The Michael J. Fox Foundation for Parkinson’s Research Strategy to Advance Therapeutic Development of PINK1 and Parkin

{kind=link}
{kind=link}
{kind=link}
Abstract
1. Background
2. Mitochondrial Dysfunction in PD
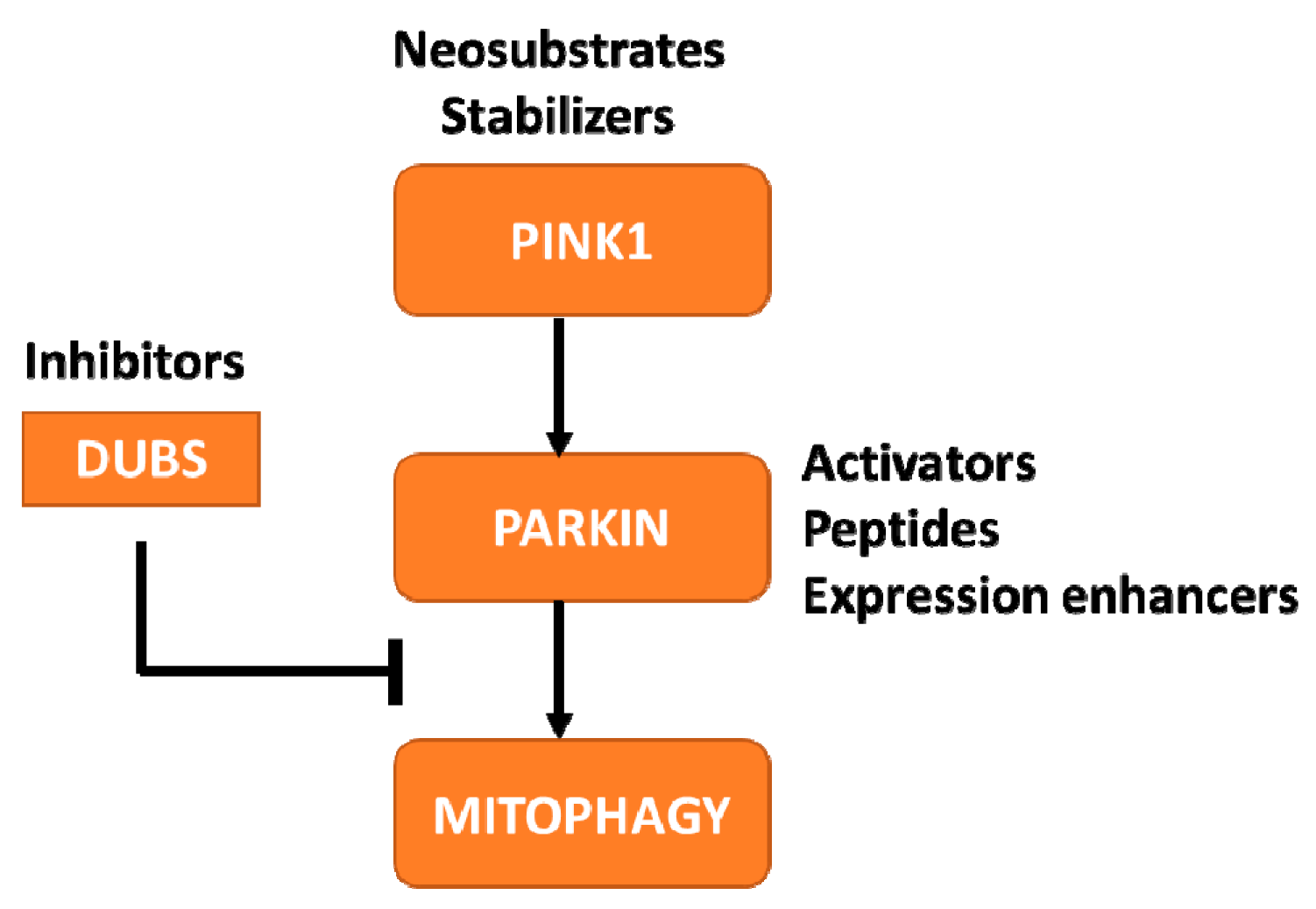
3. PINK1 and Parkin PD
- Increase understanding of the biological function of PINK1 and Parkin in PD
- Facilitate the development of PINK1-Parkin pathway-targeting therapies
- Develop, optimize and validate biomarker assays to assess PINK1-Parkin pathway activation
- Develop, characterize and distribute preclinical tools and animal models to facilitate PD research
3.1. Increase Understanding of the Biological Function of PINK1 and Parkin in PD
3.2. Facilitate the Development of PINK1-Parkin Pathway-Targeting Therapies
3.3. Develop, Optimize and Validate Biomarker Assays to Assess PINK1-Parkin Pathway Activation
3.4. Develop, Characterize and Distribute Preclinical Tools and Animal Models to Facilitate PD Research
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Langston, J.W. The Parkinson’s complex: Parkinsonism is just the tip of the iceberg. Ann. Neurol. 2006, 59, 591–596. [Google Scholar] [CrossRef]
- Paisan-Ruiz, C.; Jain, S.; Evans, E.W.; Gilks, W.P.; Simon, J.; van der Brug, M.; Lopez de Munain, A.; Aparicio, S.; Gil, A.M.; Khan, N.; et al. Cloning of the gene containing mutations that cause PARK8-linked Parkinson’s disease. Neuron 2004, 44, 595–600. [Google Scholar] [CrossRef] [PubMed]
- Zimprich, A.; Biskup, S.; Leitner, P.; Lichtner, P.; Farrer, M.; Lincoln, S.; Kachergus, J.; Hulihan, M.; Uitti, R.J.; Calne, D.B.; et al. Mutations in LRRK2 cause autosomal-dominant parkinsonism with pleomorphic pathology. Neuron 2004, 44, 601–607. [Google Scholar] [CrossRef] [PubMed]
- Stafa, K.; Tsika, E.; Moser, R.; Musso, A.; Glauser, L.; Jones, A.; Biskup, S.; Xiong, Y.; Bandopadhyay, R.; Dawson, V.L.; et al. Functional interaction of Parkinson’s disease-associated LRRK2 with members of the dynamin GTPase superfamily. Hum. Mol. Genet. 2014, 23, 2055–2077. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Yan, M.H.; Fujioka, H.; Liu, J.; Wilson-Delfosse, A.; Chen, S.G.; Perry, G.; Casadesus, G.; Zhu, X. LRRK2 regulates mitochondrial dynamics and function through direct interaction with DLP1. Hum. Mol. Genet. 2012, 21, 1931–1944. [Google Scholar] [CrossRef] [PubMed]
- Greggio, E.; Jain, S.; Kingsbury, A.; Bandopadhyay, R.; Lewis, P.; Kaganovich, A.; van der Brug, M.P.; Beilina, A.; Blackinton, J.; Thomas, K.J.; et al. Kinase activity is required for the toxic effects of mutant LRRK2/dardarin. Neurobiol. Dis. 2006, 23, 329–341. [Google Scholar] [CrossRef] [PubMed]
- Steger, M.; Tonelli, F.; Ito, G.; Davies, P.; Trost, M.; Vetter, M.; Wachter, S.; Lorentzen, E.; Duddy, G.; Wilson, S.; et al. Phosphoproteomics reveals that Parkinson’s disease kinase LRRK2 regulates a subset of Rab GTPases. Elife 2016, 5. [Google Scholar] [CrossRef] [PubMed]
