The Ensemble of Conformations of Antifreeze Glycoproteins (AFGP8): A Study Using Nuclear Magnetic Resonance Spectroscopy


Abstract
1. Introduction
2. Materials and Methods
2.1. Protein Samples
2.2. NMR Spectroscopy
2.3. Ensemble Characterization
3. Results
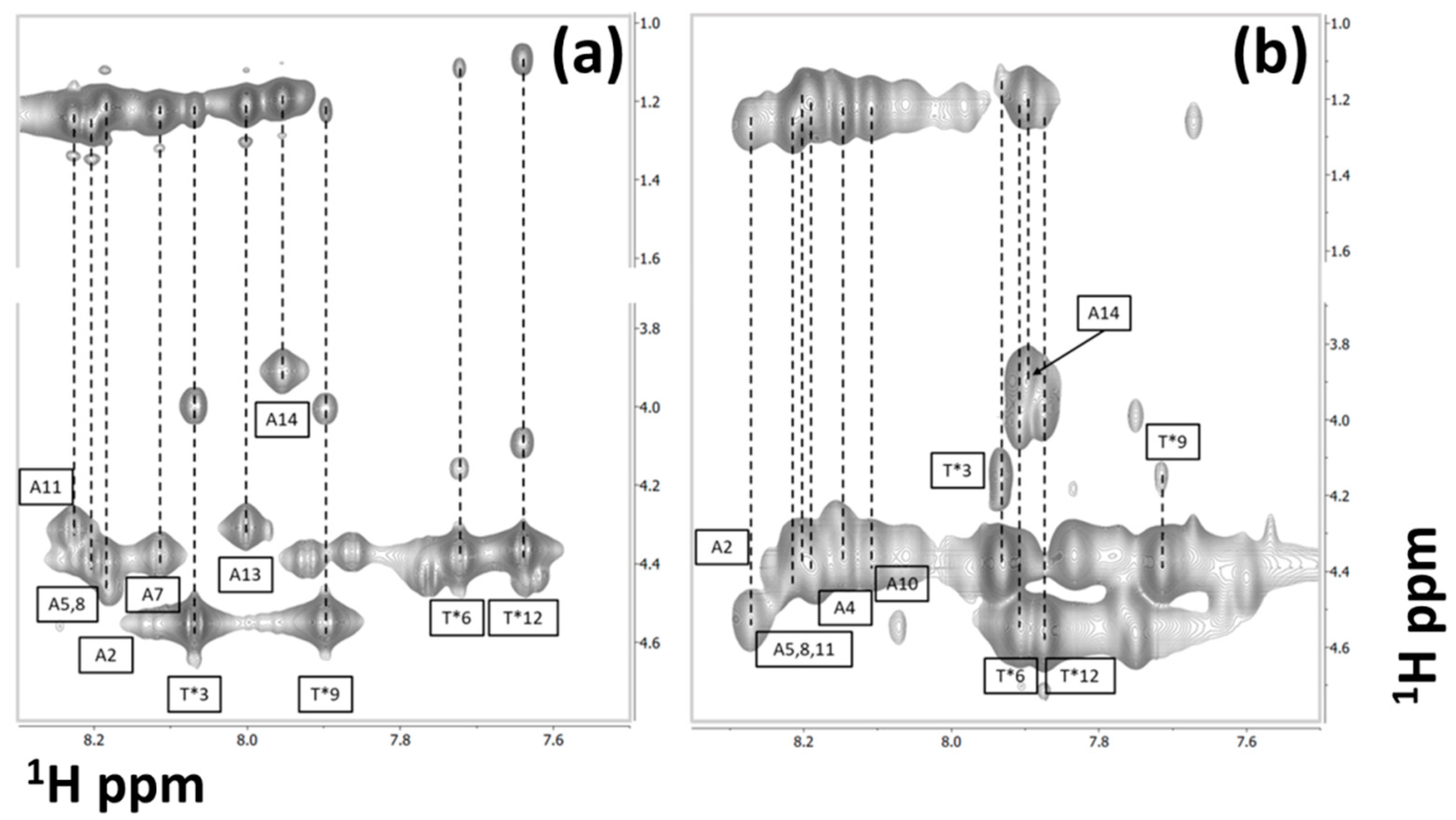
3.1. Sequence-Specific Chemical Shift Assignments
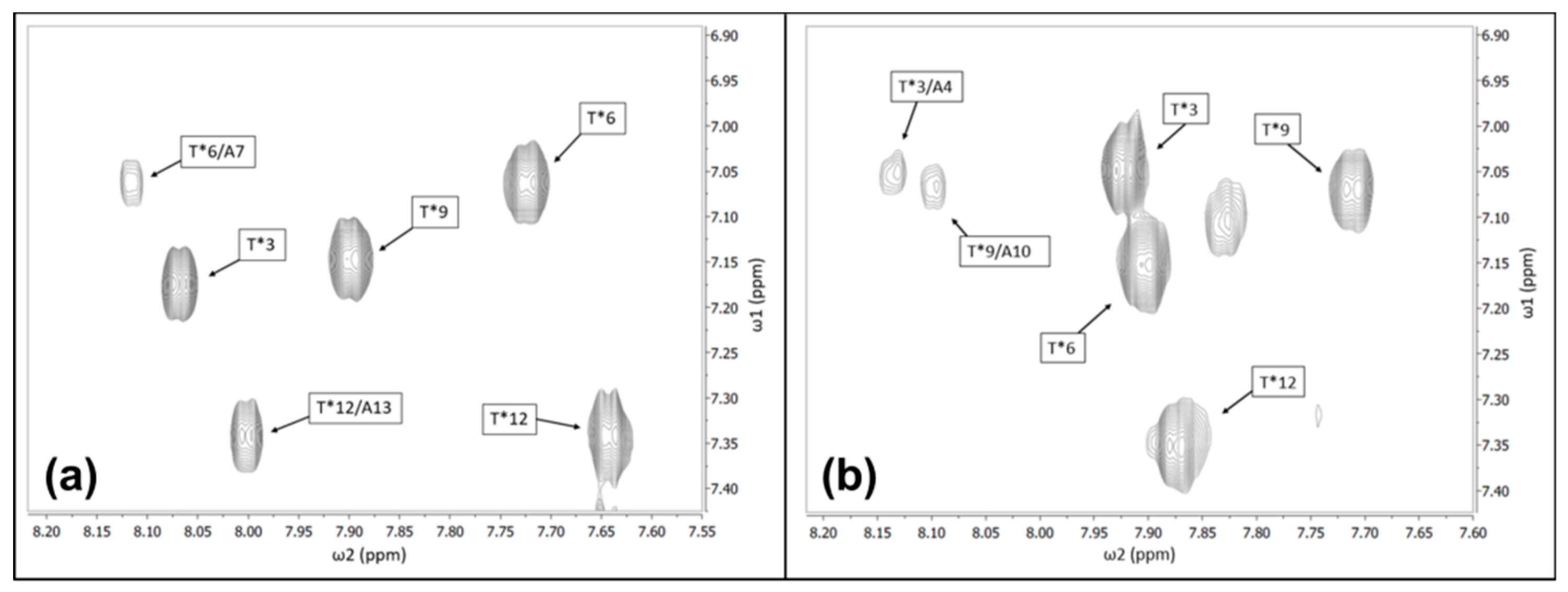
3.2. Chemical Shift Assignments of the Carbohydrate Side Chains
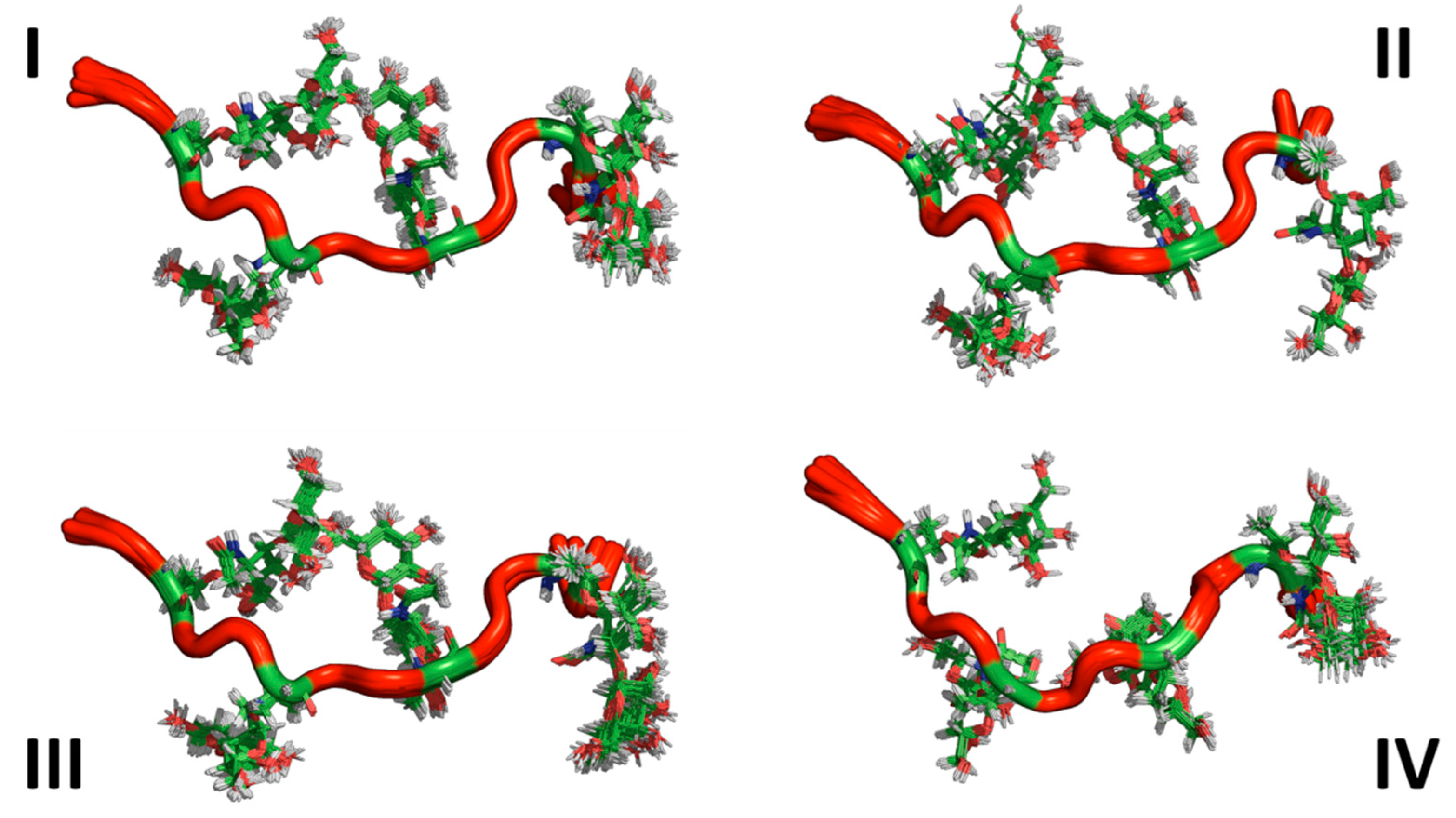
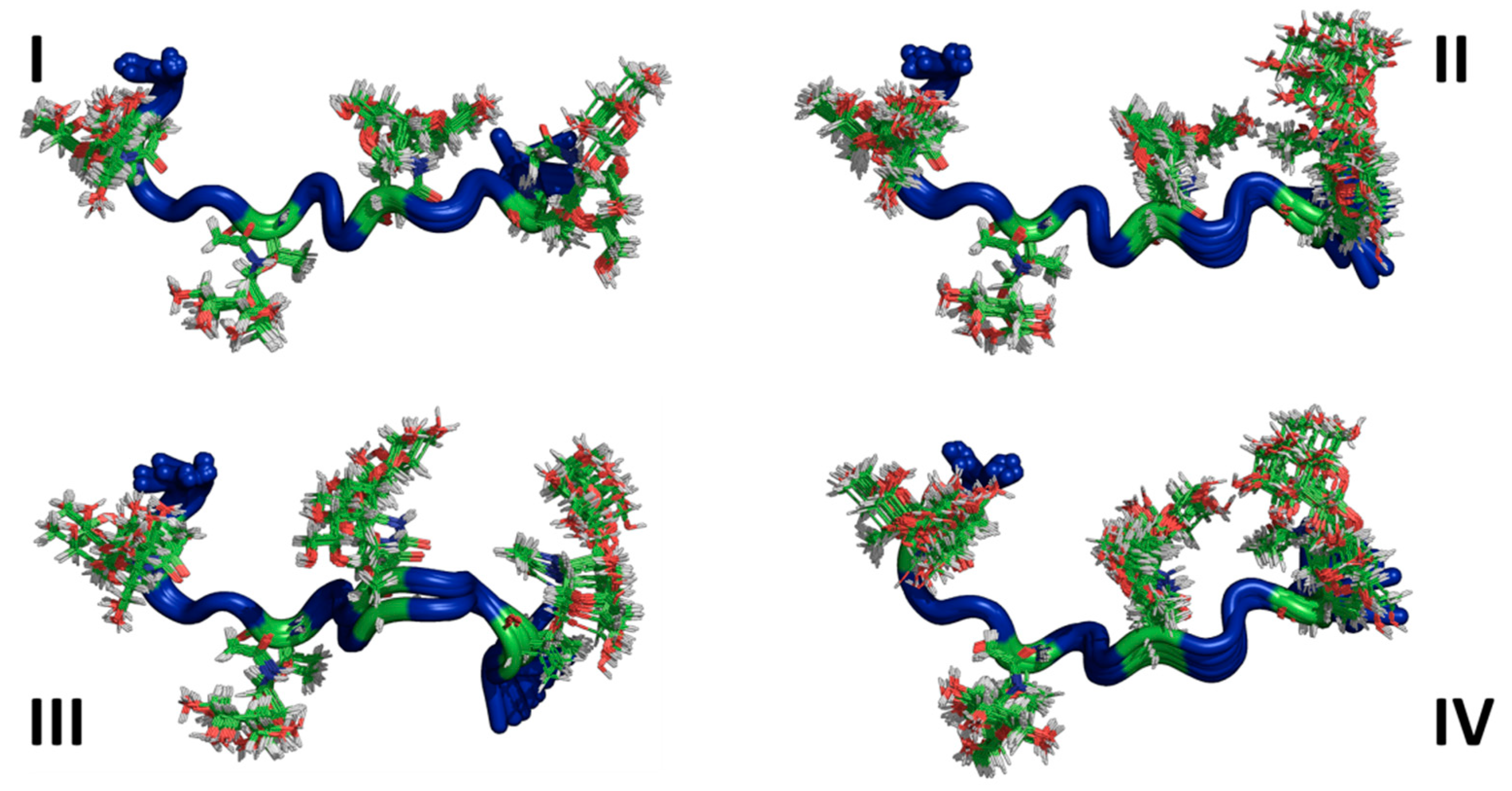
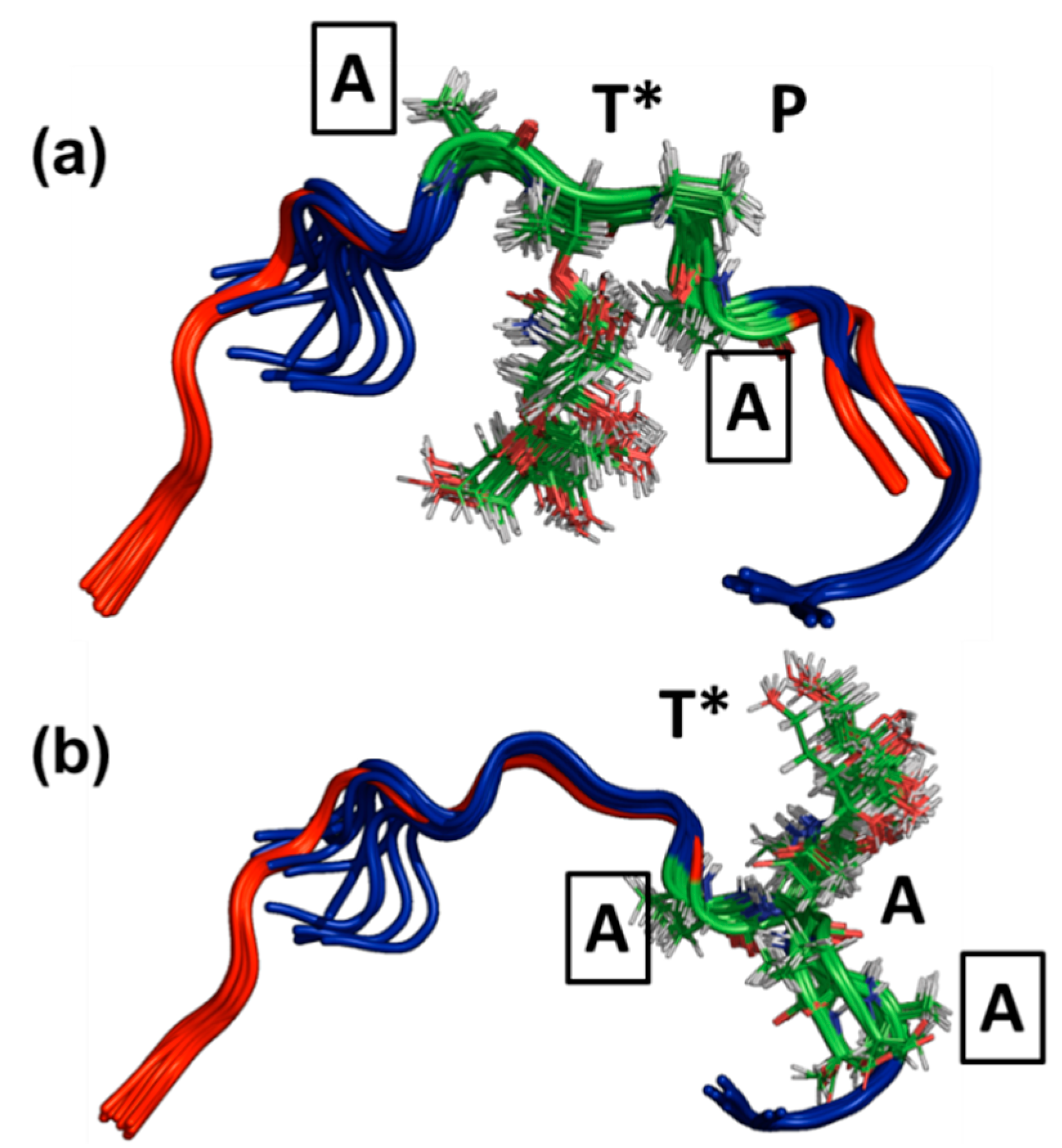
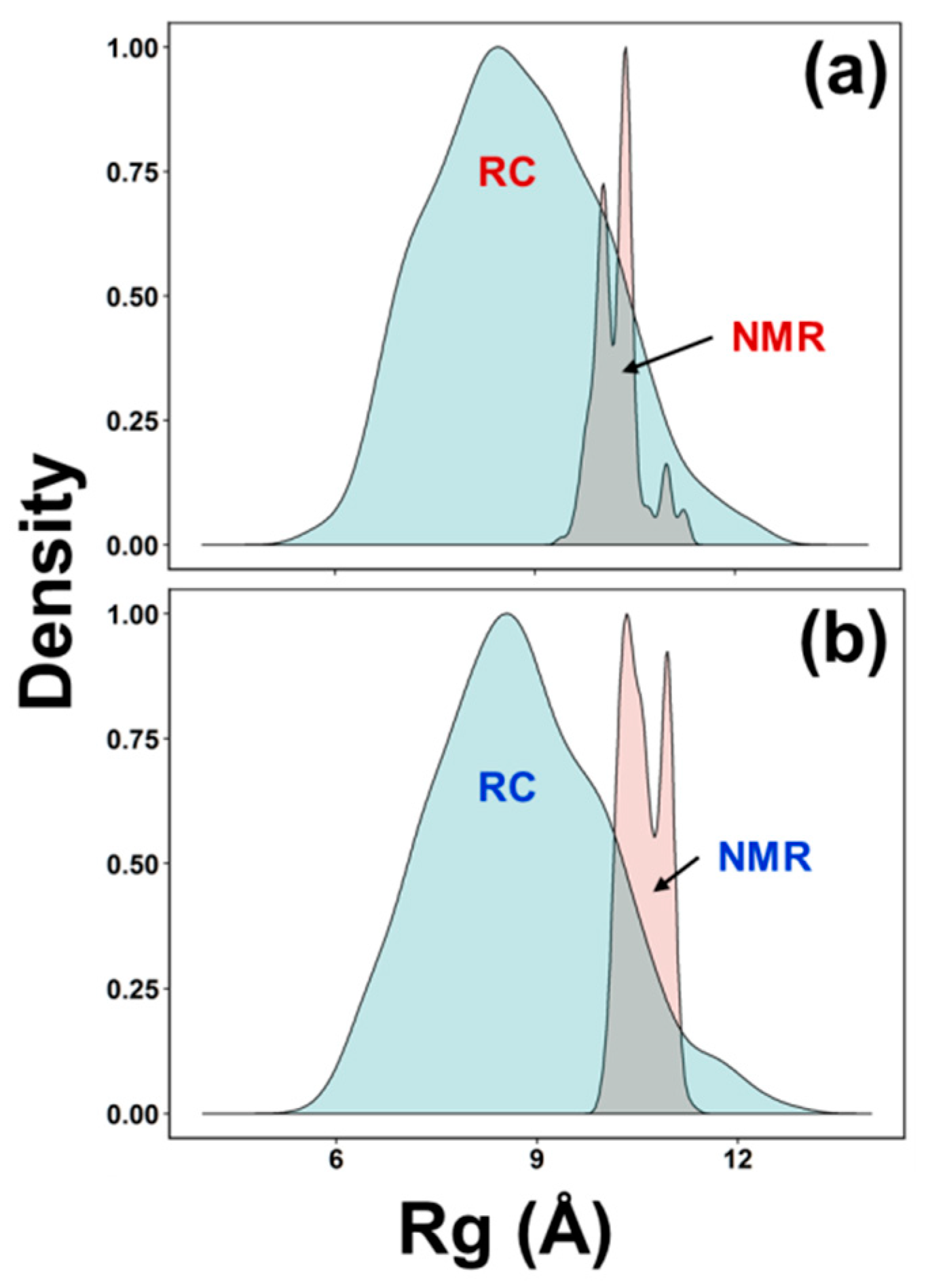
3.3. Ensemble Characterization
3.4. AFGP8-BS and AFGP8-TB Had a Distinct Ensemble in Solution State
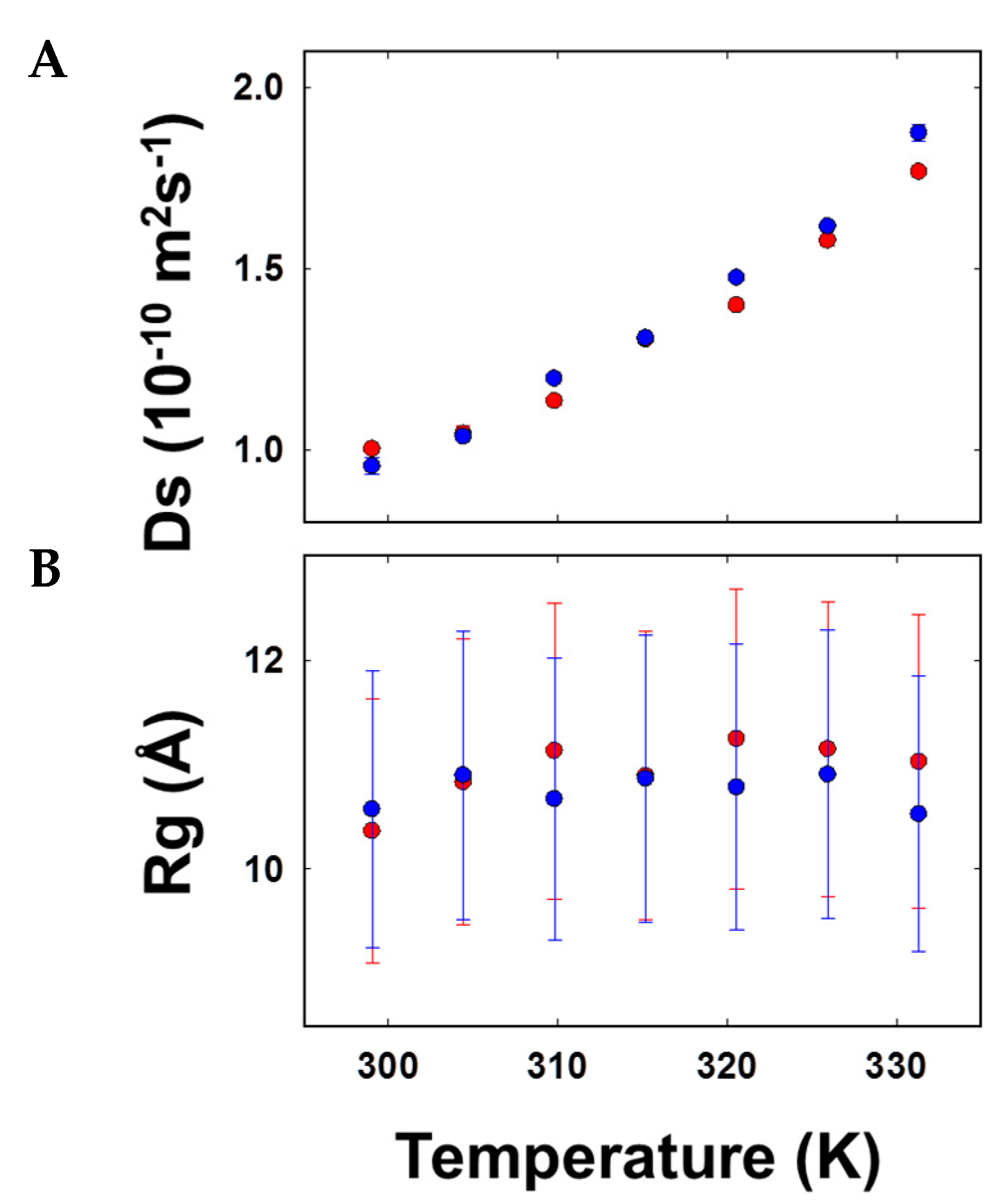
3.5. Size Estimations through Diffusion Coefficient Measures of AFGP8-BS and AFGP8-TB Suggested Comparable Compactness
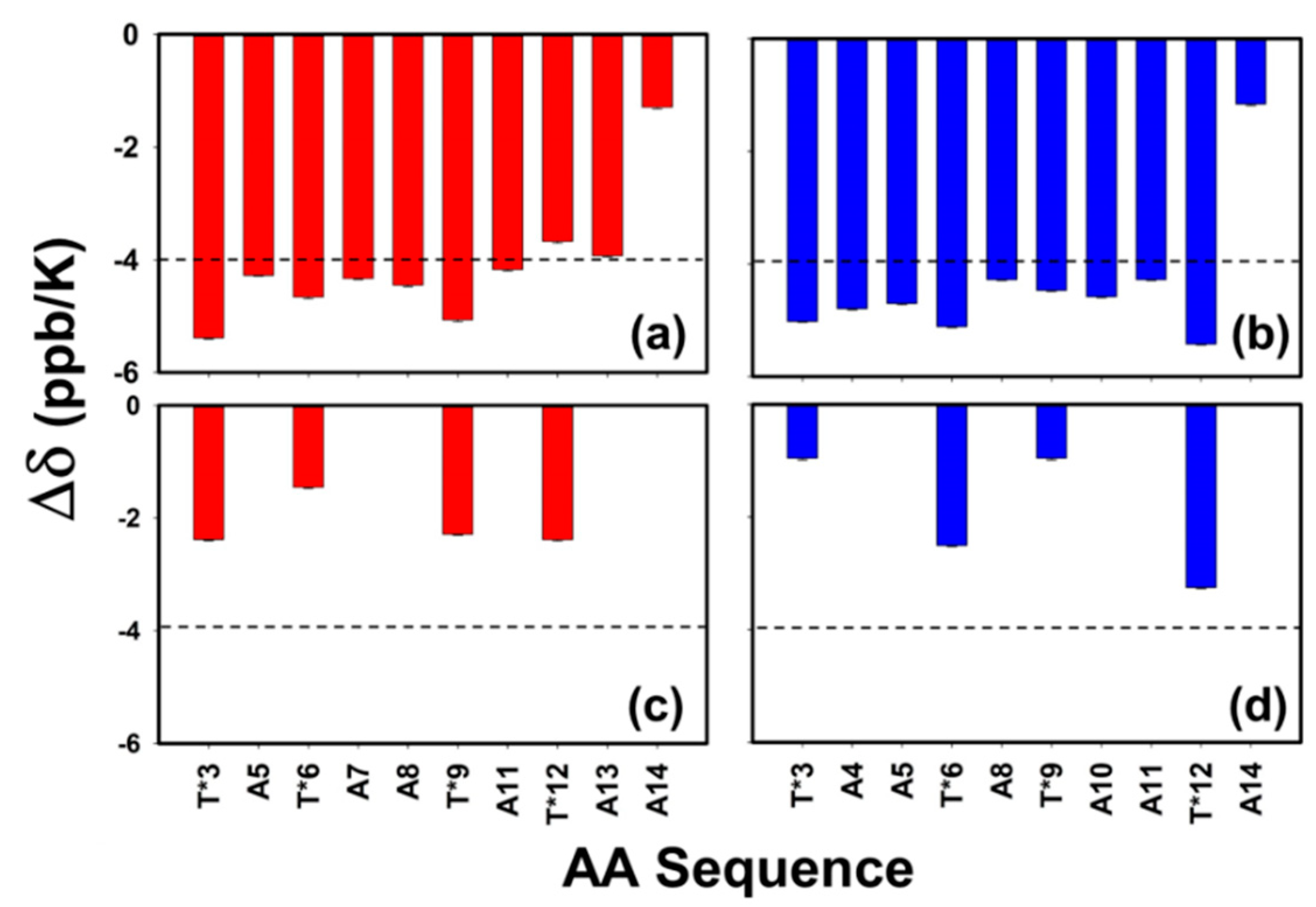
3.6. Role of Backbone to Side-Chain Hydrogen Bonding
4. Discussion
4.1. Integral Role of Proline Residues in the Short AFGPs
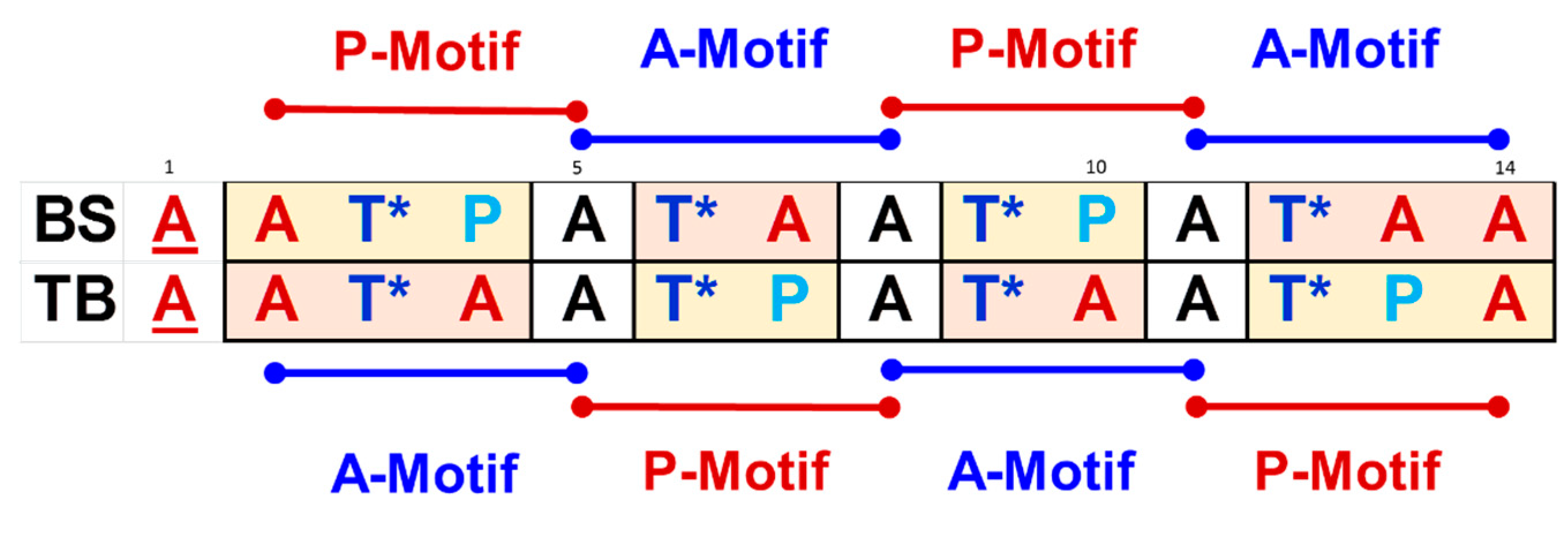
4.2. The Significance of Sequential Motifs
4.3. N-Acetyl Functional Group of Disaccharides to Backbone Hydrogen Bonding
4.4. AFGPs as Intrinsically Disordered Proteins: Effectors and Entropic Chains
4.5. Inferences about AFGP8 in Native Conditions: Water
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Gordon, M.S.; Amdur, B.H.; Scholander, P.F. Freezing Resistance in Some Northern Fishes. Biol. Bull. 1962, 122, 52–62. [Google Scholar] [CrossRef]
- Scholander, P.F.; van Dam, L.; Kanwisher, J.W.; Hammel, H.T.; Gordon, M.S. Supercooling and osmoregulation in arctic fish. J. Cell. Compar. Phys. 1957, 49, 5–24. [Google Scholar] [CrossRef]
- DeVries, A.L.; Komatsu, S.K.; Feeney, R.E. Chemical and physical properties of freezing point-depressing glycoproteins from Antarctic fishes. J. Biol. Chem. 1970, 245, 2901–2908. [Google Scholar] [PubMed]
- DeVries, A.L.; Wohlschlag, D.E. Freezing resistance in some Antarctic fishes. Science 1969, 163, 1073–1075. [Google Scholar] [CrossRef] [PubMed]
- Osuga, D.T.; Ward, F.C.; Yeh, Y.; Feeney, R.E. Cooperative Functioning between Antifreeze Glycoproteins. J. Biol. Chem. 1978, 253, 6669–6672. [Google Scholar] [PubMed]
- Feeney, R.E.; Yeh, Y. Antifreeze proteins from fish bloods. Adv. Protein. Chem. 1978, 32, 191–282. [Google Scholar] [PubMed]
- Yeh, Y.; Feeney, R.E. Antifreeze Proteins: Structures and Mechanisms of Function. Chem. Rev. 1996, 96, 601–618. [Google Scholar] [CrossRef] [PubMed]
- Knight, C.A.; Devries, A.L. Melting inhibition and superheating of ice by an antifreeze glycopeptide. Science 1989, 245, 505–507. [Google Scholar] [CrossRef]
