Allopurinol Prevents the Lipogenic Response Induced by an Acute Oral Fructose Challenge in Short-Term Fructose Fed Rats

 ,
,  ,
,
Abstract
1. Introduction
2. Materials and Methods
2.1. Effect of an Acute Fructose/Glucose Load in the Liver
2.2. Plasma and Liver Uric Acid
2.3. Mitochondria Isolation
2.3.1. Mitochondrial Oxygen Consumption
2.3.2. Aconitase Activity
2.4. Protein Extraction and Immunoblotting
2.5. Statistical Analysis
3. Results
3.1. General Parameters and Renal Function
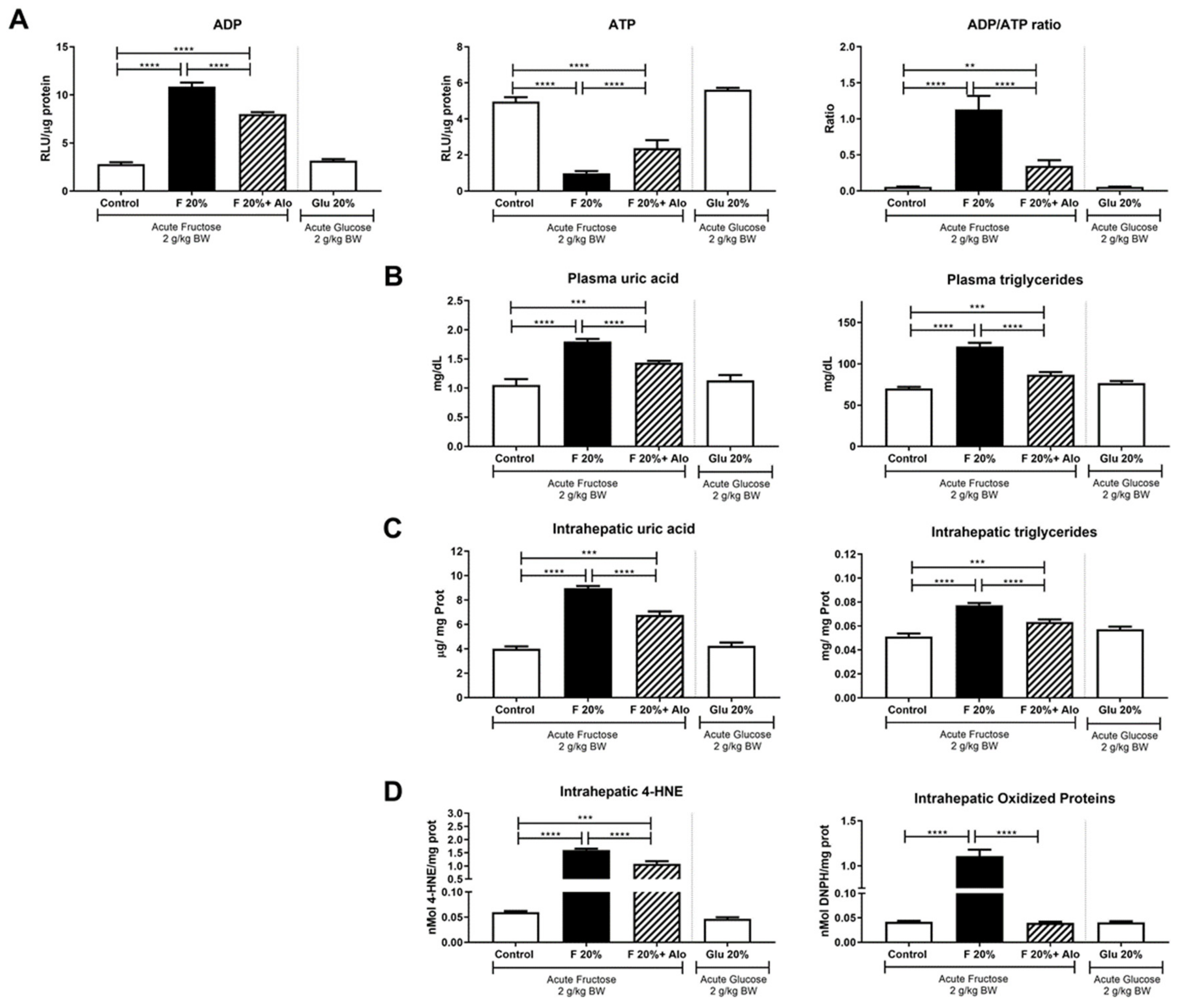
3.2. Allopurinol Prevented Acute ATP Depletion
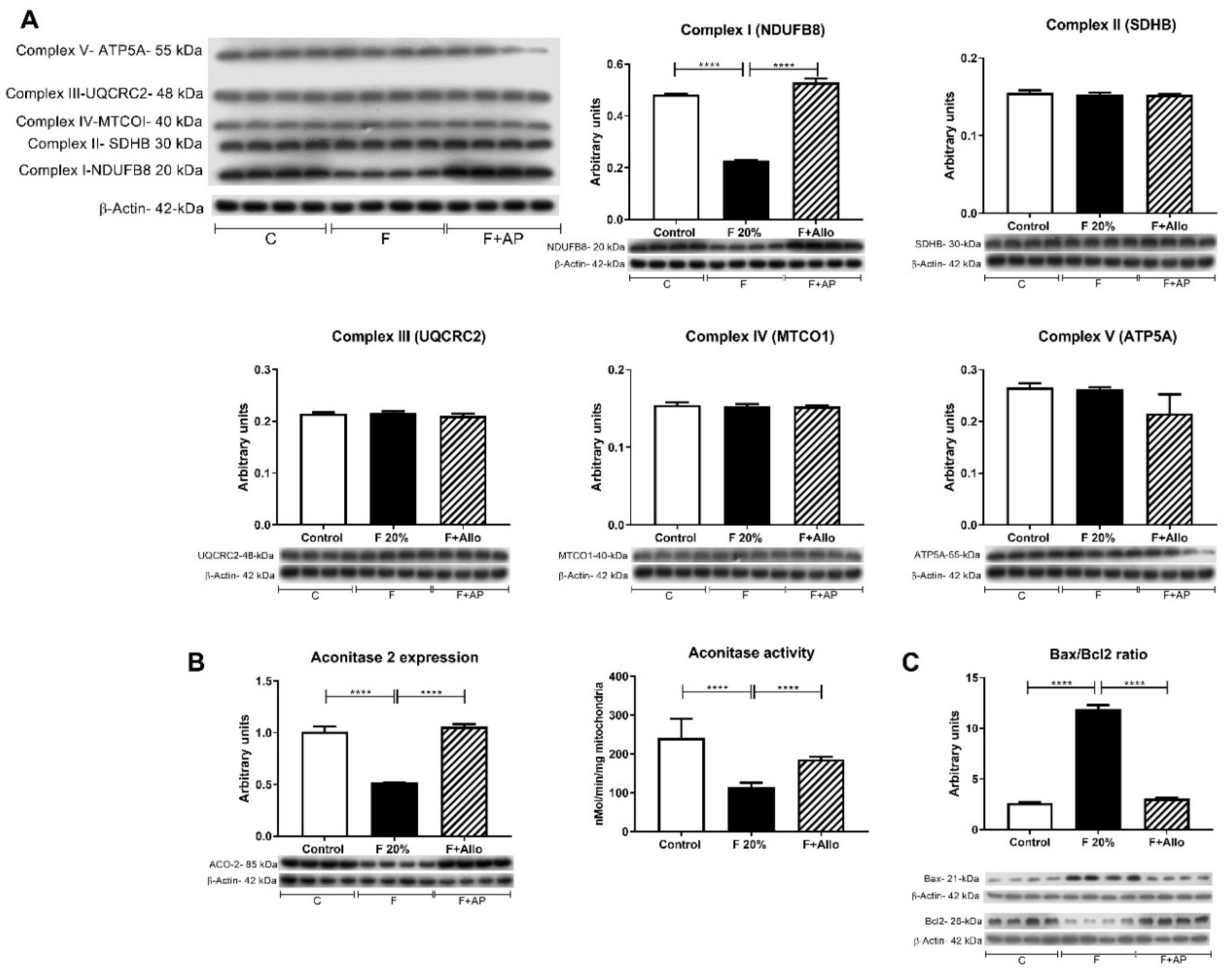
3.3. Allopurinol Prevented Hepatic Mitochondria Uncoupling
3.4. Allopurinol Prevented Apoptosis
3.5. Immunoblotting for KHK and XO
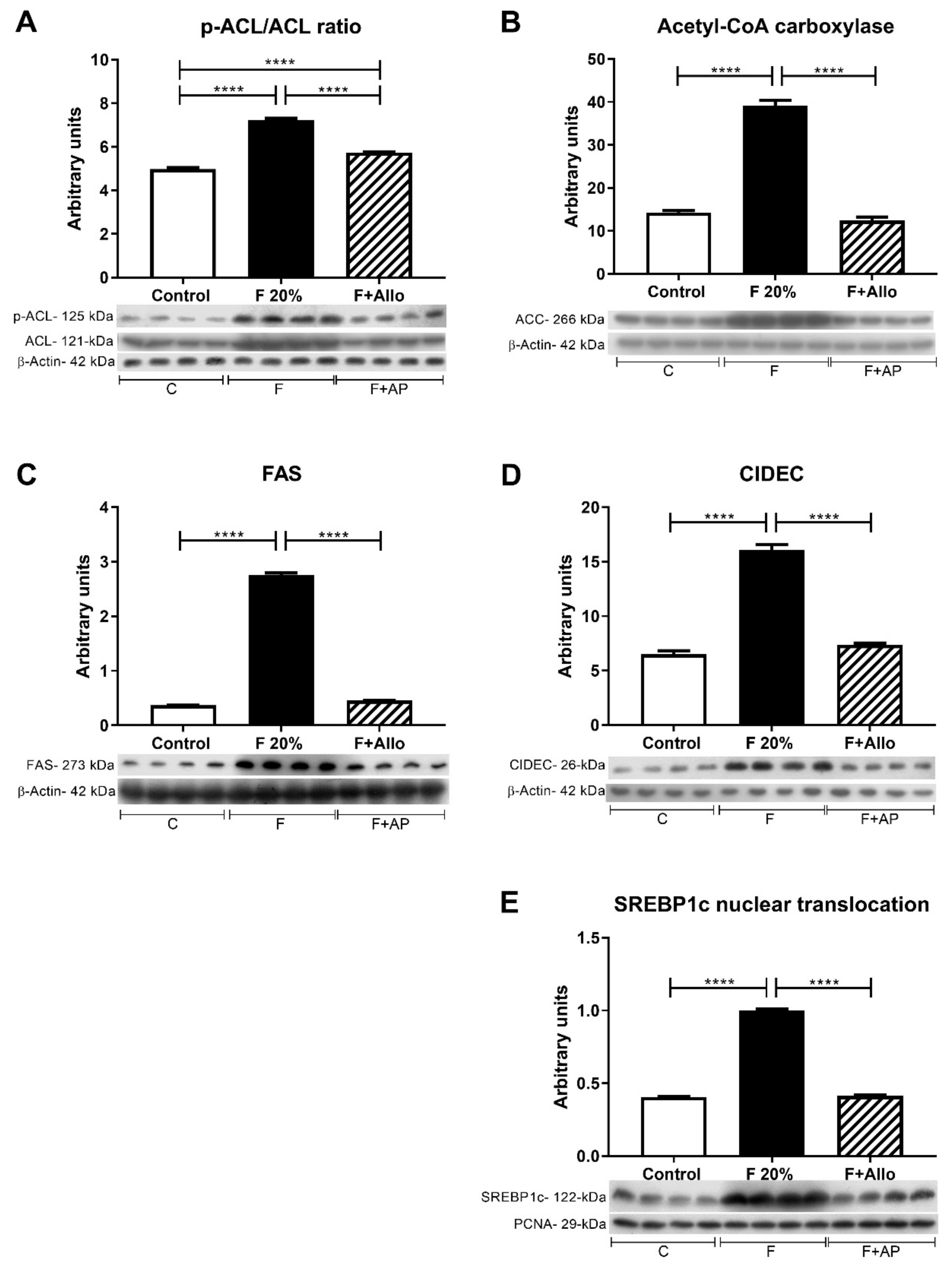
3.6. Immunoblotting for Markers of Lipid Metabolism after an Acute Load of Fructose
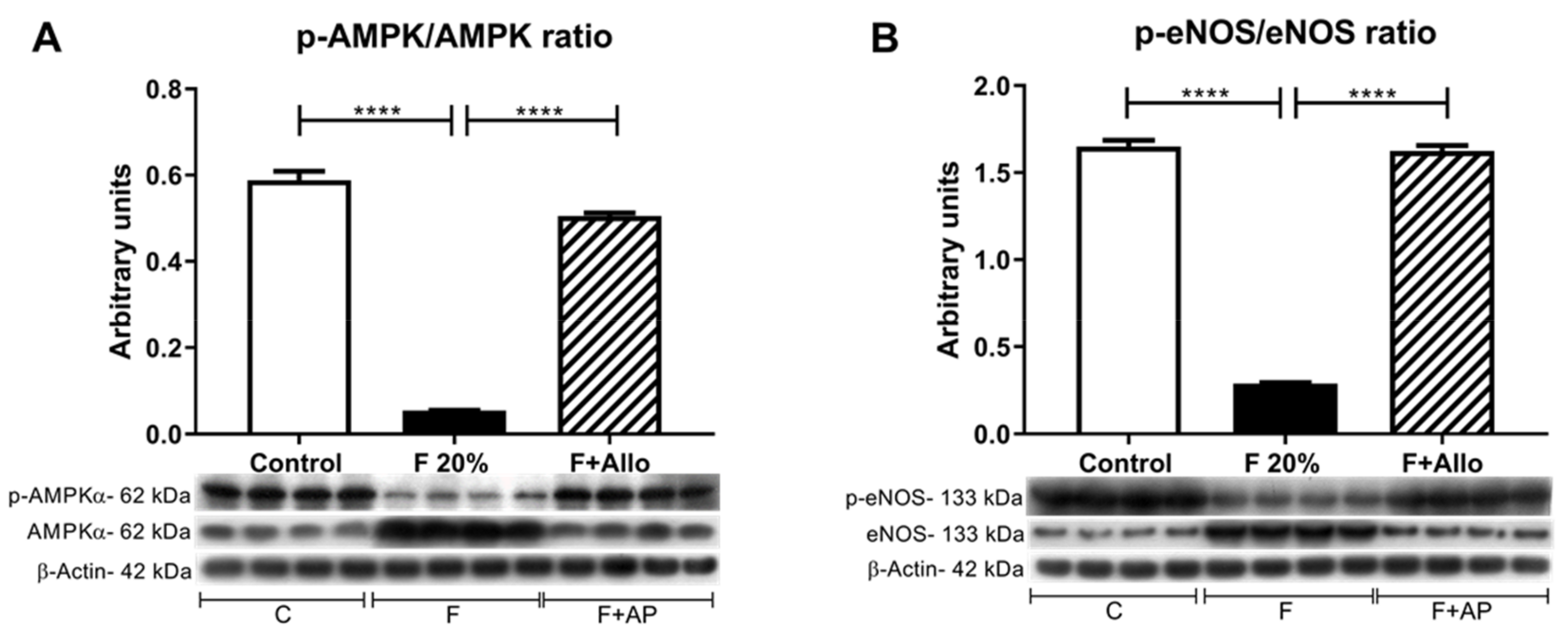
3.7. Immunobloting for p-AMPk/AMPk Ratio and p-eNOS/eNOS Ratio
4. Discussion
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Johnson, R.J.; Segal, M.S.; Sautin, Y.; Nakagawa, T.; Feig, D.I.; Kang, D.H.; Gersch, M.S.; Benner, S.; Sanchez-Lozada, L.G. Potential role of sugar (fructose) in the epidemic of hypertension, obesity and the metabolic syndrome, diabetes, kidney disease, and cardiovascular disease. Am. J. Clin. Nutr. 2007, 86, 899–906. [Google Scholar] [PubMed]
- Hallfrisch, J. Metabolic effects of dietary fructose. FASEB J. 1990, 4, 2652–2660. [Google Scholar] [CrossRef] [PubMed]
- Hallfrisch, J.; Ellwood, K.; Michaelis, O.E.; Reiser, S.; Prather, E.S. Plasma fructose, uric acid, and inorganic phosphorus responses of hyperinsulinemic men fed fructose. J. Am. Coll. Nutr. 1986, 5, 61–68. [Google Scholar] [CrossRef] [PubMed]
