Inhibition of Oncogenic Kinases: An In Vitro Validated Computational Approach Identified Potential Multi-Target Anticancer Compounds

 ,
,  , ,
, ,  ,
,
Abstract
:
1. Introduction
2. Material and Methods
2.1. Computational Methods
2.1.1. Protein Dataset Preparation
2.1.2. Ligand Preparation
2.1.3. Virtual Screening
2.1.4. Molecular Dynamics Simulations
2.1.5. Comparative Docking
2.2. MM-GBSA Calculations Using AMBER
2.3. Experimental Procedures: In Vitro Cytotoxicity
2.3.1. Materials
2.3.2. MTT Assay
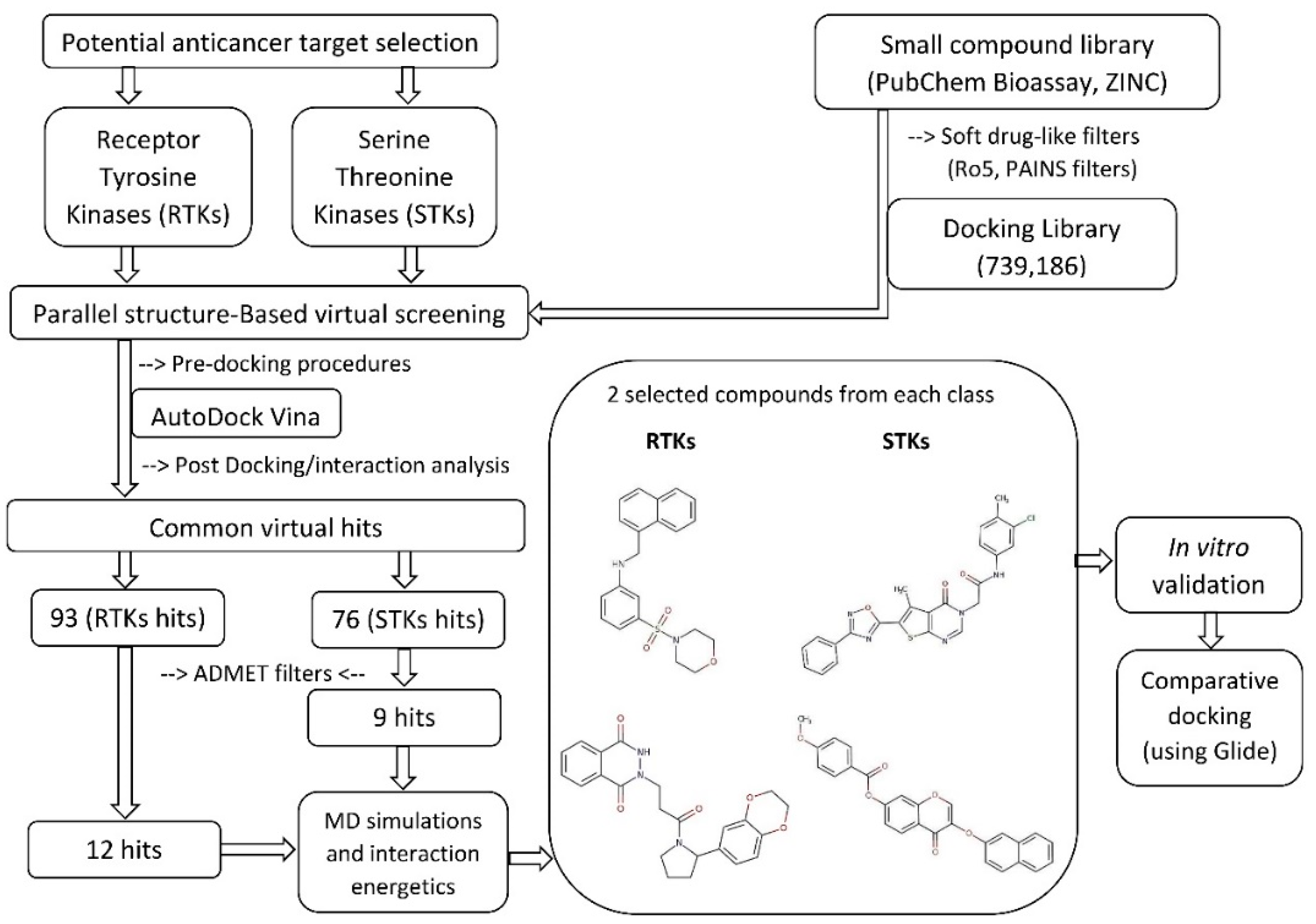
3. Results and Discussion
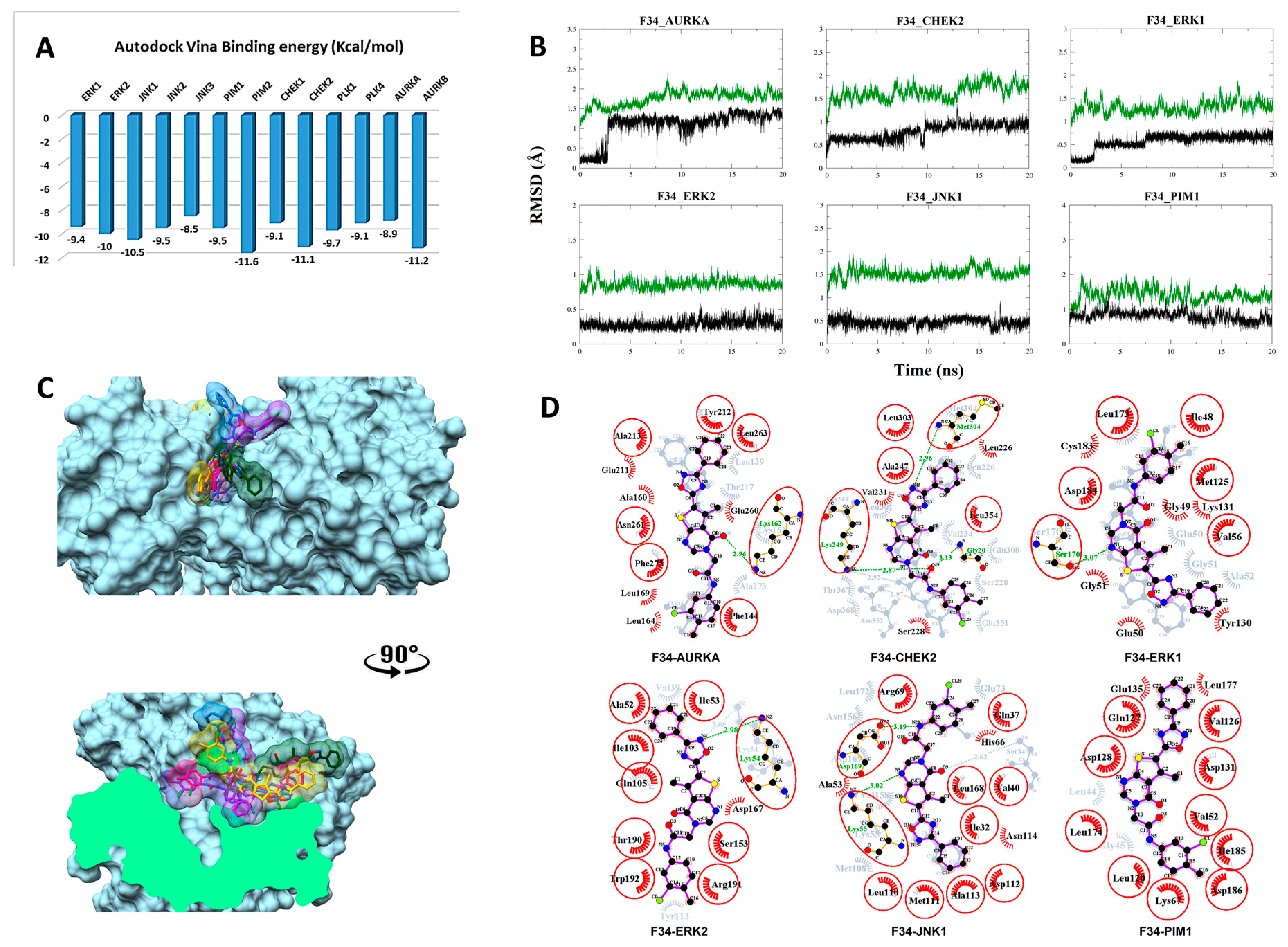
3.1. Structure-Based Virtual Screening
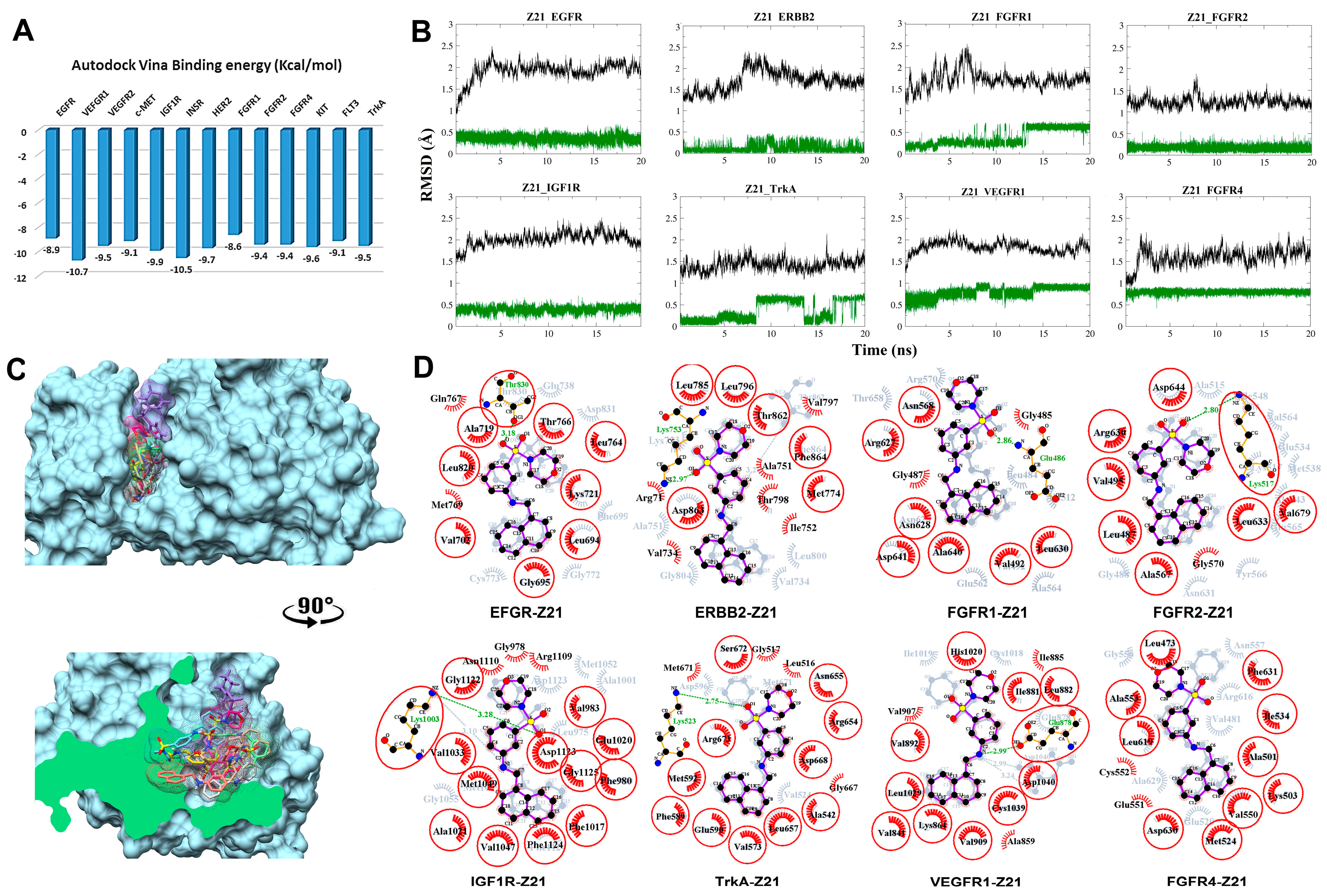
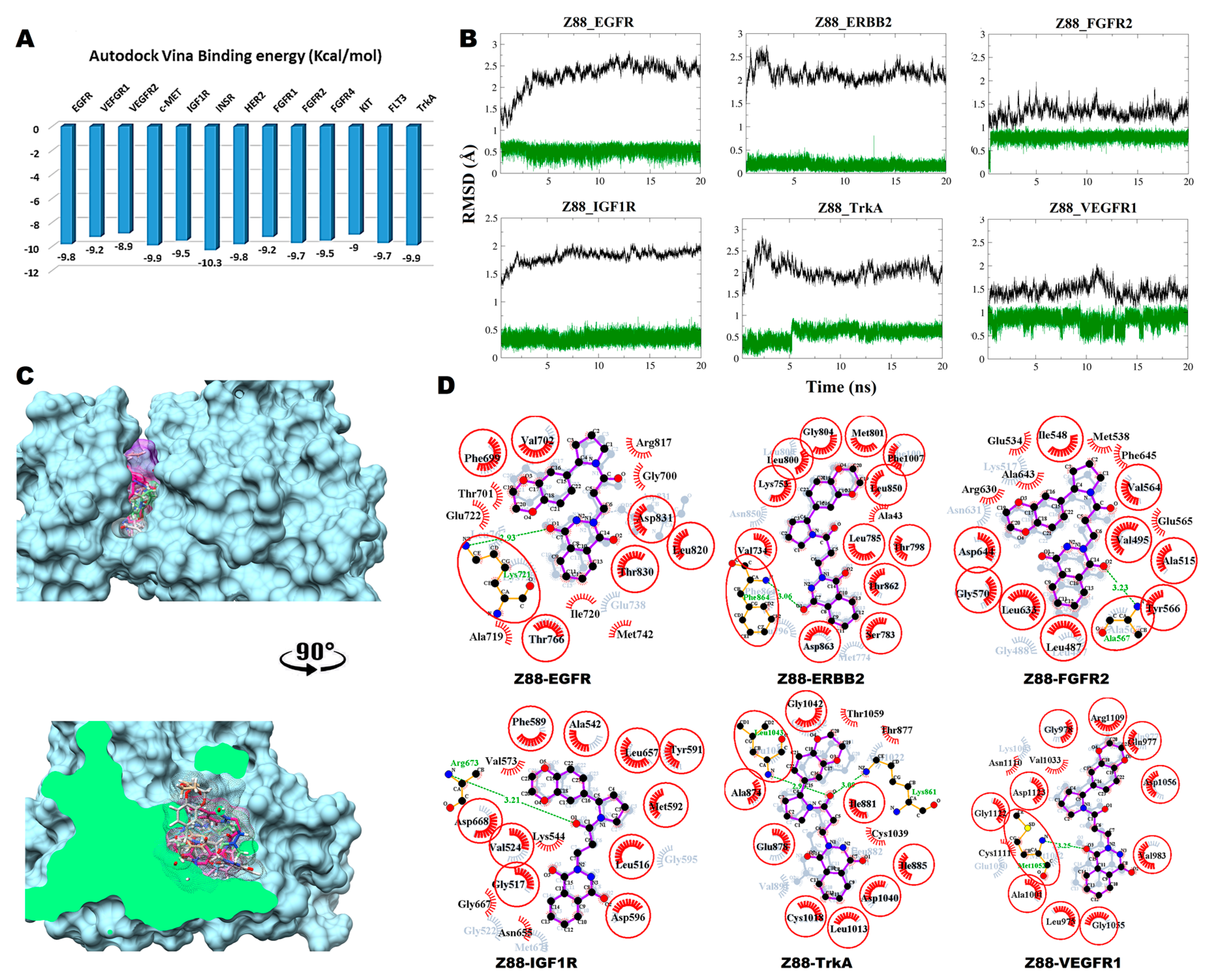
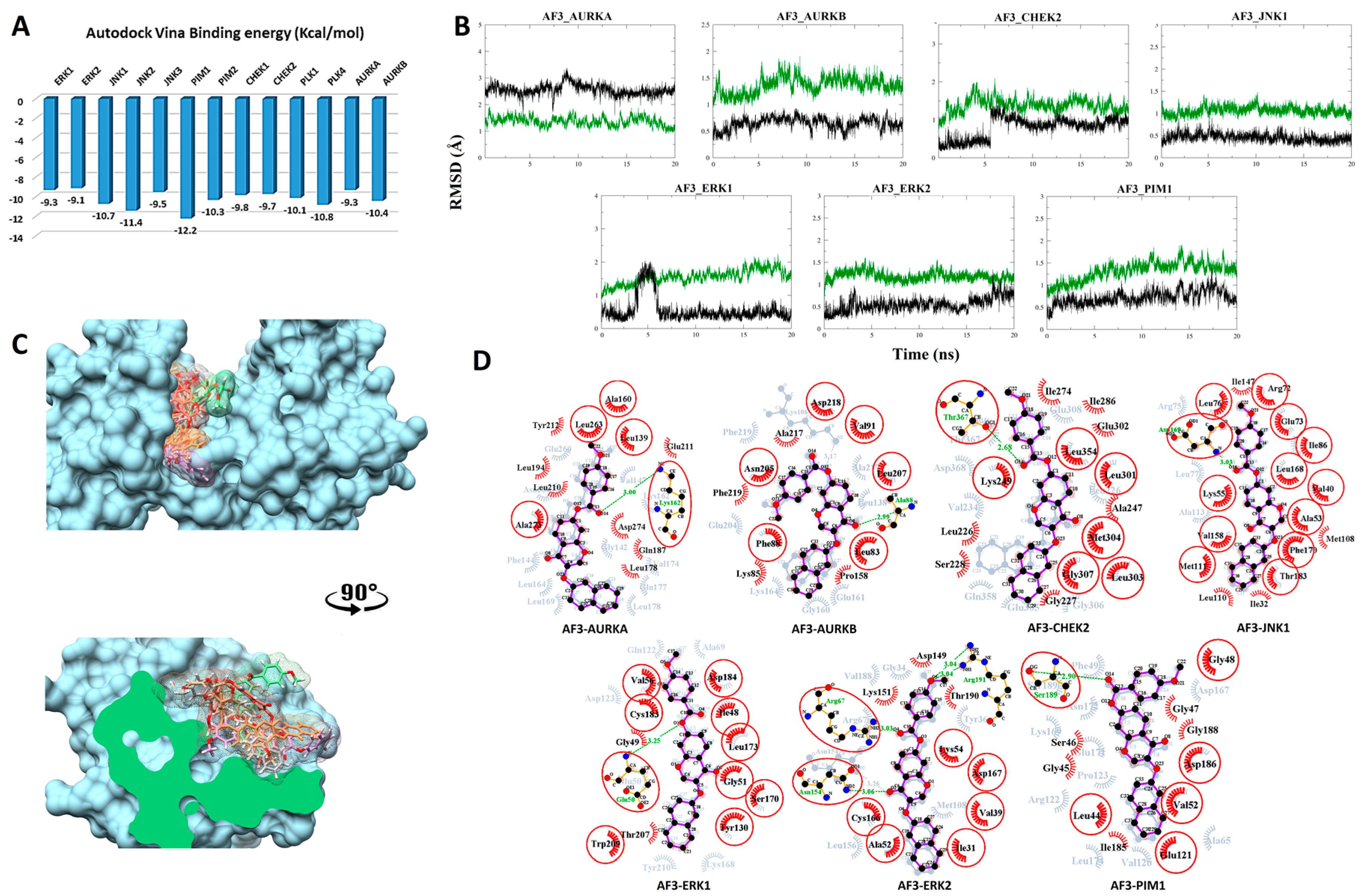
3.2. Molecular Interactions with RTKs and STKs
3.2.1. Compound Z21 with RTKs
3.2.2. Compound Z88 with RTKs
3.2.3. Compound AF3 with SKTs
3.2.4. Compound F34 with STKs
3.3. Estimated Total Binding Free Energy (MM-GBSA) Calculations
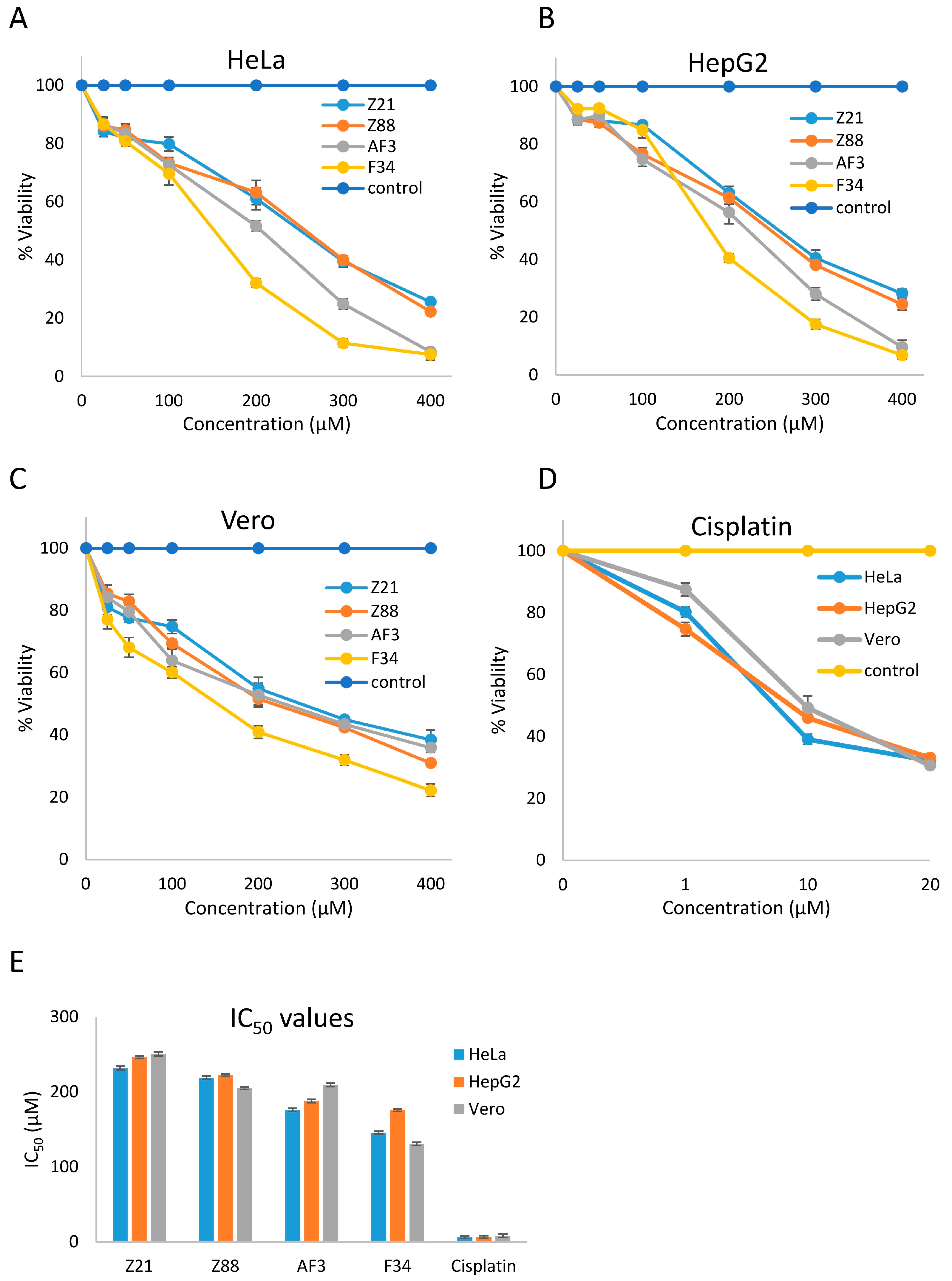
3.4. In Vitro Cytotoxicity
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Hanahan, D.; Weinberg, R.A. The hallmarks of cancer. Cell 2000, 100, 57–70. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed]
- Fleuren, E.D.; Zhang, L.; Wu, J.; Daly, R.J. The kinome’at large’in cancer. Nat. Rev. Cancer 2016, 16, 83. [Google Scholar] [CrossRef]
- Braconi Quintaje, S.; Orchard, S. The annotation of both human and mouse kinomes in UniProtKB/Swiss-Prot: One small step in manual annotation, one giant leap for full comprehension of genomes. Mol. Cell. Proteomics 2008, 7, 1409–1419. [Google Scholar] [CrossRef]
- Manning, G.; Whyte, D.B.; Martinez, R.; Hunter, T.; Sudarsanam, S. The protein kinase complement of the human genome. Science 2002, 298, 1912–1934. [Google Scholar] [CrossRef]
- Cross, T.G.; Scheel-Toellner, D.; Henriquez, N.V.; Deacon, E.; Salmon, M.; Lord, J.M. Serine/threonine protein kinases and apoptosis. Exp. Cell Res. 2000, 256, 34–41. [Google Scholar] [CrossRef] [PubMed]
- van der Geer, P.; Hunter, T.; Lindberg, R.A. Receptor protein-tyrosine kinases and their signal transduction pathways. Annu. Rev. Cell Biol. 1994, 10, 251–337. [Google Scholar] [CrossRef] [PubMed]
- Macdonald, S.G.; Crews, C.M.; Wu, L.E.L.I.A.; Driller, J.; Clark, R.; Erikson, R.L.; McCormick, F. Reconstitution of the Raf-1-MEK-ERK signal transduction pathway in vitro. Mol. Cell. Biol. 1993, 13, 6615–6620. [Google Scholar] [CrossRef]
- Eriksson, A.; Siegbahn, A.; Westermark, B.; Heldin, C.; Claesson-Welsh, L. PDGF alpha-and beta-receptors activate unique and common signal transduction pathways. EMBO J. 1992, 11, 543–550. [Google Scholar] [CrossRef] [PubMed]
- Fischer, O.M.; Hart, S.; Gschwind, A.; Ullrich, A. EGFR signal transactivation in cancer cells. Biochem. Soc. Trans. 2003, 31, 1203–1208. [Google Scholar] [CrossRef]
- Malumbres, M.; Barbacid, M. Cell cycle kinases in cancer. Curr. Opin. Genet. Dev. 2007, 17, 60–65. [Google Scholar] [CrossRef]
- Nicholson, R.; Gee, J.; Harper, M. EGFR and cancer prognosis. Eur. J. Cancer 2001, 37, 9–15. [Google Scholar] [CrossRef]
- Blume-Jensen, P.; Hunter, T. Oncogenic kinase signalling. Nature 2001, 411, 355. [Google Scholar] [CrossRef] [PubMed]
- Mantovani, A.; Allavena, P.; Sica, A.; Balkwill, F. Cancer-related inflammation. Nature 2008, 454, 436. [Google Scholar] [CrossRef] [PubMed]
- Biamonti, G.; Catillo, M.; Pignataro, D.; Montecucco, A.; Ghigna, C. The Alternative Splicing Side of Cancer; Seminars in cell & developmental biology Ser. 32; Elsevier: Amsterdam, The Netherlands, 2014. [Google Scholar]
- Belfiore, A.; Malaguarnera, R. Insulin receptor and cancer. Endocr. Relat. Cancer 2011, 18, R125–R147. [Google Scholar] [CrossRef]
- Xia, Z.; Dickens, M.; Raingeaud, J.; Davis, R.J.; Greenberg, M.E. Opposing effects of ERK and JNK-p38 MAP kinases on apoptosis. Science 1995, 270, 1326–1331. [Google Scholar] [CrossRef]