- Guaitoli, G.; Raimondi, F.; Gilsbach, B.K.; Gomez-Llorente, Y.; Deyaert, E.; Renzi, F.; Li, X.; Schaffner, A.; Jagtap, P.K.; Boldt, K.; et al. Structural model of the dimeric Parkinson’s protein LRRK2 reveals a compact architecture involving distant interdomain contacts. Proc. Natl. Acad. Sci. USA 2016, 113, E4357–E4366. [Google Scholar] [CrossRef] [PubMed]
- Zhang, P.; Fan, Y.; Ru, H.; Wang, L.; Magupalli, V.G.; Taylor, S.S.; Alessi, D.R.; Wu, H. Crystal structure of the WD40 domain dimer of LRRK2. Proc. Natl. Acad. Sci. USA 2019, 116, 1579–1584. [Google Scholar] [CrossRef]
- Baptista, M.A.S.; Merchant, K.; Barrett, T.; Bryce, D.K.; Ellis, M.; Estrada, A.A.; Fell, M.J.; Fiske, B.K.; Fuji, R.N.; Galatsis, P.; et al. LRRK2 kinase inhibitors induce a reversible effect in the lungs of non-human primates with no measurable pulmonary deficits. bioRxiv 2018. [Google Scholar]
- Di Maio, R.; Hoffman, E.K.; Rocha, E.M.; Keeney, M.T.; Sanders, L.H.; De Miranda, B.R.; Zharikov, A.; Van Laar, A.; Stepan, A.F.; Lanz, T.A.; et al. LRRK2 activation in idiopathic Parkinson’s disease. Sci. Transl. Med. 2018, 10. [Google Scholar] [CrossRef] [PubMed]
- Kluss, J.H.; Mamais, A.; Cookson, M.R. LRRK2 links genetic and sporadic Parkinson’s disease. Biochem. Soc. Trans. 2019, 47, 651–661. [Google Scholar] [CrossRef] [PubMed]
- Langston, J.W.; Ballard, P.; Tetrud, J.W.; Irwin, I. Chronic Parkinsonism in humans due to a product of meperidine-analog synthesis. Science 1983, 219, 979–980. [Google Scholar] [CrossRef] [PubMed]
- Langston, J.W.; Ballard, P.A., Jr. Parkinson’s disease in a chemist working with 1-methyl-4-phenyl-1,2,5,6-tetrahydropyridine. N. Engl. J. Med. 1983, 309, 310. [Google Scholar] [PubMed]
- Schapira, A.H.; Cooper, J.M.; Dexter, D.; Clark, J.B.; Jenner, P.; Marsden, C.D. Mitochondrial complex I deficiency in Parkinson’s disease. J. Neurochem. 1990, 54, 823–827. [Google Scholar] [CrossRef] [PubMed]
- Przedborski, S.; Jackson-Lewis, V.; Naini, A.B.; Jakowec, M.; Petzinger, G.; Miller, R.; Akram, M. The parkinsonian toxin 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP): A technical review of its utility and safety. J. Neurochem. 2001, 76, 1265–1274. [Google Scholar] [CrossRef] [PubMed]
- Mizuno, Y.; Ohta, S.; Tanaka, M.; Takamiya, S.; Suzuki, K.; Sato, T.; Oya, H.; Ozawa, T.; Kagawa, Y. Deficiencies in complex I subunits of the respiratory chain in Parkinson’s disease. Biochem. Biophys. Res. Commun. 1989, 163, 1450–1455. [Google Scholar] [CrossRef]
- Schapira, A.H.; Cooper, J.M.; Dexter, D.; Jenner, P.; Clark, J.B.; Marsden, C.D. Mitochondrial complex I deficiency in Parkinson’s disease. Lancet 1989, 1, 1269. [Google Scholar] [CrossRef]
