The Role of Protein Tyrosine Phosphatase (PTP)-1B in Cardiovascular Disease and Its Interplay with Insulin Resistance



Abstract

1. Introduction
2. The Endothelium
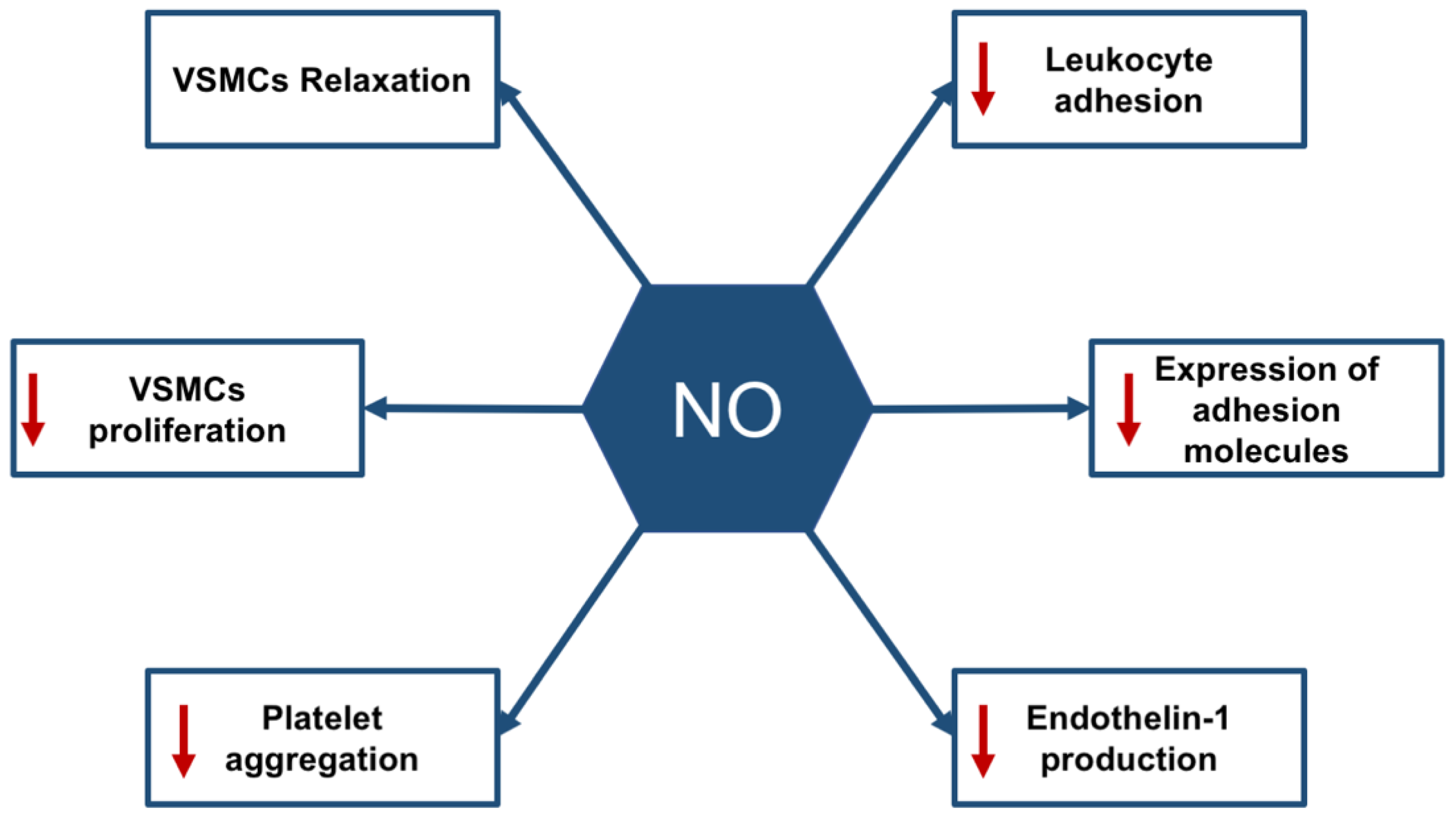
2.1. The Physiological Roles of the Endothelium
2.2. Endothelial Dysfunction and Diabetes
3. Protein Tyrosine Phosphatase 1B
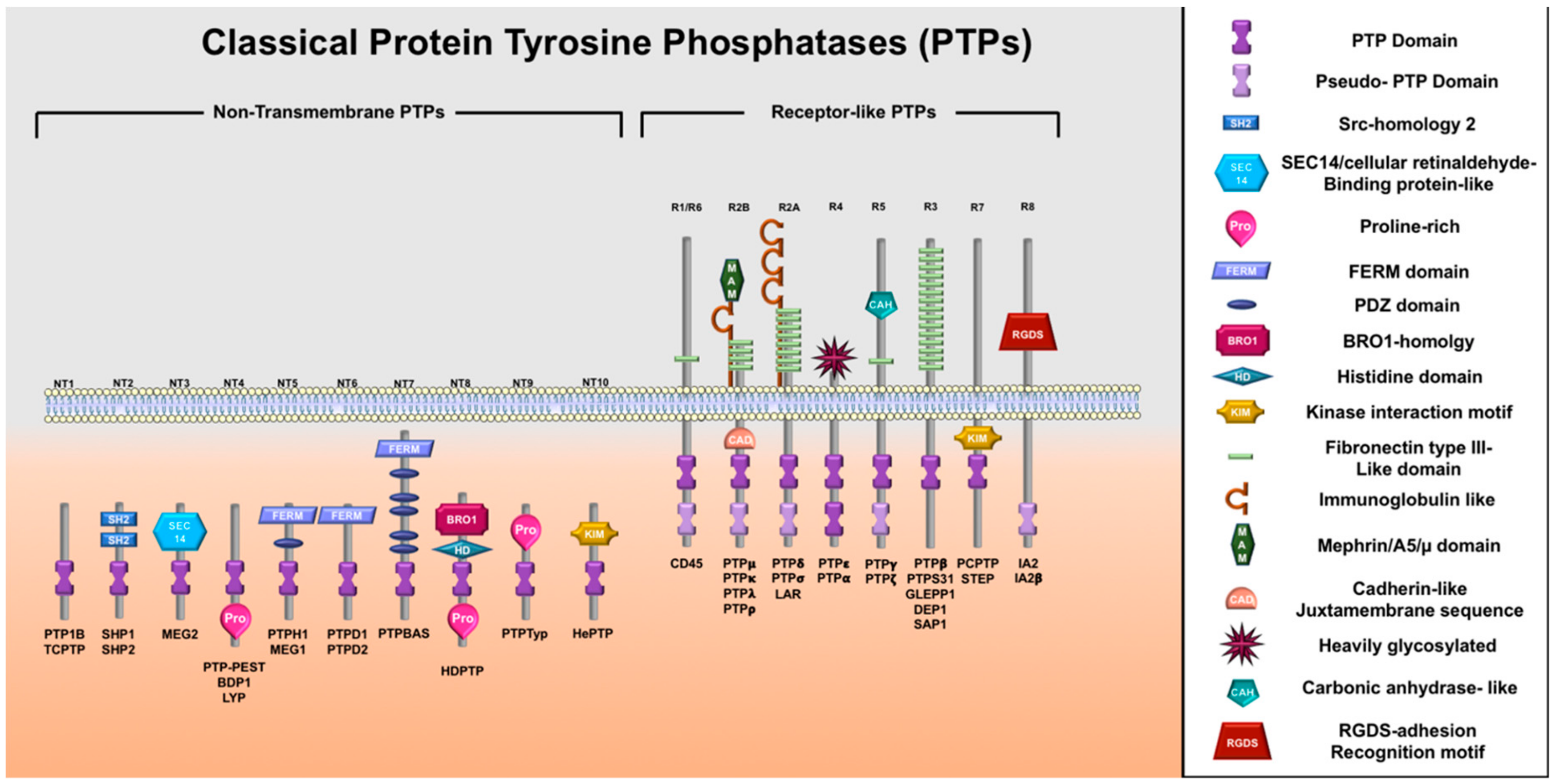
3.1. Protein Tyrosine Phosphatases Superfamily Family and PTP1B
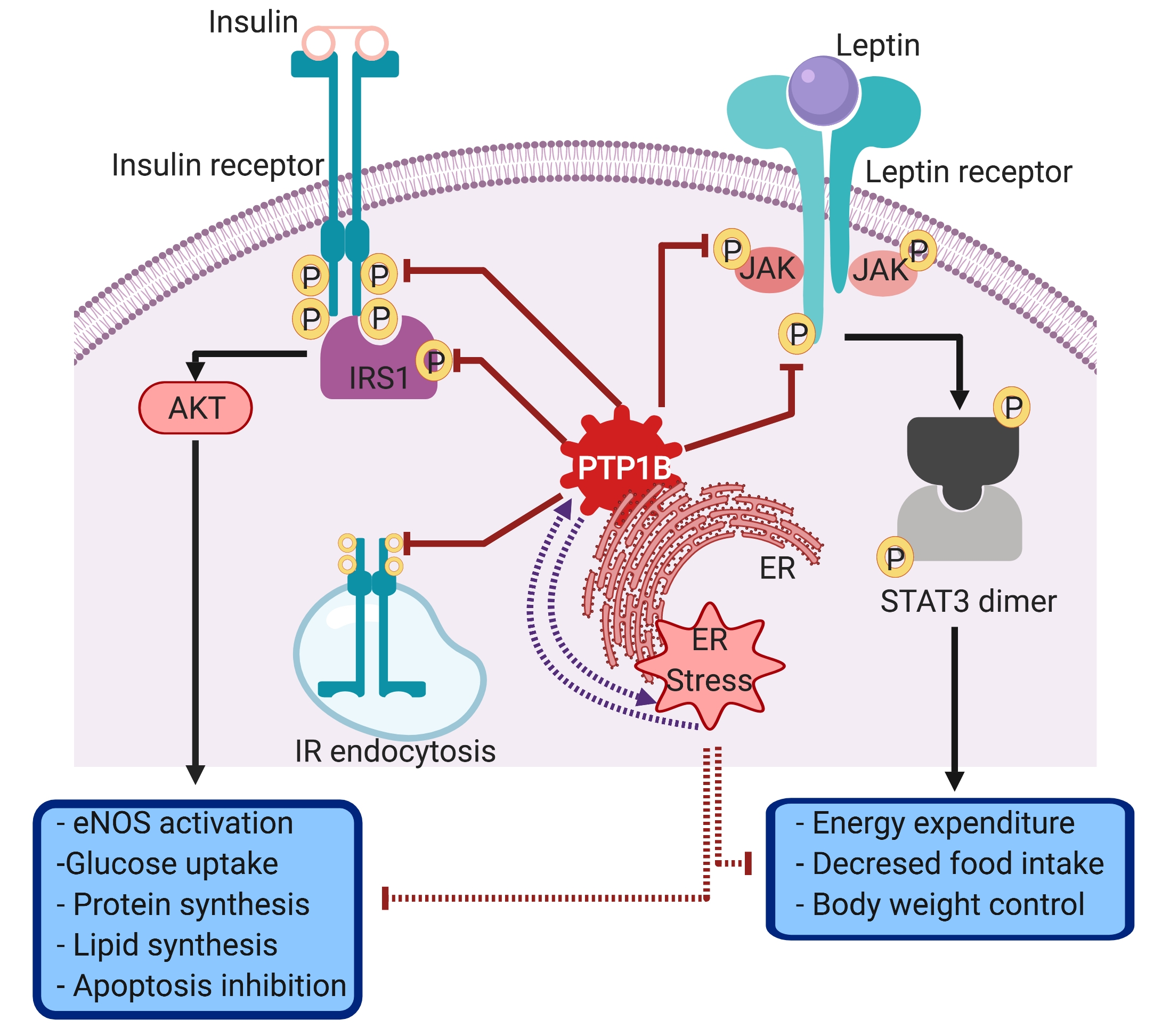
3.2. PTP1B Intracellular Localization and Molecular Substrates
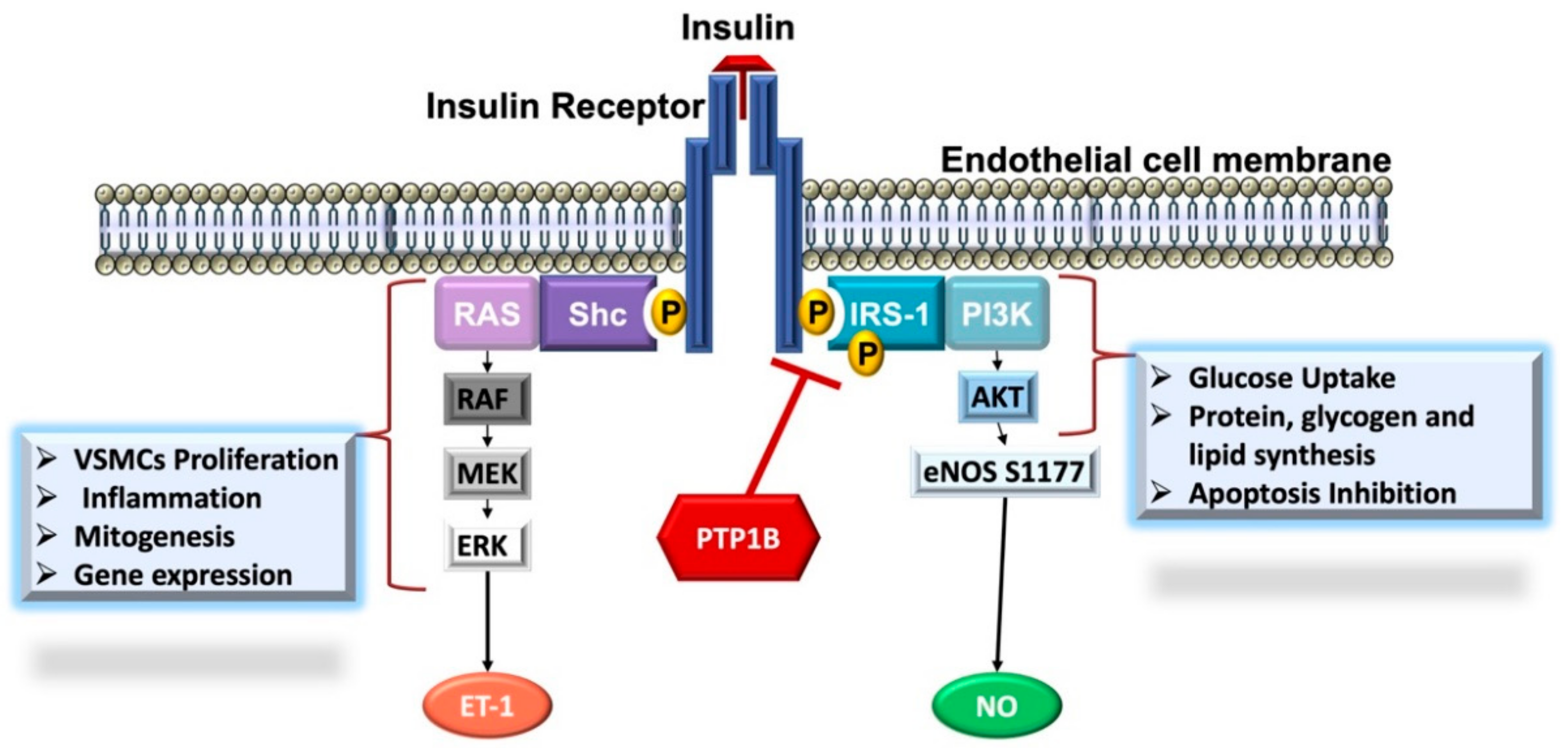
3.3. The Involvement of PTP1B in the Pathophysiology of Cardio-Metabolic Diseases
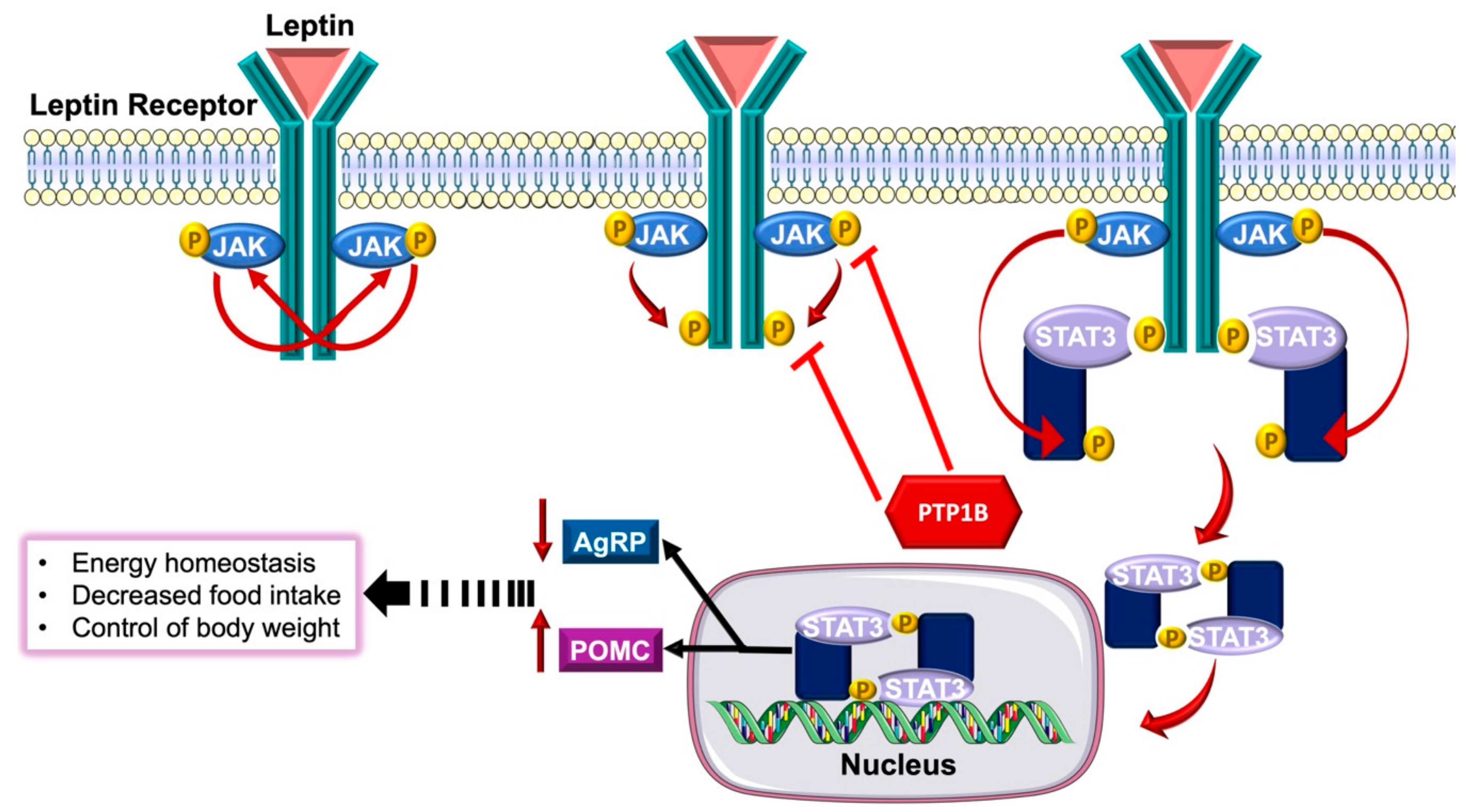
3.3.1. The Role of PTP1B in Obesity and Insulin Resistance
3.3.2. PTP1B and Cardiovascular Complications
PTP1B and Endothelial Dysfunction
PTP1B and Vascular Inflammation
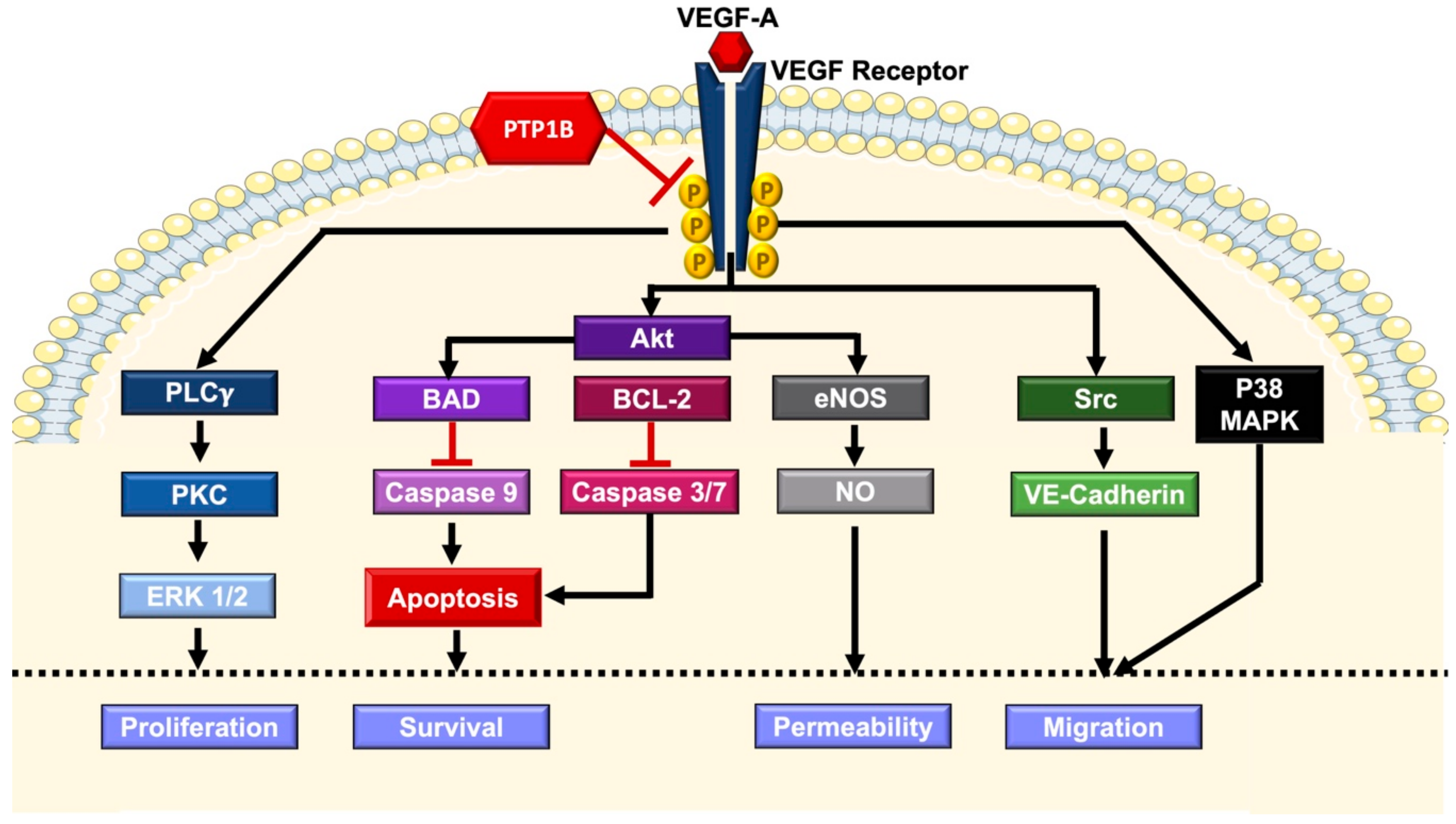
PTP1B and Altered Angiogenesis
3.3.3. Endoplasmic Reticulum Stress as a Possible Bridging Link Between PTP1B, Insulin Resistance and Cardiovascular Dysfunction
4. PTP1B as a Potential Therapeutic Target
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- WHO. Obesity and Overweight Fact Sheet; WHO: Geneva, Switzerland, 2018. [Google Scholar]
- Mandviwala, T.; Khalid, U.; Deswal, A. Obesity and cardiovascular disease: A risk factor or a risk marker? Curr. Atheroscler. Rep. 2016, 18, 21. [Google Scholar] [CrossRef] [PubMed]
- Navarro Díaz, M. Consequences of morbid obesity on the kidney. Where are we going? Clin. Kidney J. 2016, 9, 782–787. [Google Scholar] [CrossRef] [PubMed]
- Waring, J.F.; Ciurlionis, R.; Clampit, J.E.; Morgan, S.; Gum, R.J.; Jolly, R.A.; Kroeger, P.; Frost, L.; Trevillyan, J.; Zinker, B.A.; et al. PTP1B antisense-treated mice show regulation of genes involved in lipogenesis in liver and fat. Mol. Cell. Endocrinol. 2003, 203, 155–168. [Google Scholar] [CrossRef]
- Shoelson, S.E.; Herrero, L.; Naaz, A. Obesity, inflammation, and insulin resistance. Gastroenterology 2007, 132, 2169–2180. [Google Scholar] [CrossRef] [PubMed]
- Chawla, A.; Chawla, R.; Jaggi, S. Microvasular and macrovascular complications in diabetes mellitus: Distinct or continuum? Ind. J. Endocrinol. Metab. 2016, 20, 546–551. [Google Scholar] [CrossRef] [PubMed]
- International Diabetes Federation. IDF Diabetes Atlas, 8th ed.; International Diabetes Federation: Brussels, Belgium, 2017. [Google Scholar]
- Ismail-Beigi, F. Pathogenesis and glycemic management of type 2 diabetes mellitus: A physiological approach. Arch. Iran. Med. (AIM) 2012, 15, 239–246. [Google Scholar]
- Schalkwijk, C.; Stehouwer, C.D.A. Vascular complications in diabetes mellitus: The role of endothelial dysfunction. Clin. Sci. 2005, 109, 143. [Google Scholar] [CrossRef] [PubMed]
- WHO. Diabetes; WHO: Geneva, Switzerland, 2018. [Google Scholar]
- Patel, T.P.; Rawal, K.; Bagchi, A.K.; Akolkar, G.; Bernardes, N.; Dias, D.d.S.; Gupta, S.; Singal, P.K. Insulin resistance: An additional risk factor in the pathogenesis of cardiovascular disease in type 2 diabetes. Heart Fail. Rev. 2016, 21, 11–23. [Google Scholar] [CrossRef] [PubMed]
- Cersosimo, E.; DeFronzo, R.A. Insulin resistance and endothelial dysfunction: The road map to cardiovascular diseases. Diabetes Metab. Res. Rev. 2006, 22, 423–436. [Google Scholar] [CrossRef]
- Maamoun, H.; Abdelsalam, S.S.; Zeidan, A.; Korashy, H.M.; Agouni, A. Endoplasmic reticulum stress: A critical molecular driver of endothelial dysfunction and cardiovascular disturbances associated with diabetes. Int. J. Mol. Sci. 2019, 20, 1658. [Google Scholar] [CrossRef]
- Maamoun, H.; Benameur, T.; Pintus, G.; Munusamy, S.; Agouni, A. Crosstalk between oxidative stress and endoplasmic reticulum (ER) stress in endothelial dysfunction and aberrant angiogenesis associated with diabetes: A focus on the protective roles of heme oxygenase (HO)-1. Front. Physiol. 2019, 10, 70. [Google Scholar] [CrossRef] [PubMed]
- Galic, S.; Klingler-Hoffmann, M.; Fodero-Tavoletti, M.T.; Puryer, M.A.; Meng, T.-C.; Tonks, N.K.; Tiganis, T. Regulation of insulin receptor signaling by the protein tyrosine phosphatase TCPTP. Mol. Cell. Biol. 2003, 23, 2096–2108. [Google Scholar] [CrossRef] [PubMed]