- Parrizas, M.; Saltiel, A.R.; LeRoith, D. Insulin-like growth factor 1 inhibits apoptosis using the phosphatidylinositol 3′-kinase and mitogen-activated protein kinase pathways. J. Biol. Chem. 1997, 272, 154–161. [Google Scholar] [CrossRef] [PubMed]
- Kennedy, S.G.; Wagner, A.J.; Conzen, S.D.; Jordan, J.; Bellacosa, A.; Tsichlis, P.N.; Hay, N. The PI 3-kinase/Akt signaling pathway delivers an anti-apoptotic signal. Genes Dev. 1997, 11, 701–713. [Google Scholar] [CrossRef]
- Knight, Z.A.; Lin, H.; Shokat, K.M. Targeting the cancer kinome through polypharmacology. Nat. Rev. Cancer 2010, 10, 130. [Google Scholar] [CrossRef]
- Wu, P.; Nielsen, T.E.; Clausen, M.H. FDA-approved small-molecule kinase inhibitors. Trends Pharmacol. Sci. 2015, 36, 422–439. [Google Scholar] [CrossRef] [PubMed]
- Otto, T.; Sicinski, P. Cell cycle proteins as promising targets in cancer therapy. Nat. Rev. Cancer 2017, 17, 93. [Google Scholar] [CrossRef]
- Turkson, J. The Molecular Basis of Human Cancer; Springer: New York, NY, USA, 2017; pp. 695–707. [Google Scholar]
- Cohen, P. Protein kinases—The major drug targets of the twenty-first century? Nat. Rev. Drug Discov. 2002, 1, 309. [Google Scholar] [CrossRef]
- Goldstein, D.M.; Gray, N.S.; Zarrinkar, P.P. High-throughput kinase profiling as a platform for drug discovery. Nat. Rev. Drug Discov. 2008, 7, 391. [Google Scholar] [CrossRef]
- Zhang, J.; Yang, P.L.; Gray, N.S. Targeting cancer with small molecule kinase inhibitors. Nat. Rev. Cancer 2009, 9, 28. [Google Scholar] [CrossRef]
- Ferreira, L.G.; dos Santos, R.N.; Oliva, G.; Andricopulo, A.D. Molecular docking and structure-based drug design strategies. Molecules 2015, 20, 13384–13421. [Google Scholar] [CrossRef]
- Ripphausen, P.; Nisius, B.; Bajorath, J. State-of-the-art in ligand-based virtual screening. Drug Discov. Today 2011, 16, 372–376. [Google Scholar] [CrossRef]
- Mirza, M.U.; Vanmeert, M.; Ali, A.; Iman, K.; Froeyen, M.; Idrees, M. Perspectives towards antiviral drug discovery against Ebola virus. J. Med. Virol. 2018. [Google Scholar] [CrossRef]
- Ekins, S.; Mestres, J.; Testa, B. In silico pharmacology for drug discovery: Applications to targets and beyond. Br. J. Pharmacol. 2007, 152, 21–37. [Google Scholar] [CrossRef]
- Muegge, I.; Oloff, S. Advances in virtual screening. Drug Discov. Today 2006, 3, 405–411. [Google Scholar] [CrossRef]
- Cavasotto, C.N.; Orry, W.; Andrew, J. Ligand docking and structure-based virtual screening in drug discovery. Curr. Top. Med. Chem. 2007, 7, 1006–1014. [Google Scholar] [CrossRef]
- Kapetanovic, I. Computer-aided drug discovery and development (CADDD): In silico-chemico-biological approach. Chem. Biol. Interact. 2008, 171, 165–176. [Google Scholar] [CrossRef] [PubMed]
- Ferdous, S.; Mirza, M.U.; Saeed, U. Docking studies reveal phytochemicals as the long searched anticancer drugs for breast cancer. Int. J. Comput. Appl. 2013, 67, 1–5. [Google Scholar] [CrossRef]
- Mirza, M.U.; Mirza, A.H.; Ghori, N.U.; Ferdous, S. Glycyrrhetinic acid and E. resveratroloside act as potential plant derived compounds against dopamine receptor D3 for Parkinson’s disease: A pharmacoinformatics study. Drug Des. Dev. Ther. 2014, 9, 187–198. [Google Scholar] [CrossRef]
- Sliwoski, G.; Kothiwale, S.; Meiler, J.; Lowe, E.W., Jr. Computational methods in drug discovery. Pharmacol. Rev. 2013, 66, 334–395. [Google Scholar] [CrossRef]
- Mirza, M.U.; Ghori, N.U.; Ikram, N.; Adil, A.R.; Manzoor, S. Pharmacoinformatics approach for investigation of alternative potential hepatitis C virus nonstructural protein 5B inhibitors. Drug Des. Dev. Ther. 2015, 9, 1825–1841. [Google Scholar] [CrossRef]
- Kumar, A.; Zhang, K.Y. Hierarchical virtual screening approaches in small molecule drug discovery. Methods 2015, 71, 26–37. [Google Scholar] [CrossRef] [PubMed]
- de Ruyck, J.; Brysbaert, G.; Blossey, R.; Lensink, M.F. Molecular docking as a popular tool in drug design, an in silico travel. Adv. Appl. Bioinform Chem. 2016, 9, 1–11. [Google Scholar] [CrossRef]
- De Vivo, M.; Masetti, M.; Bottegoni, G.; Cavalli, A. Role of molecular dynamics and related methods in drug discovery. J. Med. Chem. 2016, 59, 4035–4061. [Google Scholar] [CrossRef]
