- Leedale, J.; Herrmann, A.; Bagnall, J.; Fercher, A.; Papkovsky, D.; Sée, V.; Bearon, R.N. Modeling the dynamics of hypoxia inducible factor-1α (HIF-1α) within single cells and 3D cell culture systems. Math. Biosci. 2014, 258, 33–43. [Google Scholar] [CrossRef] [PubMed]
- Bagnall, J.; Leedale, J.; Taylor, S.E.; Spiller, D.G.; White, M.R.H.; Sharkey, K.J.; Bearon, R.N.; Sée, V. Tight control of hypoxia-inducible factor-α transient dynamics is essential for cell survival in hypoxia. J. Biol. Chem. 2014, 289, 5549–5564. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Cao, S.; Xu, Y. Population dynamics inside cancer biomass driven by repeated hypoxia–reoxygenation cycles. Quant. Biol. 2014, 2, 85–99. [Google Scholar] [CrossRef]
- Li, M.; Tang, Y.; Yao, J. Photoacoustic tomography of blood oxygenation: A mini review. Photoacoustics 2018, 10, 65–73. [Google Scholar] [CrossRef] [PubMed]
- Papkovsky, D.B.; Dmitriev, R.I. Imaging of oxygen and hypoxia in cell and tissue samples. Cell. Mol. Life Sci. 2018, 75, 2963–2980. [Google Scholar] [CrossRef]
- Ron, A.; Deán-Ben, X.L.; Gottschalk, S.; Razansky, D. Volumetric optoacoustic imaging unveils higH–Resolution patterns of acute and cyclic hypoxia in a murine model of breast cancer. Cancer Res. 2019, canres.3769.2018. [Google Scholar] [CrossRef]
- Acosta, M.A.; Jiang, X.; Huang, P.K.; Cutler, K.B.; Grant, C.S.; Walker, G.M.; Gamcsik, M.P. A microfluidic device to study cancer metastasis under chronic and intermittent hypoxia. Biomicrofluidics 2014, 8. [Google Scholar] [CrossRef]
- Hsieh-Fu, T.; Alen, T.; Shen, A.Q.; Gang, B. Tumour-on-a-chip: Microfluidic models of tumour morphology, growth and microenvironment. J. R. Soc. Interface 2017, 14, 20170137. [Google Scholar]
- Lam, S.F.; Shirure, V.S.; Chu, Y.E.; Soetikno, A.G.; George, S.C. Microfluidic device to attain high spatial and temporal control of oxygen. PLoS ONE 2018, 13, e0209574. [Google Scholar] [CrossRef] [PubMed]
- Reiterer, M.; Colaço, R.; Emrouznejad, P.; Jensen, A.; Rundqvist, H.; Johnson, R.S.; Branco, C. Acute and chronic hypoxia differentially predispose lungs for metastases. Sci. Rep. 2019, 9, 10246. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| SN. | Cell Line | Conditions of Hypoxia | HIF-1α Stability | HIF-2α Stability | HIF-1α vs HIF-2α | Ref. |
|---|---|---|---|---|---|---|
| 1 | SK-NBE(2) | 1% O2, 4 h and 72 h | Stabilized at 4 h, absent at 72 h | Stabilized at 4 h and 72 h | Greater HIF-2α expression at 4 h and 72 h hypoxia | [42] |
| 2 | SK-NBE(2), KCN-69n | 1% and 5% O2, 2–72 h | Stabilized at 1 % O2 after 2 h then gradually decreased, undetected at 5% O2 | Stabilized at 1% and 5% O2 after 2 h then gradually increased | HIF-1α stabilized under acute hypoxia, HIF-2α stabilized under chronic hypoxia | [38] |
| 3 | T24 and J82 | 1% O2, 0–48 h | Stabilized at 6 h, then gradually decreased | Stabilized at 6 h, then gradually increased | HIF-1α stabilized under acute hypoxia, HIF-2α stabilized under chronic hypoxia | [52] |
| 4 | SK-N-BE(2)C, IMR32 SK-N-ER, SH-SY5Y | 1% O2, 24 h and 72 h | Stabilized at 24 h | Stabilized at 24 h and 72 h | HIF-1α stabilized under acute hypoxia, HIF-2α stabilized under chronic hypoxia | [43] |
