NMDA Receptor Opening and Closing—Transitions of a Molecular Machine Revealed by Molecular Dynamics

 , ,
, ,  and
and {kind=link}
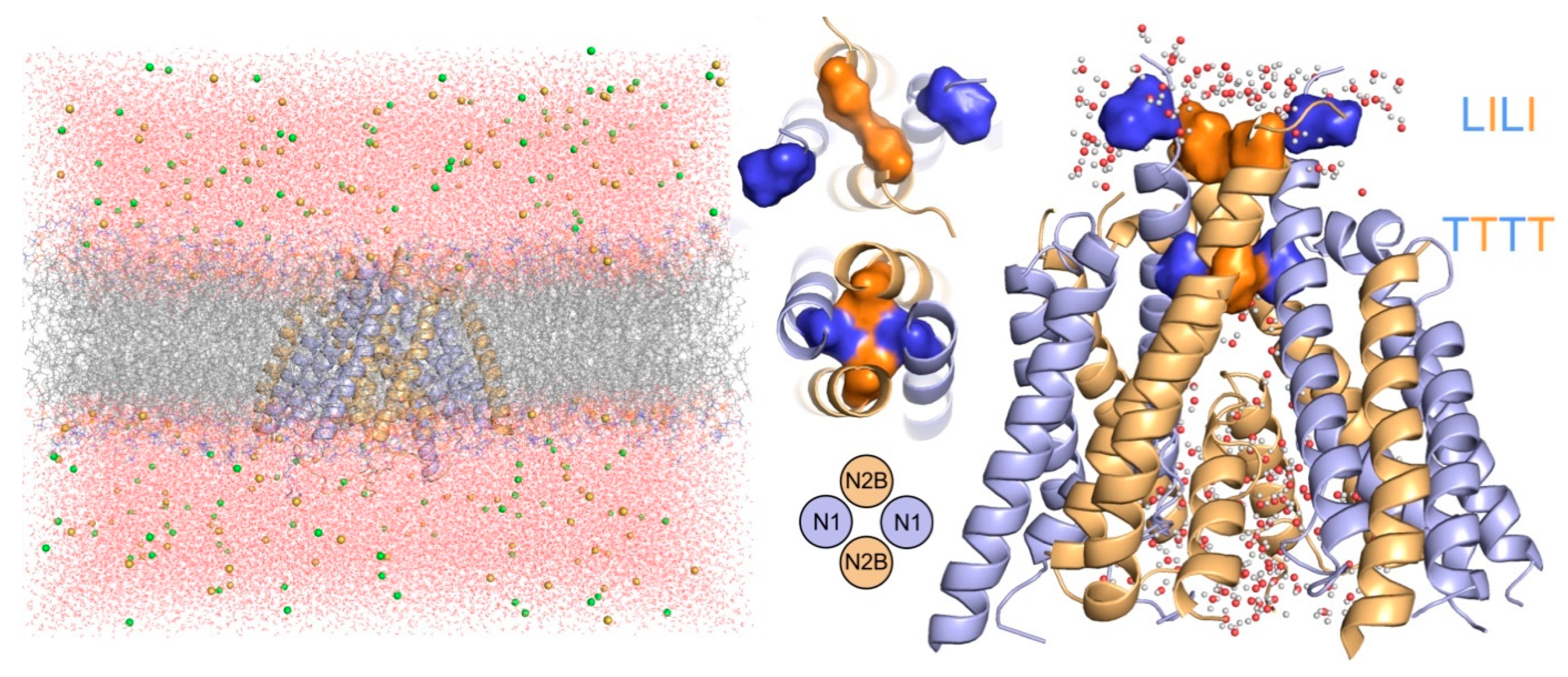{kind=link}
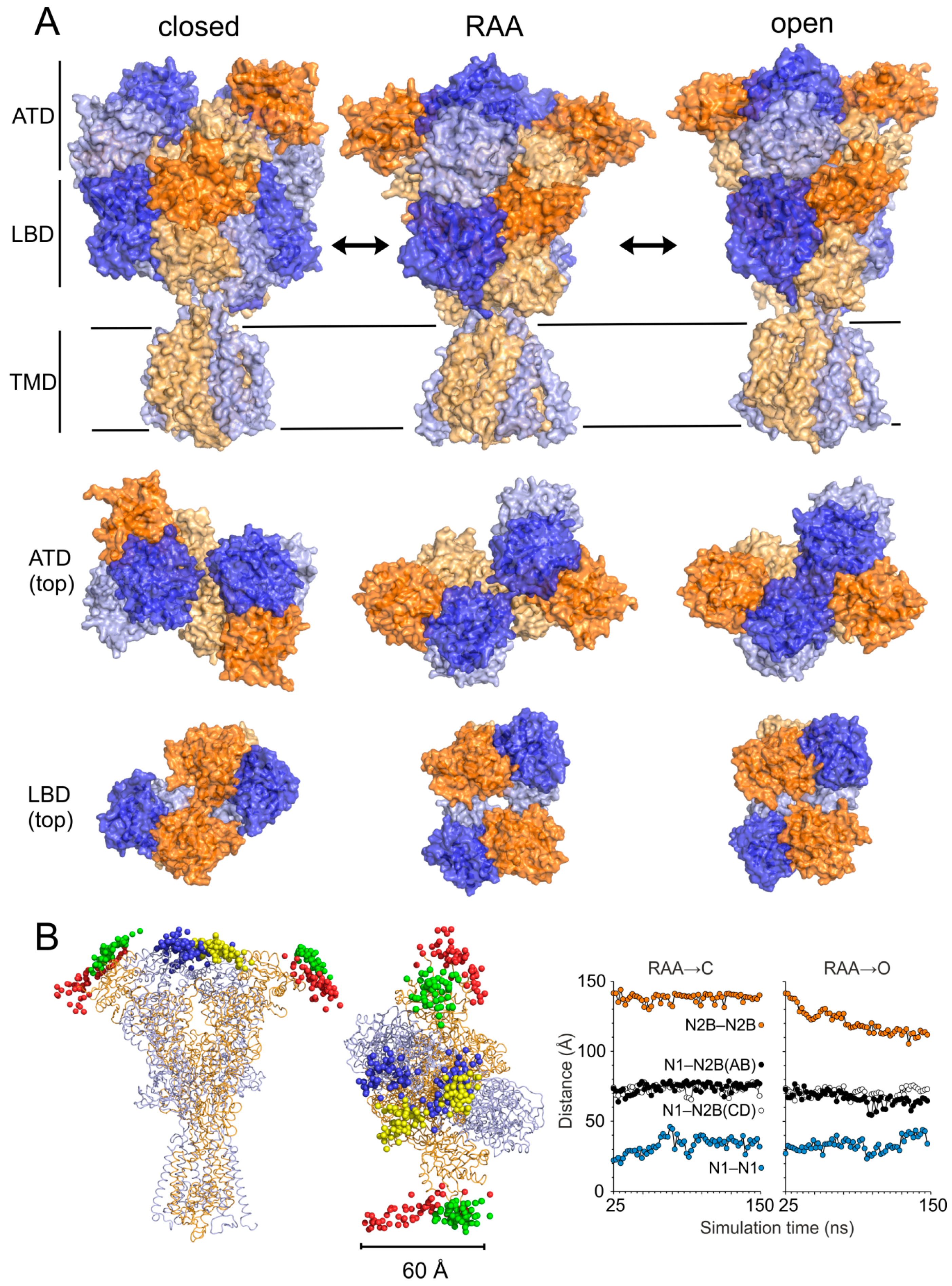{kind=link}
{kind=link}
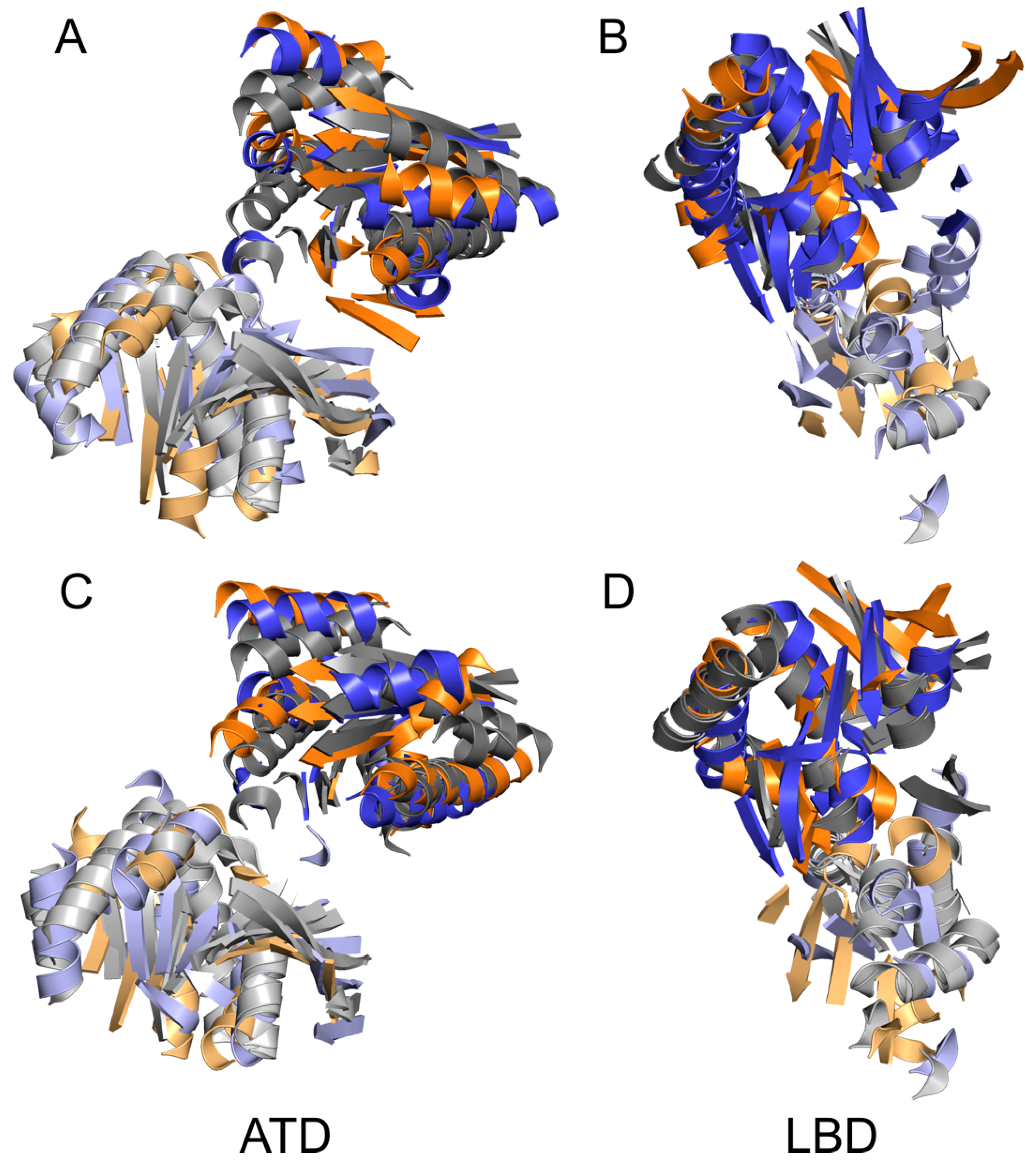{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
3. Results
3.1. Relaxation of the Crystal-Based Homology Model (NMDAR in the Agonist Activated Receptor State)
3.2. Modeling of the Open State
3.3. Modeling of the Closed State
4. Discussion
4.1. Mechanism of NMDAR Ion Channel Opening and Closing
4.2. Common and Different Structural Features Derived from the Three Independent Simulations
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Watkins, J.C.; Evans, R.H. Excitatory amino acid transmitters. Annu. Rev. Pharmacol. Toxicol. 1981, 21, 165–204. [Google Scholar] [CrossRef] [PubMed]
- Moriyoshi, K.; Masu, M.; Ishii, T.; Shigemoto, R.; Mizuno, N.; Nakanishi, S. Molecular cloning and characterization of the rat NMDA receptor. Nature 1991, 354, 31–37. [Google Scholar] [CrossRef] [PubMed]
- Kutsuwada, T.; Kashiwabuchi, N.; Mori, H.; Sakimura, K.; Kushiya, E.; Araki, K.; Meguro, H.; Masaki, H.; Kumanishi, T.; Arakawa, M.; et al. Molecular diversity of the NMDA receptor channel. Nature 1992, 358, 36–41. [Google Scholar] [CrossRef] [PubMed]
- Meguro, H.; Mori, H.; Araki, K.; Kushiya, E.; Kutsuwada, T.; Yamazaki, M.; Kumanishi, T.; Arakawa, M.; Sakimura, K.; Mishina, M. Functional characterization of a heteromeric NMDA receptor channel expressed from cloned cdnas. Nature 1992, 357, 70–74. [Google Scholar] [CrossRef]
- Monyer, H.; Sprengel, R.; Schoepfer, R.; Herb, A.; Higuchi, M.; Lomeli, H.; Burnashev, N.; Sakmann, B.; Seeburg, P.H. Heteromeric NMDA receptors: Molecular and functional distinction of subtypes. Science 1992, 256, 1217–1221. [Google Scholar] [CrossRef] [PubMed]