- Iman, K.; Mirza, M.U.; Mazhar, N.; Vanmeert, M.; Irshad, I.; Kamal, M.A. In silico Structure-based Identification of Novel Acetylcholinesterase Inhibitors Against Alzheimer’s Disease. CNS Neurol. Disord.-Drug Targets 2018, 17, 54–68. [Google Scholar] [CrossRef]
- Shoichet, B.K. Virtual screening of chemical libraries. Nature 2004, 432, 862. [Google Scholar] [CrossRef]
- Orry, A.J.; Abagyan, R.A.; Cavasotto, C.N. Structure-based development of target-specific compound libraries. Drug Discov. Today 2006, 11, 261–266. [Google Scholar] [CrossRef]
- Gozalbes, R.; Simon, L.; Froloff, N.; Sartori, E.; Monteils, C.; Baudelle, R. Development and experimental validation of a docking strategy for the generation of kinase-targeted libraries. J. Med. Chem. 2008, 51, 3124–3132. [Google Scholar] [CrossRef]
- Cavasotto, C.N.; Ortiz, M.A.; Abagyan, R.A.; Piedrafita, F.J. In silico identification of novel EGFR inhibitors with antiproliferative activity against cancer cells. Bioorg. Med. Chem. Lett. 2006, 16, 1969–1974. [Google Scholar] [CrossRef]
- Mahajan, P.; Chashoo, G.; Gupta, M.; Kumar, A.; Singh, P.P.; Nargotra, A. Fusion of structure and ligand based methods for identification of novel CDK2 inhibitors. J. Chem. Inf. Model. 2017, 57, 1957–1969. [Google Scholar] [CrossRef]
- Foloppe, N.; Fisher, L.M.; Howes, R.; Potter, A.; Robertson, A.G.; Surgenor, A.E. Identification of chemically diverse Chk1 inhibitors by receptor-based virtual screening. Bioorg. Med. Chem. 2006, 14, 4792–4802. [Google Scholar] [CrossRef]
- Thomas, M.P.; McInnes, C.; Fischer, P.M. Protein structures in virtual screening: A case study with CDK2. J. Med. Chem. 2006, 49, 92–104. [Google Scholar] [CrossRef]
- Golub, A.; Yakovenko, O.Y.; Prykhod’Ko, A.O.; Lukashov, S.S.; Bdzhola, V.G.; Yarmoluk, S.M. Evaluation of 4, 5, 6, 7-tetrahalogeno-1H-isoindole-1, 3 (2H)-diones as inhibitors of human protein kinase CK2. Biochim. Biophys. Acta 2008, 1784, 143–149. [Google Scholar] [CrossRef]
- Taylor, P.; Blackburn, E.; Sheng, Y.G.; Harding, S.; Hsin, K.Y.; Kan, D.; Shave, S.; Walkinshaw, M.D. Ligand discovery and virtual screening using the program LIDAEUS. Br. J. Pharmacol. 2008, 153, S55–S67. [Google Scholar] [CrossRef]
- Patel, D.S.; Bharatam, P.V. New leads for selective GSK-3 inhibition: Pharmacophore mapping and virtual screening studies. J. Comput. Aided Mol. Des. 2006, 20, 55–66. [Google Scholar] [CrossRef]
- Palanki, M.S.; Akiyama, H.; Campochiaro, P.; Cao, J.; Chow, C.P.; Dellamary, L.; Doukas, J.; Fine, R.; Gritzen, C.; Hood, J.D.; et al. Development of prodrug 4-chloro-3-(5-methyl-3-{[4-(2-pyrrolidin-1-ylethoxy) phenyl] amino}-1, 2, 4-benzotriazin-7-yl) phenyl benzoate (TG100801): A topically administered therapeutic candidate in clinical trials for the treatment of age-related macular degeneration. J. Med. Chem. 2008, 51, 1546–1559. [Google Scholar]
- Spicer, J.A.; Rewcastle, G.W.; Kaufman, M.D.; Black, S.L.; Plummer, M.S.; Denny, W.A.; Quin, J.; Shahripour, A.B.; Barrett, S.D.; Whitehead, C.E.; et al. 4-anilino-5-carboxamido-2-pyridone derivatives as noncompetitive inhibitors of mitogen-activated protein kinase kinase. J. Med. Chem. 2007, 50, 5090–5102. [Google Scholar] [CrossRef] [PubMed]
- Angell, R.M.; Angell, T.D.; Bamborough, P.; Bamford, M.J.; Chung, C.W.; Cockerill, S.G.; Flack, S.S.; Jones, K.L.; Laine, D.I.; Longstaff, T.; et al. Biphenyl amide p38 kinase inhibitors 4: DFG-in and DFG-out binding modes. Bioorg. Med. Chem. Lett. 2008, 18, 4433–4437. [Google Scholar] [CrossRef] [PubMed]
- Deng, X.; Wang, H.Y.; Zhao, Y.L.; Xiang, M.L.; Jiang, P.D.; Cao, Z.X.; Zheng, Y.-Z.; Luo, S.-D.; Yu, L.-T.; Wei, Y.-Q.; et al. Pharmacophore Modelling and Virtual Screening for Identification of New Aurora-A Kinase Inhibitors. Chem. Biol. Drug Des. 2008, 71, 533–539. [Google Scholar] [CrossRef]
- Vyas, V.K.; Ghate, M.; Goel, A. Pharmacophore modeling, virtual screening, docking and in silico ADMET analysis of protein kinase B (PKB β) inhibitors. J. Mol. Graph. Model. 2013, 42, 17–25. [Google Scholar] [CrossRef]