- Bawden, S.J.; Stephenson, M.C.; Ciampi, E.; Hunter, K.; Marciani, L.; Macdonald, I.A.; Aithal, G.P.; Morris, P.G.; Gowland, P.A. Investigating the effects of an oral fructose challenge on hepatic ATP reserves in healthy volunteers: A (31)P MRS study. Clin. Nutr. 2016, 35, 645–649. [Google Scholar] [CrossRef] [PubMed]
- Khitan, Z.; Kim, D. Fructose: A Key Factor in the Development of Metabolic Syndrome and Hypertension. J. Nutr. Metab. 2013, 2013, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Roncal-Jimenez, C.A.; Lanaspa, M.A.; Rivard, C.J.; Nakagawa, T.; Sanchez-Lozada, L.G.; Jalal, D.; Andres-Hernando, A.; Tanabe, K.; Madero, M.; Li, N.; et al. Sucrose induces fatty liver and pancreatic inflammation in male breeder rats independent of excess energy intake. Metabolism 2011, 60, 1259–1270. [Google Scholar] [CrossRef] [PubMed]
- Cortez-Pinto, H.; Chatham, J.; Chacko, V.P.; Arnold, C.; Rashid, A.; Diehl, A.M. Alterations in liver ATP homeostasis in human nonalcoholic steatohepatitis: A pilot study. JAMA 1999, 282, 1659–1664. [Google Scholar] [CrossRef]
- Nakagawa, T.; Hu, H.; Zharikov, S.; Tuttle, K.R.; Short, R.A.; Glushakova, O.; Ouyang, X.; Feig, D.I.; Block, E.R.; Herrera-Acosta, J.; et al. A causal role for uric acid in fructose-induced metabolic syndrome. Am. J. Physiol. Ren. Physiol. 2006, 290, F625–F631. [Google Scholar] [CrossRef]
- Sanchez-Lozada, L.G.; Tapia, E.; Bautista-Garcia, P.; Soto, V.; vila-Casado, C.; Vega-Campos, I.P.; Nakagawa, T.; Zhao, L.; Franco, M.; Johnson, R.J. Effects of febuxostat on metabolic and renal alterations in rats with fructose-induced metabolic syndrome. AJP Ren. Physiol. 2008, 294, F710–F718. [Google Scholar] [CrossRef]
- Perez-Pozo, S.E.; Schold, J.; Nakagawa, T.; Sanchez-Lozada, L.G.; Johnson, R.J.; Lillo, J.L. Excessive fructose intake induces the features of metabolic syndrome in healthy adult men: role of uric acid in the hypertensive response. Int. J. Obes. (Lond) 2010, 34, 454–461. [Google Scholar] [CrossRef]