- Elchebly, M.; Payette, P.; Michaliszyn, E.; Cromlish, W.; Collins, S.; Loy, A.L.; Normandin, D.; Cheng, A.; Himms-Hagen, J.; Chan, C.-C.; et al. Increased insulin sensitivity and obesity resistance in mice lacking the protein tyrosine phosphatase-1B gene. Science 1999, 283, 1544–1548. [Google Scholar] [CrossRef] [PubMed]
- Rajendran, P.; Rengarajan, T.; Thangavel, J.; Nishigaki, Y.; Sakthisekaran, D.; Sethi, G.; Nishigaki, I. The vascular endothelium and human diseases. Int. J. Biol. Sci. 2013, 9, 1057–1069. [Google Scholar] [CrossRef] [PubMed]
- Cines, D.B.; Pollak, E.S.; Buck, C.A.; Loscalzo, J.; Zimmerman, G.A.; McEver, R.P.; Pober, J.S.; Wick, T.M.; Konkle, B.A.; Schwartz, B.S.; et al. Endothelial cells in physiology and in the pathophysiology of vascular disorders. Blood 1998, 91, 3527. [Google Scholar] [PubMed]
- Zhang, H.-N.; Xu, Q.-Q.; Thakur, A.; Alfred, M.O.; Chakraborty, M.; Ghosh, A.; Yu, X.-B. Endothelial dysfunction in diabetes and hypertension: Role of microRNAs and long non-coding RNAs. Life Sci. 2018, 213, 258–268. [Google Scholar] [CrossRef] [PubMed]
- Cahill, P.A.; Redmond, E.M. Vascular endothelium—Gatekeeper of vessel health. Atherosclerosis 2016, 248, 97–109. [Google Scholar] [CrossRef]
- Michiels, C. Endothelial cell functions. J. Cell. Physiol. 2003, 196, 430–443. [Google Scholar] [CrossRef]
- Moncada, S.; Higgs, E.A. The discovery of nitric oxide and its role in vascular biology. Br. J. Pharmacol. 2006, 147, S193–S201. [Google Scholar] [CrossRef]
- Roberts, A.C.; Porter, K.E. Cellular and molecular mechanisms of endothelial dysfunction in diabetes. Diabetes Vasc. Dis. Res. 2013, 10, 472–482. [Google Scholar] [CrossRef]
- Jamwal, S.; Sharma, S. Vascular endothelium dysfunction: A conservative target in metabolic disorders. Inflamm. Res. 2018, 67, 391–405. [Google Scholar] [CrossRef] [PubMed]
- Potenza, M.A.; Gagliardi, S.; Nacci, C.; Carratu, M.R.; Montagnani, M. Endothelial dysfunction in diabetes: From mechanisms to therapeutic targets. Curr. Med. Chem. 2009, 16, 94–112. [Google Scholar] [CrossRef] [PubMed]
- Muniyappa, R.; Iantorno, M.; Quon, M.J. An integrated view of insulin resistance and endothelial dysfunction. Endocrinol. Metab. Clin. N. Am. 2008, 37, 685–711. [Google Scholar] [CrossRef] [PubMed]
- Okon, E.B.; Chung, A.W.Y.; Rauniyar, P.; Padilla, E.; Tejerina, T.; McManus, B.M.; Luo, H.; van Breemen, C. Compromised arterial function in human type 2 diabetic patients. Diabetes 2005, 54, 2415. [Google Scholar] [CrossRef] [PubMed]
- Duncan, E.R.; Crossey, P.A.; Walker, S.; Anilkumar, N.; Poston, L.; Douglas, G.; Ezzat, V.A.; Wheatcroft, S.B.; Shah, A.M.; Kearney, M.I. Effect of endothelium-specific insulin resistance on endothelial function in vivo. Diabetes 2008, 57, 3307–3314. [Google Scholar] [CrossRef] [PubMed]
- Thiebaut, P.-A.; Besnier, M.; Gomez, E.; Richard, V. Role of protein tyrosine phosphatase 1B in cardiovascular diseases. J. Mol. Cell. Cardiol. 2016, 101, 50–57. [Google Scholar] [CrossRef] [PubMed]
- Koren, S.; Fantus, I.G. Inhibition of the protein tyrosine phosphatase PTP1B: Potential therapy for obesity, insulin resistance and type-2 diabetes mellitus. Best Pract. Res. Clin. Endocrinol. Metab. 2007, 21, 621–640. [Google Scholar] [CrossRef]
- Hale, A.J.; ter Steege, E.; den Hertog, J. Recent advances in understanding the role of protein-tyrosine phosphatases in development and disease. Dev. Biol. 2017, 428, 283–292. [Google Scholar] [CrossRef]
- Li, L.; Dixon, J.E. Form, function, and regulation of protein tyrosine phosphatases and their involvement in human diseases. Semin. Immunol. 2000, 12, 75–84. [Google Scholar] [CrossRef]
- Stauffacher, C.V.; Charbonneau, H. Protein tyrosine phosphatases. In Principles of Molecular Regulation; Conn, P.M., Means, A.R., Eds.; Humana Press: Totowa, NJ, USA, 2000; pp. 323–347. [Google Scholar]
- Zhong-Yin, Z. Protein tyrosine phosphatases: Structure and function, substrate specificity, and inhibitor development. Ann. Rev. Pharmacol. Toxicol. 2002, 42, 209. [Google Scholar] [CrossRef]
- Tonks, N.K.; Diltz, C.D.; Fischer, E.H. Purification of the major protein-tyrosine-phosphatases of human placenta. J. Biol. Chem. 1988, 263, 6722–6730. [Google Scholar] [PubMed]