- Cziko, P.A.; DeVries, A.L.; Evans, C.W.; Cheng, C.H. Antifreeze protein-induced superheating of ice inside Antarctic notothenioid fishes inhibits melting during summer warming. Proc. Natl. Acad. Sci. USA 2014, 111, 14583–14588. [Google Scholar] [CrossRef]
- Bar Dolev, M.; Braslavsky, I.; Davies, P.L. Ice-Binding Proteins and Their Function. Annu. Rev. Biochem. 2016, 85, 515–542. [Google Scholar] [CrossRef]
- Raymond, J.A.; DeVries, A.L. Adsorption inhibition as a mechanism of freezing resistance in polar fishes. Proc. Natl. Acad. Sci. USA 1977, 74, 2589–2593. [Google Scholar] [CrossRef] [PubMed]
- Davies, P.L.; Hew, C.L. Biochemistry of fish antifreeze proteins. FASEB J. 1990, 4, 2460–2468. [Google Scholar] [CrossRef] [PubMed]
- Hew, C.L.; Yang, D.S. Protein interaction with ice. Eur. J. Biochem. 1992, 203, 33–42. [Google Scholar] [CrossRef] [PubMed]
- Mochizuki, K.; Molinero, V. Antifreeze Glycoproteins Bind Reversibly to Ice via Hydrophobic Groups. J. Am. Chem. Soc. 2018, 140, 4803–4811. [Google Scholar] [CrossRef] [PubMed]
- Budke, C.; Dreyer, A.; Jaeger, J.; Gimpel, K.; Berkemeier, T.; Bonin, A.S.; Nagel, L.; Plattner, C.; DeVries, A.L.; Sewald, N.; et al. Quantitative Efficacy Classification of Ice Recrystallization Inhibition Agents. Cryst. Growth Des. 2014, 14, 4285–4294. [Google Scholar] [CrossRef]
- Davies, P.L.; Sykes, B.D. Antifreeze proteins. Curr. Opin. Struct. Biol. 1997, 7, 828–834. [Google Scholar] [CrossRef]
- De Vries, A.L. Antifreeze glycopeptides and peptides: Interactions with ice and water. Methods Enzymol. 1986, 127, 293–303. [Google Scholar]
- Duman, J.G. Antifreeze and ice nucleator proteins in terrestrial arthropods. Annu. Rev. Physiol. 2001, 63, 327–357. [Google Scholar] [CrossRef]
- Feeney, R.E.; Burcham, T.S.; Yeh, Y. Antifreeze glycoproteins from polar fish blood. Annu. Rev. Biophys. Biophys. Chem. 1986, 15, 59–78. [Google Scholar] [CrossRef]
- Feeney, R.E.; Yeh, Y. Antifreeze proteins: Current status and possible food uses. Trends Food Sci. Technol. 1998, 9, 102–106. [Google Scholar] [CrossRef]
- Graether, S.P.; Sykes, B.D. Cold survival in freeze-intolerant insects: The structure and function of beta-helical antifreeze proteins. Eur. J. Biochem. 2004, 271, 3285–3296. [Google Scholar] [CrossRef] [PubMed]
- Harding, M.M.; Anderberg, P.I.; Haymet, A.D.J. ‘Antifreeze’ glycoproteins from polar fish. Eur. J. Biochem. 2003, 270, 1381–1392. [Google Scholar] [CrossRef]
- Harding, M.M.; Ward, L.G.; Haymet, A.D.J. Type I ‘antifreeze’ proteins—Structure-activity studies and mechanisms of ice growth inhibition [Review]. Eur. J. Biochem. 1999, 264, 653–665. [Google Scholar] [CrossRef] [PubMed]
