Exploring Solanum tuberosum Epoxide Hydrolase Internal Architecture by Water Molecules Tracking

 , ,
, ,  ,
, 
Abstract
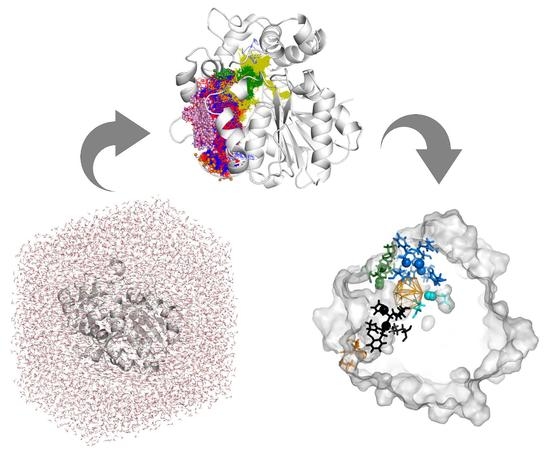
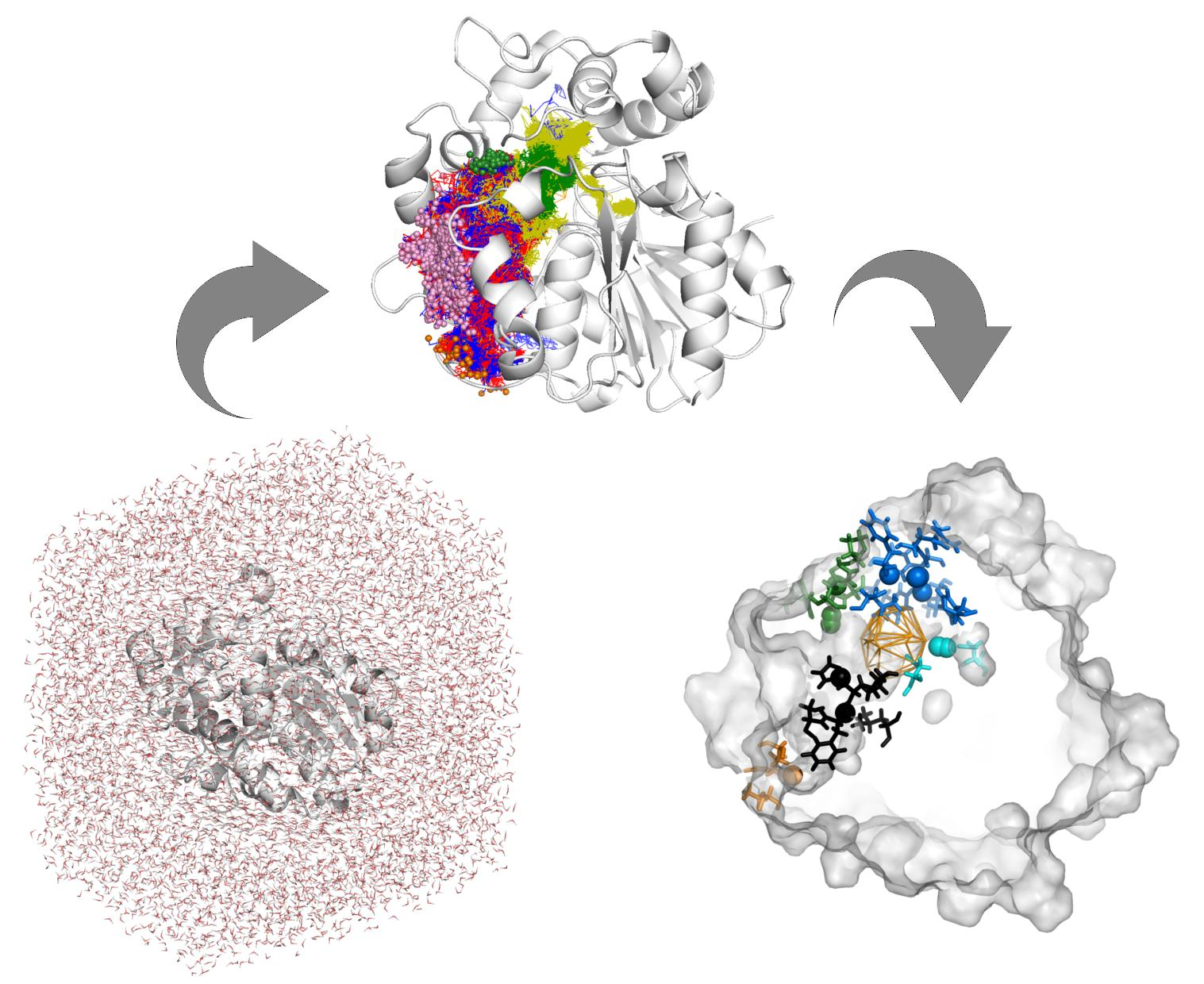
1. Introduction
2. Materials and Methods
2.1. Protein Preparation
2.2. Molecular Dynamics Simulations
2.3. Tunnel Identification
2.4. Water Tracking, Hot-Spot and Pocket Detection
2.5. Evolutionary Analysis
3. Results
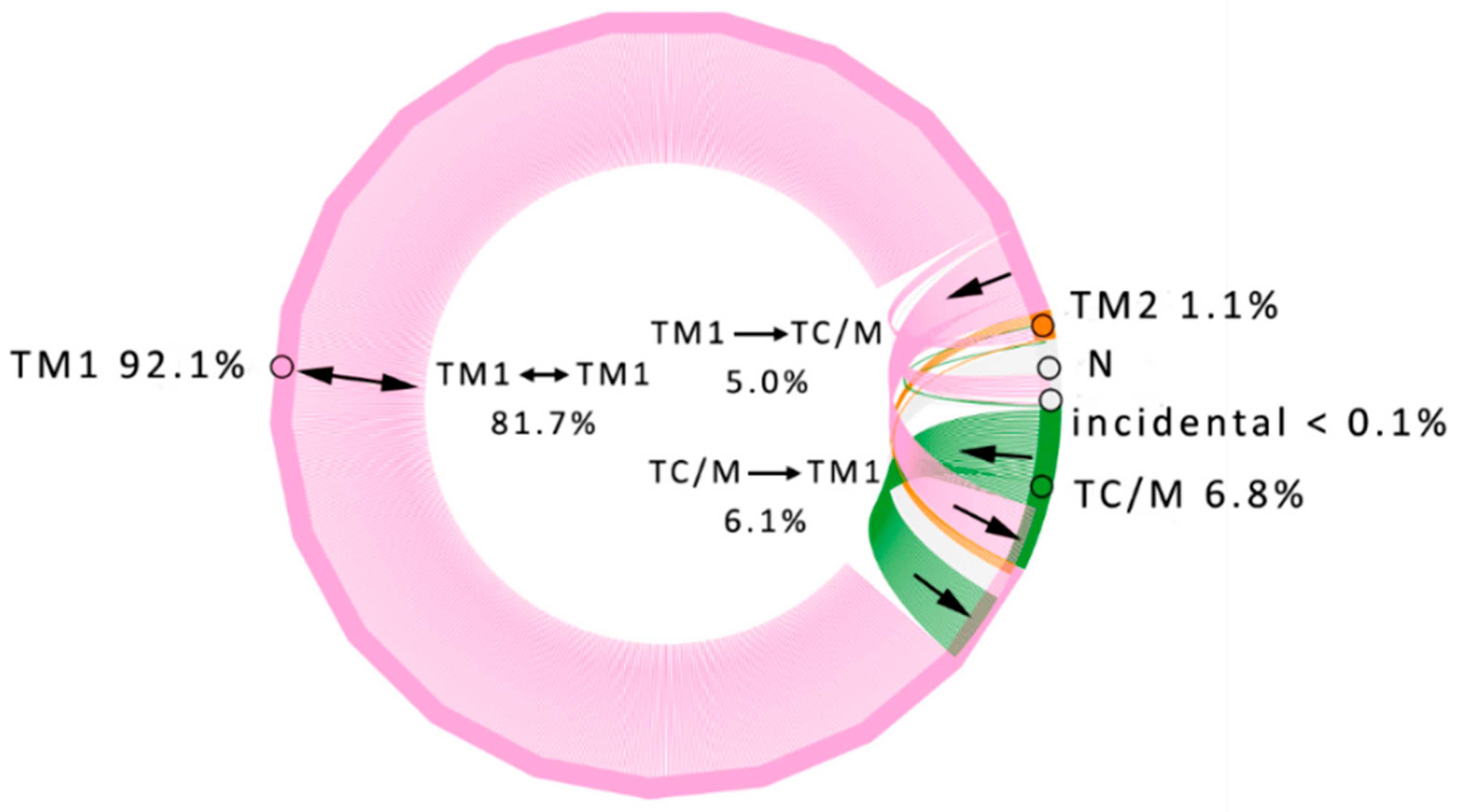
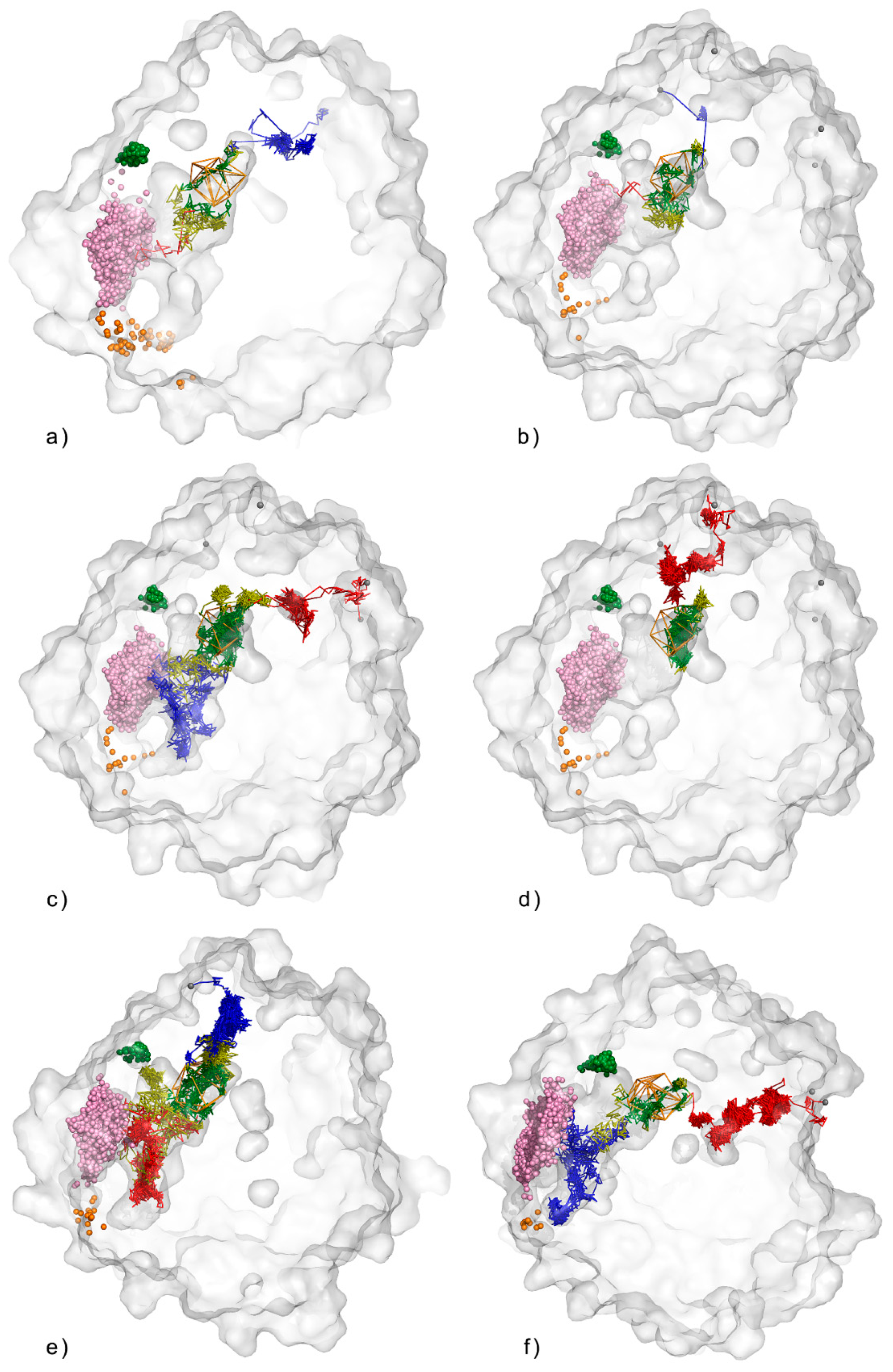
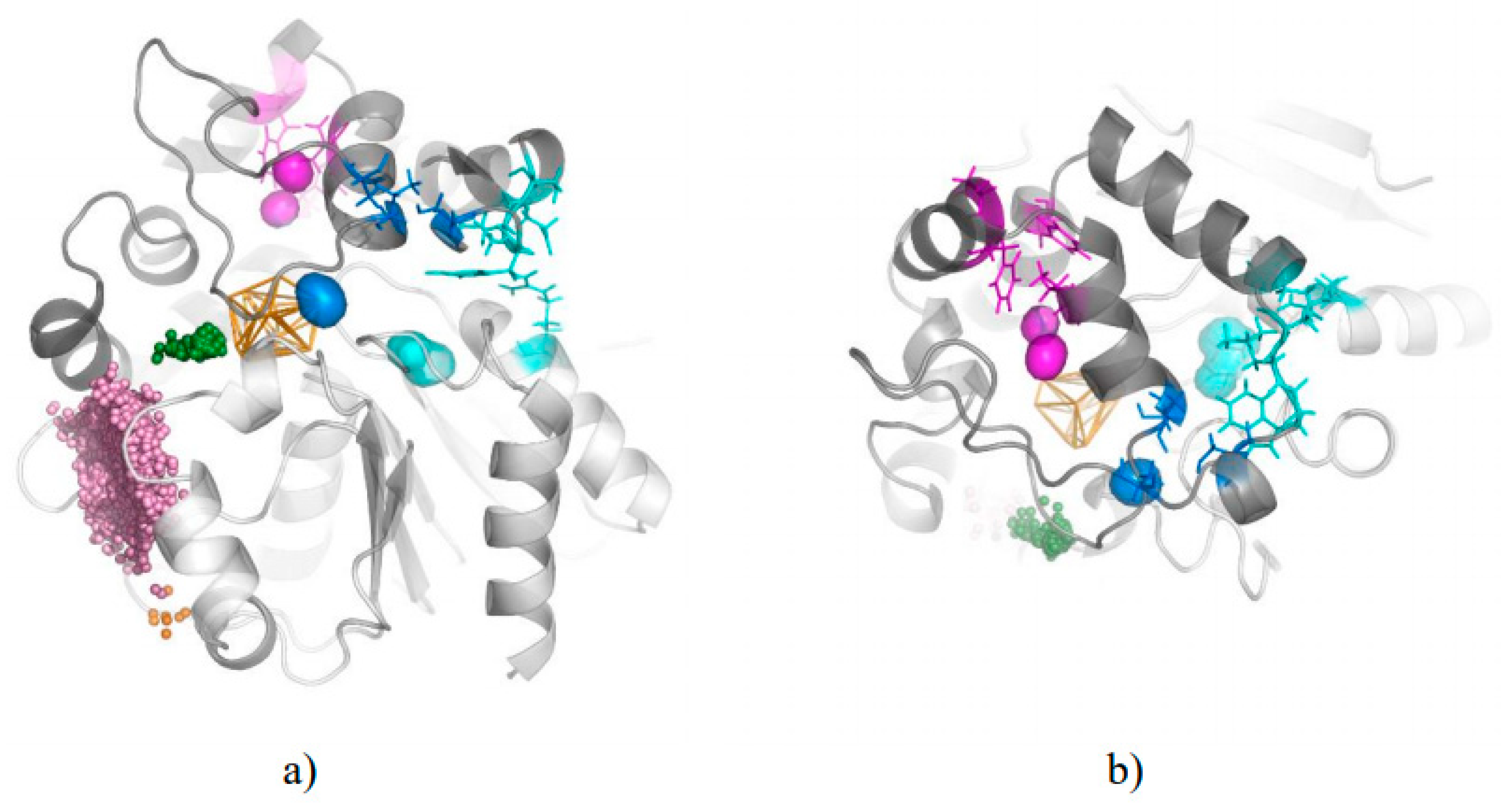
3.1. Water Trajectories—Tunnels and Incidental Pathways
3.2. Cavities and Pockets