- Lanaspa, M.A.; Sanchez-Lozada, L.G.; Choi, Y.J.; Cicerchi, C.; Kanbay, M.; Roncal-Jimenez, C.A.; Ishimoto, T.; Li, N.; Marek, G.; Duranay, M.; et al. Uric acid induces hepatic steatosis by generation of mitochondrial oxidative stress: potential role in fructose-dependent and -independent fatty liver. J. Biol. Chem. 2012, 287, 40732–40744. [Google Scholar] [CrossRef] [PubMed]
- Lanaspa, M.A.; Sanchez-Lozada, L.G.; Cicerchi, C.; Li, N.; Roncal-Jimenez, C.A.; Ishimoto, T.; Le, M.; Garcia, G.E.; Thomas, J.B.; Rivard, C.J.; et al. Uric acid stimulates fructokinase and accelerates fructose metabolism in the development of fatty liver. PLoS ONE 2012, 7, e47948. [Google Scholar] [CrossRef] [PubMed]
- Wan, X.; Xu, C.; Lin, Y.; Lu, C.; Li, D.; Sang, J.; He, H.; Liu, X.; Li, Y.; Yu, C. Uric acid regulates hepatic steatosis and insulin resistance through the NLRP3 inflammasome-dependent mechanism. J. Hepatol. 2016, 64, 925–932. [Google Scholar] [CrossRef] [PubMed]
- Sanchez-Lozada, L.G.; Andres-Hernando, A.; Garcia-Arroyo, F.E.; Cicerchi, C.; Li, N.; Kuwabara, M.; Roncal-Jimenez, C.A.; Johnson, R.J.; Lanaspa, M.A. Uric acid activates aldose reductase and the polyol pathway for endogenous fructose and fat production causing development of fatty liver in rats. J. Biol. Chem. 2019, 294, 4272–4281. [Google Scholar] [CrossRef]
- Luyckx, F.H.; Lefebvre, P.J.; Scheen, A.J. Non-alcoholic steatohepatitis: association with obesity and insulin resistance, and influence of weight loss. Diabetes Metab. 2000, 26, 98–106. [Google Scholar]
- Lim, J.S.; Mietus-Snyder, M.; Valente, A.; Schwarz, J.M.; Lustig, R.H. The role of fructose in the pathogenesis of NAFLD and the metabolic syndrome. Nat. Rev. Gastroenterol. Hepatol. 2010, 7, 251–264. [Google Scholar] [CrossRef]