- Fletcher, G.L.; Hew, C.L.; Davies, P.L. Antifreeze proteins of teleost fishes. Annu. Rev. Physiol. 2001, 63, 359–390. [Google Scholar] [CrossRef] [PubMed]
- Graether, S.P. Biochemistry and Function of Antifreeze Proteins; Nova Science: Hauppauge, NY, USA, 2010. [Google Scholar]
- Krishnan, V.V.; Yeh, Y. Structure and Functional Dynamics of Antifreeze Glycoproteins. In Biochemistry and Function of Antifreeze Proteins; Graether, S.P., Ed.; Nova Science: Hauppauge, NY, USA, 2010; pp. 105–140. [Google Scholar]
- Urbanczyk, M.; Gora, J.; Latajka, R.; Sewald, N. Antifreeze glycopeptides: From structure and activity studies to current approaches in chemical synthesis. Amino Acids 2017, 49, 209–222. [Google Scholar] [CrossRef] [PubMed]
- Lane, A.N.; Hays, L.M.; Feeney, R.E.; Crowe, L.M.; Crowe, J.H. Conformational and dynamic properties of a 14 residue antifreeze glycopeptide from Antarctic cod. Protein Sci. 1998, 7, 1555–1563. [Google Scholar] [CrossRef] [PubMed]
- Lane, A.N.; Hays, L.M.; Tsvetkova, N.; Feeney, R.E.; Crowe, L.M.; Crowe, J.H. Comparison of the solution conformation and dynamics of antifreeze glycoproteins from Antarctic fish. Biophys. J. 2000, 78, 3195–3207. [Google Scholar] [CrossRef]
- Tachibana, Y.; Fletcher, G.L.; Fujitani, N.; Tsuda, S.; Monde, K.; Nishimura, S. Antifreeze glycoproteins: Elucidation of the structural motifs that are essential for antifreeze activity. Angew. Chem. (Int. Ed.) 2004, 43, 856–862. [Google Scholar] [CrossRef]
- LeBel, R.G.; Goring, D.A.I. Density, Viscosity, Refractive Index, and Hygroscopicity of Mixtures of Water and Dimethyl Sulfoxide. J. Chem. Eng. Data 1962, 7, 100–101. [Google Scholar] [CrossRef]
- Eto, T.K.; Rubinsky, B. Antifreeze glycoproteins increase solution viscosity. Biochem. Biophys. Res. Commun. 1993, 197, 927–931. [Google Scholar] [CrossRef]
- Heisel, K.A.; Krishnan, V.V. NMR based solvent exchange experiments to understand the conformational preference of intrinsically disordered proteins using FG-nucleoporin peptide as a model. Biopolymers 2014, 102, 69–77. [Google Scholar] [CrossRef] [PubMed]
- Rucker, S.P.; Shaka, A.J. Broadband homonuclear cross polarization in 2D NMR using DIPSI-2. Mol. Phys. 1989, 68, 509–517. [Google Scholar] [CrossRef]
- Delaglio, F.; Grzesiek, S.; Vuister, G.W.; Zhu, G.; Pfeifer, J.; Bax, A. NMRPipe: A multidimensional spectral processing system based on UNIX pipes. J. Biomol. NMR 1995, 6, 277–293. [Google Scholar] [CrossRef] [PubMed]
- Goddard, T.D.; Kneller, D.G. SPARKY 3; University of California: San Francisco, CA, USA, 2008. [Google Scholar]