- Tonks, N.K.; Diltz, C.D.; Fischer, E.H. Characterization of the major protein-tyrosine-phosphatases of human placenta. J. Biol. Chem. 1988, 263, 6731–6737. [Google Scholar] [PubMed]
- Chernoff, J.; Schievella, A.R.; Jost, C.A.; Erikson, R.L.; Neel, B.G. Cloning of a cDNA for a major human protein-tyrosine-phosphatase. Proc. Natl. Acad. Sci. USA 1990, 87, 2735–2739. [Google Scholar] [CrossRef] [PubMed]
- Anderie, I.; Schulz, I.; Schmid, A. Characterization of the C-terminal ER membrane anchor of PTP1B. Experimental Cell Res. 2007, 313, 3189–3197. [Google Scholar] [CrossRef] [PubMed]
- Frangioni, J.V.; Beahm, P.H.; Shifrin, V.; Jost, C.A.; Neel, B.G. The nontransmembrane tyrosine phosphatase PTP-1B localizes to the endoplasmic reticulum via its 35 amino acid C-terminal sequence. Cell 1992, 68, 545–560. [Google Scholar] [CrossRef]
- Tonks, N.K. PTP1B: From the sidelines to the front lines! FEBS Lett. 2003, 546, 140–148. [Google Scholar] [CrossRef]
- Bandyopadhyay, D.; Kusari, A.; Kenner, K.A.; Liu, F.; Chernoff, J.; Gustafson, T.A.; Kusari, J. Protein-tyrosine phosphatase 1B complexes with the insulin receptor in vivo and is tyrosine-phosphorylated in the presence of insulin. J. Biol. Chem. 1997, 272, 1639–1645. [Google Scholar] [CrossRef]
- Gu, F.; Dubé, N.; Kim, J.W.; Cheng, A.; Ibarra-Sanchez, M.d.J.; Tremblay, M.L.; Boisclair, Y.R. Protein tyrosine phosphatase 1B attenuates growth hormone-mediated JAK2-STAT signaling. Mol. Cell. Biol. 2003, 23, 3753–3762. [Google Scholar] [CrossRef]
- Qian, J.; Fulton, D. Post-translational regulation of endothelial nitric oxide synthase in vascular endothelium. Front. Physiol. 2013, 4, 347. [Google Scholar] [CrossRef]
- Fleming, I.; Fisslthaler, B.; Dimmeler, S.; Kemp, B.E.; Busse, R. Phosphorylation of Thr(495) regulates Ca(2+)/calmodulin-dependent endothelial nitric oxide synthase activity. Circ. Res. 2001, 88, E68–E75. [Google Scholar] [CrossRef]
- Fisslthaler, B.; Dimmeler, S.; Hermann, C.; Busse, R.; Fleming, I. Phosphorylation and activation of the endothelial nitric oxide synthase by fluid shear stress. Acta Physiol. Scand. 2000, 168, 81–88. [Google Scholar] [CrossRef] [PubMed]
- Fulton, D.; Church, J.E.; Ruan, L.; Li, C.; Sood, S.G.; Kemp, B.E.; Jennings, I.G.; Venema, R.C. Src kinase activates endothelial nitric-oxide synthase by phosphorylating Tyr-83. J. Biol. Chem. 2005, 280, 35943–35952. [Google Scholar] [CrossRef] [PubMed]
- Fulton, D.; Ruan, L.; Sood, S.G.; Li, C.; Zhang, Q.; Venema, R.C. Agonist-stimulated endothelial nitric oxide synthase activation and vascular relaxation. Role of eNOS phosphorylation at Tyr83. Circ. Res. 2008, 102, 497–504. [Google Scholar] [CrossRef] [PubMed]
- Loot, A.E.; Schreiber, J.G.; Fisslthaler, B.; Fleming, I. Angiotensin II impairs endothelial function via tyrosine phosphorylation of the endothelial nitric oxide synthase. J. Exp. Med. 2009, 206, 2889–2896. [Google Scholar] [CrossRef] [PubMed]
- Bibli, S.I.; Zhou, Z.; Zukunft, S.; Fisslthaler, B.; Andreadou, I.; Szabo, C.; Brouckaert, P.; Fleming, I.; Papapetropoulos, A. Tyrosine phosphorylation of eNOS regulates myocardial survival after an ischaemic insult: Role of PYK2. Cardiovasc. Res. 2017, 113, 926–937. [Google Scholar] [CrossRef] [PubMed]
- Gogiraju, R.; Schroeter, M.R.; Bochenek, M.L.; Hubert, A.; Munzel, T.; Hasenfuss, G.; Schafer, K. Endothelial deletion of protein tyrosine phosphatase-1B protects against pressure overload-induced heart failure in mice. Cardiovasc. Res. 2016, 111, 204–216. [Google Scholar] [CrossRef]
- Agouni, A.; Tual-Chalot, S.; Chalopin, M.; Duluc, L.; Mody, N.; Martinez, M.C.; Andriantsitohaina, R.; Delibegovic, M. Hepatic protein tyrosine phosphatase 1B (PTP1B) deficiency protects against obesity-induced endothelial dysfunction. Biochem. Pharmacol. 2014, 92, 607–617. [Google Scholar] [CrossRef]
- Gomez, E.; Vercauteren, M.; Kurtz, B.; Ouvrard-Pascaud, A.; Mulder, P.; Henry, J.-P.; Besnier, M.; Waget, A.; Hooft Van Huijsduijnen, R.; Tremblay, M.L.; et al. Reduction of heart failure by pharmacological inhibition or gene deletion of protein tyrosine phosphatase 1B. J. Mol. Cell. Cardiol. 2012, 52, 1257–1264. [Google Scholar] [CrossRef]