- Gangwal, R.P.; Das, N.R.; Thanki, K.; Damre, M.V.; Dhoke, G.V.; Sharma, S.S.; Jain, S.; Sangamwar, A.T. Identification of p38α MAP kinase inhibitors by pharmacophore based virtual screening. J. Mol. Graph. Model. 2014, 49, 18–24. [Google Scholar] [CrossRef]
- He, L.; Dai, R.; Zhang, X.R.; Gao, S.Y.; He, Y.Y.; Wang, L.B.; Gao, X.; Yang, L.Q. Ligand-based 3D pharmacophore design, virtual screening and molecular docking for novel p38 MAPK inhibitors. Med. Chem. Res. 2015, 24, 797–809. [Google Scholar] [CrossRef]
- Katari, S.K.; Natarajan, P.; Swargam, S.; Kanipakam, H.; Pasala, C.; Umamaheswari, A. Inhibitor design against JNK1 through e-pharmacophore modeling docking and molecular dynamics simulations. J. Recept. Signal Trans. 2016, 36, 558–571. [Google Scholar] [CrossRef]
- Chaudhari, P.; Bari, S. In silico exploration of c-KIT inhibitors by pharmaco-informatics methodology: Pharmacophore modeling, 3D QSAR, docking studies, and virtual screening. Mol. Divers. 2016, 20, 41–53. [Google Scholar] [CrossRef] [PubMed]
- Zou, F.; Yang, Y.; Ma, T.; Xi, J.; Zhou, J.; Zha, X. Identification of novel MEK1 inhibitors by pharmacophore and docking based virtual screening. Med. Chem. Res. 2017, 26, 701–713. [Google Scholar] [CrossRef]
- Sudha, A.; Srinivasan, P.; Rameshthangam, P. Exploration of potential EGFR inhibitors: A combination of pharmacophore-based virtual screening, atom-based 3D-QSAR and molecular docking analysis. J. Recept. Signal Trans. 2015, 35, 137–148. [Google Scholar] [CrossRef]
- Tian, Y.; Zhou, L.; Li, X.; Zhou, S.; Yong, R.; Zhong, L. In silico Studies Toward the Discovery of Novel Type-II Inhibitors of TrkA: Pharmacophore-based 3D-QSAR Modeling, Database Screening and Molecular Docking. Lett. Drug Des. Discov. 2016, 13, 526–538. [Google Scholar] [CrossRef]
- Zhou, N.; Xu, Y.; Liu, X.; Wang, Y.; Peng, J.; Luo, X.; Zheng, M.; Chen, K.; Jian, H. Combinatorial pharmacophore-based 3D-QSAR analysis and virtual screening of FGFR1 inhibitors. Int. J. Mol. Sci. 2015, 16, 13407–13426. [Google Scholar] [CrossRef]
- Vrontaki, E.; Melagraki, G.; Voskou, S.; Phylactides, M.; Mavromoustakos, T.; Kleanthous, M.; Afantitis, A. Development of a predictive pharmacophore model and a 3D-QSAR study for an in silico screening of new potent Bcr-Abl kinase inhibitors. Mini Rev. Med. Chem. 2017, 17, 188–204. [Google Scholar] [CrossRef]
- Asati, V.; Bharti, S.K.; Budhwani, A.K. 3D-QSAR and virtual screening studies of thiazolidine-2, 4-dione analogs: Validation of experimental inhibitory potencies towards PIM-1 kinase. J. Mol. Struct. 2017, 1133, 278–293. [Google Scholar] [CrossRef]
- Smalley, K.S.; Haass, N.K.; Brafford, P.A.; Lioni, M.; Flaherty, K.T.; Herlyn, M. Multiple signaling pathways must be targeted to overcome drug resistance in cell lines derived from melanoma metastases. Mol. Cancer Ther. 2006, 5, 1136–1144. [Google Scholar] [CrossRef] [PubMed]
- Müller, R. Crosstalk of oncogenic and prostanoid signaling pathways. J. Cancer Res. Clin. Oncol. 2004, 130, 429–444. [Google Scholar] [CrossRef]
- Pilpel, Y.; Sudarsanam, P.; Church, G.M. Identifying regulatory networks by combinatorial analysis of promoter elements. Nat. Genet. 2001, 29, 153. [Google Scholar] [CrossRef]
- Sergina, N.V.; Rausch, M.; Wang, D.; Blair, J.; Hann, B.; Shokat, K.M.; Moasser, M.M. Escape from HER-family tyrosine kinase inhibitor therapy by the kinase-inactive HER3. Nature 2007, 445, 437. [Google Scholar] [CrossRef]
- Force, T.; Krause, D.S.; Van Etten, R.A. Molecular mechanisms of cardiotoxicity of tyrosine kinase inhibition. Nat. Rev. Cancer 2007, 7, 332. [Google Scholar] [CrossRef] [PubMed]
- Overall, C.M.; Kleifeld, O. Validating matrix metalloproteinases as drug targets and anti-targets for cancer therapy. Nat. Rev. Cancer 2006, 6, 227. [Google Scholar] [CrossRef]
- Larder, B.A.; Kemp, S.D.; Harrigan, P.R. Potential mechanism for sustained antiretroviral efficacy of AZT-3TC combination therapy. Science 1995, 269, 696–699. [Google Scholar] [CrossRef]