| 5 | PC-3, DU145, LNCaP | 1% O2, 2–24 h | Stabilized at 0.5–6 h, absent at 24 h | NA | HIF-1α active during acute hypoxia | [53] |
| 6 | MCF7 | 1% O2, 4–72 h | Stabilized at 4–8 h, decreased after 24 h | Stabilized at 24 h | HIF-1α stabilized under acute hypoxia, HIF-2α stabilized under chronic hypoxia | [54] |
| 7 | A549 cells | 0.5% O2, 4 h and 12 h | Stabilized at 4 h, then gradually decreased | Stabilized at 4–12 h | HIF-1α stabilized under acute hypoxia, HIF-2α stabilized under chronic hypoxia | [41] |
| 8 | HEK-293, MCF7, MDA-MB-231, MCF10A | 1% O2,0–72 h | Stabilized at 4–16 h, then gradually decreased | NA | NA | [49] |
| SN. | Cell Line | Intermittent Hypoxia (IH) (O2 Level, H-Duration, R-Duration, no. of Cycles) | Chronic /Continuous Hypoxia (CH) (O2 Level, H-Duration) | HIF-1α Stability | IH vs CH | Mechanism of HIF-1α Activation | Ref. |
|---|---|---|---|---|---|---|---|
| 1 | EAhy9, HUVEC | 1%, 1 h, 30 min, 4 | 1%, 5.5 h | Stabilized during hypoxia and degraded during reoxygenation | Greater migration and tubulogenesis of endothelial cells under IH | NA | [44] |
| 2 | HUVEC | <1%, 1 h, 30 min, 3 | <1%, 3 h | Progressively@ stabilized and accumulated during hypoxia; degraded during reoxygenation | Higher stability and activity of HIF-1α under IH | Mitochondrial respiration/ PI3K/AKT | [55] |
| 3 | A549 | 1%, 2 h, 2 h, multiple cycles for 6 h | 1%, 6 h | Stabilized, highest HIF-1α levels after third hypoxia period of IH | NA | NOX1/NRF2 | [56] |
| 4 | HUVE, BAOEC | 0.5–1%, 1 h, 30 min, 3 | <1%, 3 h and 6 h | Progressively@ stabilized and accumulated during hypoxia; degraded during reoxygenation | NA | NA | [57] |
| 5 | U87 | 0.5–1%, 1 h, 30 min, 3 | 0.5–1%, 4 h | Stabilized after 3 H-R cycles | Prolonged HIF-1α activity under IH | ROS dependent | [58] |
| 6 | PC12 | 1.5%, 30 s, 4 min, 60 | 1.5%, 1 h | Stabilized after 60 H-R cycles | Higher HIF-1α activity under IH | Transactivation by CaM kinase | [59] |
| 7 | U251, U87 | 0.5–1%, 1 h, 30 min, 3 | 0.5–1%, 3 h | Stabilized after 3 H-R cycles | Greater induction of Bcl-XL by HIF-1α under IH | ROS dependent | [60] |
| 8 | EAhy9, HMEC-1 | 1%,1 h, 30 min, 4 | 1%, 5.5 h | Progressively@ increase in phosphorylated HIF-1α. Highest expression at the 4th hypoxia period | PKA mediated phosphorylation of HIF-1α under IH | PKA | [63] |
| 9 | NB1691 | 1%, 24 h, 24 h, 10 | 1%, 24 h | Stabilized after 10 H-R cycles | Greater HIF-1α and HIF-2α stabilization under IH | NA | [71] |
| 10 | SGC-7901 | 1%, 12 h, 12 h, for 168 h | 1%, for 168 h | Stabilized between 48 h and 168 h | Greater nuclear HIF-1α intensity, GLUT-1 and OCT-4 expression under IH | NA | [72] |
| 11 | Panc-1, BxPC-3 | 1%, 12 h, 12 h, 5 | NA | Highest levels at 72 h | NA | NA | [73] |
| 12 | MDA-MB-231 | 1%, 12 h, 12 h, 2 | 1%, 48 h | Stabilized during hypoxia and degraded during reoxygenation | Greater migration under IH | NA | [74] |
| 13 | U87, GBM8401 | 0.5–1%, 1 h, 30 min, 3 | 1%, 4 h | Stabilized (assayed after 3 cycles of H-R) | Greater stability and activity of HIF-1α under IH | NA | [75] |
| 14 | HCT116 | 5 min 59 mmHg O2, 5 min 0 mm Hg, for 6 h or 18 h | 4 mmHg O2, 6 h or 18 h | Stabilized (assayed after 6 h of H-R) | Greater stability of HIF-1α under CH | NA | [76] |