- Sucher, N.J.; Akbarian, S.; Chi, C.L.; Leclerc, C.L.; Awobuluyi, M.; Deitcher, D.L.; Wu, M.K.; Yuan, J.P.; Jones, E.G.; Lipton, S.A. Developmental and regional expression pattern of a novel NMDA receptor-like subunit (NMDAR-L) in the rodent brain. J. Neurosci. 1995, 15, 6509–6520. [Google Scholar] [CrossRef] [PubMed]
- Lynch, M.A. Long-term potentiation and memory. Physiol. Rev. 2004, 84, 87–136. [Google Scholar] [CrossRef]
- Malenka, R.C.; Bear, M.F. Ltp and ltd: An embarrassment of riches. Neuron 2004, 44, 5–21. [Google Scholar] [CrossRef]
- Bouvier, G.; Larsen, R.S.; Rodriguez-Moreno, A.; Paulsen, O.; Sjostrom, P.J. Towards resolving the presynaptic NMDA receptor debate. Curr. Opin. Neurobiol. 2018, 51, 1–7. [Google Scholar] [CrossRef]
- Choi, D.W. Ionic dependence of glutamate neurotoxicity. J. Neurosci. 1987, 7, 369–379. [Google Scholar] [CrossRef]
- Dingledine, R.; Borges, K.; Bowie, D.; Traynelis, S.F. The glutamate receptor ion channels. Pharmacol. Rev. 1999, 51, 7–61. [Google Scholar]
- Tarabeux, J.; Kebir, O.; Gauthier, J.; Hamdan, F.F.; Xiong, L.; Piton, A.; Spiegelman, D.; Henrion, E.; Millet, B.; Team, S.D.; et al. Rare mutations in N-methyl-d-aspartate glutamate receptors in autism spectrum disorders and schizophrenia. Transl. Psychiatry 2011, 1, e55. [Google Scholar] [CrossRef] [PubMed]
- Soto, D.; Altafaj, X.; Sindreu, C.; Bayes, A. Glutamate receptor mutations in psychiatric and neurodevelopmental disorders. Commun. Integr. Biol. 2014, 7, e27887. [Google Scholar] [CrossRef] [PubMed]
- MacDermott, A.B.; Mayer, M.L.; Westbrook, G.L.; Smith, S.J.; Barker, J.L. NMDA-receptor activation increases cytoplasmic calcium concentration in cultured spinal cord neurones. Nature 1986, 321, 519–522. [Google Scholar] [CrossRef] [PubMed]
- Lester, R.A.; Jahr, C.E. NMDA channel behavior depends on agonist affinity. J. Neurosci. 1992, 12, 635–643. [Google Scholar] [CrossRef] [PubMed]