- Asgharpour, A.; Cazanave, S.C.; Pacana, T.; Seneshaw, M.; Vincent, R.; Banini, B.A.; Kumar, D.P.; Daita, K.; Min, H.K.; Mirshahi, F.; et al. A diet-induced animal model of non-alcoholic fatty liver disease and hepatocellular cancer. J. Hepatol. 2016, 65, 579–588. [Google Scholar] [CrossRef]
- Sundborn, G.; Thornley, S.; Merriman, T.R.; Lang, B.; King, C.; Lanaspa, M.A.; Johnson, R.J. Are Liquid Sugars Different from Solid Sugar in Their Ability to Cause Metabolic Syndrome? Obesity 2019, 27, 879–887. [Google Scholar] [CrossRef]
- Togo, J.; Hu, S.; Li, M.; Niu, C.; Speakman, J.R. Impact of dietary sucrose on adiposity and glucose homeostasis in C57BL/6J mice depends on mode of ingestion: liquid or solid. Mol. Metab. 2019, 27, 22–32. [Google Scholar] [CrossRef]
- Gordish, K.L.; Kassem, K.M.; Ortiz, P.A.; Beierwaltes, W.H. Moderate (20%) fructose-enriched diet stimulates salt-sensitive hypertension with increased salt retention and decreased renal nitric oxide. Physiol. Rep. 2017, 5. [Google Scholar] [CrossRef]
- Chavez, E.; Briones, R.; Michel, B.; Bravo, C.; Jay, D. Evidence for the involvement of dithiol groups in mitochondrial calcium transport: studies with cadmium. Arch. Biochem. Biophys. 1985, 242, 493–497. [Google Scholar] [CrossRef]
- Correa, F.; Garcia, N.; Robles, C.; Martinez-Abundis, E.; Zazueta, C. Relationship between oxidative stress and mitochondrial function in the post-conditioned heart. J. Bioenerg. Biomembr. 2008, 40, 599–606. [Google Scholar] [CrossRef] [PubMed]
- Hausladen, A.; Fridovich, I. Superoxide and peroxynitrite inactivate aconitases, but nitric oxide does not. J. Biol. Chem. 1994, 269, 29405–29408. [Google Scholar] [PubMed]
- Rippe, J.M.; Angelopoulos, T.J. Added sugars and risk factors for obesity, diabetes and heart disease. Int. J. Obes. (London) 2016, 40, S22–S27. [Google Scholar] [CrossRef] [PubMed]