- Vercauteren, M.; Remy, E.; Devaux, C.; Dautreaux, B.; Henry, J.-P.; Bauer, F.; Mulder, P.; Hooft van Huijsduijnen, R.; Bombrun, A.; Thuillez, C.; et al. Improvement of peripheral endothelial dysfunction by protein tyrosine phosphatase inhibitors in heart failure. Circulation 2006, 114, 2498–2507. [Google Scholar] [CrossRef]
- Villalobos-Labra, R.; Subiabre, M.; Toledo, F.; Pardo, F.; Sobrevia, L. Endoplasmic reticulum stress and development of insulin resistance in adipose, skeletal, liver, and foetoplacental tissue in diabesity. Mol. Aspects Med. 2018, 66, 49–61. [Google Scholar] [CrossRef]
- Reaven, G.M. Insulin resistance: The link between obesity and cardiovascular disease. Med. Clin. N. Am. 2011, 95, 875–892. [Google Scholar] [CrossRef] [PubMed]
- Kenner, K.A.; Anyanwu, E.; Olefsky, J.M.; Kusari, J. Protein-tyrosine phosphatase 1B Is a negative regulator of insulin- and insulin-like growth factor-I-stimulated signaling. J. Biol. Chem. 1996, 271, 19810–19816. [Google Scholar] [CrossRef] [PubMed]
- Dadke, S.; Kusari, J.; Chernoff, J. Down-regulation of insulin signaling by protein-tyrosine phosphatase 1B is mediated by an N-terminal binding region. J. Biol. Chem. 2000, 275, 23642–23647. [Google Scholar] [CrossRef] [PubMed]
- Kuga, G.K.; Muñoz, V.R.; Gaspar, R.C.; Nakandakari, S.C.B.R.; da Silva, A.S.R.; Botezelli, J.D.; Leme, J.A.C.d.A.; Gomes, R.J.; de Moura, L.P.; Cintra, D.E.; et al. Impaired insulin signaling and spatial learning in middle-aged rats: The role of PTP1B. Exp. Gerontol. 2018, 104, 66–71. [Google Scholar] [CrossRef] [PubMed]
- Sugiyama, M.; Banno, R.; Mizoguchi, A.; Tominaga, T.; Tsunekawa, T.; Onoue, T.; Hagiwara, D.; Ito, Y.; Morishita, Y.; Iwama, S.; et al. PTP1B deficiency improves hypothalamic insulin sensitivity resulting in the attenuation of AgRP mRNA expression under high-fat diet conditions. Biochem. Biophys. Res. Commun. 2017, 488, 116–121. [Google Scholar] [CrossRef]
- Panzhinskiy, E.; Ren, J.; Nair, S. Protein tyrosine phosphatase 1B and insulin resistance: Role of endoplasmic reticulum stress/reactive oxygen species/nuclear factor kappa B axis. PLoS ONE 2013, 8, e77228. [Google Scholar] [CrossRef]
- Klaman, L.D.; Boss, O.; Peroni, O.D.; Kim, J.K.; Martino, J.L.; Zabolotny, J.M.; Moghal, N.; Lubkin, M.; Kim, Y.-B.; Sharpe, A.H.; et al. Increased energy expenditure, decreased adiposity, and tissue-specific insulin sensitivity in protein-tyrosine phosphatase 1b-deficient mice. Mol. Cell. Biol. 2000, 20, 5479–5489. [Google Scholar] [CrossRef]
- Ye, Z.; Liu, G.; Guo, J.; Su, Z. Hypothalamic endoplasmic reticulum stress as a key mediator of obesity-induced leptin resistance. Obes. Rev. 2018, 19, 770–785. [Google Scholar] [CrossRef]
- Zabolotny, J.M.; Bence-Hanulec, K.K.; Stricker-Krongrad, A.; Haj, F.; Wang, Y.; Minokoshi, Y.; Kim, Y.-B.; Elmquist, J.K.; Tartaglia, L.A.; Kahn, B.B.; et al. PTP1B regulates leptin signal transduction in vivo. Dev. Cell 2002, 2, 489–495. [Google Scholar] [CrossRef]
- Ali, M.I.; Ketsawatsomkron, P.; Belin de Chantemele, E.J.; Mintz, J.D.; Muta, K.; Salet, C.; Black, S.M.; Tremblay, M.L.; Fulton, D.J.; Marrero, M.B.; et al. Deletion of protein tyrosine phosphatase 1b improves peripheral insulin resistance and vascular function in obese, leptin-resistant mice via reduced oxidant tone. Circ. Res. 2009, 105, 1013–1022. [Google Scholar] [CrossRef]
- González-Rodríguez, A.; Mas Gutierrez, J.A.; Sanz-González, S.; Ros, M.; Burks, D.J.; Valverde, A.M. Inhibition of PTP1B restores IRS1-mediated hepatic insulin signaling in IRS2-deficient mice. Diabetes 2010, 59, 588–599. [Google Scholar] [CrossRef] [PubMed]
- Bence, K.K.; Delibegovic, M.; Xue, B.; Gorgun, C.Z.; Hotamisligil, G.S.; Neel, B.G.; Kahn, B.B. Neuronal PTP1B regulates body weight, adiposity and leptin action. Nat. Med. 2006, 12, 917. [Google Scholar] [CrossRef] [PubMed]
- Banno, R.; Zimmer, D.; De Jonghe, B.C.; Atienza, M.; Rak, K.; Yang, W.; Bence, K.K. PTP1B and SHP2 in POMC neurons reciprocally regulate energy balance in mice. J. Clin. Invest. 2010, 120, 720–734. [Google Scholar] [CrossRef] [PubMed]
- Tsou, R.C.; Zimmer, D.J.; De Jonghe, B.C.; Bence, K.K. Deficiency of PTP1B in leptin receptor-expressing neurons leads to decreased body weight and adiposity in mice. Endocrinology 2012, 153, 4227–4237. [Google Scholar] [CrossRef] [PubMed]
