Beyond Chelation: EDTA Tightly Binds Taq DNA Polymerase, MutT and dUTPase and Directly Inhibits dNTPase Activity

 , , , ,
, , , ,  and
and 
Abstract
1. Introduction
2. Materials and Methods
2.1. Reagents
2.2. ICP-OES
2.3. Photometric Enzyme Activity Measurement
2.4. Thermofluor Stability Assay
2.5. Protein and Ligand Preparation Procedure for Molecular Docking
2.6. Molecular Docking
2.7. Determination of the Enzyme-Ligand Interaction Network
2.8. Isothermal Titration Calorimetry (ITC) Measurement
2.9. Figures
3. Results
3.1. Assessment of the Mg2+ Content of the Assay Solutions
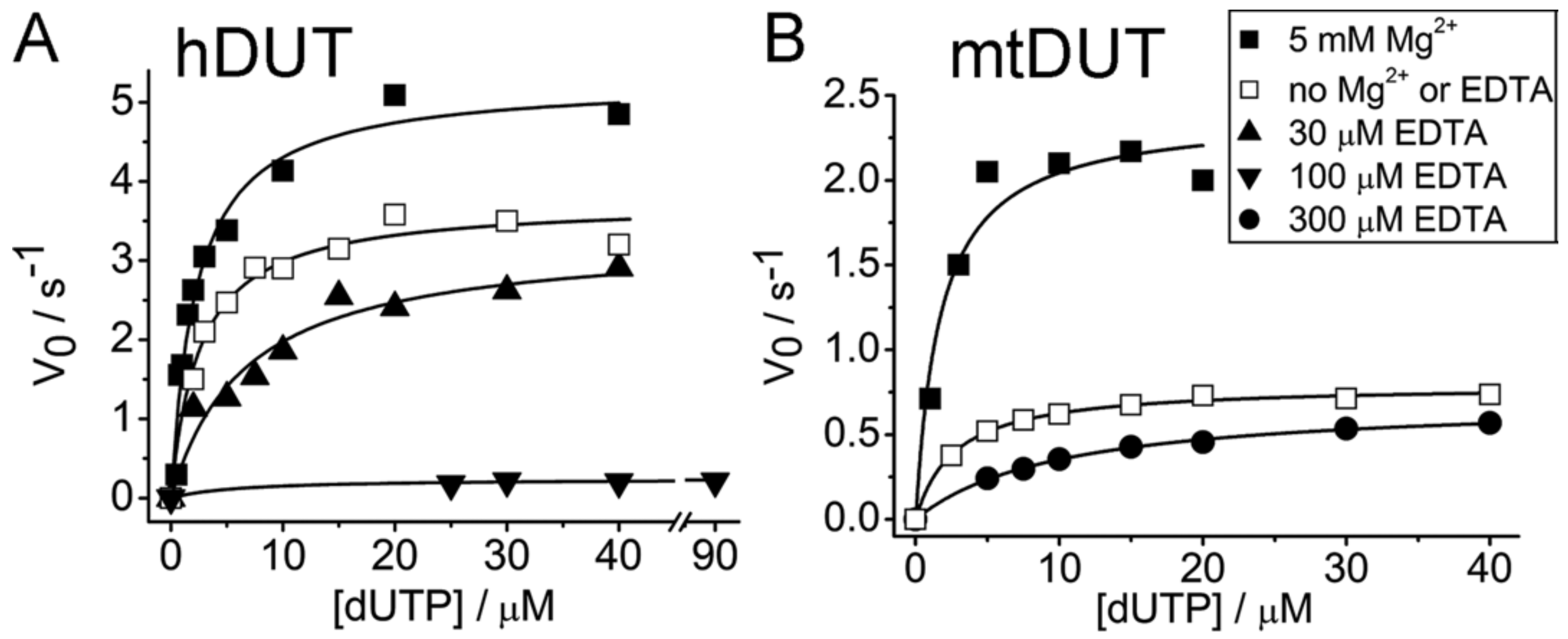
3.2. EDTA Decreases Enzyme Activity
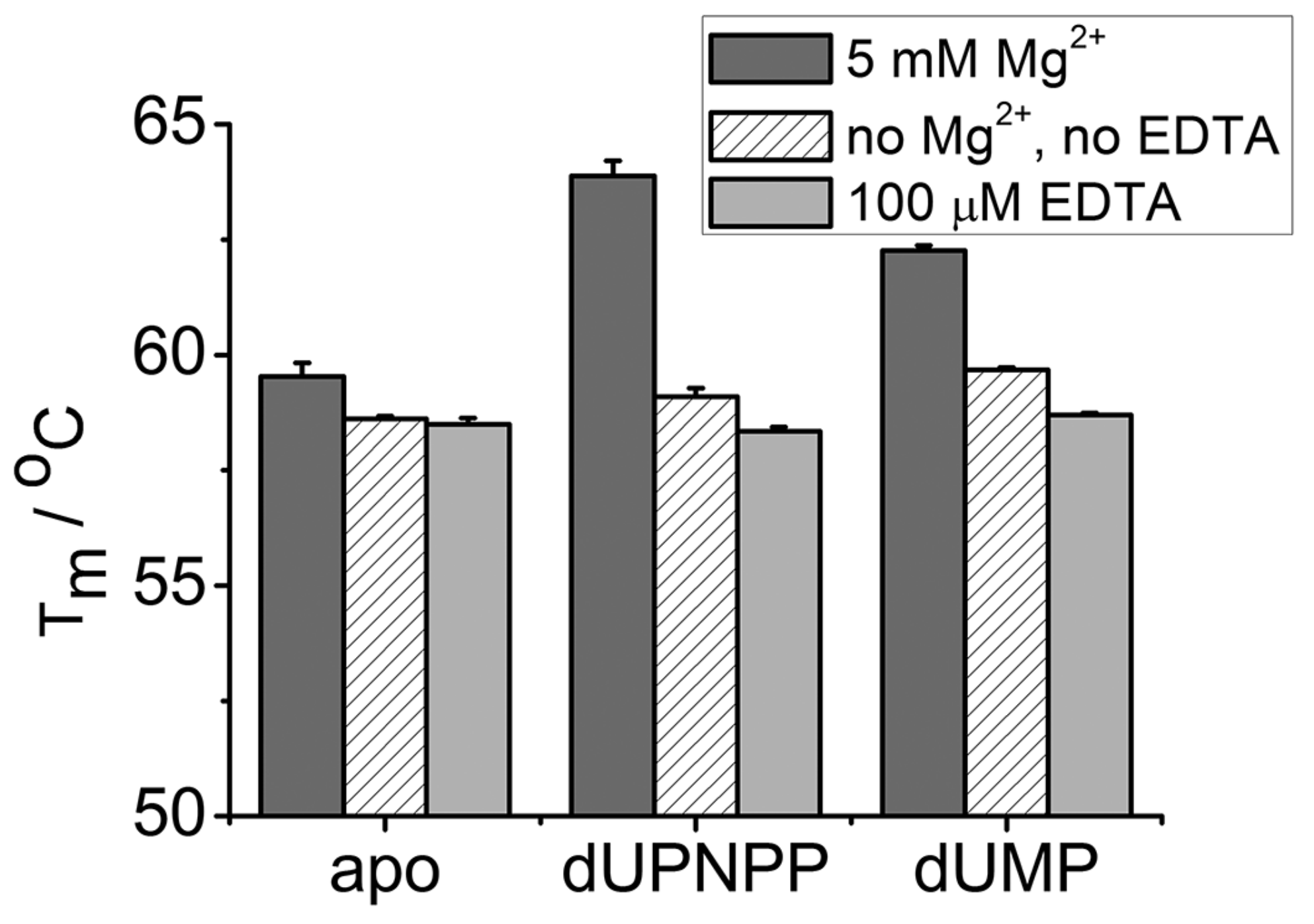
3.3. EDTA Does Not Destabilize the Enzyme
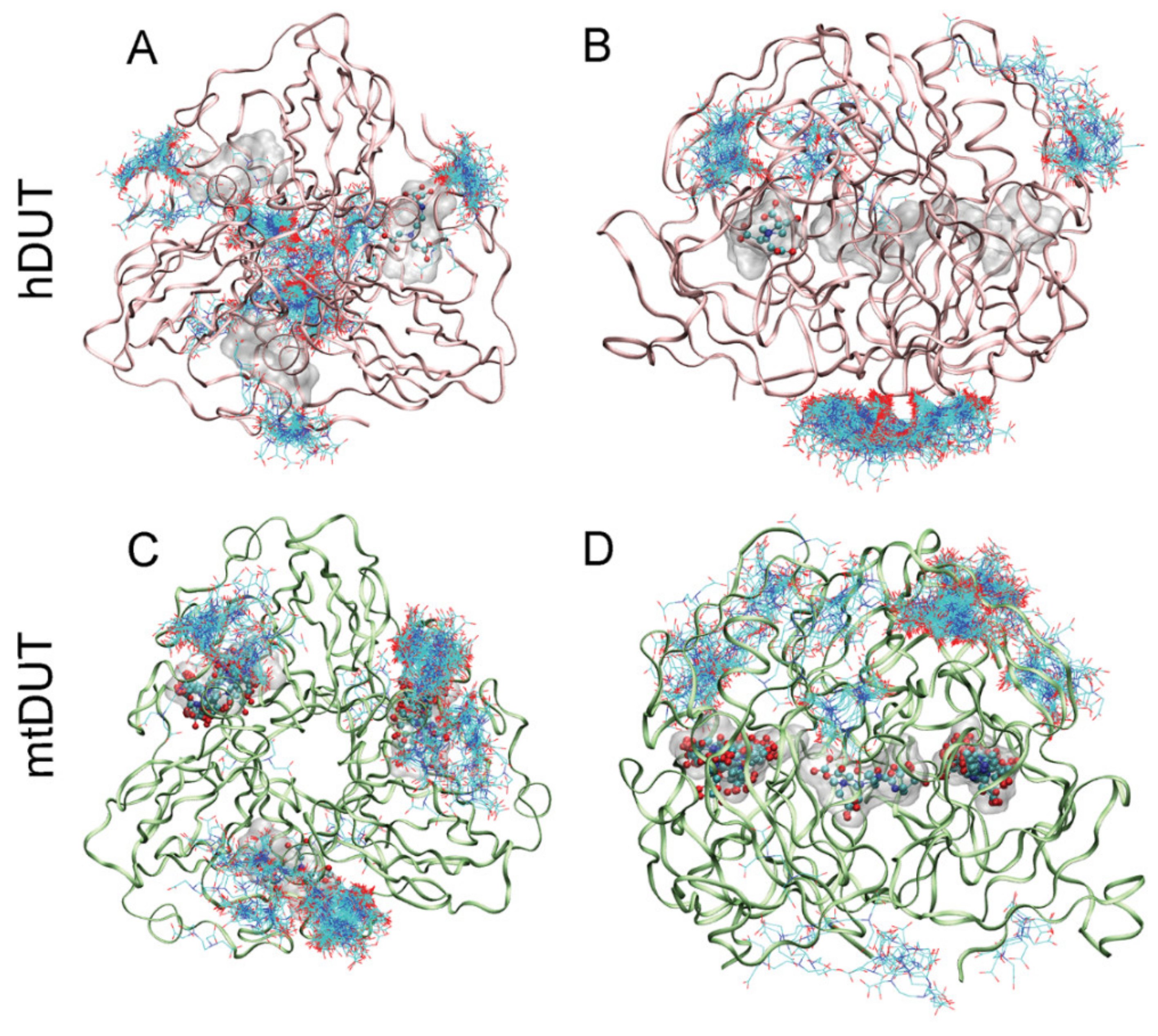
3.4. Blind Docking Indicates EDTA Binding to the Surface and the Active Site of Both dUTPases
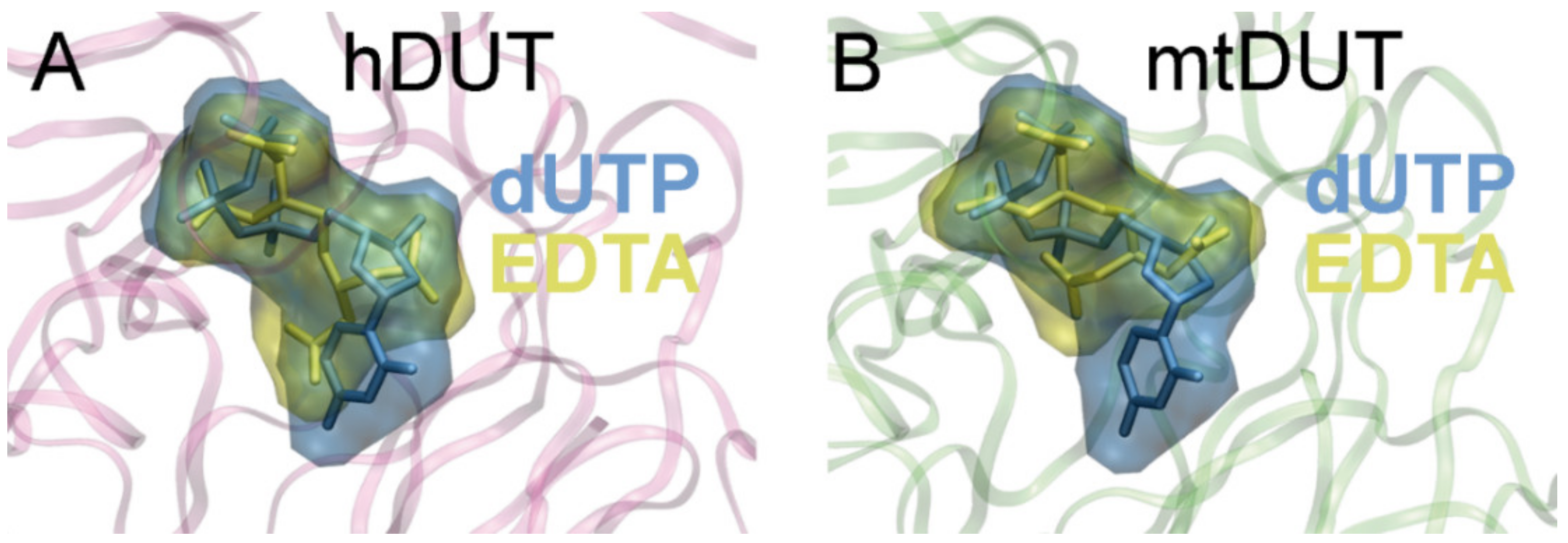
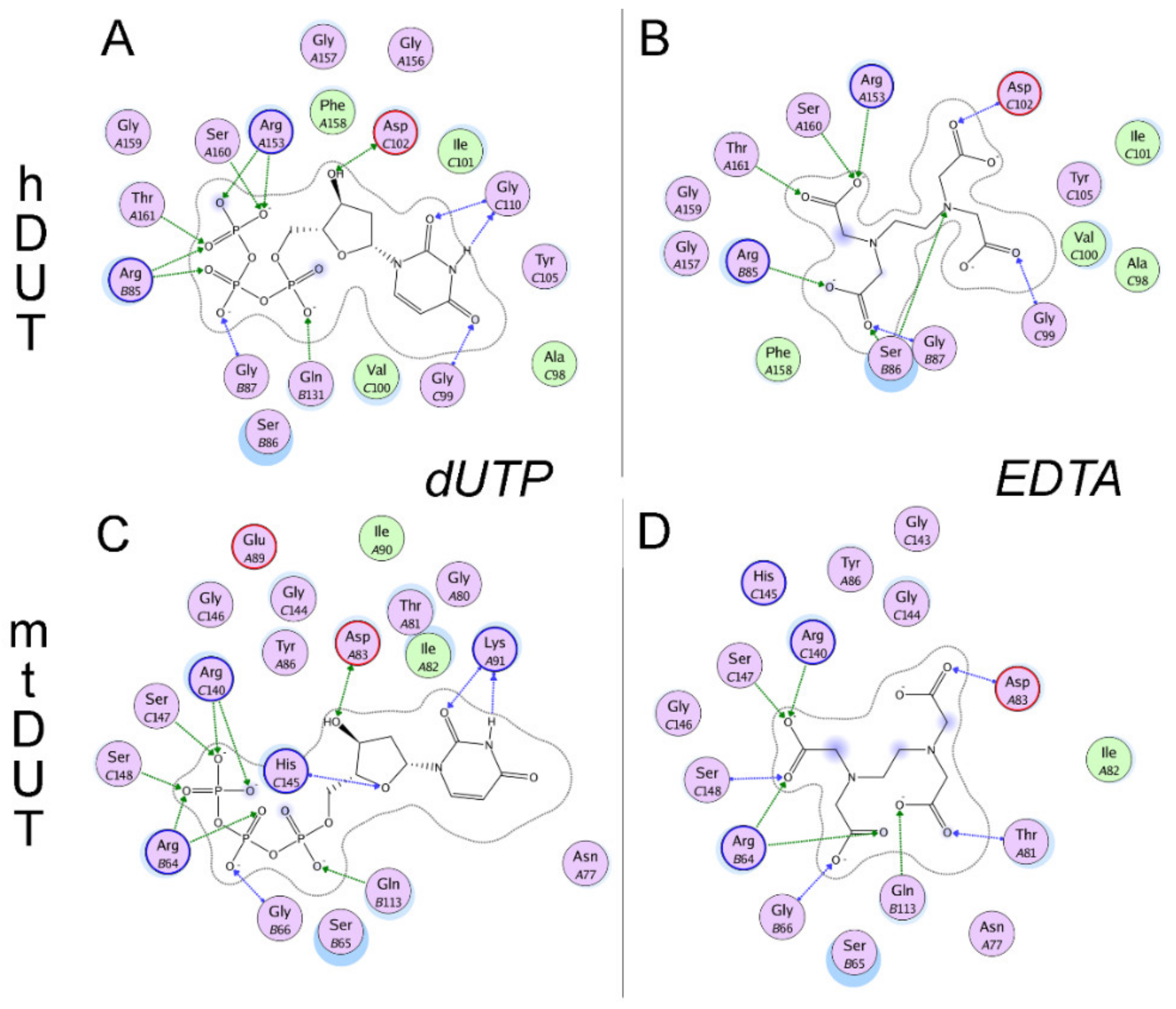
3.5. Molecular Docking Suggests That Nucleotide and EDTA Share Common Interaction Points Within the Active Site
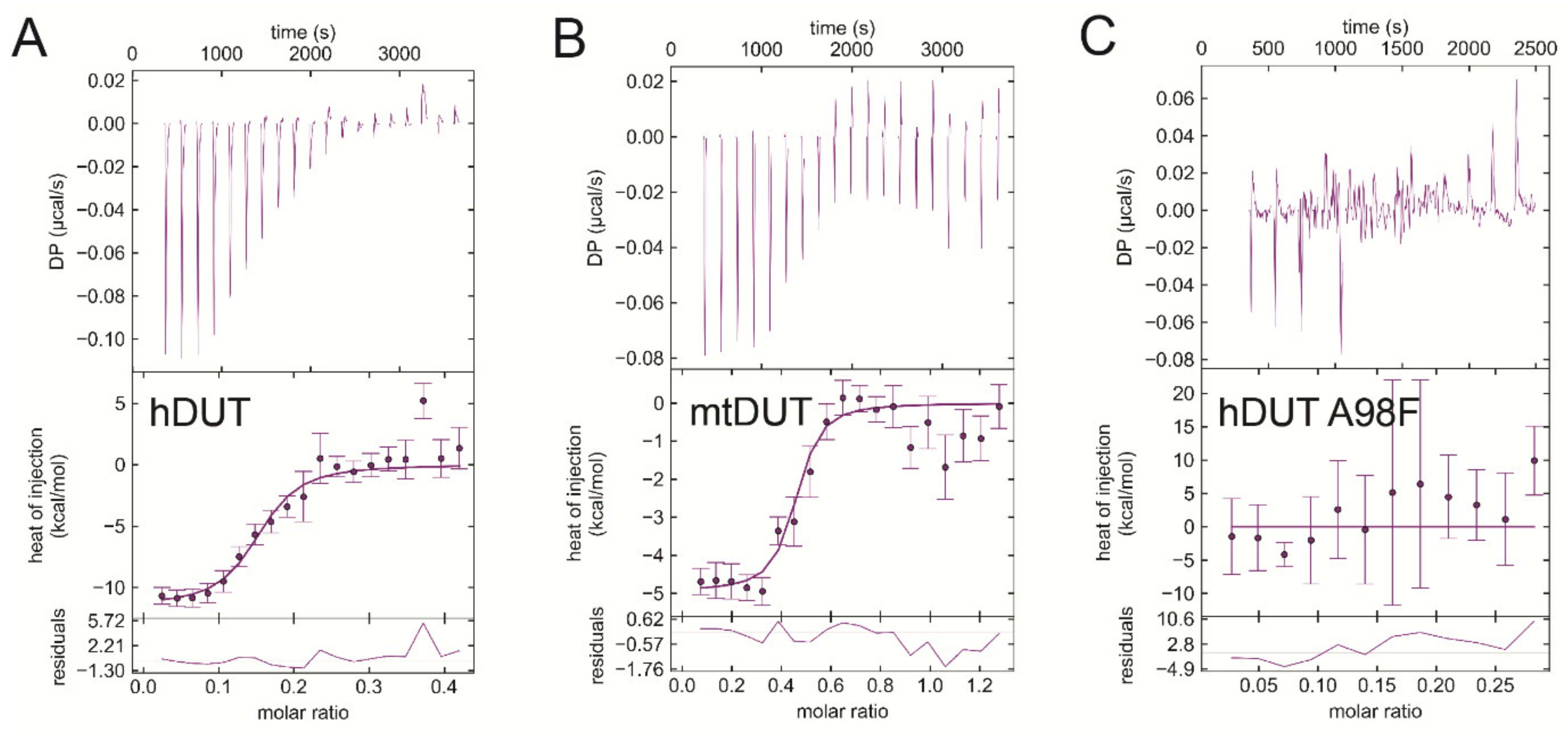
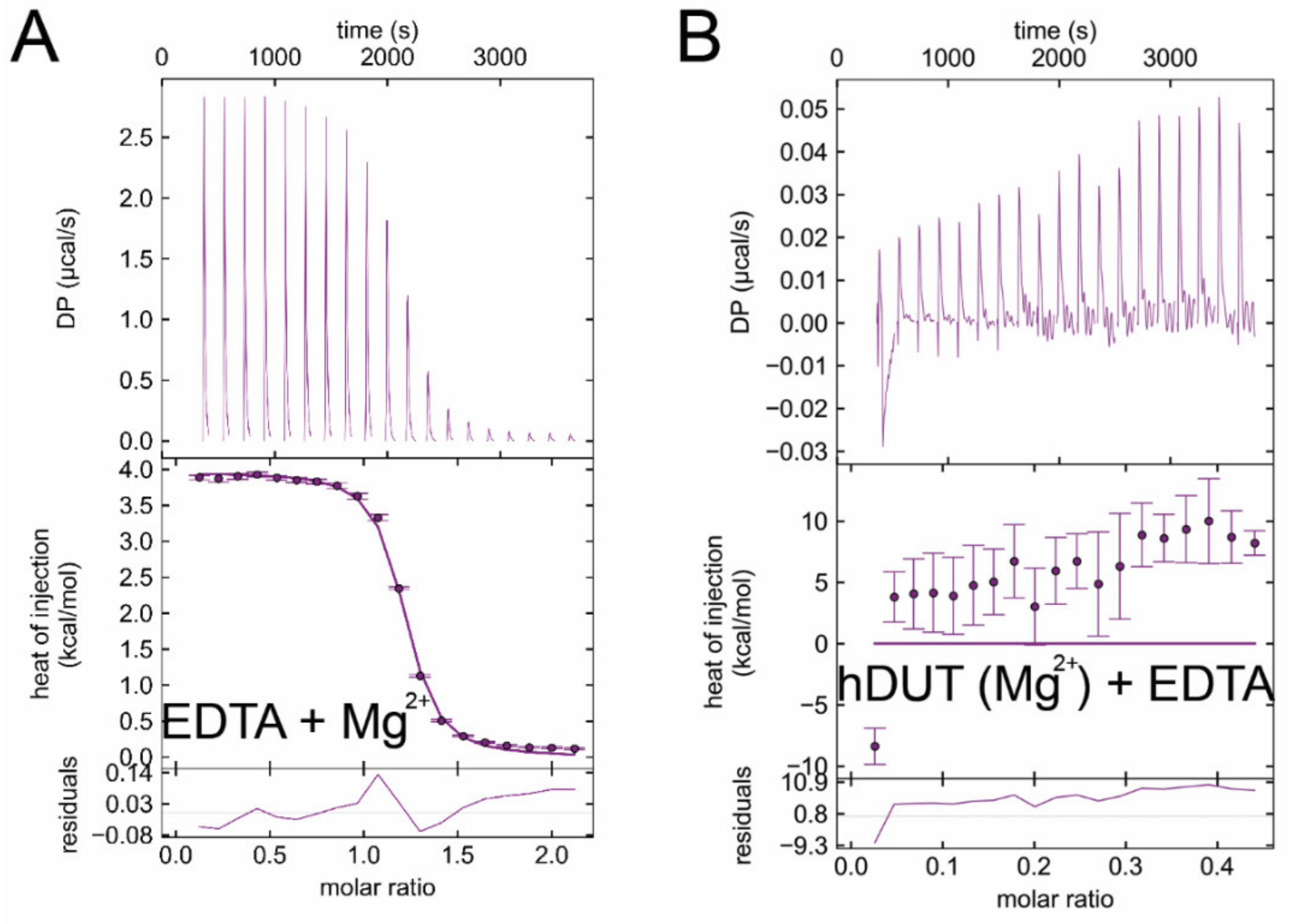
3.6. Isothermal Titration Calorimetry Data Directly Prove EDTA Binding to the Nucleotide Binding Pocket of dUTPase
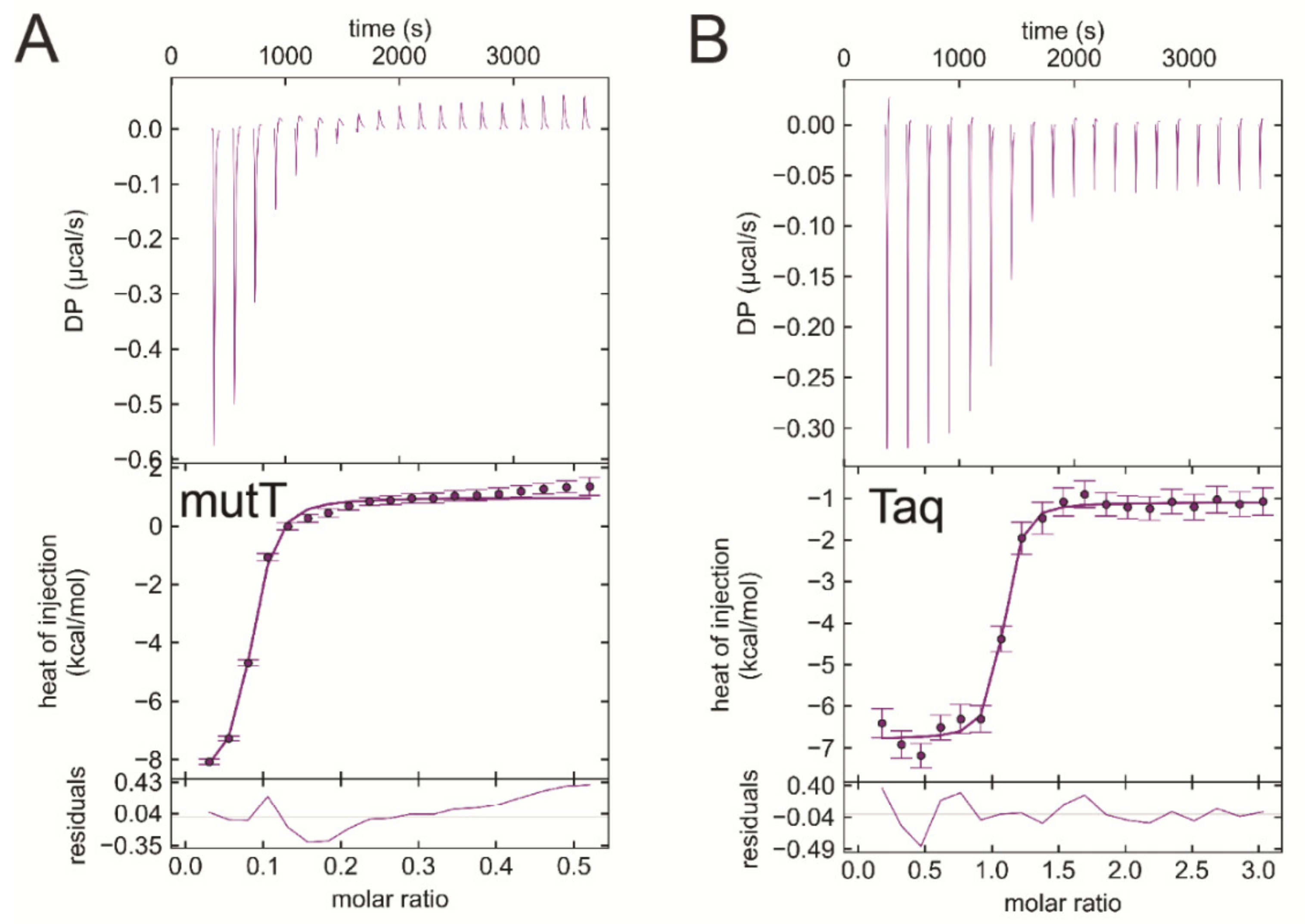
3.7. EDTA Binds to Taq Polymerase and MutT, Two Additional dNTP Processing Enzymes
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Jorgensen, P.L.; Håkansson, K.O.; Karlish, S.J.D. Structure and Mechanism of Na, K-ATPase: Functional Sites and Their Interactions. Annu. Rev. Physiol. 2003, 65, 817–849. [Google Scholar] [CrossRef] [PubMed]
- Sprang, S.R. G Protein Mechanisms: Insights from Structural Analysis. Annu. Rev. Biochem. 1997, 66, 639–678. [Google Scholar] [CrossRef] [PubMed]