- Lee, W.; Tonelli, M.; Markley, J.L. NMRFAM-SPARKY: Enhanced software for biomolecular NMR spectroscopy. Bioinformatics 2015, 31, 1325–1327. [Google Scholar] [CrossRef] [PubMed]
- Wu, D.; Chen, A.; Johnson, C.S. An improved diffusion-ordered spectroscopy experiment incorporating bipolar-gradient pulses. J. Magn. Reson. Ser. A 1995, 115, 260–264. [Google Scholar] [CrossRef]
- Jerschow, A.; Müller, N. Suppression of Convection Artifacts in Stimulated-Echo Diffusion Experiments. Double-Stimulated-Echo Experiments. J. Magn. Reson. 1997, 125, 372–375. [Google Scholar] [CrossRef]
- Castanar, L.; Poggetto, G.D.; Colbourne, A.A.; Morris, G.A.; Nilsson, M. The GNAT: A new tool for processing NMR data. Magn. Reson. Chem. MRC 2018, 56, 546–558. [Google Scholar] [CrossRef] [PubMed]
- Evans, R.; Dal Poggetto, G.; Nilsson, M.; Morris, G.A. Improving the Interpretation of Small Molecule Diffusion Coefficients. Anal. Chem. 2018, 90, 3987–3994. [Google Scholar] [CrossRef] [PubMed]
- Gierer, A.; Wirtz, K. Molekulare Theorie der Mikroreibung. Z. Für Nat. A 1953, 8, 532. [Google Scholar] [CrossRef]
- Guntert, P. Automated NMR structure calculation with CYANA. Methods Mol. Biol. 2004, 278, 353–378. [Google Scholar] [CrossRef]
- Guntert, P.; Mumenthaler, C.; Wuthrich, K. Torsion angle dynamics for NMR structure calculation with the new program DYANA. J. Mol. Biol. 1997, 273, 283–298. [Google Scholar] [CrossRef] [PubMed]
- Shen, Y.; Bax, A. Protein structural information derived from NMR chemical shift with the neural network program TALOS-N. Methods Mol. Biol. 2015, 1260, 17–32. [Google Scholar] [CrossRef] [PubMed]
- Shen, Y.; Delaglio, F.; Cornilescu, G.; Bax, A. TALOS+: A hybrid method for predicting protein backbone torsion angles from NMR chemical shifts. J. Biomol. NMR 2009, 44, 213–223. [Google Scholar] [CrossRef] [PubMed]
- Hanwell, M.D.; Curtis, D.E.; Lonie, D.C.; Vandermeersch, T.; Zurek, E.; Hutchison, G.R. Avogadro: An advanced semantic chemical editor, visualization, and analysis platform. J. Cheminform. 2012, 4, 17. [Google Scholar] [CrossRef] [PubMed]
- Guex, N.; Peitsch, M.C. SWISS-MODEL and the Swiss-Pdb Viewer: An environment for comparative protein modeling. Electrophoresis 1997, 18, 2714–2723. [Google Scholar] [CrossRef]
- Kelley, L.A.; Gardner, S.P.; Sutcliffe, M.J. An automated approach for clustering an ensemble of NMR-derived protein structures into conformationally related subfamilies. Protein Eng. 1996, 9, 1063. [Google Scholar] [CrossRef]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera—A visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef]