- Bender, A.; Krishnan, K.J.; Morris, C.M.; Taylor, G.A.; Reeve, A.K.; Perry, R.H.; Jaros, E.; Hersheson, J.S.; Betts, J.; Klopstock, T.; et al. High levels of mitochondrial DNA deletions in substantia nigra neurons in aging and Parkinson disease. Nat. Genet. 2006, 38, 515–517. [Google Scholar] [CrossRef] [PubMed]
- Park, J.S.; Davis, R.L.; Sue, C.M. Mitochondrial Dysfunction in Parkinson’s Disease: New Mechanistic Insights and Therapeutic Perspectives. Curr. Neurol. Neurosci. Rep. 2018, 18, 21. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; Turnbull, D.M.; Reeve, A.K. Mitochondrial Dysfunction in Parkinson’s Disease-Cause or Consequence? Biology 2019, 8, 38. [Google Scholar] [CrossRef] [PubMed]
- Puschmann, A. Monogenic Parkinson’s disease and parkinsonism: Clinical phenotypes and frequencies of known mutations. Parkinsonism Relat. Disord. 2013, 19, 407–415. [Google Scholar] [CrossRef] [PubMed]
- Hilker, R.; Klein, C.; Ghaemi, M.; Kis, B.; Strotmann, T.; Ozelius, L.J.; Lenz, O.; Vieregge, P.; Herholz, K.; Heiss, W.D.; et al. Positron emission tomographic analysis of the nigrostriatal dopaminergic system in familial parkinsonism associated with mutations in the parkin gene. Ann. Neurol. 2001, 49, 367–376. [Google Scholar] [CrossRef] [PubMed]
- Puschmann, A.; Fiesel, F.C.; Caulfield, T.R.; Hudec, R.; Ando, M.; Truban, D.; Hou, X.; Ogaki, K.; Heckman, M.G.; James, E.D.; et al. Heterozygous PINK1 p.G411S increases risk of Parkinson’s disease via a dominant-negative mechanism. Brain 2017, 140, 98–117. [Google Scholar] [CrossRef] [PubMed]
- Weissbach, A.; Baumer, T.; Pramstaller, P.P.; Bruggemann, N.; Tadic, V.; Chen, R.; Klein, C.; Munchau, A. Abnormal premotor-motor interaction in heterozygous Parkin- and Pink1 mutation carriers. Clin. Neurophysiol. 2017, 128, 275–280. [Google Scholar] [CrossRef] [PubMed]
- Klein, C.; Lohmann-Hedrich, K.; Rogaeva, E.; Schlossmacher, M.G.; Lang, A.E. Deciphering the role of heterozygous mutations in genes associated with parkinsonism. Lancet Neurol. 2007, 6, 652–662. [Google Scholar] [CrossRef]
- Kasten, M.; Hartmann, C.; Hampf, J.; Schaake, S.; Westenberger, A.; Vollstedt, E.J.; Balck, A.; Domingo, A.; Vulinovic, F.; Dulovic, M.; et al. Genotype-Phenotype Relations for the Parkinson’s Disease Genes Parkin, PINK1, DJ1: MDSGene Systematic Review. Mov. Disord. 2018, 33, 730–741. [Google Scholar] [CrossRef]
- Pramstaller, P.P.; Schlossmacher, M.G.; Jacques, T.S.; Scaravilli, F.; Eskelson, C.; Pepivani, I.; Hedrich, K.; Adel, S.; Gonzales-McNeal, M.; Hilker, R.; et al. Lewy body Parkinson’s disease in a large pedigree with 77 Parkin mutation carriers. Ann. Neurol. 2005, 58, 411–422. [Google Scholar] [CrossRef]