- Abdelmalek, M.F.; Lazo, M.; Horska, A.; Bonekamp, S.; Lipkin, E.W.; Balasubramanyam, A.; Bantle, J.P.; Johnson, R.J.; Diehl, A.M.; Clark, J.M. Higher dietary fructose is associated with impaired hepatic adenosine triphosphate homeostasis in obese individuals with type 2 diabetes. Hepatology 2012, 56, 952–960. [Google Scholar] [CrossRef]
- Burant, C.F.; Saxena, M. Rapid reversible substrate regulation of fructose transporter expression in rat small intestine and kidney. Am. J. Physiol. 1994, 267, G71–G79. [Google Scholar] [CrossRef]
- Miyamoto, K.; Hase, K.; Takagi, T.; Fujii, T.; Taketani, Y.; Minami, H.; Oka, T.; Nakabou, Y. Differential responses of intestinal glucose transporter mRNA transcripts to levels of dietary sugars. Biochem. J. 1993, 295, 211–215. [Google Scholar] [CrossRef]
- Sullivan, J.S.; Le, M.T.; Pan, Z.; Rivard, C.; Love-Osborne, K.; Robbins, K.; Johnson, R.J.; Sokol, R.J.; Sundaram, S.S. Oral fructose absorption in obese children with non-alcoholic fatty liver disease. Pediatr. Obes. 2015, 10, 188–195. [Google Scholar] [CrossRef]
- Madero, M.; Rodriguez Castellanos, F.E.; Jalal, D.; Villalobos-Martin, M.; Salazar, J.; Vazquez-Rangel, A.; Johnson, R.J.; Sanchez-Lozada, L.G. A pilot study on the impact of a low fructose diet and allopurinol on clinic blood pressure among overweight and prehypertensive subjects: a randomized placebo controlled trial. J. Am. Soc. Hypertens. 2015, 9, 837–844. [Google Scholar] [CrossRef]
- Jensen, T.; Abdelmalek, M.F.; Sullivan, S.; Nadeau, K.J.; Green, M.; Roncal, C.; Nakagawa, T.; Kuwabara, M.; Sato, Y.; Kang, D.H.; et al. Fructose and sugar: A major mediator of non-alcoholic fatty liver disease. J. Hepatol. 2018, 68, 1063–1075. [Google Scholar] [CrossRef]