- Delibegovic, M.; Bence, K.K.; Mody, N.; Hong, E.-G.; Ko, H.J.; Kim, J.K.; Kahn, B.B.; Neel, B.G. Improved glucose homeostasis in mice with muscle-specific deletion of protein-tyrosine phosphatase 1B. Mol. Cell. Biol. 2007, 27, 7727–7734. [Google Scholar] [CrossRef] [PubMed]
- Delibegovic, M.; Zimmer, D.; Kauffman, C.; Rak, K.; Hong, E.-G.; Cho, Y.-R.; Kim, J.K.; Kahn, B.B.; Neel, B.G.; Bence, K.K. Liver-specific deletion of protein-tyrosine phosphatase 1B (PTP1B) improves metabolic syndrome and attenuates diet-induced endoplasmic reticulum stress. Diabetes 2009, 58, 590–599. [Google Scholar] [CrossRef] [PubMed]
- Agouni, A.; Mody, N.; Owen, C.; Czopek, A.; Zimmer, D.; Bentires-Alj, M.; Bence, K.K.; Delibegović, M. Liver-specific deletion of protein tyrosine phosphatase (PTP) 1B improves obesity- and pharmacologically-induced endoplasmic reticulum stress. Biochem. J. 2011, 438, 369–378. [Google Scholar] [CrossRef] [PubMed]
- Owen, C.L.; Lees, E.K.; Grant, L.; Zimmer, D.J.; Mody, N.; Bence, K.K.; Delibegovic, M. Inducible liver-specific knockdown of protein tyrosine phosphatase 1B improves glucose and lipid homeostasis in adult mice. Diabetologia 2013, 56, 2286–2296. [Google Scholar] [CrossRef]
- Owen, C.; Czopek, A.; Agouni, A.; Grant, L.; Judson, R.; Lees, E.K.; McIlroy, G.D.; Göransson, O.; Welch, A.; Bence, K.K.; et al. Adipocyte-specific protein tyrosine phosphatase 1B deletion increases lipogenesis, adipocyte cell size and is a minor regulator of glucose homeostasis. PLoS ONE 2012, 7, e32700. [Google Scholar] [CrossRef]
- Kaszubska, W.; Falls, H.D.; Schaefer, V.G.; Haasch, D.; Frost, L.; Hessler, P.; Kroeger, P.E.; White, D.W.; Jirousek, M.R.; Trevillyan, J.M. Protein tyrosine phosphatase 1B negatively regulates leptin signaling in a hypothalamic cell line. Mol. Cell. Endocrinol. 2002, 195, 109–118. [Google Scholar] [CrossRef]
- Zabolotny, J.M.; Haj, F.G.; Kim, Y.-B.; Kim, H.-J.; Shulman, G.I.; Kim, J.K.; Neel, B.G.; Kahn, B.B. Transgenic overexpression of protein-tyrosine phosphatase 1B in muscle causes insulin resistance, but overexpression with leukocyte antigen-related phosphatase does not additively impair insulin action. J. Biol. Chem. 2004, 279, 24844–24851. [Google Scholar] [CrossRef] [PubMed]
- Haj, F.G.; Zabolotny, J.M.; Kim, Y.-B.; Kahn, B.B.; Neel, B.G. Liver-specific protein-tyrosine phosphatase 1B (PTP1B) Re-expression alters glucose homeostasis of PTP1B−/−Mice. J. Biol. Chem. 2005, 280, 15038–15046. [Google Scholar] [CrossRef] [PubMed]
- Hamilton, S.J.; Watts, G.F. Endothelial dysfunction in diabetes: Pathogenesis, significance, and treatment. Rev. Diabet. Stud. RDS 2013, 10, 133–156. [Google Scholar] [CrossRef] [PubMed]
- Grandl, G.; Wolfrum, C. Hemostasis, endothelial stress, inflammation, and the metabolic syndrome. Semin. Immunopathol. 2018, 40, 215–224. [Google Scholar] [CrossRef] [PubMed]
- Libby, P.; Ridker Paul, M.; Maseri, A. Inflammation and atherosclerosis. Circulation 2002, 105, 1135–1143. [Google Scholar] [CrossRef] [PubMed]
- Coquerel, D.; Neviere, R.; Delile, E.; Mulder, P.; Marechal, X.; Montaigne, D.; Renet, S.; Remy-Jouet, I.; Gomez, E.; Henry, J.P.; et al. Gene deletion of protein tyrosine phosphatase 1B protects against sepsis-induced cardiovascular dysfunction and mortality. Arterioscler. Thromb. Vasc. Biol. 2014, 34, 1032–1044. [Google Scholar] [CrossRef] [PubMed]
- Grant, L.; Shearer, K.D.; Czopek, A.; Lees, E.K.; Owen, C.; Agouni, A.; Workman, J.; Martin-Granados, C.; Forrester, J.V.; Wilson, H.M.; et al. Myeloid-cell protein tyrosine phosphatase-1B deficiency in mice protects against high-fat diet and lipopolysaccharide-induced inflammation, hyperinsulinemia, and endotoxemia through an IL-10 STAT3-dependent mechanism. Diabetes 2014, 63, 456. [Google Scholar] [CrossRef]
- Thompson, D.; Morrice, N.; Grant, L.; Le Sommer, S.; Ziegler, K.; Whitfield, P.; Mody, N.; Wilson, H.M.; Delibegovic, M. Myeloid protein tyrosine phosphatase 1B (PTP1B) deficiency protects against atherosclerotic plaque formation in the ApoE(−/−) mouse model of atherosclerosis with alterations in IL10/AMPKalpha pathway. Mol. Metab. 2017, 6, 845–853. [Google Scholar] [CrossRef]