- R Core Team. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2013. [Google Scholar]
- Grant, B.J.; Rodrigues, A.P.; ElSawy, K.M.; McCammon, J.A.; Caves, L.S. Bio3d: An R package for the comparative analysis of protein structures. Bioinformatics 2006, 22, 2695–2696. [Google Scholar] [CrossRef]
- Her, C. Determination of the Solution Structure of Antifreeze Glycoprotein Fraction 8 (AFGP8) in Deuterated Dimethyl Sulfoxide (DMSO) Using Nuclear Magnetic Resonance (NMR) Spectroscopy; California State University: Fresno, CA, USA, 2018. [Google Scholar]
- Inglis, S.R.; McGann, M.J.; Price, W.S.; Harding, M.M. Diffusion NMR studies on fish antifreeze proteins and synthetic analogues. FEBS Lett. 2006, 580, 3911–3915. [Google Scholar] [CrossRef]
- Krishnan, V.V.; Fink, W.H.; Feeney, R.E.; Yeh, Y. Translational dynamics of antifreeze glycoprotein in supercooled water. Biophys. Chem. 2004, 110, 223–230. [Google Scholar] [CrossRef][Green Version]
- Iqbal, M.; Balaram, P. Aggregation of apolar peptides in organic solvents. Concentration dependence of 1H-nmr parameters for peptide NH groups in 310 helical decapeptide fragment of suzukacillin. Biopolymers 1982, 21, 1427–1433. [Google Scholar] [CrossRef]
- Cierpicki, T.; Otlewski, J. Amide proton temperature coefficients as hydrogen bond indicators in proteins. J. Biomol. NMR 2001, 21, 249–261. [Google Scholar] [CrossRef] [PubMed]
- Mimura, Y.; Yamamoto, Y.; Inoue, Y.; Chûjô, R. NMR study of interaction between sugar and peptide moieties in mucin-type model glycopeptides. Int. J. Biol. Macromol. 1992, 14, 242–248. [Google Scholar] [CrossRef]
- Mandumpal, J.B.; Kreck, C.A.; Mancera, R.L. A molecular mechanism of solvent cryoprotection in aqueous DMSO solutions. Phys. Chem. Chem. Phys. 2011, 13, 3839–3842. [Google Scholar] [CrossRef]
- Sydykov, B.; Oldenhof, H.; Sieme, H.; Wolkers, W.F. Storage stability of liposomes stored at elevated subzero temperatures in DMSO/sucrose mixtures. PLoS ONE 2018, 13, e0199867. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Saez, N.; Peregrina, J.M.; Corzana, F. Principles of mucin structure: Implications for the rational design of cancer vaccines derived from MUC1-glycopeptides. Chem. Soc. Rev. 2017, 46, 7154–7175. [Google Scholar] [CrossRef]
- Dunker, A.K.; Lawson, J.D.; Brown, C.J.; Williams, R.M.; Romero, P.; Oh, J.S.; Oldfield, C.J.; Campen, A.M.; Ratliff, C.M.; Hipps, K.W.; et al. Intrinsically disordered protein. J. Mol. Graph. Model. 2001, 19, 26–59. [Google Scholar] [CrossRef]
- Tompa, P.; Kovacs, D. Intrinsically disordered chaperones in plants and animals. Biochem. Cell Biol. Biochim. Et Biol. Cell. 2010, 88, 167–174. [Google Scholar] [CrossRef]