- Iacobucci, G.J.; Popescu, G.K. Kinetic models for activation and modulation of NMDA receptor subtypes. Curr. Opin. Physiol. 2018, 2, 114–122. [Google Scholar] [CrossRef]
- Johnson, J.W.; Ascher, P. Glycine potentiates the NMDA response in cultured mouse brain neurons. Nature 1987, 325, 529–531. [Google Scholar] [CrossRef]
- Benveniste, M.; Clements, J.; Vyklicky, L., Jr.; Mayer, M.L. A kinetic analysis of the modulation of N-methyl-d-aspartic acid receptors by glycine in mouse cultured hippocampal neurones. J. Physiol. 1990, 428, 333–357. [Google Scholar] [CrossRef]
- Rosenmund, C.; Westbrook, G.L. Calcium-induced actin depolymerization reduces NMDA channel activity. Neuron 1993, 10, 805–814. [Google Scholar] [CrossRef]
- Vyklicky, L., Jr. Calcium-mediated modulation of N-methyl-d-aspartate (NMDA) responses in cultured rat hippocampal neurones. J. Physiol. 1993, 470, 575–600. [Google Scholar] [CrossRef]
- Popescu, G.; Robert, A.; Howe, J.R.; Auerbach, A. Reaction mechanism determines NMDA receptor response to repetitive stimulation. Nature 2004, 430, 790–793. [Google Scholar] [CrossRef] [PubMed]
- Banke, T.G.; Traynelis, S.F. Activation of NR1/NR2B NMDA receptors. Nat. Neurosci. 2003, 6, 144–152. [Google Scholar] [CrossRef] [PubMed]
- Popescu, G.; Auerbach, A. Modal gating of NMDA receptors and the shape of their synaptic response. Nat. Neurosci. 2003, 6, 476–483. [Google Scholar] [CrossRef]
- Erreger, K.; Dravid, S.M.; Banke, T.G.; Wyllie, D.J.; Traynelis, S.F. Subunit-specific gating controls rat NR1/NR2A and NR1/NR2B NMDA channel kinetics and synaptic signalling profiles. J. Physiol. 2005, 563, 345–358. [Google Scholar] [CrossRef] [PubMed]
- Amico-Ruvio, S.A.; Popescu, G.K. Stationary gating of GluN1/GluN2B receptors in intact membrane patches. Biophys. J. 2010, 98, 1160–1169. [Google Scholar] [CrossRef] [PubMed]