- Keith, C.T.; Borisy, A.A.; Stockwell, B.R. Multicomponent therapeutics for networked systems. Nat. Rev. Drug Discov. 2005, 4, 71. [Google Scholar] [CrossRef]
- Krug, M.; Hilgeroth, A. Recent advances in the development of multi-kinase inhibitors. Mini Rev. Med. Chem. 2008, 8, 1312–1327. [Google Scholar] [CrossRef]
- Adrian, G.L.; Marcel, V.; Robert, B.G.; Richard, T. A comparison of physicochemical property profiles of marketed oral drugs and orally bioavailable anti-cancer protein kinase inhibitors in clinical development. Curr. Top. Med. Chem. 2007, 7, 1408–1422. [Google Scholar] [CrossRef]
- Hopkins, A.L. Network pharmacology: The next paradigm in drug discovery. Nat. Chem. Biol. 2008, 4, 682. [Google Scholar] [CrossRef]
- Medina-Franco, J.L.; Giulianotti, M.A.; Welmaker, G.S.; Houghten, R.A. Shifting from the single to the multitarget paradigm in drug discovery. Drug Discov. Today 2013, 18, 495–501. [Google Scholar] [CrossRef]
- Yousuf, Z.; Iman, K.; Iftikhar, N.; Mirza, M.U. Structure-based virtual screening and molecular docking for the identification of potential multi-targeted inhibitors against breast cancer. Breast Cancer Targets Ther. 2017, 9, 447. [Google Scholar] [CrossRef]
- Ou-Yang, S.S.; Lu, J.Y.; Kong, X.Q.; Liang, Z.J.; Luo, C.; Jiang, H. Computational drug discovery. Acta Pharmacol. Sin. 2012, 33, 1131. [Google Scholar] [CrossRef]
- Tropsha, A.; Bajorath, J. Computational methods for drug discovery and design. J. Med. Chem. 2016, 59, 1. [Google Scholar] [CrossRef]
- Dar, A.C.; Das, T.K.; Shokat, K.M.; Cagan, R.L. Chemical genetic discovery of targets and anti-targets for cancer polypharmacology. Nature 2012, 486, 80. [Google Scholar] [CrossRef]
- Jalencas, X.; Mestres, J. On the origins of drug polypharmacology. MedChemComm 2013, 4, 80–87. [Google Scholar] [CrossRef]
- Zhang, W.; Pei, J.; Lai, L. Computational multitarget drug design. J. Chem. Inf. Model. 2017, 57, 403–412. [Google Scholar] [CrossRef]
- Berman, H.M.; Westbrook, J.; Feng, Z.; Gilliland, G.; Bhat, T.N.; Weissig, H.; Shindyalov, I.N.; Bourne, P.E. The Protein Data Bank. Nucleic Acids Res. 2000, 28, 235–242. [Google Scholar] [CrossRef]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera—A visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef]
- Mirza, M.U.; Ikram, N. Integrated computational approach for virtual hit identification against Ebola viral proteins VP35 and VP40. Int. J. Mol. Sci. 2016, 17, 1748. [Google Scholar] [CrossRef]
- Wang, Y.; Bryant, S.H.; Cheng, T.; Wang, J.; Gindulyte, A.; Shoemaker, B.A.; Thiessen, P.A.; He, S.; Zhang, J. PubChem BioAssay: 2017 update. Nucleic Acids Res. 2016, 45, D955–D963. [Google Scholar] [CrossRef]
- Irwin, J.J.; Shoichet, B.K. ZINC—A free database of commercially available compounds for virtual screening. J. Chem. Inf. Model. 2005, 45, 177–182. [Google Scholar] [CrossRef]
- Lipinski, C.A.; Lombardo, F.; Dominy, B.W.; Feeney, P.J. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Deliv. Rev. 1997, 23, 3–25. [Google Scholar] [CrossRef]
- Hughes, J.D.; Blagg, J.; Price, D.A.; Bailey, S.; DeCrescenzo, G.A.; Devraj, R.V.; Ellsworth, E.; Fobian, Y.M.; Gibbs, M.E.; Gilles, R.W.; et al. Physiochemical drug properties associated with in vivo toxicological outcomes. Bioorg. Med. Chem. Lett. 2008, 18, 4872–4875. [Google Scholar] [CrossRef]
- Gleeson, M.P. Generation of a set of simple, interpretable ADMET rules of thumb. J. Med. Chem. 2008, 51, 817–834. [Google Scholar] [CrossRef]
- Veber, D.F.; Johnson, S.R.; Cheng, H.Y.; Smith, B.R.; Ward, K.W.; Kopple, K.D. Molecular properties that influence the oral bioavailability of drug candidates. J. Med. Chem. 2002, 45, 2615–2623. [Google Scholar] [CrossRef]
- O’Boyle, N.M.; Banck, M.; James, C.A.; Morley, C.; Vandermeersch, T.; Hutchison, G.R. Open Babel: An open chemical toolbox. J. Cheminf. 2011, 3, 33. [Google Scholar] [CrossRef]
- Egan, W.J.; Merz, K.M.; Baldwin, J.J. Prediction of drug absorption using multivariate statistics. J. Med. Chem. 2000, 43, 3867–3877. [Google Scholar] [CrossRef]
- Oldendorf, W.H. Lipid solubility and drug penetration of the blood brain barrier. Proc. Soc. Exp. Biol. Med. 1974, 147, 813–816. [Google Scholar] [CrossRef]