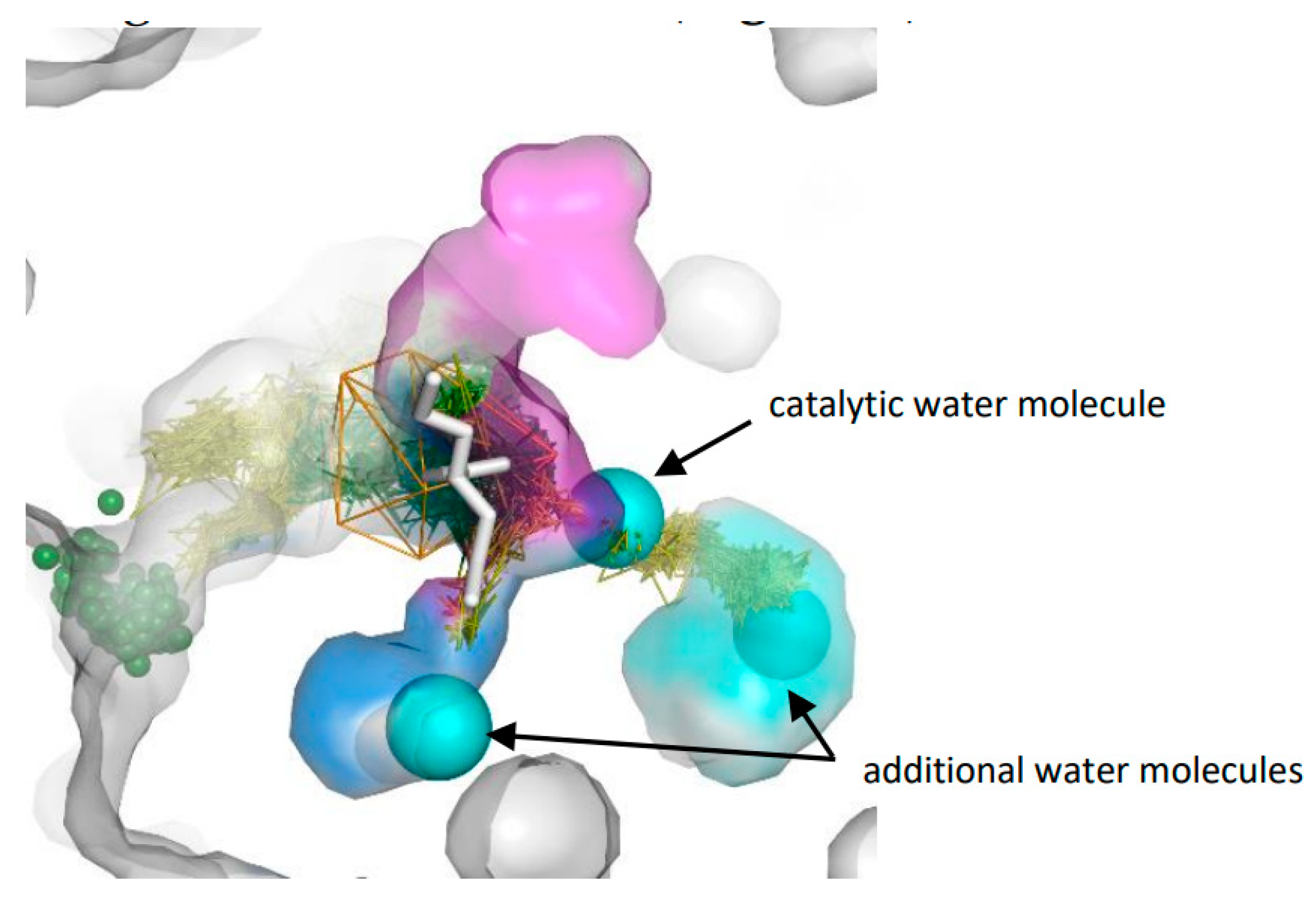
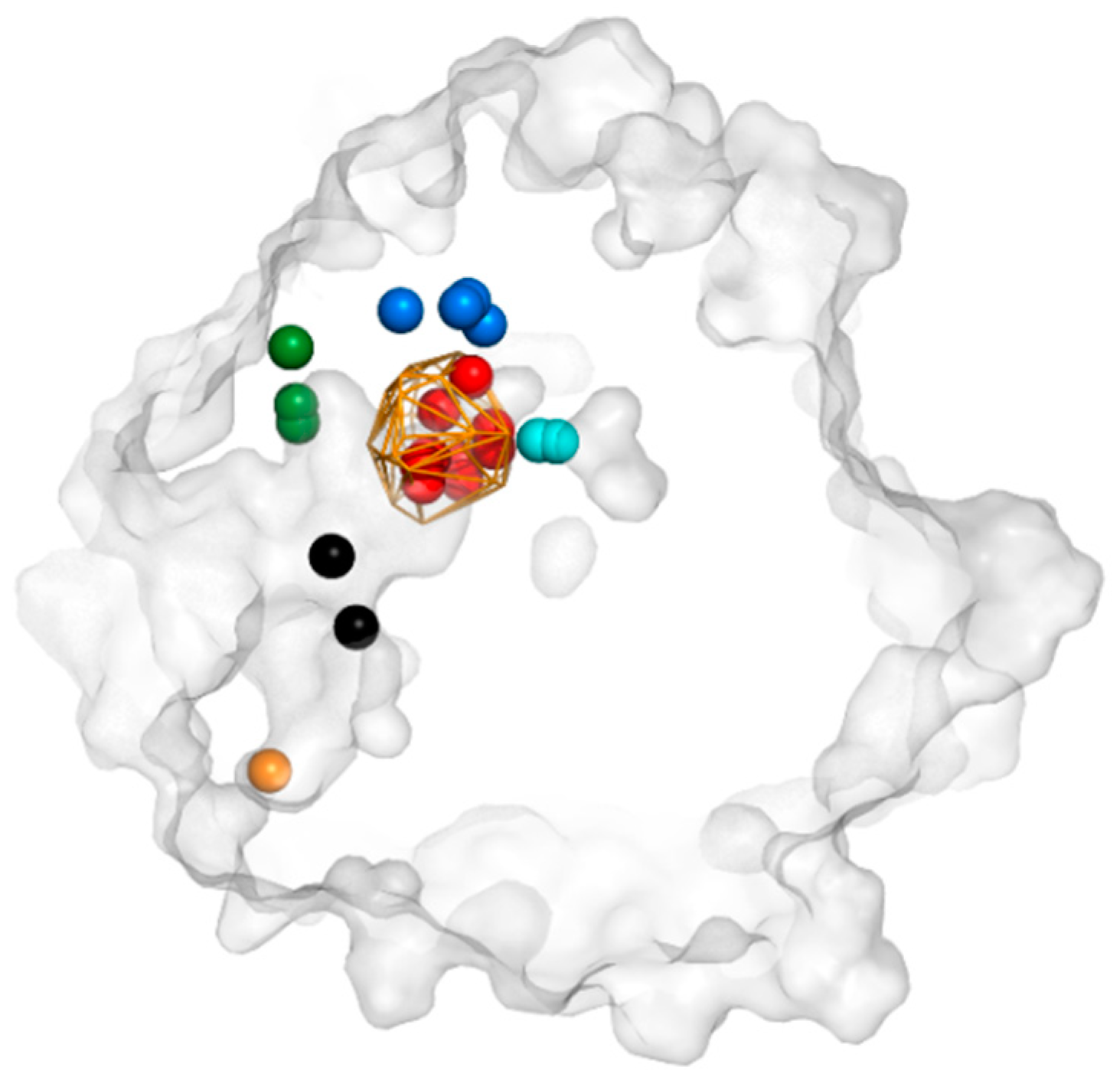
3.3. Trapped Water Molecules—Hot-Spot Identification
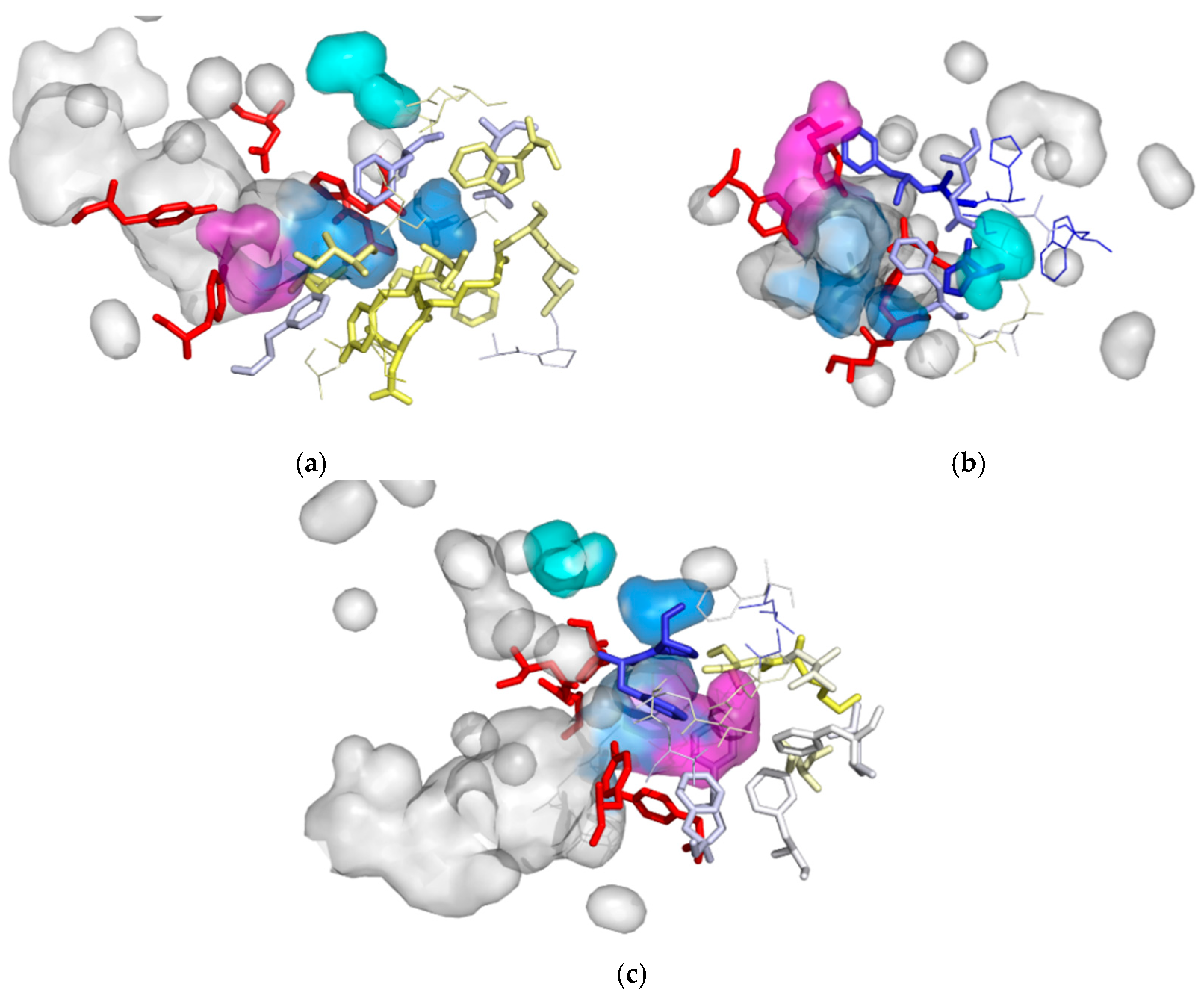
3.4. Evolutionary Analysis
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Biedermannová, L.; Prokop, Z.; Gora, A.; Chovancová, E.; Kovács, M.; Damborský, J.; Wade, R.C. A single mutation in a tunnel to the active site changes the mechanism and kinetics of product release in haloalkane dehalogenase LinB. J. Biol. Chem. 2012, 287, 29062–29074. [Google Scholar] [CrossRef] [PubMed]