| SN. | Cell Line/Mouse Model | Intermittent Hypoxia | Chronic Hypoxia | Effect of IH | Ref. |
|---|---|---|---|---|---|
| 1 | A-07 xenograft model # | 8% O2, 10 min H; 10 min R, 12 cycles, once per day, 7 days per week till tumor volume reached 100 µm | NA | Increased angiogenesis, perfusion, vascular density | [81] |
| A-07 $ | 10–100 ppm O2, 30 min H, 30 min R, 6 cycles | 10–10 ppm O2, 6 h | Increased VEGF secretion but no effect on lung metastasis | ||
| 2 | EAhy926, HUVEC, BAOEC $ | 0.5–1% O2, 1 h H; 30 min R, 3–4 cycles | 1% O2, 5.5 h | Increased migration and tubulogenesis, increased survival under proapoptotic stimuli | [57], [44] |
| SN. | Cell line/Mouse Model | Intermittent Hypoxia | Chronic Hypoxia | Effect of IH | Important Markers | Ref. |
|---|---|---|---|---|---|---|
| 1 | MDA-MB-231 and BCM2 $ | 1% O2, 7 days H, 1–3 weeks R, 3 cycles | NA | Expansion of stem like cancer cells (CD44+/CD24-/ESA+) with high tumor initiating capability, metastasis and EMT | CH44, CD24, ESA, CDH1, SNAIL, SLUG, TWIST, miR200c, miR205 | [92] |
| 2 | NB1691 $ | 1% O2, 24 h H, 24 h R, 1, 5 or 10 cycles | NA | Enhanced stem like properties with suppressed differentiation | VEGF, OCT4, CD133, ID-2, HES1, c-Kit, Notch1, NPY, HASH-1, dHAND, Neu N, NF-M | [71] |
| 3 | SGC-7901 $ | 1% O2, 12 h H, 12 h R, 48 h | 1% O2, 48 h | Increased stem-like/progenitor properties with enhanced self-renewal, invasion and EMT | GLUT1, CDH1, α-SMA, OCT4 | [72] |
| 4 | Panc-1 and BxPC-3 $ | 1% O2, 12 h H, 12 h R, 5 cycles | NA | Increased stem like cells with increased EMT, invasion, migration and autophagy | CD133, CDH1, Vimentin, CDH2, OCT4, SOX2, Beclin-1, ATG-5, LC3-II, LC3-1 | [73] |
| 5 | MCF-7 and HUVEC $ | 1% O2, 8 h hypoxia, 3 times a week, multiple shots | 1% O2, 72 h, once per week | Expansion of stem like population with elevated chemoresistance and capability to induce angiogenesis | CD44, CD24, VEGF | [93] |
| 6 | MDA-MB-231 # | 1% O2, 12 h H, 12 h R, 10 cycles | 1% O2, 48 h | Increased migration and vimentin expression | Vim. | [74] |
| 7 | DAOY, D283 and HMEC # | 1% O2, 48 h H, 48 h R, 18–20 cycles | 1% O2, 48 h | Enhanced EMT, cell invasion, migration and angiogenesis | SNAIL, Vim., CDH2, CDH1, Zo-1 | [94] |
| 8 | CNE1 and CNE2 # | 0.1% O2, 8 h H, 2 to 8 h R | NA | Increased cell proliferation and decreased invasion | NA | [95] |
| 9 | KHT murine fibrosarcoma # | 2–7% breathing O2, 10 min H, 10 min R, 12 cycles, 7 days per week | 5–7% O2 for 2 h | Greater spontaneous lung metastases | NA | [61] |
| 10 | ME-180 xenograft mouse model # | 7% breathing O2, 10 min H, 10 min R, 12 cycles, 21 days | NA | Greater lymph node metastasis and reduced tumor growth | NA | [96] |
| 11 | PyMT-WT Luciferase/Cherry cells # | 1% O2, 24 h H, 24 h R, 9 days | 1%O2, 9 days | Higher tumor initiating capability and metastatic potential | VEGF, MMP2, MMP9, HIF1, Aldh1, Pai, ELF5, GATA3, CH24, CH44, CD14, SCA1 | [97] |
| SN. | Cell Line/Mouse Model | Intermittent Hypoxia | Chronic Hypoxia | Effect of IH | IH vs CH | Ref. |
|---|---|---|---|---|---|---|
| 1 | U251 and U87 $ | 0.5–1% O2, 1 h H; 30 min R; 3 cycles | 0.5–1% O2, 3 h | Resistance to temozolomide treatment mediated by Bcl-xL | Greater chemoresistance under IH | [60] |