- Xu, C.; Wan, X.; Xu, L.; Weng, H.; Yan, M.; Miao, M.; Sun, Y.; Xu, G.; Dooley, S.; Li, Y.; et al. Xanthine oxidase in non-alcoholic fatty liver disease and hyperuricemia: One stone hits two birds. J. Hepatol. 2015, 62, 1412–1419. [Google Scholar] [CrossRef] [PubMed]
- Choi, Y.-J.; Shin, H.-S.; Choi, H.; Park, J.-W.; Jo, I.; Oh, E.-S.; Lee, K.-Y.; Lee, B.-H.; Johnson, R.J.; Kang, D.-H. Uric acid induces fat accumulation via generation of endoplasmic reticulum stress and SREBP-1c activation in hepatocytes. Lab. Investig. 2014, 94, 1114–1125. [Google Scholar] [CrossRef] [PubMed]
- Choi, Y.; Abdelmegeed, M.A.; Song, B.J. Diet high in fructose promotes liver steatosis and hepatocyte apoptosis in C57BL/6J female mice: Role of disturbed lipid homeostasis and increased oxidative stress. Food Chem. Toxicol. 2017, 103, 111–121. [Google Scholar] [CrossRef] [PubMed]
- Sekiya, M.; Hiraishi, A.; Touyama, M.; Sakamoto, K. Oxidative stress induced lipid accumulation via SREBP1c activation in HepG2 cells. Biochem. Biophys. Res. Commun. 2008, 375, 602–607. [Google Scholar] [CrossRef]
- Matsusue, K. A physiological role for fat specific protein 27/cell death-inducing DFF45-like effector C in adipose and liver. Biol. Pharm. Bull 2010, 33, 346–350. [Google Scholar] [CrossRef]
- Matsusue, K.; Kusakabe, T.; Noguchi, T.; Takiguchi, S.; Suzuki, T.; Yamano, S.; Gonzalez, F.J. Hepatic steatosis in leptin-deficient mice is promoted by the PPARgamma target gene Fsp27. Cell Metab. 2008, 7, 302–311. [Google Scholar] [CrossRef]
- Liu, K.; Zhou, S.; Kim, J.Y.; Tillison, K.; Majors, D.; Rearick, D.; Lee, J.H.; Fernandez-Boyanapalli, R.F.; Barricklow, K.; Houston, M.S.; et al. Functional analysis of FSP27 protein regions for lipid droplet localization, caspase-dependent apoptosis, and dimerization with CIDEA. Am. J. Physiol. Endocrinol. Metab. 2009, 297, E1395–E1413. [Google Scholar] [CrossRef]