- Clark, I.E.; Dodson, M.W.; Jiang, C.; Cao, J.H.; Huh, J.R.; Seol, J.H.; Yoo, S.J.; Hay, B.A.; Guo, M. Drosophila pink1 is required for mitochondrial function and interacts genetically with parkin. Nature 2006, 441, 1162–1166. [Google Scholar] [CrossRef]
- Park, J.; Lee, S.B.; Lee, S.; Kim, Y.; Song, S.; Kim, S.; Bae, E.; Kim, J.; Shong, M.; Kim, J.M.; et al. Mitochondrial dysfunction in Drosophila PINK1 mutants is complemented by parkin. Nature 2006, 441, 1157–1161. [Google Scholar] [CrossRef]
- Narendra, D.; Tanaka, A.; Suen, D.F.; Youle, R.J. Parkin is recruited selectively to impaired mitochondria and promotes their autophagy. J. Cell Biol. 2008, 183, 795–803. [Google Scholar] [CrossRef] [PubMed]
- Yi, W.; MacDougall, E.J.; Tang, M.Y.; Krahn, A.I.; Gan-Or, Z.; Trempe, J.F.; Fon, E.A. The Landscape of Parkin Variants Reveals Pathogenic Mechanisms and Therapeutic Targets in Parkinson’s Disease. Hum. Mol. Genet. 2019. [Google Scholar] [CrossRef] [PubMed]
- Wood-Kaczmar, A.; Gandhi, S.; Yao, Z.; Abramov, A.Y.; Miljan, E.A.; Keen, G.; Stanyer, L.; Hargreaves, I.; Klupsch, K.; Deas, E.; et al. PINK1 is necessary for long term survival and mitochondrial function in human dopaminergic neurons. PLoS ONE 2008, 3, e2455. [Google Scholar] [CrossRef]
- Yasuda, T.; Hayakawa, H.; Nihira, T.; Ren, Y.R.; Nakata, Y.; Nagai, M.; Hattori, N.; Miyake, K.; Takada, M.; Shimada, T.; et al. Parkin-mediated protection of dopaminergic neurons in a chronic MPTP-minipump mouse model of Parkinson disease. J. Neuropathol. Exp. Neurol. 2011, 70, 686–697. [Google Scholar] [CrossRef] [PubMed]
- Yasuda, T.; Miyachi, S.; Kitagawa, R.; Wada, K.; Nihira, T.; Ren, Y.R.; Hirai, Y.; Ageyama, N.; Terao, K.; Shimada, T.; et al. Neuronal specificity of alpha-synuclein toxicity and effect of Parkin co-expression in primates. Neuroscience 2007, 144, 743–753. [Google Scholar] [CrossRef] [PubMed]
- Riley, B.E.; Lougheed, J.C.; Callaway, K.; Velasquez, M.; Brecht, E.; Nguyen, L.; Shaler, T.; Walker, D.; Yang, Y.; Regnstrom, K.; et al. Structure and function of Parkin E3 ubiquitin ligase reveals aspects of RING and HECT ligases. Nat. Commun. 2013, 4, 1982. [Google Scholar] [CrossRef] [PubMed]
- Spratt, D.E.; Martinez-Torres, R.J.; Noh, Y.J.; Mercier, P.; Manczyk, N.; Barber, K.R.; Aguirre, J.D.; Burchell, L.; Purkiss, A.; Walden, H.; et al. A molecular explanation for the recessive nature of parkin-linked Parkinson’s disease. Nat. Commun. 2013, 4, 1983. [Google Scholar] [CrossRef] [PubMed]
- Trempe, J.F.; Sauve, V.; Grenier, K.; Seirafi, M.; Tang, M.Y.; Menade, M.; Al-Abdul-Wahid, S.; Krett, J.; Wong, K.; Kozlov, G.; et al. Structure of parkin reveals mechanisms for ubiquitin ligase activation. Science 2013, 340, 1451–1455. [Google Scholar] [CrossRef] [PubMed]
- Wauer, T.; Komander, D. Structure of the human Parkin ligase domain in an autoinhibited state. EMBO J. 2013, 32, 2099–2112. [Google Scholar] [CrossRef] [PubMed]