- Baell, J.B.; Holloway, G.A. New substructure filters for removal of pan assay interference compounds (PAINS) from screening libraries and for their exclusion in bioassays. J. Med. Chem. 2010, 53, 2719–2740. [Google Scholar] [CrossRef]
- Brenk, R.; Schipani, A.; James, D.; Krasowski, A.; Gilbert, I.H.; Frearson, J.; Wyatt, P.G. Lessons learnt from assembling screening libraries for drug discovery for neglected diseases. ChemMedChem 2008, 3, 435–444. [Google Scholar] [CrossRef]
- Walters, W.P.; Stahl, M.T.; Murcko, M.A. Virtual screening—An overview. Drug Discov. Today 1998, 3, 160–178. [Google Scholar] [CrossRef]
- Walters, W.P.; Murcko, A.A.; Murcko, M.A. Recognizing molecules with drug-like properties. Curr. Opin. Chem. Biol. 1999, 3, 384–387. [Google Scholar] [CrossRef]
- Herbert, J.; Augereau, J.; Gleye, J.; Maffrand, J. Chelerythrine is a potent and specific inhibitor of protein kinase C. Biochem. Biophys. Res. Commun. 1990, 172, 993–999. [Google Scholar] [CrossRef]
- Chresta, C.M.; Davies, B.R.; Hickson, I.; Harding, T.; Cosulich, S.; Critchlow, S.E.; Vincent, J.P.; Ellston, R.; Jones, D.; Sini, P.; et al. AZD8055 is a potent, selective, and orally bioavailable ATP-competitive mammalian target of rapamycin kinase inhibitor with in vitro and in vivo antitumor activity. Cancer Res. 2010, 70, 288–298. [Google Scholar] [CrossRef]
- Yu, K.; Toral-Barza, L.; Shi, C.; Zhang, W.G.; Lucas, J.; Shor, B.; Kim, J.; Verheijen, J.; Curran, K.; Malwitz, D.J.; et al. Biochemical, cellular, and in vivo activity of novel ATP-competitive and selective inhibitors of the mammalian target of rapamycin. Cancer Res. 2009, 69, 6232–6240. [Google Scholar] [CrossRef]
- Moore, J.D.; Staniszewska, A.; Shaw, T.; D’Alessandro, J.; Davis, B.; Surgenor, A.; Baker, L.; Matassova, N.; Murra, J.; Macia, A.; et al. VER-246608, a novel pan-isoform ATP competitive inhibitor of pyruvate dehydrogenase kinase, disrupts Warburg metabolism and induces context-dependent cytostasis in cancer cells. Oncotarget 2014, 5, 12862–12876. [Google Scholar] [CrossRef]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef]
- Gaillard, T. Evaluation of AutoDock and AutoDock Vina on the CASF-2013 benchmark. J. Chem. Inf. Model. 2018, 58, 1697–1706. [Google Scholar] [CrossRef]
- Kiss, R.; Sandor, M.; Szalai, F.A. http://Mcule.com: A public web service for drug discovery. J. Cheminf. 2012, 4, P17. [Google Scholar] [CrossRef]
- Cheng, F.; Li, W.; Zhou, Y.; Shen, J.; Wu, Z.; Liu, G.; Lee, P.W.; Tang, Y. admetSAR: A comprehensive source and free tool for assessment of chemical ADMET properties. J. Chem. Inf. Model 2012, 52, 3099–3105. [Google Scholar] [CrossRef]
- Lee, S.; Lee, I.H.; Kim, H.J.; Chang, G.S.; Chung, J.E.; No, K.T. The PreADME Approach: Web-based program for rapid prediction of physico-chemical, drug absorption and drug-like properties. In EuroQSAR 2002 Designing Drugs and Crop Protectants: Processes, Problems and Solutions; Blackwell Science Inc.: Hoboken, NJ, USA, 2003; Volume 2003, pp. 418–420. [Google Scholar]
- Cheng, A.; Merz, K.M. Prediction of aqueous solubility of a diverse set of compounds using quantitative structure− property relationships. J. Med. Chem. 2003, 46, 3572–3580. [Google Scholar] [CrossRef]
- Susnow, R.G.; Dixon, S.L. Use of robust classification techniques for the prediction of human cytochrome P450 2D6 inhibition. J. Chem. Inf. Comput. Sci. 2003, 43, 1308–1315. [Google Scholar] [CrossRef]
- Hengstler, J.G.; Oesch, F. Encyclopedia of Genetics; Brenner, S., Miller, J.H., Eds.; Academic Press: New York, NY, USA, 2001; pp. 51–54. [Google Scholar]
- Laskowski, R.A.; Swindells, M.B. LigPlot+: Multiple ligand–protein interaction diagrams for drug discovery. J. Chem. Inf. Model 2011, 51, 2778–2786. [Google Scholar] [CrossRef]
- Case, C.D.A.; Betz, R.M.; Cerutti, D.S.; Cheatham, T.E., III; Darden, T.A.; Duke, R.E.; Giese, T.J.; Gohlke, H.; Goetz, A.W.; Homeyer, N.; et al. AMBER 2016, University of California: San Francisco, CA, USA, 2016.