- Kokkonen, P.; Bednar, D.; Dockalova, V.; Prokop, Z.; Damborsky, J. Conformational Changes Allow Processing of Bulky Substrates by a Haloalkane Dehalogenase with a Small and Buried Active Site. J. Biol. Chem. 2018. [Google Scholar] [CrossRef] [PubMed]
- Prokop, Z.; Gora, A.; Brezovsky, J.; Chaloupkova, R.; Stepankova, V.; Damborsky, J. Engineering of Protein Tunnels: The Keyhole—Lock—Key Model for Catalysis by Enzymes with Buried Active Sites. Protein Eng. Handb. 2012, 3, 421–464. [Google Scholar]
- Kingsley, L.J.; Lill, M.A. Substrate tunnels in enzymes: Structure-function relationships and computational methodology. Proteins Struct. Funct. Bioinform. 2015, 83, 599–611. [Google Scholar] [CrossRef] [PubMed]
- Kaushik, S.; Marques, S.M.; Khirsariya, P.; Paruch, K.; Libichova, L.; Brezovsky, J.; Prokop, Z.; Chaloupkova, R.; Damborsky, J. Impact of the access tunnel engineering on catalysis is strictly ligand-specific. FEBS J. 2018, 285, 1456–1476. [Google Scholar] [CrossRef] [PubMed]
- Dyson, P.J.; Jessop, P.G. Solvent effects in catalysis: Rational improvements of catalysts: Via manipulation of solvent interactions. Catal. Sci. Technol. 2016, 6, 3302–3316. [Google Scholar] [CrossRef]
- Yang, L.; Dordick, J.S.; Garde, S. Hydration of enzyme in nonaqueous media is consistent with solvent dependence of its activity. Biophys. J. 2004, 87, 812–821. [Google Scholar] [CrossRef] [PubMed]