- Thompson, D.; Morrice, N.; Grant, L.; Le Sommer, S.; Lees, E.K.; Mody, N.; Wilson, H.M.; Delibegovic, M. Pharmacological inhibition of protein tyrosine phosphatase 1B protects against atherosclerotic plaque formation in the LDLR(−/−) mouse model of atherosclerosis. Clin. Sci. 2017, 131, 2489–2501. [Google Scholar] [CrossRef]
- Delile, E.; Neviere, R.; Thiebaut, P.A.; Maupoint, J.; Mulder, P.; Coquerel, D.; Renet, S.; Rieusset, J.; Richard, V.; Tamion, F. reduced insulin resistance contributes to the beneficial effect of protein tyrosine phosphatase-1B deletion in a mouse model of sepsis. Shock 2017, 48, 355–363. [Google Scholar] [CrossRef]
- Zabolotny, J.M.; Kim, Y.-B.; Welsh, L.A.; Kershaw, E.E.; Neel, B.G.; Kahn, B.B. Protein-tyrosine phosphatase 1B expression is induced by inflammation in vivo. J. Biol. Chem. 2008, 283, 14230–14241. [Google Scholar] [CrossRef] [PubMed]
- Peach, C.J.; Mignone, V.W.; Arruda, M.A.; Alcobia, D.C.; Hill, S.J.; Kilpatrick, L.E.; Woolard, J. Molecular pharmacology of VEGF-A Isoforms: binding and signalling at VEGFR2. Int. J. Mol. Sci. 2018, 19, 1264. [Google Scholar] [CrossRef] [PubMed]
- Caporarello, N.; Lupo, G.; Olivieri, M.; Cristaldi, M.; Cambria, M.T.; Salmeri, M.; Anfuso, C.D. Classical VEGF, Notch and Ang signalling in cancer angiogenesis, alternative approaches and future directions (review). Mol. Med. Rep. 2017, 16, 4393–4402. [Google Scholar] [CrossRef] [PubMed]
- Koch, S.; Claesson-Welsh, L. Signal transduction by vascular endothelial growth factor receptors. Cold Spring Harbor Perspect. Med. 2012, 2, a006502. [Google Scholar] [CrossRef] [PubMed]
- Lanahan, A.A.; Lech, D.; Dubrac, A.; Zhang, J.; Zhuang, Z.W.; Eichmann, A.; Simons, M. PTP1b Is a physiologic regulator of vascular endothelial growth factor signaling in endothelial cells. Circulation 2014, 130, 902. [Google Scholar] [CrossRef] [PubMed]
- Besnier, M.; Galaup, A.; Nicol, L.; Henry, J.-P.; Coquerel, D.; Gueret, A.; Mulder, P.; Brakenhielm, E.; Thuillez, C.; Germain, S.; et al. Enhanced angiogenesis and increased cardiac perfusion after myocardial infarction in protein tyrosine phosphatase 1B-deficient mice. FASEB J. 2014, 28, 3351–3361. [Google Scholar] [CrossRef]
- Kandadi, M.R.; Panzhinskiy, E.; Roe, N.D.; Nair, S.; Hu, D.; Sun, A. Deletion of protein tyrosine phosphatase 1B rescues against myocardial anomalies in high fat diet-induced obesity: Role of AMPK-dependent autophagy. Biochim. Biophys. Acta (BBA) Mol. Basis Dis. 2015, 1852, 299–309. [Google Scholar] [CrossRef]
- Zhang, J.; Li, L.; Li, J.; Liu, Y.; Zhang, C.-Y.; Zhang, Y.; Zen, K. Protein tyrosine phosphatase 1B impairs diabetic wound healing through vascular endothelial growth factor receptor 2 dephosphorylation. Arterioscler. Thromb. Vasc. Biol. 2015, 35, 163–174. [Google Scholar] [CrossRef]
- Bailey, D.; Barreca, C.; O’Hare, P. Trafficking of the bZIP transmembrane transcription factor CREB-H into alternate pathways of ERAD and stress-regulated intramembrane proteolysis. Traffic 2007, 8, 1796–1814. [Google Scholar] [CrossRef]
- Ron, D.; Hubbard, S.R. How IRE1 reacts to ER stress. Cell 2008, 132, 24–26. [Google Scholar] [CrossRef]
- Özcan, U.; Cao, Q.; Yilmaz, E.; Lee, A.-H.; Iwakoshi, N.N.; Özdelen, E.; Tuncman, G.; Görgün, C.; Glimcher, L.H.; Hotamisligil, G.S. Endoplasmic reticulum stress links obesity, insulin action, and type 2 diabetes. Science 2004, 306, 457–461. [Google Scholar] [CrossRef] [PubMed]
- Andreozzi, F.; Laratta, E.; Procopio, C.; Hribal, M.L.; Sciacqua, A.; Perticone, M.; Miele, C.; Perticone, F.; Sesti, G. Interleukin-6 impairs the insulin signaling pathway, promoting production of nitric oxide in human umbilical vein endothelial cells. Mol. Cell. Biol. 2007, 27, 2372–2383. [Google Scholar] [CrossRef] [PubMed]
- Civelek, M.; Manduchi, E.; Riley, R.J.; Stoeckert, C.J., Jr.; Davies, P.F. Chronic endoplasmic reticulum stress activates unfolded protein response in arterial endothelium in regions of susceptibility to atherosclerosis. Circ. Res. 2009, 105, 453–461. [Google Scholar] [CrossRef] [PubMed]