| 2 | U87 and GBM8401 $ | 0.5–1% O2, 1 h H, 30 min R, 3 cycles | 1% O2 for 4 h | Resistance to doxorubicin and BCNU treatment mediated by ABCB1 | Greater chemoresistance under IH | [75] |
| 3 | TLT xenograft model # | 7% O2, 1 h H, 30 min R, 3 cycles | 7% O2, 3 h | Increased radioresistance and tumor regrowth | Greater tumor cell and vasculature Radioresistance under IH | [57] |
| FsaII and B16-F10 $ | <1% O2, 1 h H, 30 min R, 3 cycles | NA | Increased radioresistance | NA | ||
| 4 | A549 and NCI-H446 $ | 0.1% O2, 24 h H, 72 h R, 20 cycles | 0.1% O2, 16 h | Radioresistance promoted by increased S phase proliferation | Greater radioresistance shown by IH conditioned cells | [62] |
| 5 | U87 cells $ | 0.5–1% O2, 1 h H, 30 min R, 3 cycles | 0.5–1% O2 for 4 h | Increased radioresistance | Greater radioresistance shown by IH conditioned cells | [58] |
| U87 xenograft with regulatable HIF-1 # | 7% O2, 1 h H, 30 min R, 3 cycles | 7% O2, 4 h | Increased radioresistance and tumor regrowth | Greater surviving fraction and tumor regrowth in mice treated with IH | ||
| 6 | GBM8401 and U251 $ | 0.5–1% O2, 10 min H, 10 min R, 12 cycles | 0.5–1% O2, 4 h | Radioresistance mediated by NOX4 | Greater NOX4 induction and ROS production under IH | [118] |
| 7 | U373-MG and HCT116 $ | <0.02% O2, 1 h H, 1 h R, 2–5 cycles | NA | Radioresistance mediated by PERK/eIF2a | NA | [119] |
| 8 | NCI-H460, DU145 and T98G $ | <1% O2, 48 h H, 120 h R, 16 and 25 cycles | NA | Radioresistance mediated by SLC25A1 | NA | [120] |
| 19 | NCI-H460, DU145 and T98G $ | <1% O2, 48 h H, 120 h R, 16 and 25 cycles | NA | Radioresistance mediated by SLC25A10 | NA | [121] |
| 10 | NCH-H460, DU145 and T98G $ | <0.1% O2, 48 h H, 120 h R, 16 or 25 cycles | NA | Increased radioresistance mediated by GOT1 | NA | [122] |
| SN. | Cell Line/Mouse Model | Intermittent Hypoxia | Chronic Hypoxia | Effect of IH | Ref. |
|---|---|---|---|---|---|
| 1 | EAhy926 and HUVEC $ | 1% O2, 1 h H, 30 min R, 4 cycles | 1% O2, 6 h | Amplified proangiogenic phenotype, higher THP-1 monocyte adhesion through NF-κB | [127] |
| LLC mouse model # | 7% O2, 1 h H, 30 min R, 3 cycles | NA | Enhanced inflammation in tumors with increased PTGS-2, IL-6, CXCL1 and macrophage inflammatory protein | ||
| 2 | SUM149PT $ | 0.2% O2, 1-day H, 3 days R, 15 cycles | NA | NF-κB mediated enhanced expression of prometastatic and proangiogenic factors | [128] |
| 3 | TC1 mouse model $ | Decreasing oxygen pressure (pO2-84 mmHg) for 4 h followed by slow recovery under normal air, 30 days | NA | Increased protumor effects of TAMs mediated by NRP-1, increase in NRP-1 levels and infiltration of CD206+ macrophages in tumor | [125] |
| SN. | Cell Line/Mouse Model | Intermittent Hypoxia | Chronic Hypoxia | Effect of IH | Ref. |
|---|---|---|---|---|---|
| 1 | OSA mouse model of melanoma (B16F10) # | 5% O2, 20 s H, 40 s R, 60 cycles per h, 6 h per day for 14 days | NA | Enhanced tumor growth | [138] |
| 2 | 5TGM1 $ | 1.5% O2, 30 s H, 4 min R, 60 cycle | 1.5% O2, 1 h | Reduced growth and proliferation | [135] |
| OSA mouse model of myeloma resistant cells (5TGM1) # | 10% O2, 2 min H, 2 min R, 12cycles per h, 10 h/day, for 4 weeks | NA | Increased multiple myeloma progression with bone marrow engraftment | ||