- Tomkinson, A.E.; Vijayakumar, S.; Pascal, J.M.; Ellenberger, T. DNA Ligases: Structure, Reaction Mechanism, and Function. Chem. Rev. 2006, 106, 687–699. [Google Scholar] [CrossRef] [PubMed]
- Rothwell, P.J.; Waksman, G. Structure and mechanism of DNA polymerases. Advances Protein Chem. 2005, 71, 401–440. [Google Scholar] [PubMed]
- Mildvan, A.S.; Xia, Z.; Azurmendi, H.F.; Saraswat, V.; Legler, P.M.; Massiah, M.A.; Gabelli, S.B.; Bianchet, M.A.; Kang, L.-W.; Amzel, L.M. Structures and mechanisms of Nudix hydrolases. Arch. Biochem. Biophys. 2005, 433, 129–143. [Google Scholar] [CrossRef] [PubMed]
- Vértessy, B.G.; Tóth, J. Keeping uracil out of DNA: physiological role, structure and catalytic mechanism of dUTPases. Acc. Chem. Res. 2009, 42, 97–106. [Google Scholar] [CrossRef]
- Pecsi, I.; Leveles, I.; Harmat, V.; Vertessy, B.G.; Toth, J. Aromatic stacking between nucleobase and enzyme promotes phosphate ester hydrolysis in dUTPase. Nucleic Acids Res. 2010, 38, 7179–7186. [Google Scholar] [CrossRef]
- Mol, C.D.; Harris, J.M.; McIntosh, E.M.; Tainer, J.A. Human dUTP pyrophosphatase: uracil recognition by a beta hairpin and active sites formed by three separate subunits. Structure 1996, 4, 1077–1092. [Google Scholar] [CrossRef]
- Kovári, J.; Barabás, O.; Takács, E.; Békési, A.; Dubrovay, Z.; Pongrácz, V.; Zagyva, I.; Imre, T.; Szabó, P.; Vértessy, B.G. Altered Active Site Flexibility and a Structural Metal-binding Site in Eukaryotic dUTPase. J. Biol. Chem. 2004, 279, 17932–17944. [Google Scholar] [CrossRef]
- Freeman, L.; Buisson, M.; Tarbouriech, N.; Van der Heyden, A.; Labbé, P.; Burmeister, W.P. The Flexible Motif V of Epstein-Barr Virus Deoxyuridine 5′-Triphosphate Pyrophosphatase Is Essential for Catalysis. J. Biol. Chem. 2009, 284, 25280–25289. [Google Scholar] [CrossRef]