- Gladkova, C.; Maslen, S.L.; Skehel, J.M.; Komander, D. Mechanism of parkin activation by PINK1. Nature 2018, 559, 410–414. [Google Scholar] [CrossRef] [PubMed]
- Sauve, V.; Sung, G.; Soya, N.; Kozlov, G.; Blaimschein, N.; Miotto, L.S.; Trempe, J.F.; Lukacs, G.L.; Gehring, K. Mechanism of parkin activation by phosphorylation. Nat. Struct. Mol. Biol. 2018, 25, 623–630. [Google Scholar] [CrossRef]
- Rasool, S.; Trempe, J.F. New insights into the structure of PINK1 and the mechanism of ubiquitin phosphorylation. Crit. Rev. Biochem. Mol. Biol. 2018, 53, 515–534. [Google Scholar] [CrossRef] [PubMed]
- Kumar, A.; Tamjar, J.; Waddell, A.D.; Woodroof, H.I.; Raimi, O.G.; Shaw, A.M.; Peggie, M.; Muqit, M.M.; van Aalten, D.M. Structure of PINK1 and mechanisms of Parkinson’s disease-associated mutations. Elife 2017, 6. [Google Scholar] [CrossRef]
- Rasool, S.; Soya, N.; Truong, L.; Croteau, N.; Lukacs, G.L.; Trempe, J.F. PINK1 autophosphorylation is required for ubiquitin recognition. EMBO Rep. 2018, 19. [Google Scholar] [CrossRef]
- McWilliams, T.G.; Prescott, A.R.; Allen, G.F.; Tamjar, J.; Munson, M.J.; Thomson, C.; Muqit, M.M.; Ganley, I.G. mito-QC illuminates mitophagy and mitochondrial architecture in vivo. J. Cell Biol. 2016, 214, 333–345. [Google Scholar] [CrossRef] [PubMed]
- Ordureau, A.; Sarraf, S.A.; Duda, D.M.; Heo, J.M.; Jedrychowski, M.P.; Sviderskiy, V.O.; Olszewski, J.L.; Koerber, J.T.; Xie, T.; Beausoleil, S.A.; et al. Quantitative proteomics reveal a feedforward mechanism for mitochondrial PARKIN translocation and ubiquitin chain synthesis. Mol. Cell 2014, 56, 360–375. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.; Stevens, D.A.; Kang, S.U.; Jiang, H.; Lee, Y.I.; Ko, H.S.; Scarffe, L.A.; Umanah, G.E.; Kang, H.; Ham, S.; et al. PINK1 Primes Parkin-Mediated Ubiquitination of PARIS in Dopaminergic Neuronal Survival. Cell Rep. 2017, 18, 918–932. [Google Scholar] [CrossRef]
- Shin, J.H.; Ko, H.S.; Kang, H.; Lee, Y.; Lee, Y.I.; Pletinkova, O.; Troconso, J.C.; Dawson, V.L.; Dawson, T.M. PARIS (ZNF746) repression of PGC-1alpha contributes to neurodegeneration in Parkinson’s disease. Cell 2011, 144, 689–702. [Google Scholar] [CrossRef]
- Stevens, D.A.; Lee, Y.; Kang, H.C.; Lee, B.D.; Lee, Y.I.; Bower, A.; Jiang, H.; Kang, S.U.; Andrabi, S.A.; Dawson, V.L.; et al. Parkin loss leads to PARIS-dependent declines in mitochondrial mass and respiration. Proc. Natl. Acad. Sci. USA 2015, 112, 11696–11701. [Google Scholar] [CrossRef]
- Mouton-Liger, F.; Rosazza, T.; Sepulveda-Diaz, J.; Ieang, A.; Hassoun, S.M.; Claire, E.; Mangone, G.; Brice, A.; Michel, P.P.; Corvol, J.C.; et al. Parkin deficiency modulates NLRP3 inflammasome activation by attenuating an A20-dependent negative feedback loop. Glia 2018, 66, 1736–1751. [Google Scholar] [CrossRef]