- Maier, J.A.; Martinez, C.; Kasavajhala, K.; Wickstrom, L.; Hauser, K.E.; Simmerling, C. ff14SB: Improving the accuracy of protein side chain and backbone parameters from ff99SB. J. Chem. Theory Comput. 2015, 11, 3696–3713. [Google Scholar] [CrossRef]
- Wang, J.; Wolf, R.M.; Caldwell, J.W.; Kollman, P.A.; Case, D.A. Development and testing of a general amber force field. J. Comput. Chem. 2004, 25, 1157–1174. [Google Scholar] [CrossRef]
- Jorgensen, W.L.; Chandrasekhar, J.; Madura, J.D.; Impey, R.W.; Klein, M.L. Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 1983, 79, 926–935. [Google Scholar] [CrossRef]
- Essmann, U.; Perera, L.; Berkowitz, M.L.; Darden, T.; Lee, H.; Pedersen, L.G. A smooth particle mesh Ewald method. J. Chem. Phys. 1995, 103, 8577–8593. [Google Scholar] [CrossRef]
- Ryckaert, J.; Ciccotti, G.; Berendsen, H.J. Numerical integration of the cartesian equations of motion of a system with constraints: Molecular dynamics of n-alkanes. J. Comput. Phys. 1977, 23, 327–341. [Google Scholar] [CrossRef]
- Roe, D.R.; Cheatham, T.E., III. PTRAJ and CPPTRAJ: Software for processing and analysis of molecular dynamics trajectory data. J. Chem. Theory Comput. 2013, 9, 3084–3095. [Google Scholar] [CrossRef]
- Chen, Y. Beware of docking! Trends Pharmacol. Sci. 2015, 36, 78–95. [Google Scholar] [CrossRef]
- Jain, A.N. Scoring functions for protein-ligand docking. Curr. Protein Peptide Sci. 2006, 7, 407–420. [Google Scholar]
- Halgren, T.A.; Murphy, R.B.; Friesner, R.A.; Beard, H.S.; Frye, L.L.; Pollard, W.T.; Banks, J.L. Glide: A new approach for rapid, accurate docking and scoring. 2. Enrichment factors in database screening. J. Med. Chem. 2004, 47, 1750–1759. [Google Scholar] [CrossRef]
- Friesner, R.A.; Banks, J.L.; Murphy, R.B.; Halgren, T.A.; Klicic, J.J.; Mainz, D.T.; Repasky, M.P.; Knoll, E.H.; Shelley, M.; Perry, J.K.; et al. Glide: A new approach for rapid, accurate docking and scoring. 1. Method and assessment of docking accuracy. J. Med. Chem. 2004, 47, 1739–1749. [Google Scholar] [CrossRef]
- Eldridge, M.D.; Murray, C.W.; Auton, T.R.; Paolini, G.V.; Mee, R.P. Empirical scoring functions: I. The development of a fast empirical scoring function to estimate the binding affinity of ligands in receptor complexes. J. Comput. Aided Mol. Des. 1997, 11, 425–445. [Google Scholar] [CrossRef]
- Friesner, R.A.; Murphy, R.B.; Repasky, M.P.; Frye, L.L.; Greenwood, J.R.; Halgren, T.A.; Sanschagrin, P.C.; Mainz, D.T. Extra precision glide: Docking and scoring incorporating a model of hydrophobic enclosure for protein—Ligand complexes. J. Med. Chem. 2006, 49, 6177–6196. [Google Scholar] [CrossRef]
- Li, J.; Abel, R.; Zhu, K.; Cao, Y.; Zhao, S.; Friesner, R.A. The VSGB 2.0 model: A next generation energy model for high resolution protein structure modeling. Proteins Struct. Funct. Bioinf. 2011, 79, 2794–2812. [Google Scholar] [CrossRef] [PubMed]
- Lyne, P.D.; Lamb, M.L.; Saeh, J.C. Accurate prediction of the relative potencies of members of a series of kinase inhibitors using molecular docking and MM-GBSA scoring. J. Med. Chem. 2006, 49, 4805–4808. [Google Scholar] [CrossRef] [PubMed]
- Das, D.; Koh, Y.; Tojo, Y.; Ghosh, A.K.; Mitsuya, H. Prediction of potency of protease inhibitors using free energy simulations with polarizable quantum mechanics-based ligand charges and a hybrid water model. J. Chem. Inf. Model. 2009, 49, 2851–2862. [Google Scholar] [CrossRef]
- Siddiquee, K.; Zhang, S.; Guida, W.C.; Blaskovich, M.A.; Greedy, B.; Lawrence, H.R.; Yip, M.L.R.; Jove, R.; McLaughlin, M.M.; Lawrence, N.J.; et al. Selective chemical probe inhibitor of Stat3, identified through structure-based virtual screening, induces antitumor activity. Proc. Natl. Acad. Sci. USA 2007, 104, 7391–7396. [Google Scholar] [CrossRef] [PubMed]
- Mahajan, P.; Suri, N.; Mehra, R.; Gupta, M.; Kumar, A.; Singh, S.K.; Nargotra, A. Discovery of novel small molecule EGFR inhibitory leads by structure and ligand-based virtual screening. Med. Chem. Res. 2017, 26, 74–92. [Google Scholar] [CrossRef]
- Reddy, R.H.; Kim, H.; Cha, S.; Lee, B.; Kim, Y.J. Structure-Based Virtual Screening of Protein Tyrosine Phosphatase Inhibitors: Significance, Challenges, and Solutions. J. Microbiol. Biotechnol. 2017, 27, 878–895. [Google Scholar]
- Oluić, J.; Nikolic, J.; Vucicevic, J.; Gagic, Z.; Filipic, S.; Agbaba, D. QSAR modeling and structure based virtual screening of new PI3K/mTOR inhibitors as potential anticancer agents. In CMBEBIH 2017; Springer: Singapore, 2017; pp. 379–383. [Google Scholar]
- Sharma, K.; Patidar, K.; Ali, M.A.; Patil, P.; Goud, H.; Hussain, T.; Nayarisseri, A.; Singh, S.K. Structure-Based Virtual Screening for the Identification of High Affinity Compounds as Potent VEGFR2 Inhibitors for the Treatment of Renal Cell Carcinoma. Curr. Top. Med. Chem. 2018, 18, 2174–2185. [Google Scholar] [CrossRef]
- Heinrich, M.C.; Corless, C.L.; Demetri, G.D.; Blanke, C.D.; Von Mehren, M.; Joensuu, H.; McGreevey, L.S.; Chen, C.-J.; Van den Abbeele, A.D.; Druker, B.J.; et al. Kinase mutations and imatinib response in patients with metastatic gastrointestinal stromal tumor. J. Clin. Oncol. 2003, 21, 4342–4349. [Google Scholar] [CrossRef]
- Heinrich, M.C.; Corless, C.L.; Duensing, A.; McGreevey, L.; Chen, C.J.; Joseph, N.; Singer, S.; Griffith, D.J.; Haley, A.; Town., A.; et al. PDGFRA activating mutations in gastrointestinal stromal tumors. Science 2003, 299, 708–710. [Google Scholar] [CrossRef] [PubMed]
- Carvalho, I.; Milanezi, F.; Martins, A.; Reis, R.M.; Schmitt, F. Overexpression of platelet-derived growth factor receptor α in breast cancer is associated with tumour progression. Breast Cancer Res. 2005, 7, R788. [Google Scholar] [CrossRef]
- Naoe, T.; Kiyoi, H. Normal and oncogenic FLT3. Cell Mol. Life Sci. 2004, 61, 2932–2938. [Google Scholar] [CrossRef]
- Rini, B.I.; Small, E.J. Biology and clinical development of vascular endothelial growth factor–targeted therapy in renal cell carcinoma. J. Clin. Oncol. 2005, 23, 1028–1043. [Google Scholar] [CrossRef]
- Berndt, N.; Karim, R.M.; Schönbrunn, E. Advances of small molecule targeting of kinases. Curr. Opin. Chem. Biol. 2017, 39, 126–132. [Google Scholar] [CrossRef]
- Faivre, S.; Djelloul, S.; Raymond, E. New paradigms in anticancer therapy: Targeting multiple signaling pathways with kinase inhibitors. Semin. Oncol. 2006, 33, 407–420. [Google Scholar] [CrossRef]
- Kane, R.C.; Farrell, A.T.; Saber, H.; Tang, S.; Williams, G.; Jee, J.M.; Liang, C.; Booth, B.; Chidambaram, N.; Morse, D.; et al. Sorafenib for the treatment of advanced renal cell carcinoma. Clin. Cancer Res. 2006, 12, 7271–7278. [Google Scholar] [CrossRef]
- Llovet, J.M.; Ricci, S.; Mazzaferro, V.; Hilgard, P.; Gane, E.; Blanc, J.F.; de Oliveira, A.C.; Santoro, A.; Raoul, J.-L.; Forner, A.; et al. Sorafenib in advanced hepatocellular carcinoma. N. Engl. J. Med. 2008, 359, 378–390. [Google Scholar] [CrossRef]
- Chow, L.Q.; Eckhardt, S.G. Sunitinib: From rational design to clinical efficacy. J. Clin. Oncol. 2007, 25, 884–896. [Google Scholar] [CrossRef]