- Mustafi, D.; Bekesi, A.; Vertessy, B.G.; Makinen, M.W. Catalytic and structural role of the metal ion in dUTP pyrophosphatase. Proc. Natl. Acad. Sci. USA 2003, 100, 5670–5675. [Google Scholar] [CrossRef]
- Kovári, J.; Barabás, O.; Varga, B.; Békési, A.; Tölgyesi, F.; Fidy, J.; Nagy, J.; Vértessy, B.G. Methylene substitution at the α–β bridging position within the phosphate chain of dUDP profoundly perturbs ligand accommodation into the dUTPase active site. Proteins Struct. Funct. Bioinforma. 2008, 71, 308–319. [Google Scholar] [CrossRef] [PubMed]
- Nord, J.; Larsson, G.; Kvassman, J.O.; Rosengren, A.M.; Nyman, P.O. dUTPase from the retrovirus equine infectious anemia virus: specificity, turnover and inhibition. FEBS Lett. 1997, 414, 271–274. [Google Scholar] [PubMed]
- Larsson, G.; Nyman, P.O.; Kvassman, J.O. Kinetic characterization of dUTPase from Escherichia coli. J. Biol. Chem. 1996, 271, 24010–24016. [Google Scholar] [CrossRef] [PubMed]
- Oliveros, M.; García-Escudero, R.; Alejo, A.; Viñuela, E.; Salas, M.L.; Salas, J. African swine fever virus dUTPase is a highly specific enzyme required for efficient replication in swine macrophages. J. Virol. 1999, 73, 8934–8943. [Google Scholar] [PubMed]
- Nagy, G.N.; Leveles, I.; Vértessy, B.G. Preventive DNA repair by sanitizing the cellular (deoxy)nucleoside triphosphate pool. FEBS J. 2014, 281, 4207–4223. [Google Scholar] [CrossRef] [PubMed]
- Sang, P.B.; Varshney, U. Biochemical Properties of MutT2 Proteins from Mycobacterium tuberculosis and M. smegmatis and Their Contrasting Antimutator Roles in Escherichia coli. J. Bacteriol. 2013, 195, 1552–1560. [Google Scholar] [CrossRef] [PubMed]
- Varga, B.; Barabás, O.; Kovári, J.; Tóth, J.; Hunyadi-Gulyás, E.; Klement, E.; Medzihradszky, K.F.; Tölgyesi, F.; Fidy, J.; Vértessy, B.G. Active site closure facilitates juxtaposition of reactant atoms for initiation of catalysis by human dUTPase. FEBS Lett. 2007, 581, 4783–4788. [Google Scholar] [CrossRef]