- Sliter, D.A.; Martinez, J.; Hao, L.; Chen, X.; Sun, N.; Fischer, T.D.; Burman, J.L.; Li, Y.; Zhang, Z.; Narendra, D.P.; et al. Parkin and PINK1 mitigate STING-induced inflammation. Nature 2018, 561, 258–262. [Google Scholar] [CrossRef] [PubMed]
- Matheoud, D.; Sugiura, A.; Bellemare-Pelletier, A.; Laplante, A.; Rondeau, C.; Chemali, M.; Fazel, A.; Bergeron, J.J.; Trudeau, L.E.; Burelle, Y.; et al. Parkinson’s Disease-Related Proteins PINK1 and Parkin Repress Mitochondrial Antigen Presentation. Cell 2016, 166, 314–327. [Google Scholar] [CrossRef] [PubMed]
- Rakovic, A.; Ziegler, J.; Martensson, C.U.; Prasuhn, J.; Shurkewitsch, K.; Konig, P.; Paulson, H.L.; Klein, C. PINK1-dependent mitophagy is driven by the UPS and can occur independently of LC3 conversion. Cell Death Differ. 2018. [Google Scholar] [CrossRef] [PubMed]
- Vos, M.; Geens, A.; Bohm, C.; Deaulmerie, L.; Swerts, J.; Rossi, M.; Craessaerts, K.; Leites, E.P.; Seibler, P.; Rakovic, A.; et al. Cardiolipin promotes electron transport between ubiquinone and complex I to rescue PINK1 deficiency. J. Cell Biol. 2017, 216, 695–708. [Google Scholar] [CrossRef] [PubMed]
- Miller, S.; Muqit, M.M.K. Therapeutic approaches to enhance PINK1/Parkin mediated mitophagy for the treatment of Parkinson’s disease. Neurosci. Lett. 2019, 705, 7–13. [Google Scholar] [CrossRef] [PubMed]
- Durcan, T.M.; Fon, E.A. USP8 and PARK2/parkin-mediated mitophagy. Autophagy 2015, 11, 428–429. [Google Scholar] [CrossRef]
- Von Stockum, S.; Sanchez-Martinez, A.; Corra, S.; Chakraborty, J.; Marchesan, E.; Locatello, L.; Da Re, C.; Cusumano, P.; Caicci, F.; Ferrari, V.; et al. Inhibition of the deubiquitinase USP8 corrects a Drosophila PINK1 model of mitochondria dysfunction. Life Sci. Alliance 2019, 2. [Google Scholar] [CrossRef]
- Cornelissen, T.; Haddad, D.; Wauters, F.; Van Humbeeck, C.; Mandemakers, W.; Koentjoro, B.; Sue, C.; Gevaert, K.; De Strooper, B.; Verstreken, P.; et al. The deubiquitinase USP15 antagonizes Parkin-mediated mitochondrial ubiquitination and mitophagy. Hum. Mol. Genet. 2014, 23, 5227–5242. [Google Scholar] [CrossRef]
- Bingol, B.; Tea, J.S.; Phu, L.; Reichelt, M.; Bakalarski, C.E.; Song, Q.; Foreman, O.; Kirkpatrick, D.S.; Sheng, M. The mitochondrial deubiquitinase USP30 opposes parkin-mediated mitophagy. Nature 2014, 510, 370–375. [Google Scholar] [CrossRef]
- Liang, J.R.; Martinez, A.; Lane, J.D.; Mayor, U.; Clague, M.J.; Urbe, S. USP30 deubiquitylates mitochondrial Parkin substrates and restricts apoptotic cell death. EMBO Rep. 2015, 16, 618–627. [Google Scholar] [CrossRef]
- Kluge, A.F.; Lagu, B.R.; Maiti, P.; Jaleel, M.; Webb, M.; Malhotra, J.; Mallat, A.; Srinivas, P.A.; Thompson, J.E. Novel highly selective inhibitors of ubiquitin specific protease 30 (USP30) accelerate mitophagy. Bioorg. Med. Chem. Lett. 2018, 28, 2655–2659. [Google Scholar] [CrossRef] [PubMed]