- Burcham, T.S.; Osuga, D.T.; Rao, B.N.; Bush, C.A.; Feeney, R.E. Purification and primary sequences of the major arginine-containing antifreeze glycopeptides from the fish Eleginus gracilis. J. Biol. Chem. 1986, 261, 6384–6389. [Google Scholar]
- Burcham, T.S.; Osuga, D.T.; Yeh, Y.; Feeney, R.E. A kinetic description of antifreeze glycoprotein activity. J. Biol. Chem. 1986, 261, 6390–6397. [Google Scholar]
- Van der Lee, R.; Buljan, M.; Lang, B.; Weatheritt, R.J.; Daughdrill, G.W.; Dunker, A.K.; Fuxreiter, M.; Gough, J.; Gsponer, J.; Jones, D.T.; et al. Classification of intrinsically disordered regions and proteins. Chem. Rev. 2014, 114, 6589–6631. [Google Scholar] [CrossRef] [PubMed]
- Jorda, J.; Kajava, A.V. Protein Homorepeats: Sequences, Structures, Evolution, and Functions. In Advances in Protein Chemistry and Structural Biology; McPherson, A., Ed.; Academic Press: San Diego, CA, USA, 2010; Volume 79, pp. 59–88. [Google Scholar]
- Ebbinghaus, S.; Meister, K.; Born, B.; DeVries, A.L.; Gruebele, M.; Havenith, M. Antifreeze glycoprotein activity correlates with long-range protein-water dynamics. J. Am. Chem. Soc. 2010, 132, 12210–12211. [Google Scholar] [CrossRef] [PubMed]
- Mallajosyula, S.S.; Vanommeslaeghe, K.; MacKerell, A.D., Jr. Perturbation of long-range water dynamics as the mechanism for the antifreeze activity of antifreeze glycoprotein. J. Phys. Chem. B 2014, 118, 11696–11706. [Google Scholar] [CrossRef] [PubMed]
- Wolfe, J.; Bryant, G.; Koster, K.L. What is ‘unfreezable water’, how unfreezable is it, and how much is there? Cryo Lett. 2002, 23, 157–166. [Google Scholar]
- Pandey, P.; Mallajosyula, S.S. Elucidating the role of key structural motifs in antifreeze glycoproteins. Phys. Chem. Chem. Phys. 2019, 21, 3903–3917. [Google Scholar] [CrossRef] [PubMed]
- Meister, K.; DeVries, A.L.; Bakker, H.J.; Drori, R. Antifreeze Glycoproteins Bind Irreversibly to Ice. J. Am. Chem. Soc. 2018, 140, 9365–9368. [Google Scholar] [CrossRef]
- Furukawa, Y.; Nagashima, K.; Nakatsubo, S.I.; Yoshizaki, I.; Tamaru, H.; Shimaoka, T.; Sone, T.; Yokoyama, E.; Zepeda, S.; Terasawa, T.; et al. Oscillations and accelerations of ice crystal growth rates in microgravity in presence of antifreeze glycoprotein impurity in supercooled water. Sci. Rep. 2017, 7, 43157. [Google Scholar] [CrossRef]
- Malenkov, G. Liquid water and ices: Understanding the structure and physical properties. J. Phys. Condens. Matter 2009, 21, 283101. [Google Scholar] [CrossRef]