- Szabó, J.E.; Takács, E.; Merényi, G.; Vértessy, B.G.; Tóth, J. Trading in cooperativity for specificity to maintain uracil-free DNA. Sci. Rep. 2016, 6, 24219. [Google Scholar] [CrossRef][Green Version]
- Varga, B.; Barabás, O.; Takács, E.; Nagy, N.; Nagy, P.; Vértessy, B.G. Active site of mycobacterial dUTPase: structural characteristics and a built-in sensor. Biochem. Biophys. Res. Commun. 2008, 373, 8–13. [Google Scholar] [CrossRef]
- Papp-Kádár, V.; Balázs, Z.; Vékey, K.; Ozohanics, O.; Vértessy, B.G. Mass spectrometry-based analysis of macromolecular complexes of Staphylococcus aureus uracil-DNA glycosylase and its inhibitor reveals specific variations due to naturally occurring mutations. FEBS Open Bio 2019, 9, 420–427. [Google Scholar] [PubMed]
- Bartha-Vári, J.H.; Toşa, M.I.; Irimie, F.-D.; Weiser, D.; Boros, Z.; Vértessy, B.G.; Paizs, C.; Poppe, L. Immobilization of Phenylalanine Ammonia-Lyase on Single-Walled Carbon Nanotubes for Stereoselective Biotransformations in Batch and Continuous-Flow Modes. ChemCatChem 2015, 7, 1122–1128. [Google Scholar] [CrossRef] [PubMed]
- Papp-Kádár, V.; Balázs, Z.; Nagy, G.N.; Juhász, T.; Liliom, K.; Vértessy, B.G. Functional analysis on a naturally occurring variant of the Staphylococcus Aureus uracil DNA Glycosylase inhibitor. Period. Polytech. Chem. Eng. 2018, 62, 51–56. [Google Scholar] [CrossRef]
- Bata, Z.; Qian, R.; Roller, A.; Horak, J.; Bencze, L.C.; Paizs, C.; Hammerschmidt, F.; Vértessy, B.G.; Poppe, L. A Methylidene Group in the Phosphonic Acid Analogue of Phenylalanine Reverses the Enantiopreference of Binding to Phenylalanine Ammonia-Lyases. Adv. Synth. Catal. 2017, 359, 2109–2120. [Google Scholar] [CrossRef]