- Stepankova, V.; Khabiri, M.; Brezovsky, J.; Pavelka, A.; Sykora, J.; Amaro, M.; Minofar, B.; Prokop, Z.; Hof, M.; Ettrich, R.; et al. Expansion of Access Tunnels and Active-Site Cavities Influence Activity of Haloalkane Dehalogenases in Organic Cosolvents. ChemBioChem 2013, 14, 890–897. [Google Scholar] [CrossRef] [PubMed]
- Persson, F.; Halle, B. Transient access to the protein interior: Simulation versus NMR. J. Am. Chem. Soc. 2013, 135, 8735–8748. [Google Scholar] [CrossRef] [PubMed]
- Gora, A.; Brezovsky, J.; Damborsky, J. Gates of enzymes. Chem. Rev. 2013, 113, 5871–5923. [Google Scholar] [CrossRef] [PubMed]
- Marques, S.; Brezovsky, J.; Damborsky, J. Role of Tunnels and Gates in Enzymatic Catalysis. Underst. Enzym. 2016, 421–463. [Google Scholar] [CrossRef]
- Raushel, F.M.; Thoden, J.B.; Holden, H.M. Enzymes with molecular tunnels. Acc. Chem. Res. 2003, 36, 539–548. [Google Scholar] [CrossRef] [PubMed]
- Cojocaru, V.; Winn, P.J.; Wade, R.C. The Ins and Outs of Cytochrome P450s. Biochim. Biophys. Acta 2007, 1770, 390–401. [Google Scholar] [CrossRef] [PubMed]
- Fan, Y.; Lund, L.; Shao, Q.; Gao, Y.Q.; Raushel, F.M. A combined theoretical and experimental study of the