| 3 | HCT116 $ | 5 min 59 mmHg O2, 5 min 0mm Hg, 6 h or 18 h | 4 mmHg O2, 6 h or 18 h | Stabilization and activation of HIF-1α in dose dependent manner | [76] |
| 4 | RENCA, HAEC $ | 1% O2, 30 s H, 30 s R, 24 h | NA | Increased VEGF secretion by RENCA cells | [143] |
| OSA mouse model of kidney adenocarcinoma (RENCA cells) # | 5% O2, 20 s H, 40 s R, 60 cycles per h, 6 h per day for 14 days | NA | Increased angiogenesis and macrophage infiltration | ||
| 5 | B16F10 and TC1 $ | 5% O2, 30 min H, 30 min R, 48 h | NA | Increased tumor proliferation in presence of RAW 264.7 | [140] |
| OSA mouse model of lung cancer (TC1) # | 6% FiO2, 90 s H, 90 s R, 20 cycles per h, 12 h per day for 4 weeks | NA | Shift from M1 to M2 phenotype which increased invasion and migration of tumor cells | ||
| 6 | OSA mouse model of lung cance (TC1) # | 6% FiO2, 90 s H, 90 s R, 20 cycles per h, 12 h per day for 4 weeks | NA | Higher macrophage infiltration, differential effect of IH on adipose tissue | [142] |
| 7 | LLC1 and RAW 264.7 $ | 1% O2, 30 s H, 30 s R, 24 h | NA | Increased PGE2 secretion | [144] |
| OSA mouse model of lung cancer (LLC1) # | 5% O2, 20 s H, 40 s R, 60 cycles per h, 6 h per day till tumor was palpable | NA | COX-2 dependent shift from M1 to M2 phenotype, higher growth and invasion of tumor cells | ||
| 8 | Monocytes derived from OSA patients and healthy individuals $ | 3% O2, 5 min H, 10 min R, 12 cycles, 4 h | NA | Tumor promoting environment mediated by VEGF in HIF-1α dependent manner | [145] |
| 9 | Monocytes and T cells derived from healthy individuals $ | 3% O2, 5 min H, 10 min R, 12 cycles, 4 h | NA | PD-L1 overexpressed in monocytes, PD-1 overexpressed in CD8+ T cells | [146] |
| OSA mice model # | 5% O2, 20 s H, 40 s R, 60 cycles per h, 6 h per day till tumor was palpable | NA | PD-L1 overexpressed in monocytes, PD-1 overexpressed in CD8+ T cells | ||
| 10 | OSA mouse model of lung cancer (LLC) # | 6% O2, 70 s H, 50 s R, 8 h per day, for 5 weeks | NA | Increased expression of HIF-1α and PD-L1 with positive correlation | [147] |
| 11 | OSA mouse model of lung cancer (TC1) # | 6% FiO2, 90 s H, 90 s R, 20 cycles per h, 12 h per day till the tumor is palpable | NA | Increased tumor growth and invasion, reduced granzyme-B producing CD8+ cells, increased OCT4+ CSC population | [148] |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Saxena, K.; Jolly, M.K. Acute vs. Chronic vs. Cyclic Hypoxia: Their Differential Dynamics, Molecular Mechanisms, and Effects on Tumor Progression. Biomolecules 2019, 9, 339. https://doi.org/10.3390/biom9080339
Saxena K, Jolly MK. Acute vs. Chronic vs. Cyclic Hypoxia: Their Differential Dynamics, Molecular Mechanisms, and Effects on Tumor Progression. Biomolecules. 2019; 9(8):339. https://doi.org/10.3390/biom9080339
Chicago/Turabian StyleSaxena, Kritika, and Mohit Kumar Jolly. 2019. "Acute vs. Chronic vs. Cyclic Hypoxia: Their Differential Dynamics, Molecular Mechanisms, and Effects on Tumor Progression" Biomolecules 9, no. 8: 339. https://doi.org/10.3390/biom9080339
APA StyleSaxena, K., & Jolly, M. K. (2019). Acute vs. Chronic vs. Cyclic Hypoxia: Their Differential Dynamics, Molecular Mechanisms, and Effects on Tumor Progression. Biomolecules, 9(8), 339. https://doi.org/10.3390/biom9080339