- Takács, B.; Billington, N.; Gyimesi, M.; Kintses, B.; Málnási-Csizmadia, A.; Knight, P.J.; Kovács, M. Myosin complexed with ADP and blebbistatin reversibly adopts a conformation resembling the start point of the working stroke. Proc. Natl. Acad. Sci. USA 2010, 107, 6799–6804. [Google Scholar] [CrossRef]
- Szabó, J.E.; Németh, V.; Papp-Kádár, V.; Nyíri, K.; Leveles, I.; Bendes, A.Á.; Zagyva, I.; Róna, G.; Pálinkás, H.L.; Besztercei, B.; et al. Highly potent dUTPase inhibition by a bacterial repressor protein reveals a novel mechanism for gene expression control. Nucleic Acids Res. 2014, 42, 11912–11920. [Google Scholar] [CrossRef]
- Li, H.; Robertson, A.D.; Jensen, J.H. Very fast empirical prediction and rationalization of protein pKa values. Proteins Struct. Funct. Bioinforma. 2005, 61, 704–721. [Google Scholar] [CrossRef]
- Bas, D.C.; Rogers, D.M.; Jensen, J.H. Very fast prediction and rationalization of pKa values for protein-ligand complexes. Proteins 2008, 73, 765–783. [Google Scholar] [CrossRef]
- Dolinsky, T.J.; Nielsen, J.E.; McCammon, J.A.; Baker, N.A. PDB2PQR: an automated pipeline for the setup of Poisson-Boltzmann electrostatics calculations. Nucleic Acids Res. 2004, 32, W665–W667. [Google Scholar] [CrossRef]
- Cherrier, M.V.; Martin, L.; Cavazza, C.; Jacquamet, L.; Lemaire, D.; Gaillard, J.; Fontecilla-Camps, J.C. Crystallographic and spectroscopic evidence for high affinity binding of FeEDTA(H2O)- to the periplasmic nickel transporter NikA. J. Am. Chem. Soc. 2005, 127, 10075–10082. [Google Scholar] [CrossRef]
- Chemical Computing Group Inc. MOE (The Molecular Operating Environment) Version 2007.09, software available from Chemical Computing Group Inc., 1010 Sherbrooke Street West, Suite 910, Montreal, Canada H3A 2R7. 2007. Available online: http://www.chemcomp.com (accessed on 1 May 2013).