- Hansen, K.B.; Yi, F.; Perszyk, R.E.; Furukawa, H.; Wollmuth, L.P.; Gibb, A.J.; Traynelis, S.F. Structure, function, and allosteric modulation of NMDA receptors. J. Gen. Physiol. 2018, 150, 1081–1105. [Google Scholar] [CrossRef] [PubMed]
- Karakas, E.; Furukawa, H. Crystal structure of a heterotetrameric NMDA receptor ion channel. Science 2014, 344, 992–997. [Google Scholar] [CrossRef]
- Lee, C.H.; Lu, W.; Michel, J.C.; Goehring, A.; Du, J.; Song, X.; Gouaux, E. NMDA receptor structures reveal subunit arrangement and pore architecture. Nature 2014, 511, 191–197. [Google Scholar] [CrossRef] [PubMed]
- Ladislav, M.; Cerny, J.; Krusek, J.; Horak, M.; Balik, A.; Vyklicky, L. The lili motif of M3-S2 linkers is a component of the NMDA receptor channel gate. Front. Mol. Neurosci. 2018, 11, 113. [Google Scholar] [CrossRef] [PubMed]
- Yao, Y.; Belcher, J.; Berger, A.J.; Mayer, M.L.; Lau, A.Y. Conformational analysis of NMDA receptor GluN1, GluN2, and GluN3 ligand-binding domains reveals subtype-specific characteristics. Structure 2013, 21, 1788–1799. [Google Scholar] [CrossRef]
- Dai, J.; Zhou, H.X. Reduced curvature of ligand-binding domain free-energy surface underlies partial agonism at NMDA receptors. Structure 2015, 23, 228–236. [Google Scholar] [CrossRef] [PubMed]
- Dai, J.; Zhou, H.X. Semiclosed conformations of the ligand-binding domains of NMDA receptors during stationary gating. Biophys. J. 2016, 111, 1418–1428. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Dai, J.; Zhou, H.X. An NMDA receptor gating mechanism developed from md simulations reveals molecular details underlying subunit-specific contributions. Biophys. J. 2013, 104, 2170–2181. [Google Scholar] [CrossRef] [PubMed]
- Vyklicky, V.; Krausova, B.; Cerny, J.; Balik, A.; Zapotocky, M.; Novotny, M.; Lichnerova, K.; Smejkalova, T.; Kaniakova, M.; Korinek, M.; et al. Block of NMDA receptor channels by endogenous neurosteroids: Implications for the agonist induced conformational states of the channel vestibule. Sci. Rep. 2015, 5, 10935. [Google Scholar] [CrossRef] [PubMed]