- Sheldon, R.D.; Padilla, J.; Jenkins, N.T.; Laughlin, M.H.; Rector, R.S. Chronic NOS inhibition accelerates NAFLD progression in an obese rat model. Am. J. Physiol. Gastrointest. Liver Physiol. 2015, 308, G540–G549. [Google Scholar] [CrossRef]
- You, M.; Matsumoto, M.; Pacold, C.M.; Cho, W.K.; Crabb, D.W. The role of AMP-activated protein kinase in the action of ethanol in the liver. Gastroenterology 2004, 127, 1798–1808. [Google Scholar] [CrossRef]
- Xu, Y.; Gu, Y.; Liu, G.; Zhang, F.; Li, J.; Liu, F.; Zhang, Z.; Ye, J.; Li, Q. Cidec promotes the differentiation of human adipocytes by degradation of AMPKalpha through ubiquitin-proteasome pathway. BBA General Subj. 2015, 1850, 2552–2562. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Control | F 20% | F 20% + AP | |
|---|---|---|---|
| Mean food intake, g/d | 18 ± 1 | 14 ± 2 * | 14 ± 1 * |
| Mean fluid intake, mL/d | 35 ± 3 | 58 ± 5 * | 53 ± 10 * |
| Delta Body weight, g | −6 ± 7 | 8 ± 8 | −17 ± 19 ° |
| Fasting plasma uric acid (mg/dL) | 1 ± 0.28 | 0.73 ± 0.21 | 0.89 ± 0.28 |
| Fasting plasma TG (mg/dL) | 80 ± 18 | 83 ± 25 | 74 ± 34 |
| Fasting intrahepatic uric acid (µg UA/mg prot) | 5.3 ± 1 | 4.8 ± 1.1 | 2.5 ±0.2 *° |
| Fasting intrahepatic TG (mg TG/mg prot) | 0.06 ± 0.01 | 0.04 ± 0.009 | 0.04 ± 0.006 |
| Uprot, mg/16 h | 14 ± 3 | 13 ± 2 | 16 ± 6 |
| CrCl, mL/min | 1.26 ± 0.2 | 1.41 ± 0.4 | 1.59 ± 0.4 |
| Control | F 20% | F 20% + AP | |
|---|---|---|---|
| Malate/Glutamate | |||
| State 3, ng atoms O/min/mg prot | 38 ± 11 | 45 ± 5 | 45 ± 9 |
| State 4, ng atoms O/min/mg prot | 8 ± 0.6 | 13 ± 1.3 * | 7 ± 1.4 ° |
| Respiratory control rate, S3/S4 | 5.4 ± 1.6 | 3.4 ± 0.4 * | 6.3 ± 0.2 ° |
| ADP/O2, nMol ADP/atoms O/min | 2.8 ± 0.4 | 2.6 ± 0.3 | 3.6 ± 0.1 *° |
| CCCP, ng AtO/min/mg prot | 56 ± 12 | 65 ± 11 | 38 ± 9 *° |
| Succinate/Rotenone | |||
| State 3, ng atoms O/min/mg prot | 62 ± 15 | 66 ± 6 | 64 ± 9 |