- Halgren, T.A. Merck molecular force field. I. Basis, form, scope, parameterization, and performance of MMFF94. J. Comput. Chem. 1996, 17, 490–519. [Google Scholar] [CrossRef]
- Halgren, T.A. Merck molecular force field. II. MMFF94 van der Waals and electrostatic parameters for intermolecular interactions. J. Comput. Chem. 1996, 17, 520–552. [Google Scholar] [CrossRef]
- Halgren, T.A. Merck molecular force field. III. Molecular geometries and vibrational frequencies for MMFF94. J. Comput. Chem. 1996, 17, 553–586. [Google Scholar] [CrossRef]
- Halgren, T.A.; Nachbar, R.B. Merck molecular force field. IV. conformational energies and geometries for MMFF94. J. Comput. Chem. 1996, 17, 587–615. [Google Scholar] [CrossRef]
- Halgren, T.A. Merck molecular force field. V. Extension of MMFF94 using experimental data, additional computational data, and empirical rules. J. Comput. Chem. 1996, 17, 616–641. [Google Scholar] [CrossRef]
- Gasteiger, J.; Marsili, M. Iterative partial equalization of orbital electronegativity—a rapid access to atomic charges. Tetrahedron 1980, 36, 3219–3228. [Google Scholar] [CrossRef]
- Morris, G.M.; Huey, R.; Lindstrom, W.; Sanner, M.F.; Belew, R.K.; Goodsell, D.S.; Olson, A.J. AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility. J. Comput. Chem. 2009, 30, 2785–2791. [Google Scholar] [CrossRef] [PubMed]
- Morris, G.M.; Goodsell, D.S.; Halliday, R.S.; Huey, R.; Hart, W.E.; Belew, R.K.; Olson, A.J. Automated docking using a Lamarckian genetic algorithm and an empirical binding free energy function. J. Comput. Chem. 1998, 19, 1639–1662. [Google Scholar] [CrossRef]
- Hetényi, C.; van der Spoel, D. Efficient docking of peptides to proteins without prior knowledge of the binding site. Protein Sci. 2002, 11, 1729–1737. [Google Scholar] [CrossRef]
- Hetényi, C.; van der Spoel, D. Blind docking of drug-sized compounds to proteins with up to a thousand residues. FEBS Lett. 2006, 580, 1447–1450. [Google Scholar] [CrossRef]
- Clark, A.M.; Labute, P. 2D depiction of protein-ligand complexes. J. Chem. Inf. Model. 2007, 47, 1933–1944. [Google Scholar] [CrossRef] [PubMed]
- Róna, G.; Marfori, M.; Borsos, M.; Scheer, I.; Takács, E.; Tóth, J.; Babos, F.; Magyar, A.; Erdei, A.; Bozóky, Z.; et al. Phosphorylation adjacent to the nuclear localization signal of human dUTPase abolishes nuclear import: structural and mechanistic insights. Acta Crystallogr. D. Biol. Crystallogr. 2013, 69, 2495–2505. [Google Scholar] [CrossRef] [PubMed]
- Keller, S.; Vargas, C.; Zhao, H.; Piszczek, G.; Brautigam, C.A.; Schuck, P. High-precision isothermal titration calorimetry with automated peak-shape analysis. Anal. Chem. 2012, 84, 5066–5073. [Google Scholar] [CrossRef] [PubMed]
- Zhao, H.; Piszczek, G.; Schuck, P. SEDPHAT—A platform for global ITC analysis and global multi-method analysis of molecular interactions. Methods 2015, 76, 137–148. [Google Scholar] [CrossRef] [PubMed]
- Brautigam, C.A.; Zhao, H.; Vargas, C.; Keller, S.; Schuck, P. Integration and global analysis of isothermal titration calorimetry data for studying macromolecular interactions. Nat. Protoc. 2016, 11, 882–894. [Google Scholar] [CrossRef] [PubMed]
- Brautigam, C.A. Calculations and Publication-Quality Illustrations for Analytical Ultracentrifugation Data. Methods Enzymol. 2015, 562, 109–133. [Google Scholar] [PubMed]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: visual molecular dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef]
- Schrödinger LLC. The PyMOL Molecular Graphics System, Version 1.2r3pre. Available online: http://www.schrodinger.com/pymol (accessed on 19 October 2018).
- Zhang, W.; Truttmann, A.C.; Lüthi, D.; McGuigan, J.A. Apparent Mg2+-adenosine 5-triphosphate dissociation constant measured with Mg2+ macroelectrodes under conditions pertinent to 31P NMR ionized magnesium determinations. Anal. Biochem. 1997, 251, 246–250. [Google Scholar] [CrossRef]
- Tóth, J.; Varga, B.; Kovács, M.; Málnási-Csizmadia, A.; Vértessy, B.G. Kinetic Mechanism of Human dUTPase, an Essential Nucleotide Pyrophosphatase Enzyme. J. Biol. Chem. 2007, 282, 33572–33582. [Google Scholar] [CrossRef]
- Lopata, A.; Jambrina, P.G.; Sharma, P.K.; Brooks, B.R.; Toth, J.; Vertessy, B.G.; Rosta, E. Mutations Decouple Proton Transfer from Phosphate Cleavage in the dUTPase Catalytic Reaction. ACS Catal. 2015, 5, 3225–3237. [Google Scholar] [CrossRef]
- Sanner, M.F.; Olson, A.J.; Spehner, J.C. Reduced surface: An efficient way to compute molecular surfaces. Biopolymers 1996, 38, 305–320. [Google Scholar] [CrossRef]