- Zheng, W.; Wen, H.; Iacobucci, G.J.; Popescu, G.K. Probing the structural dynamics of the NMDA receptor activation by coarse-grained modeling. Biophys. J. 2017, 112, 2589–2601. [Google Scholar] [CrossRef]
- Pang, X.; Zhou, H.X. Structural modeling for the open state of an NMDA receptor. J. Struct. Biol. 2017, 200, 369–375. [Google Scholar] [CrossRef]
- Palmai, Z.; Houenoussi, K.; Cohen-Kaminsky, S.; Tchertanov, L. How does binding of agonist ligands control intrinsic molecular dynamics in human NMDA receptors? PLoS ONE 2018, 13, e0201234. [Google Scholar] [CrossRef]
- Zhang, J.B.; Chang, S.; Xu, P.; Miao, M.; Wu, H.; Zhang, Y.; Zhang, T.; Wang, H.; Zhang, J.; Xie, C.; et al. Structural basis of the proton sensitivity of human GluN1-GluN2A NMDA receptors. Cell Rep. 2018, 25, 3582–3590. [Google Scholar] [CrossRef]
- Omotuyi, O.I.; Ueda, H. Molecular dynamics study-based mechanism of nefiracetam-induced NMDA receptor potentiation. Comput. Biol. Chem. 2015, 55, 14–22. [Google Scholar] [CrossRef]
- Song, X.; Jensen, M.O.; Jogini, V.; Stein, R.A.; Lee, C.H.; McHaourab, H.S.; Shaw, D.E.; Gouaux, E. Mechanism of NMDA receptor channel block by MK-801 and memantine. Nature 2018, 556, 515–519. [Google Scholar] [CrossRef]
- Sinitskiy, A.V.; Pande, V.S. Simulated dynamics of glycans on ligand-binding domain of NMDA receptors reveals strong dynamic coupling between glycans and protein core. J. Chem. Theory Comput. 2017, 13, 5496–5505. [Google Scholar] [CrossRef] [PubMed]
- Sinitskiy, A.V.; Stanley, N.H.; Hackos, D.H.; Hanson, J.E.; Sellers, B.D.; Pande, V.S. Computationally discovered potentiating role of glycans on NMDA receptors. Sci. Rep. 2017, 7, 44578. [Google Scholar] [CrossRef]
- Hu, C.; Chen, W.; Myers, S.J.; Yuan, H.; Traynelis, S.F. Human GRIN2B variants in neurodevelopmental disorders. J. Pharmacol. Sci. 2016, 132, 115–121. [Google Scholar] [CrossRef] [PubMed]