| State 4, ng atoms O/min/mg prot | 12 ± 3 | 16 ± 1 | 16 ± 5 |
| Respiratory control rate, S3/S4 | 5.1 ± 1 | 4.3 ± 0.3 | 4.3 ± 0.9 |
| ADP/O2, nMol ADP/atoms O/min | 1.6 ± 0.2 | 1.4 ± 0.02 | 1.7 ± 0.2 ° |
| CCCP, ng AtO/min/mg prot | 126 ± 28 | 132 ± 18 | 124 ± 21 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
García-Arroyo, F.E.; Monroy-Sánchez, F.; Muñoz-Jiménez, I.; Gonzaga, G.; Andrés-Hernando, A.; Zazueta, C.; Juárez-Rojas, J.G.; Lanaspa, M.A.; Johnson, R.J.; Sánchez-Lozada, L.G. Allopurinol Prevents the Lipogenic Response Induced by an Acute Oral Fructose Challenge in Short-Term Fructose Fed Rats. Biomolecules 2019, 9, 601. https://doi.org/10.3390/biom9100601
García-Arroyo FE, Monroy-Sánchez F, Muñoz-Jiménez I, Gonzaga G, Andrés-Hernando A, Zazueta C, Juárez-Rojas JG, Lanaspa MA, Johnson RJ, Sánchez-Lozada LG. Allopurinol Prevents the Lipogenic Response Induced by an Acute Oral Fructose Challenge in Short-Term Fructose Fed Rats. Biomolecules. 2019; 9(10):601. https://doi.org/10.3390/biom9100601
Chicago/Turabian StyleGarcía-Arroyo, Fernando E., Fabiola Monroy-Sánchez, Itzel Muñoz-Jiménez, Guillermo Gonzaga, Ana Andrés-Hernando, Cecilia Zazueta, J. Gabriel Juárez-Rojas, Miguel A. Lanaspa, Richard J. Johnson, and L. Gabriela Sánchez-Lozada. 2019. "Allopurinol Prevents the Lipogenic Response Induced by an Acute Oral Fructose Challenge in Short-Term Fructose Fed Rats" Biomolecules 9, no. 10: 601. https://doi.org/10.3390/biom9100601
APA StyleGarcía-Arroyo, F. E., Monroy-Sánchez, F., Muñoz-Jiménez, I., Gonzaga, G., Andrés-Hernando, A., Zazueta, C., Juárez-Rojas, J. G., Lanaspa, M. A., Johnson, R. J., & Sánchez-Lozada, L. G. (2019). Allopurinol Prevents the Lipogenic Response Induced by an Acute Oral Fructose Challenge in Short-Term Fructose Fed Rats. Biomolecules, 9(10), 601. https://doi.org/10.3390/biom9100601