- Barabás, O.; Pongrácz, V.; Kovári, J.; Wilmanns, M.; Vértessy, B.G. Structural insights into the catalytic mechanism of phosphate ester hydrolysis by dUTPase. J. Biol. Chem. 2004, 279, 42907–42915. [Google Scholar] [CrossRef] [PubMed]
- Barabás, O.; Németh, V.; Bodor, A.; Perczel, A.; Rosta, E.; Kele, Z.; Zagyva, I.; Szabadka, Z.; Grolmusz, V.I.; Wilmanns, M.; et al. Catalytic mechanism of α-phosphate attack in dUTPase is revealed by X-ray crystallographic snapshots of distinct intermediates, 31P-NMR spectroscopy and reaction path modelling. Nucleic Acids Res. 2013, 41, 10542–10555. [Google Scholar] [CrossRef] [PubMed]
- Hirmondo, R.; Lopata, A.; Suranyi, E.V.; Vertessy, B.G.; Toth, J. Differential control of dNTP biosynthesis and genome integrity maintenance by the dUTPase superfamily enzymes. Sci. Rep. 2017, 7, 6043. [Google Scholar] [CrossRef] [PubMed]
- Pecsi, I.; Szabo, J.E.; Adams, S.D.; Simon, I.; Sellers, J.R.; Vertessy, B.G.; Toth, J. Nucleotide pyrophosphatase employs a P-loop-like motif to enhance catalytic power and NDP/NTP discrimination. Proc. Natl. Acad. Sci. 2011, 108, 14437–14442. [Google Scholar] [CrossRef] [PubMed]
- O’Brien, L.C.; Root, H.B.; Wei, C.-C.; Jensen, D.; Shabestary, N.; De Meo, C.; Eder, D.J. M2+ •EDTA Binding Affinities: A Modern Experiment in Thermodynamics for the Physical Chemistry Laboratory. J. Chem. Educ. 2015, 92, 1547–1551. [Google Scholar] [CrossRef]
- Brandis, J.W.; Edwards, S.G.; Johnson, K.A. Slow rate of phosphodiester bond formation accounts for the strong bias that Taq DNA polymerase shows against 2’,3’-dideoxynucleotide terminators. Biochemistry 1996, 35, 2189–2200. [Google Scholar] [CrossRef] [PubMed]
- Maki, H.; Sekiguchi, M. MutT protein specifically hydrolyses a potent mutagenic substrate for DNA synthesis. Lett. Nat. 1992, 355, 273–275. [Google Scholar] [CrossRef]
- Carvajal, N.; Orellana, M.S.; Bórquez, J.; Uribe, E.; López, V.; Salas, M. Non-chelating inhibition of the H101N variant of human liver arginase by EDTA. J. Inorg. Biochem. 2004, 98, 1465–1469. [Google Scholar] [CrossRef]
- Shah, M.; Grover, T.; Barr, D.; Aust, S. On the mechanism of inhibition of the veratryl alcohol oxidase activity of lignin peroxidase H2 by EDTA. J. Biol. Chem. 1992, 267, 21564–21569. [Google Scholar]
- Banerjee, R.K. EDTA inhibits peroxidase-catalyzed iodide oxidation through interaction at the iodide binding site. Biochim. Biophys. Acta - Gen. Subj. 1989, 992, 393–396. [Google Scholar] [CrossRef]
- Bhattacharyya, D.K.; Adak, S.; Bandyopadhyay, U.; Banerjee, R.K. Mechanism of inhibition of horseradish peroxidase-catalysed iodide oxidation by EDTA. Biochem. J. 1994, 298 ( Pt 2, 281–288. [Google Scholar] [CrossRef]
- Ding, Y.; Williams, R.M.; Sherman, D.H. Molecular analysis of a 4-dimethylallyltryptophan synthase from Malbranchea aurantiaca. J. Biol. Chem. 2008, 283, 16068–16076. [Google Scholar] [CrossRef]
- Kondoh, G.; Tojo, H.; Nakatani, Y.; Komazawa, N.; Murata, C.; Yamagata, K.; Maeda, Y.; Kinoshita, T.; Okabe, M.; Taguchi, R.; et al. Angiotensin-converting enzyme is a GPI-anchored protein releasing factor crucial for fertilization. Nat. Med. 2005, 11, 160–166. [Google Scholar] [CrossRef] [PubMed]
- Hast, M.A.; Fletcher, S.; Cummings, C.G.; Pusateri, E.E.; Blaskovich, M.A.; Rivas, K.; Gelb, M.H.; Van Voorhis, W.C.; Sebti, S.M.; Hamilton, A.D.; et al. Structural basis for binding and selectivity of antimalarial and anticancer ethylenediamine inhibitors to protein farnesyltransferase. Chem. Biol. 2009, 16, 181–192. [Google Scholar] [CrossRef] [PubMed]
- Fletcher, S.; Keaney, E.P.; Cummings, C.G.; Blaskovich, M.A.; Hast, M.A.; Glenn, M.P.; Chang, S.-Y.; Bucher, C.J.; Floyd, R.J.; Katt, W.P.; et al. Structure-based design and synthesis of potent, ethylenediamine-based, mammalian farnesyltransferase inhibitors as anticancer agents. J. Med. Chem. 2010, 53, 6867–6888. [Google Scholar] [CrossRef] [PubMed]
- Delarue, M.; Poch, O.; Tordo, N.; Moras, D.; Argos, P. An attempt to unify the structure of polymerases. Protein Eng. 1990, 3, 461–467. [Google Scholar] [CrossRef] [PubMed]
- Kropp, H.M.; Diederichs, K.; Marx, A. The Structure of an Archaeal B-Family DNA Polymerase in Complex with a Chemically Modified Nucleotide. Angew. Chem. Int. Ed. Engl. 2019, 58, 5457–5461. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, T.; Meshitsuka, S.; Kitagawa, S.; Abe, N.; Yamada, J.; Ishino, T.; Nakano, H.; Tsuzuki, T.; Doi, T.; Kobayashi, Y.; et al. Structural and dynamic features of the MutT protein in the recognition of nucleotides with the mutagenic 8-oxoguanine base. J. Biol. Chem. 2010, 285, 444–452. [Google Scholar] [CrossRef]