- Sekine, S.; Youle, R.J. PINK1 import regulation; a fine system to convey mitochondrial stress to the cytosol. BMC Biol. 2018, 16, 2. [Google Scholar] [CrossRef] [PubMed]
- Meissner, C.; Lorenz, H.; Hehn, B.; Lemberg, M.K. Intramembrane protease PARL defines a negative regulator of PINK1- and PARK2/Parkin-dependent mitophagy. Autophagy 2015, 11, 1484–1498. [Google Scholar] [CrossRef] [PubMed]
- Hertz, N.T.; Berthet, A.; Sos, M.L.; Thorn, K.S.; Burlingame, A.L.; Nakamura, K.; Shokat, K.M. A neo-substrate that amplifies catalytic activity of parkinson’s-disease-related kinase PINK1. Cell 2013, 154, 737–747. [Google Scholar] [CrossRef] [PubMed]
- Hasson, S.A.; Kane, L.A.; Yamano, K.; Huang, C.H.; Sliter, D.A.; Buehler, E.; Wang, C.; Heman-Ackah, S.M.; Hessa, T.; Guha, R.; et al. High-content genome-wide RNAi screens identify regulators of parkin upstream of mitophagy. Nature 2013, 504, 291–295. [Google Scholar] [CrossRef] [PubMed]
- Ivatt, R.M.; Sanchez-Martinez, A.; Godena, V.K.; Brown, S.; Ziviani, E.; Whitworth, A.J. Genome-wide RNAi screen identifies the Parkinson disease GWAS risk locus SREBF1 as a regulator of mitophagy. Proc. Natl. Acad. Sci. USA 2014, 111, 8494–8499. [Google Scholar] [CrossRef] [PubMed]
- Lefebvre, V.; Du, Q.; Baird, S.; Ng, A.C.; Nascimento, M.; Campanella, M.; McBride, H.M.; Screaton, R.A. Genome-wide RNAi screen identifies ATPase inhibitory factor 1 (ATPIF1) as essential for PARK2 recruitment and mitophagy. Autophagy 2013, 9, 1770–1779. [Google Scholar] [CrossRef]
- Potting, C.; Crochemore, C.; Moretti, F.; Nigsch, F.; Schmidt, I.; Manneville, C.; Carbone, W.; Knehr, J.; DeJesus, R.; Lindeman, A.; et al. Genome-wide CRISPR screen for PARKIN regulators reveals transcriptional repression as a determinant of mitophagy. Proc. Natl. Acad. Sci. USA 2018, 115, E180–E189. [Google Scholar] [CrossRef]
- Gautier, C.A.; Erpapazoglou, Z.; Mouton-Liger, F.; Muriel, M.P.; Cormier, F.; Bigou, S.; Duffaure, S.; Girard, M.; Foret, B.; Iannielli, A.; et al. The endoplasmic reticulum-mitochondria interface is perturbed in PARK2 knockout mice and patients with PARK2 mutations. Hum. Mol. Genet. 2016, 25, 2972–2984. [Google Scholar] [CrossRef]
- Hsieh, C.H.; Shaltouki, A.; Gonzalez, A.E.; Bettencourt da Cruz, A.; Burbulla, L.F.; St Lawrence, E.; Schule, B.; Krainc, D.; Palmer, T.D.; Wang, X. Functional Impairment in Miro Degradation and Mitophagy Is a Shared Feature in Familial and Sporadic Parkinson’s Disease. Cell Stem. Cell 2016, 19, 709–724. [Google Scholar] [CrossRef]
- Soutar, M.P.M.; Kempthorne, L.; Miyakawa, S.; Annuario, E.; Melandri, D.; Harley, J.; O’Sullivan, G.A.; Wray, S.; Hancock, D.C.; Cookson, M.R.; et al. AKT signalling selectively regulates PINK1 mitophagy in SHSY5Y cells and human iPSC-derived neurons. Sci. Rep. 2018, 8, 8855. [Google Scholar] [CrossRef] [PubMed]