- Weisberg, E.; Manley, P.W.; Breitenstein, W.; Brüggen, J.; Cowan-Jacob, S.W.; Ray, A.; Huntly, B.; Fabbro, D.; Fendrich, G.; Hall-Meyers, E.; et al. Characterization of AMN107, a selective inhibitor of native and mutant Bcr-Abl. Cancer Cell 2005, 7, 129–141. [Google Scholar] [CrossRef]
- Palmer, B.D.; Trumpp-Kallmeyer, S.; Fry, D.W.; Nelson, J.M.; Showalter, H.H.; Denny, W.A. Tyrosine kinase inhibitors. 11. Soluble analogues of pyrrolo-and pyrazoloquinazolines as epidermal growth factor receptor inhibitors: Synthesis, biological evaluation, and modeling of the mode of binding. J. Med. Chem. 1997, 40, 1519–1529. [Google Scholar] [CrossRef]
- Ishikawa, T.; Seto, M.; Banno, H.; Kawakita, Y.; Oorui, M.; Taniguchi, T.; Ohta, Y.; Tamura, T.; Nakayama, A.; Miki, H.; et al. Design and synthesis of novel human epidermal growth factor receptor 2 (HER2)/epidermal growth factor receptor (EGFR) dual inhibitors bearing a pyrrolo [3, 2-d] pyrimidine scaffold. J. Med. Chem. 2011, 54, 8030–8050. [Google Scholar] [CrossRef]
- Cheetham, G.M. Novel protein kinases and molecular mechanisms of autoinhibition. Curr. Opin. Struct. Biol. 2004, 14, 700–705. [Google Scholar] [CrossRef]
- Eathiraj, S.; Palma, R.; Hirschi, M.; Volckova, E.; Nakuci, E.; Castro, J.; Chen, C.-R.; Chan, T.C.K.; France, D.S.; Ashwell, M.A. A novel mode of protein kinase inhibition exploiting hydrophobic motifs of autoinhibited kinases: Discovery of ATP-independent inhibitors of fibroblast growth factor receptor. J. Biol. Chem. 2011, 286, 20677–20687. [Google Scholar] [CrossRef]
- Carry, J.; Clerc, F.; Minoux, H.; Schio, L.; Mauger, J.; Nair, A.; Parmantier, E.; Moigne, R.L.; Delorme, C.; Nicolas, J.-P.; et al. SAR156497, an exquisitely selective inhibitor of aurora kinases. J. Med. Chem. 2014, 58, 362–375. [Google Scholar] [CrossRef]
- Lountos, G.T.; Tropea, J.E.; Zhang, D.; Jobson, A.G.; Pommier, Y.; Shoemaker, R.H.; Waugh, D.S. Crystal structure of checkpoint kinase 2 in complex with NSC 109555, a potent and selective inhibitor. Protein Sci. 2009, 18, 92–100. [Google Scholar] [CrossRef]
- Li, B.; Cociorva, O.M.; Nomanbhoy, T.; Weissig, H.; Li, Q.; Nakamura, K.; Liyanage, M.; Zhang, M.C.; Shih, A.Y.; Aban, A.; et al. Hit-to-lead optimization and kinase selectivity of imidazo [1,2-a] quinoxalin-4-amine derived JNK1 inhibitors. Bioorg. Med. Chem. Lett. 2013, 23, 5217–5222. [Google Scholar] [CrossRef]
- Kinoshita, T.; Yoshida, I.; Nakae, S.; Okita, K.; Gouda, M.; Matsubara, M.; Yokota, K.; Ishiguro, H.; Tada, T. Crystal structure of human mono-phosphorylated ERK1 at Tyr204. Biochem. Biophys. Res. Commun. 2008, 377, 1123–1127. [Google Scholar] [CrossRef]
- Lambeng, N.; Lebon, F.; Christophe, B.; Burton, M.; De Ryck, M.; Quéré, L. Arylsulfonamides as a new class of cannabinoid CB1 receptor ligands: Identification of lead and initial SAR studies. Bioorg. Med. Chem. Lett. 2007, 17, 272–277. [Google Scholar] [CrossRef]
- Demuth, D.G.; Molleman, A. Cannabinoid signalling. Life Sci. 2006, 78, 549–563. [Google Scholar] [CrossRef] [PubMed]
- Dexheimer, T.S.; Antony, S.; Marchand, C.; Pommier, Y. Tyrosyl-DNA phosphodiesterase as a target for anticancer therapy. Anti-Cancer Agents Med. Chem. 2008, 8, 381–389. [Google Scholar] [CrossRef]
- Kumamoto, T.; Fujii, M.; Hou, D.X. Myricetin directly targets JAK1 to inhibit cell transformation. Cancer Lett. 2009, 275, 17–26. [Google Scholar] [CrossRef] [PubMed]
- Overington, J.P.; Al-Lazikani, B.; Hopkins, A.L. How many drug targets are there? Nat. Rev. Drug Discov. 2006, 5, 993. [Google Scholar] [CrossRef]
- Wang, Z.; Bhattacharya, N.; Weaver, M.; Petersen, K.; Meyer, M.; Gapter, L.; Magnuson, N.S. Pim-1: A serine/threonine kinase with a role in cell survival, proliferation, differentiation and tumorigenesis. J. Vet. Sci. 2001, 2, 167–179. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.E.; Hunt, C.R.; Chen, J.; Taylor, J.S. Biotinylated quercetin as an intrinsic photoaffinity proteomics probe for the identification of quercetin target proteins. Bioorg. Med. Chem. Lett. 2011, 19, 4710–4720. [Google Scholar] [CrossRef] [PubMed]
- Tukun, F.L.; Olberg, D.; Riss, P.; Haraldsen, I.; Kaass, A.; Klaveness, J. Recent Development of Non-Peptide GnRH Antagonists. Molecules 2017, 22, 2188. [Google Scholar] [CrossRef]
- Hara, T.; Araki, H.; Kusaka, M.; Harada, M.; Cho, N.; Suzuki, N.; Fujino, M. Suppression of a pituitary-ovarian axis by chronic oral administration of a novel non-peptide gonadotropin-releasing hormone antagonist, TAK-013, in cynomolgus monkeys. J. Clin.Endocrinol. Amp Metabol. 2003, 88, 1697–1704. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| RTK Targets | Compound | ΔGtol | STK Targets | Compound | ΔGtol |
|---|---|---|---|---|---|
| EGFR | Z88 | −33.58 | AURKA | F34 | −50.76 |
| Z21 | −48.59 | AF3 | −41.11 | ||
| FGFR1 | Z88 | −37.16 | AURKB | F34 | −45.18 |
| Z21 | −36.88 | AF3 | −39.76 | ||
| FGFR2 | Z88 | −41.01 | ERK1 | F34 | −52.65 |
| Z21 | −42.94 | AF3 | −17.99 | ||
| FGFR4 | Z88 | −36.76 | ERK2 | F34 | −42.03 |
| Z21 | −33.37 | AF3 | −38.65 | ||
| VEGFR1 | Z88 | −38.39 | JNK1 | F34 | −45.5 |
| Z21 | −41.33 | AF3 | −33.23 | ||
| ERBB2 | Z88 | −33.93 | CHEK2 | F34 | −43.11 |
| Z21 | −45.93 | AF3 | −36.27 | ||
| IGF1R | Z88 | −30.82 | PIM1 | F34 | −56.06 |
| Z21 | −42.64 | AF3 | −31.87 | ||
| TrkA | Z88 | −33.33 | |||
| Z21 | −43.39 |
| Z21 | Z88 | AF3 | F34 | Cisplatin | |
|---|---|---|---|---|---|
| HeLa | 231.44 ± 2.35 | 218.68 ± 2.04 | 175.69 ± 2.12 | 145.46 ± 2.06 | 6.02 ± 1.59 |
| HepG2 | 245.96 ± 1.93 | 222.05 ± 1.68 | 187.54 ± 2.31 | 175.48 ± 1.66 | 6.41 ± 1.54 |
| Vero | 250.24 ± 2.34 | 204.78 ± 1.78 | 209.16 ± 2.43 | 130.52 ± 2.3 | 7.99 ± 2.38 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ikram, N.; Mirza, M.U.; Vanmeert, M.; Froeyen, M.; Salo-Ahen, O.M.H.; Tahir, M.; Qazi, A.; Ahmad, S. Inhibition of Oncogenic Kinases: An In Vitro Validated Computational Approach Identified Potential Multi-Target Anticancer Compounds. Biomolecules 2019, 9, 124. https://doi.org/10.3390/biom9040124
Ikram N, Mirza MU, Vanmeert M, Froeyen M, Salo-Ahen OMH, Tahir M, Qazi A, Ahmad S. Inhibition of Oncogenic Kinases: An In Vitro Validated Computational Approach Identified Potential Multi-Target Anticancer Compounds. Biomolecules. 2019; 9(4):124. https://doi.org/10.3390/biom9040124
Chicago/Turabian StyleIkram, Nazia, Muhammad Usman Mirza, Michiel Vanmeert, Matheus Froeyen, Outi M. H. Salo-Ahen, Muhammad Tahir, Aamer Qazi, and Sarfraz Ahmad. 2019. "Inhibition of Oncogenic Kinases: An In Vitro Validated Computational Approach Identified Potential Multi-Target Anticancer Compounds" Biomolecules 9, no. 4: 124. https://doi.org/10.3390/biom9040124
APA StyleIkram, N., Mirza, M. U., Vanmeert, M., Froeyen, M., Salo-Ahen, O. M. H., Tahir, M., Qazi, A., & Ahmad, S. (2019). Inhibition of Oncogenic Kinases: An In Vitro Validated Computational Approach Identified Potential Multi-Target Anticancer Compounds. Biomolecules, 9(4), 124. https://doi.org/10.3390/biom9040124