- Zeng, L.; Zampetaki, A.; Margariti, A.; Pepe, A.E.; Alam, S.; Martin, D.; Xiao, Q.; Wang, W.; Jin, Z.-G.; Cockerill, G.; et al. Sustained activation of XBP1 splicing leads to endothelial apoptosis and atherosclerosis development in response to disturbed flow. Proc. Natl. Acad. Sci. USA 2009, 106, 8326–8331. [Google Scholar] [CrossRef] [PubMed]
- Lenna, S.; Townsend, D.M.; Tan, F.K.; Kapanadze, B.; Markiewicz, M.; Trojanowska, M.; Scorza, R. HLA-B35 upregulates endothelin-1 and downregulates endothelial nitric oxide synthase via endoplasmic reticulum stress response in endothelial cells. J. Immunol. 2010, 184, 4654–4661. [Google Scholar] [CrossRef]
- Erbay, E.; Babaev, V.R.; Mayers, J.R.; Makowski, L.; Charles, K.N.; Snitow, M.E.; Fazio, S.; Wiest, M.M.; Watkins, S.M.; Linton, M.F.; et al. Reducing endoplasmic reticulum stress through a macrophage lipid chaperone alleviates atherosclerosis. Nat. Med. 2009, 15, 1383–1391. [Google Scholar] [CrossRef] [PubMed]
- Tufanli, O.; Telkoparan Akillilar, P.; Acosta-Alvear, D.; Kocaturk, B.; Onat, U.I.; Hamid, S.M.; Cimen, I.; Walter, P.; Weber, C.; Erbay, E. Targeting IRE1 with small molecules counteracts progression of atherosclerosis. Proc. Natl. Acad. Sci. USA 2017, 114, E1395–E1404. [Google Scholar] [CrossRef]
- Gu, F.; Nguyên, D.T.; Stuible, M.; Dubé, N.; Tremblay, M.L.; Chevet, E. Protein-tyrosine phosphatase 1B potentiates IRE1 signaling during endoplasmic reticulum stress. J. Biol. Chem. 2004, 279, 49689–49693. [Google Scholar] [CrossRef]
- Wang, S.; Chen, X.; Nair, S.; Sun, D.; Wang, X.; Ren, J. Deletion of protein tyrosine phosphatase 1B obliterates endoplasmic reticulum stress-induced myocardial dysfunction through regulation of autophagy. Biochim. Biophys. Acta (BBA) Mol. Basis Dis. 2017, 1863, 3060–3074. [Google Scholar] [CrossRef]
- Bettaieb, A.; Liu, S.; Xi, Y.; Nagata, N.; Matsuo, K.; Matsuo, I.; Chahed, S.; Bakke, J.; Keilhack, H.; Tiganis, T.; et al. Differential regulation of endoplasmic reticulum stress by protein tyrosine phosphatase 1B and T cell protein tyrosine phosphatase. J. Biol. Chem. 2011, 286, 9225–9235. [Google Scholar] [CrossRef]
- Bettaieb, A.; Matsuo, K.; Matsuo, I.; Wang, S.; Melhem, R.; Koromilas, A.E.; Haj, F.G. Protein tyrosine phosphatase 1B deficiency potentiates PERK/eIF2α signaling in brown adipocytes. PLoS ONE 2012, 7, e34412. [Google Scholar] [CrossRef] [PubMed]
- Panzhinskiy, E.; Hua, Y.; Culver, B.; Ren, J.; Nair, S. Endoplasmic reticulum stress upregulates protein tyrosine phosphatase 1B and impairs glucose uptake in cultured myotubes. Diabetologia 2013, 56, 598–607. [Google Scholar] [CrossRef] [PubMed]
- Chio, C.M.; Yu, X.; Bishop, A.C. Rational design of allosteric-inhibition sites in classical protein tyrosine phosphatases. Bioorg. Med. Chem. 2015, 23, 2828–2838. [Google Scholar] [CrossRef] [PubMed]
- Barr, A.J. Protein tyrosine phosphatases as drug targets: Strategies and challenges of inhibitor development. Future Med. Chem. 2010, 2, 1563–1576. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Zhang, J.; Lu, S.; Huang, W.; Geng, L.; Shen, Q.; Zhang, J. The mechanism of allosteric inhibition of protein tyrosine phosphatase 1B. PLoS ONE 2014, 9, e97668. [Google Scholar] [CrossRef] [PubMed]
- Tiganis, T. PTP1B and TCPTP—Nonredundant phosphatases in insulin signaling and glucose homeostasis. FEBS J. 2013, 280, 445–458. [Google Scholar] [CrossRef] [PubMed]
- Vieira, M.N.N.; Lyra e Silva, N.M.; Ferreira, S.T.; De Felice, F.G. Protein tyrosine phosphatase 1B (PTP1B): A potential target for Alzheimer’s therapy? Front. Aging Neurosci. 2017, 9, 7. [Google Scholar] [CrossRef]
- Zhang, S.; Zhang, Z.-Y. PTP1B as a drug target: Recent developments in PTP1B inhibitor discovery. Drug Discov. Today 2007, 12, 373–381. [Google Scholar] [CrossRef]
- Krishnan, N.; Koveal, D.; Miller, D.H.; Xue, B.; Akshinthala, S.D.; Kragelj, J.; Jensen, M.R.; Gauss, C.-M.; Page, R.; Blackledge, M.; et al. Targeting the disordered C-terminus of PTP1B with an allosteric inhibitor. Nat. Chem. Biol. 2014, 10, 558–566. [Google Scholar] [CrossRef]
- Qin, Z.; Pandey, N.R.; Zhou, X.; Stewart, C.A.; Hari, A.; Huang, H.; Stewart, A.F.R.; Brunel, J.M.; Chen, H.-H. Functional properties of Claramine: A novel PTP1B inhibitor and insulin-mimetic compound. Biochem. Biophys. Res. Commun. 2015, 458, 21–27. [Google Scholar] [CrossRef]