- Hou, X.; Fiesel, F.C.; Truban, D.; Castanedes Casey, M.; Lin, W.L.; Soto, A.I.; Tacik, P.; Rousseau, L.G.; Diehl, N.N.; Heckman, M.G.; et al. Age- and disease-dependent increase of the mitophagy marker phospho-ubiquitin in normal aging and Lewy body disease. Autophagy 2018, 14, 1404–1418. [Google Scholar] [CrossRef] [PubMed]
- Vollstedt, E.J.; Kasten, M.; Klein, C.; Group, M.G.G.P.s.D.S. Using global team science to identify genetic Parkinson’s disease worldwide. Ann. Neurol. 2019. [Google Scholar] [CrossRef] [PubMed]
- Baptista, M.A.; Dave, K.D.; Sheth, N.P.; De Silva, S.N.; Carlson, K.M.; Aziz, Y.N.; Fiske, B.K.; Sherer, T.B.; Frasier, M.A. A strategy for the generation, characterization and distribution of animal models by The Michael J. Fox Foundation for Parkinson’s Research. Dis. Model. Mech. 2013, 6, 1316–1324. [Google Scholar] [CrossRef] [PubMed]
- Ordureau, A.; Heo, J.M.; Duda, D.M.; Paulo, J.A.; Olszewski, J.L.; Yanishevski, D.; Rinehart, J.; Schulman, B.A.; Harper, J.W. Defining roles of PARKIN and ubiquitin phosphorylation by PINK1 in mitochondrial quality control using a ubiquitin replacement strategy. Proc. Natl. Acad. Sci. USA 2015, 112, 6637–6642. [Google Scholar] [CrossRef] [PubMed]
- Pickrell, A.M.; Huang, C.H.; Kennedy, S.R.; Ordureau, A.; Sideris, D.P.; Hoekstra, J.G.; Harper, J.W.; Youle, R.J. Endogenous Parkin Preserves Dopaminergic Substantia Nigral Neurons following Mitochondrial DNA Mutagenic Stress. Neuron 2015, 87, 371–381. [Google Scholar] [CrossRef] [PubMed]



© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Padmanabhan, S.; Polinski, N.K.; Menalled, L.B.; Baptista, M.A.S.; Fiske, B.K. The Michael J. Fox Foundation for Parkinson’s Research Strategy to Advance Therapeutic Development of PINK1 and Parkin. Biomolecules 2019, 9, 296. https://doi.org/10.3390/biom9080296
Padmanabhan S, Polinski NK, Menalled LB, Baptista MAS, Fiske BK. The Michael J. Fox Foundation for Parkinson’s Research Strategy to Advance Therapeutic Development of PINK1 and Parkin. Biomolecules. 2019; 9(8):296. https://doi.org/10.3390/biom9080296
Chicago/Turabian StylePadmanabhan, Shalini, Nicole K. Polinski, Liliana B. Menalled, Marco A.S. Baptista, and Brian K. Fiske. 2019. "The Michael J. Fox Foundation for Parkinson’s Research Strategy to Advance Therapeutic Development of PINK1 and Parkin" Biomolecules 9, no. 8: 296. https://doi.org/10.3390/biom9080296
APA StylePadmanabhan, S., Polinski, N. K., Menalled, L. B., Baptista, M. A. S., & Fiske, B. K. (2019). The Michael J. Fox Foundation for Parkinson’s Research Strategy to Advance Therapeutic Development of PINK1 and Parkin. Biomolecules, 9(8), 296. https://doi.org/10.3390/biom9080296