- Wang, S.; Wen, X.; DeVries, A.L.; Bagdagulyan, Y.; Morita, A.; Golen, J.A.; Duman, J.G.; Rheingold, A.L. Molecular recognition of methyl alpha-D-mannopyranoside by antifreeze (glyco)proteins. J. Am. Chem. Soc. 2014, 136, 8973–8981. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Residue | (a) AFGP8 from Boreogadus saida | ||||||||
| HN | Hα | Hβ | Hγ | Hδ | Cα | Cβ | Cγ | Cδ | |
| A1 | - | 3.37 | 1.17 | - | - | 49.61 | 20.91 | - | - |
| A2 | 8.18 | 4.47 | 1.22 | - | - | 47.51 | 18.28 | - | - |
| T3* | 8.07 | 4.55 | 4.00 | 1.23 | - | 54.63 | 74.19 | 18.29 | - |
| P4 | - | 4.68 | 2.05; 1.88 | 1.90 | 3.63 | 58.63 | 29.10 | 23.94 | 46.74 |
| A5 | 8.20 | 4.38 | 1.26 | - | - | 47.69 | 18.11 | - | - |
| T6* | 7.72 | 4.39 | 4.16 | 1.12 | - | 55.86 | 74.73 | 17.77 | - |
| A7 | 8.12 | 4.38 | 1.23 | - | - | 47.68 | 18.28 | - | - |
| A8 | 8.19 | 4.40 | 1.22 | - | - | 47.73 | 18.28 | - | - |
| T9* | 7.90 | 4.55 | 4.00 | 1.22 | - | 54.68 | 74.20 | 18.29 | - |
| P10 | - | 4.70 | 2.05; 1.88 | 1.90 | 3.63 | 58.63 | 29.10 | 23.94 | 46.74 |
| A11 | 8.23 | 4.31 | 1.24 | - | - | 47.82 | 18.20 | - | - |
| T12* | 7.64 | 4.36 | 4.09 | 1.09 | - | 55.34 | 74.12 | 17.80 | - |
| A13 | 8.00 | 4.31 | 1.21 | - | - | 47.82 | 18.26 | - | - |
| A14 | 7.95 | 3.91 | 1.20 | - | - | 48.65 | 18.30 | - | - |
| Residue | (b) AFGP8 Pathogenia (Trematomus) borchgrevinki | ||||||||
| HN | Hα | Hβ | Hγ | Hδ | Cα | Cβ | Cγ | Cδ | |
| A1 | - | 3.38 | 1.18 | - | - | 49.53 | 20.80 | - | - |
| A2 | 8.21 | 4.53 | 1.25 | - | - | 47.55 | 18.01 | - | - |
| T3* | 7.92 | 4.38 | 4.16 | 1.13 | - | 55.87 | 74.82 | 18.29 | - |
| A4 | 8.19 | 4.37 | 1.23 | - | - | 47.53 | 18.36 | - | - |
| A5 | 8.18 | 4.41 | 1.21 | - | - | 47.79 | 17.68 | - | - |
| T6* | 7.90 | 4.55 | 4.01 | 1.22 | - | 54.58 | 74.02 | 18.64 | - |
| P7 | - | 4.48 | 2.05; 1.88 | 1.88 | 3.62 | 58.62 | 28.98 | 24.22 | 46.82 |
| A8 | 8.21 | 4.38 | 1.26 | - | - | 47.69 | 18.01 | - | - |
| T9* | 7.71 | 4.38 | 4.16 | 1.13 | - | 55.87 | 74.82 | 18.29 | - |
| A10 | 8.10 | 4.47 | 1.22 | - | - | 47.53 | 18.69 | - | - |
| A11 | 8.19 | 4.36 | 1.21 | - | - | 47.51 | 17.65 | - | - |
| T12* | 7.88 | 4.57 | 3.97 | 1.23 | - | 54.00 | 73.99 | 18.64 | - |
| P13 | - | 4.38 | 2.01; 1.86 | 1.86 | 3.61 | 59.08 | 28.98 | 24.22 | 46.82 |
| A14 | 7.86 | 3.87 | 1.20 | - | - | 48.65 | 18.30 | - | - |
| Disaccharide | (a) AFGP8 from Boreogadus saida | ||||||||||||||
| HN | HNAc | H1 | H2 | H3 | H4 | H5 | H6 | C1 | C2 | C3 | C4 | C5 | C6 | CNAc | |
| α3 | 7.18 | 1.83 | 4.88 | 4.18 | 3.64 | 3.90 | 3.71 | 3.47 | 98.5 | 47.8 | 77.8 | 67.7 | 71.2 | 60.4 | 22.8 |
| α6 | 7.06 | 1.87 | 4.73 | 4.13 | 3.59 | 3.90 | 3.68 | 3.46 | 98.5 | 47.8 | 78.3 | 67.7 | 71.2 | 60.4 | 22.8 |