- Krishnan, N.; Konidaris, K.F.; Gasser, G.; Tonks, N.K. A potent, selective, and orally bioavailable inhibitor of the protein-tyrosine phosphatase PTP1B improves insulin and leptin signaling in animal models. J. Biol. Chem. 2018, 293, 1517–1525. [Google Scholar] [CrossRef] [PubMed]
- Verma, M.; Gupta, S.J.; Chaudhary, A.; Garg, V.K. Protein tyrosine phosphatase 1B inhibitors as antidiabetic agents—A brief review. Bioorg. Chem. 2017, 70, 267–283. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Zhang, W.; Liu, X.; Yu, H.; Lu, X.; Jiao, B. Inhibitors of protein tyrosine phosphatase 1B from marine natural products. Chem. Biodivers. 2017, 14, e1600462. [Google Scholar] [CrossRef]
- Swarbrick, M.M.; Havel, P.J.; Levin, A.A.; Bremer, A.A.; Stanhope, K.L.; Butler, M.; Booten, S.L.; Graham, J.L.; McKay, R.A.; Murray, S.F.; et al. Inhibition of protein tyrosine phosphatase-1B with antisense oligonucleotides improves insulin sensitivity and increases adiponectin concentrations in monkeys. Endocrinology 2009, 150, 1670–1679. [Google Scholar] [CrossRef] [PubMed]
- Xue, W.; Tian, J.; Wang, X.S.; Xia, J.; Wu, S. Discovery of potent PTP1B inhibitors via structure-based drug design, synthesis and in vitro bioassay of Norathyriol derivatives. Bioorg. Chem. 2019, 86, 224–234. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Insulin Resistance |
| Impaired glucose tolerance |
| Type-2 diabetes |
| Abnormal Plasma Lipids |
| High total cholesterol |
| Hypertriglyceridemia |
| High apolipoprotein B |
| Lower levels of apolipoprotein A1 |
| Hemodynamics |
| Increased blood volume |
| Elevated LV wall stress |
| High arterial pressure |
| Pulmonary artery hypertension |
| Structure of the Heart |
| Remodeling of LV |
| Hypertrophy of LV |
| Hypertrophy of RV |
| Enlargement of LA |
| Cardiac Function |
| LV systolic and diastolic dysfunction |
| Failure of RV |
| Inflammatory Response |
| High levels of C-reactive protein |
| Overproduction of tumor necrosis factor (TNF)-α |
| Neuro-Hormonal |
| Hyperinsulinemia |
| Resistance to leptin and hyperleptinemia |
| Reduced adiponectin |
| Sympathetic activation |
| Vascular Tone | |
| Endothelium derived relaxing factors | Endothelium derived contracting factors |
| Nitric Oxide (NO) | Elevated triglyceride |
| Prostacyclin | Decreased apolipoprotein-A1 |
| Bradykinin | Thromboxane |
| Endothelium-derived hyperpolarizing factor | |
| Coagulation | |
| Anti-coagulants | Pro-coagulants |
| Thrombomodulin | Von Willebrand factor |
| Protein C | Factor V |
| Urokinase | Plasminogen activator inhibitor |
| Tissue plasminogen activator | Tissue factor |
| Adhesion and Permeability | |
| Vascular cell adhesion molecule 1 (VCAM-1) | Tumor necrosis factor-α (TNF-α) |
| Intracellular adhesion molecule 1 (ICAM-1) | Monocyte chemoattractant protein 1 (MCP-1) |
| Platelet-endothelial cells adhesion molecule | Cytokines |
| Differentiation and Cellular Growth | |
| Transforming growth factor-β (TGF-β) | Insulin-like growth factor 1 (IGF-1) |
| Platelet-derived growth factor (PDGF) | Basic fibroblast growth factor (BFGF) |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Abdelsalam, S.S.; Korashy, H.M.; Zeidan, A.; Agouni, A. The Role of Protein Tyrosine Phosphatase (PTP)-1B in Cardiovascular Disease and Its Interplay with Insulin Resistance. Biomolecules 2019, 9, 286. https://doi.org/10.3390/biom9070286
Abdelsalam SS, Korashy HM, Zeidan A, Agouni A. The Role of Protein Tyrosine Phosphatase (PTP)-1B in Cardiovascular Disease and Its Interplay with Insulin Resistance. Biomolecules. 2019; 9(7):286. https://doi.org/10.3390/biom9070286
Chicago/Turabian StyleAbdelsalam, Shahenda S., Hesham M. Korashy, Asad Zeidan, and Abdelali Agouni. 2019. "The Role of Protein Tyrosine Phosphatase (PTP)-1B in Cardiovascular Disease and Its Interplay with Insulin Resistance" Biomolecules 9, no. 7: 286. https://doi.org/10.3390/biom9070286
APA StyleAbdelsalam, S. S., Korashy, H. M., Zeidan, A., & Agouni, A. (2019). The Role of Protein Tyrosine Phosphatase (PTP)-1B in Cardiovascular Disease and Its Interplay with Insulin Resistance. Biomolecules, 9(7), 286. https://doi.org/10.3390/biom9070286