- Yang, Y.; Gourinath, S.; Kovács, M.; Nyitray, L.; Reutzel, R.; Himmel, D.M.; O’Neall-Hennessey, E.; Reshetnikova, L.; Szent-Györgyi, A.G.; Brown, J.H.; et al. Rigor-like structures from muscle myosins reveal key mechanical elements in the transduction pathways of this allosteric motor. Structure 2007, 15, 553–564. [Google Scholar] [CrossRef]
- Moeschler, H.J.; Schaer, J.J.; Cox, J.A. A thermodynamic analysis of the binding of calcium and magnesium ions to parvalbumin. Eur. J. Biochem. 1980, 111, 73–78. [Google Scholar] [CrossRef] [PubMed]
- Lopata, A.; Leveles, I.; Bendes, Á.Á.; Viskolcz, B.; Vértessy, B.G.; Jójárt, B.; Tóth, J. A Hidden Active Site in the Potential Drug Target Mycobacterium tuberculosis dUTPase Is Accessible through Small Amplitude Protein Conformational Changes. J. Biol. Chem. 2016, 291, 26320–26331. [Google Scholar] [CrossRef] [PubMed]
- Mónico, A.; Martínez-Senra, E.; Cañada, F.J.; Zorrilla, S.; Pérez-Sala, D. Drawbacks of Dialysis Procedures for Removal of EDTA. PLoS ONE 2017, 12, e0169843. [Google Scholar] [CrossRef] [PubMed]
- Sharpe, J.C.; London, E. Inadvertent Concentrating of EDTA by Ion Exchange Chromatography: Avoiding Artifacts That Can Interfere with Protein Purification. Anal. Biochem. 1997, 250, 124–125. [Google Scholar] [CrossRef]
- Chumanov, R.S.; Burgess, R.R. Artifact-inducing enrichment of ethylenediaminetetraacetic acid and ethyleneglycoltetraacetic acid on anion exchange resins. Anal. Biochem. 2011, 412, 34–39. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
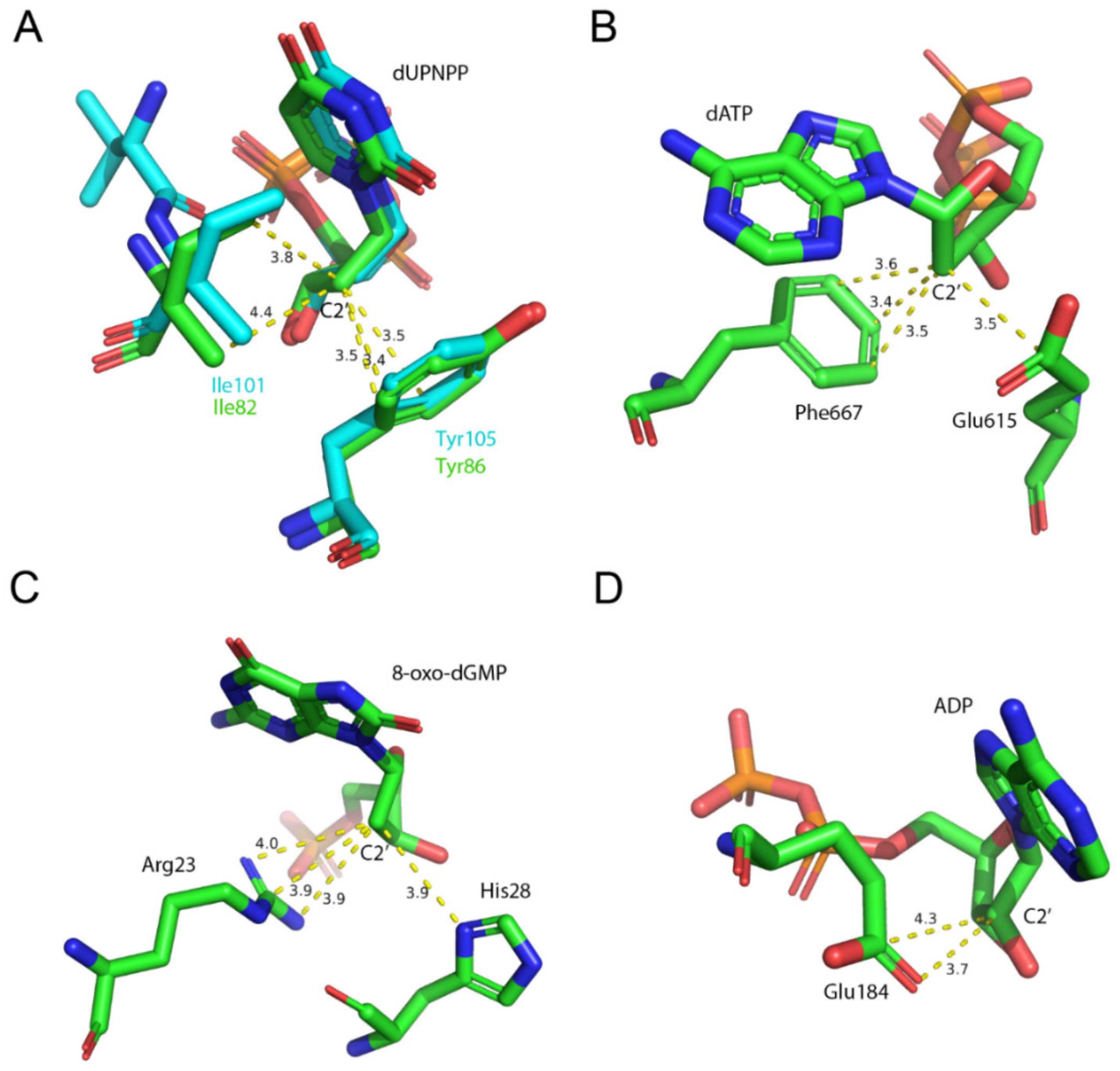{kind=link}
| hDUT | mtDUT | ||||
|---|---|---|---|---|---|
| kcat/s−1 | KM/µM | kcat/s−1 | KM/µM | ||
| 5 mM Mg2+, no EDTA | 5.27 ± 0.24 | 2.26 ± 0.33 | 5 mM Mg2+, no EDTA | 2.39 ± 0.16 | 1.72 ± 0.51 |
| No Mg2+, no EDTA | 3.71 ± 0.12 | 2.49 ± 0.37 | No Mg2+, no EDTA | 0.79 ± 0.01 | 2.70 ± 0.18 |
| 30 µM EDTA, no Mg2+ | 3.30 ± 0.26 | 6.72 ± 1.69 | 300 µM EDTA, no Mg2+ | 0.71 ± 0.01 | 10.04 ± 0.50 |
| 100 µM EDTA, no Mg2+ | 0.24 ± 0.03 | 6.16 ± 5.34 | |||
| Tm (5 mM Mg2+)/°C | Tm (No Mg2+, no EDTA)/°C | Tm (100 µM EDTA)/℃ | |
|---|---|---|---|
| apo | 59.5 ± 0.3 | 58.6 ± 0.1 | 58.5 ± 0.1 |
| dUPNPP | 63.9 ± 0.3 | 59.1 ± 0.2 | 58.3 ± 0.1 |
| dUMP | 62.3 ± 0.1 | 59.7 ± 0.1 | 58.7 ± 0.1 |
| hDUT | mtDUT | ||
|---|---|---|---|
| EDTA | dUTP | EDTA | dUTP |
| Arg85 (II) | Arg85 (II) | Arg64 (II) | Arg64 (II) |
| Ser86 (II) | |||
| Gly87 (II) | Gly87 (II) | Gly66 (II) | Gly66 (II) |
| Gly99 (III) | Gly99 (III) | ||
| Thr81 (III) | |||
| Asp102 (III) | Asp102 (III) | Asp83 (III) | Asp83 (III) |
| Gly110 (III) | Lys91 (III) | ||
| Gln131 (IV) | Gln113 (IV) | Gln113 (IV) | |
| Arg153 (V) | Arg153 (V) | Arg140 (V) | Arg140 (V) |
| His145 (V) | |||
| Ser160 (V) | Ser160 (V) | Ser147 (V) | Ser147 (V) |
| Thr161 (V) | Thr161 (V) | Ser148 (V) | Ser148 (V) |
| Titrand | Titrant | ΔH/kcal/mol | −TΔS/kcal/mol | ΔG/kcal/mol | Kd/μM |
|---|---|---|---|---|---|
| hDUT | EDTA | −11.2 ± 0.4 | 2.2 ± 0.4 | −9.0 ± 0.0 | 0.20 ± 0.009 |
| hDUT | dUPNPP | −17.4 ± 3.6 | 11.5 ± 3.7 | −5.9 ± 0.2 | 43.9 ± 14.6 |
| mtDUT | EDTA | −4.2 ± 1.0 | −5.3 ± 1.2 | −9.6 ± 0.2 | 0.078 ± 0.023 |
| mtDUT | dUPNPP | −4.5 ± 0.2 | −2.1 ± 0.4 | −6.6 ± 0.6 | 13.9 ± 11.7 |
| EDTA | Mg2+ | 4.4 ± 0.3 | −12.5 ± 0.6 | −8.0 ± 0.9 | 2.53 ± 0.18 |
| EcMutT | EDTA | −9.7 ± 0.02 | 1.3 ± 0.1 | −8.4 ± 0.1 | 0.55 ± 0.066 |
| Taq polymerase | EDTA | -6.3 ± 0.7 | −3.5 ± 0.7 | −9.8 ± 0.0 | 0.047 ± 0.001 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lopata, A.; Jójárt, B.; Surányi, É.V.; Takács, E.; Bezúr, L.; Leveles, I.; Bendes, Á.Á.; Viskolcz, B.; Vértessy, B.G.; Tóth, J. Beyond Chelation: EDTA Tightly Binds Taq DNA Polymerase, MutT and dUTPase and Directly Inhibits dNTPase Activity. Biomolecules 2019, 9, 621. https://doi.org/10.3390/biom9100621
Lopata A, Jójárt B, Surányi ÉV, Takács E, Bezúr L, Leveles I, Bendes ÁÁ, Viskolcz B, Vértessy BG, Tóth J. Beyond Chelation: EDTA Tightly Binds Taq DNA Polymerase, MutT and dUTPase and Directly Inhibits dNTPase Activity. Biomolecules. 2019; 9(10):621. https://doi.org/10.3390/biom9100621
Chicago/Turabian StyleLopata, Anna, Balázs Jójárt, Éva V. Surányi, Enikő Takács, László Bezúr, Ibolya Leveles, Ábris Á. Bendes, Béla Viskolcz, Beáta G. Vértessy, and Judit Tóth. 2019. "Beyond Chelation: EDTA Tightly Binds Taq DNA Polymerase, MutT and dUTPase and Directly Inhibits dNTPase Activity" Biomolecules 9, no. 10: 621. https://doi.org/10.3390/biom9100621
APA StyleLopata, A., Jójárt, B., Surányi, É. V., Takács, E., Bezúr, L., Leveles, I., Bendes, Á. Á., Viskolcz, B., Vértessy, B. G., & Tóth, J. (2019). Beyond Chelation: EDTA Tightly Binds Taq DNA Polymerase, MutT and dUTPase and Directly Inhibits dNTPase Activity. Biomolecules, 9(10), 621. https://doi.org/10.3390/biom9100621