| α9 | 7.15 | 1.83 | 4.88 | 4.13 | 3.61 | 3.90 | 3.71 | 3.47 | 98.5 | 47.8 | 78.2 | 67.7 | 71.2 | 60.4 | 22.8 |
| α12 | 7.34 | 1.85 | 4.79 | 4.14 | 3.65 | 3.90 | 3.68 | 3.46 | 98.5 | 47.8 | 77.8 | 67.7 | 71.2 | 60.4 | 22.8 |
| β3 | - | - | 4.24 | 3.31 | 3.27 | 3.62 | 3.34 | 3.53 | 104.1 | 70.7 | 72.8 | 67.9 | 75.1 | 60.4 | - |
| β6 | - | - | 4.19 | 3.32 | 3.27 | 3.62 | 3.34 | 3.47 | 104.1 | 70.7 | 72.8 | 67.9 | 75.1 | 60.4 | - |
| β9 | - | - | 4.21 | 3.32 | 3.27 | 3.62 | 3.34 | 3.53 | 104.1 | 70.7 | 72.8 | 67.9 | 75.1 | 60.4 | - |
| β12 | - | - | 4.29 | 3.30 | 3.27 | 3.62 | 3.34 | 3.47 | 104.1 | 70.7 | 72.8 | 67.9 | 75.1 | 60.4 | - |
| Disaccharide | (b) AFGP8 from Pathogenia (Trematomus) borchgrevinki | ||||||||||||||
| HN | HNAc | H1 | H2 | H3 | H4 | H5 | H6 | C1 | C2 | C3 | C4 | C5 | C6 | CNAc | |
| α3 | 7.05 | 1.87 | 4.72 | 4.15 | 3.62 | 3.90 | 3.68 | 3.47 | 98.5 | 47.9 | 78.2 | 67.7 | 71.3 | 60.4 | 22.8 |
| α6 | 7.15 | 1.84 | 4.88 | 4.13 | 3.61 | 3.90 | 3.72 | 3.48 | 98.5 | 48.0 | 78.2 | 67.7 | 71.3 | 60.4 | 22.8 |
| α9 | 7.07 | 1.87 | 4.72 | 4.14 | 3.58 | 3.90 | 3.68 | 3.47 | 98.5 | 47.9 | 78.3 | 67.7 | 71.3 | 60.4 | 22.8 |
| α12 | 7.35 | 1.81 | 4.91 | 4.11 | 3.64 | 3.90 | 3.72 | 3.48 | 98.5 | 48.0 | 77.9 | 67.7 | 71.3 | 60.4 | 22.8 |
| β3 | - | - | 4.23 | 3.28 | 3.27 | 3.62 | 3.34 | 3.47 | 104.0 | 70.6 | 72.8 | 67.9 | 75.1 | 60.4 | - |
| β6 | - | - | 4.21 | 3.28 | 3.27 | 3.62 | 3.34 | 3.53 | 104.0 | 70.6 | 72.8 | 67.9 | 75.1 | 60.4 | - |
| β9 | - | - | 4.20 | 3.28 | 3.27 | 3.62 | 3.34 | 3.57 | 104.0 | 70.6 | 72.8 | 67.9 | 75.1 | 60.4 | - |
| β12 | - | - | 4.27 | 3.30 | 3.30 | 3.62 | 3.34 | 3.53 | 104.0 | 70.6 | 72.8 | 67.9 | 75.1 | 60.4 | - |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Her, C.; Yeh, Y.; Krishnan, V.V. The Ensemble of Conformations of Antifreeze Glycoproteins (AFGP8): A Study Using Nuclear Magnetic Resonance Spectroscopy. Biomolecules 2019, 9, 235. https://doi.org/10.3390/biom9060235
Her C, Yeh Y, Krishnan VV. The Ensemble of Conformations of Antifreeze Glycoproteins (AFGP8): A Study Using Nuclear Magnetic Resonance Spectroscopy. Biomolecules. 2019; 9(6):235. https://doi.org/10.3390/biom9060235
Chicago/Turabian StyleHer, Cheenou, Yin Yeh, and Viswanathan V. Krishnan. 2019. "The Ensemble of Conformations of Antifreeze Glycoproteins (AFGP8): A Study Using Nuclear Magnetic Resonance Spectroscopy" Biomolecules 9, no. 6: 235. https://doi.org/10.3390/biom9060235
APA StyleHer, C., Yeh, Y., & Krishnan, V. V. (2019). The Ensemble of Conformations of Antifreeze Glycoproteins (AFGP8): A Study Using Nuclear Magnetic Resonance Spectroscopy. Biomolecules, 9(6), 235. https://doi.org/10.3390/biom9060235