- Swanger, S.A.; Chen, W.; Wells, G.; Burger, P.B.; Tankovic, A.; Bhattacharya, S.; Strong, K.L.; Hu, C.; Kusumoto, H.; Zhang, J.; et al. Mechanistic insight into NMDA receptor dysregulation by rare variants in the GluN2A and GluN2B agonist binding domains. Am. J. Hum. Genet. 2016, 99, 1261–1280. [Google Scholar] [CrossRef] [PubMed]
- Vyklicky, V.; Krausova, B.; Cerny, J.; Ladislav, M.; Smejkalova, T.; Kysilov, B.; Korinek, M.; Danacikova, S.; Horak, M.; Chodounska, H.; et al. Surface expression, function, and pharmacology of disease-associated mutations in the membrane domain of the human GluN2B subunit. Front. Mol. Neurosci. 2018, 11, 110. [Google Scholar] [CrossRef] [PubMed]
- The UniProt Consortium. Uniprot: The universal protein knowledgebase. Nucleic Acids Res. 2017, 45, D158–D169. [Google Scholar] [CrossRef] [PubMed]
- Webb, B.; Sali, A. Comparative protein structure modeling using modeller. Curr. Protoc. Bioinform. 2016, 54, 5.6.1–5.6.37. [Google Scholar] [CrossRef]
- Lazaridis, T. Effective energy function for proteins in lipid membranes. Proteins 2003, 52, 176–192. [Google Scholar] [CrossRef] [PubMed]
- Jo, S.; Kim, T.; Iyer, V.G.; Im, W. Charmm-gui: A web-based graphical user interface for charmm. J. Comput. Chem. 2008, 29, 1859–1865. [Google Scholar] [CrossRef] [PubMed]
- Brooks, B.R.; Brooks, C.L., 3rd; Mackerell, A.D., Jr.; Nilsson, L.; Petrella, R.J.; Roux, B.; Won, Y.; Archontis, G.; Bartels, C.; Boresch, S.; et al. Charmm: The biomolecular simulation program. J. Comput. Chem. 2009, 30, 1545–1614. [Google Scholar] [CrossRef]
- van der Spoel, D.; Lindahl, E.; Hess, B.; Groenhof, G.; Mark, A.E.; Berendsen, H.J.C. Gromacs: Fast, flexible, and free. J. Comput. Chem. 2005, 26, 1701–1718. [Google Scholar] [CrossRef] [PubMed]
- Schrodinger, L.L.C. The PyMOL Molecular Graphics System, Version 2.2. 2018.
- Humphrey, W.; Dalke, A.; Schulten, K. Vmd: Visual molecular dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef]
- Okonechnikov, K.; Golosova, O.; Fursov, M.; Team, U. Unipro UGENE: A unified bioinformatics toolkit. Bioinformatics 2012, 28, 1166–1167. [Google Scholar] [CrossRef] [PubMed]
- Doerr, S.; Harvey, M.J.; Noe, F.; De Fabritiis, G. HTMD: High-throughput molecular dynamics for molecular discovery. J. Chem. Theory Comput. 2016, 12, 1845–1852. [Google Scholar] [CrossRef] [PubMed]
- Scherer, M.K.; Trendelkamp-Schroer, B.; Paul, F.; Perez-Hernandez, G.; Hoffmann, M.; Plattner, N.; Wehmeyer, C.; Prinz, J.H.; Noe, F. PyEMMA 2: A software package for estimation, validation, and analysis of markov models. J. Chem. Theory Comput. 2015, 11, 5525–5542. [Google Scholar] [CrossRef]
- Maki, B.A.; Aman, T.K.; Amico-Ruvio, S.A.; Kussius, C.L.; Popescu, G.K. C-terminal domains of N-methyl-d-aspartic acid receptor modulate unitary channel conductance and gating. J. Biol. Chem. 2012, 287, 36071–36080. [Google Scholar] [CrossRef]
- Lichnerova, K.; Kaniakova, M.; Park, S.P.; Skrenkova, K.; Wang, Y.X.; Petralia, R.S.; Suh, Y.H.; Horak, M. Two N-glycosylation sites in the GluN1 subunit are essential for releasing N-methyl-d-aspartate (NMDA) receptors from the endoplasmic reticulum. J. Biol. Chem. 2015, 290, 18379–18390. [Google Scholar] [CrossRef]
- Yu, A.; Lau, A.Y. Glutamate and glycine binding to the NMDA receptor. Structure 2018, 26, 1035–1043. [Google Scholar] [CrossRef]
- Esmenjaud, J.B.; Stroebel, D.; Chan, K.; Grand, T.; David, M.; Wollmuth, L.P.; Taly, A.; Paoletti, P. An inter-dimer allosteric switch controls NMDA receptor activity. EMBO J. 2019, 38. [Google Scholar] [CrossRef]
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic local alignment search tool. J. Mol. Biol. 1990, 215, 403–410. [Google Scholar] [CrossRef]







© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Černý, J.; Božíková, P.; Balík, A.; Marques, S.M.; Vyklický, L. NMDA Receptor Opening and Closing—Transitions of a Molecular Machine Revealed by Molecular Dynamics. Biomolecules 2019, 9, 546. https://doi.org/10.3390/biom9100546
Černý J, Božíková P, Balík A, Marques SM, Vyklický L. NMDA Receptor Opening and Closing—Transitions of a Molecular Machine Revealed by Molecular Dynamics. Biomolecules. 2019; 9(10):546. https://doi.org/10.3390/biom9100546
Chicago/Turabian StyleČerný, Jiří, Paulína Božíková, Aleš Balík, Sérgio M. Marques, and Ladislav Vyklický. 2019. "NMDA Receptor Opening and Closing—Transitions of a Molecular Machine Revealed by Molecular Dynamics" Biomolecules 9, no. 10: 546. https://doi.org/10.3390/biom9100546
APA StyleČerný, J., Božíková, P., Balík, A., Marques, S. M., & Vyklický, L. (2019). NMDA Receptor Opening and Closing—Transitions of a Molecular Machine Revealed by Molecular Dynamics. Biomolecules, 9(10), 546. https://doi.org/10.3390/biom9100546