ammonia tunnel in carbamoyl phosphate synthetase. J. Am. Chem. Soc. 2009, 131, 10211–10219. [Google Scholar] [CrossRef] [PubMed]
- Magdziarz, T.; Mitusińska, K.; Gołdowska, S.; Płuciennik, A.; Stolarczyk, M.; Lugowska, M.; Góra, A. AQUA-DUCT: A ligands tracking tool. Bioinformatics 2017, 33, 2045–2046. [Google Scholar] [CrossRef] [PubMed]
- Elfström, L.T.; Widersten, M. Catalysis of potato epoxide hydrolase, StEH1. Biochem. J. 2005, 390, 633–640. [Google Scholar] [CrossRef] [PubMed]
- Mowbray, S.L.; Elfström, L.T.; Ahlgren, K.M.; Andersson, C.E.; Widersten, M. X-ray structure of potato epoxide hydrolase sheds light on substrate specificity in plant enzymes. Protein Sci. 2006, 15, 1628–1637. [Google Scholar] [CrossRef] [PubMed]
- Thomaeus, A.; Carlsson, J.; Aqvist, J.; Widersten, M. Active site of epoxide hydrolases revisited: A noncanonical residue in potato StEH1 promotes both formation and breakdown of the alkylenzyme intermediate. Biochemistry 2007, 46, 2466–2479. [Google Scholar] [CrossRef] [PubMed]
- Thomaeus, A.; Naworyta, A.; Mowbray, S.L.; Widersten, M. Removal of distal protein-water hydrogen bonds in a plant epoxide hydrolase increases catalytic turnover but decreases thermostability. Protein Sci. 2008, 17, 1275–1284. [Google Scholar] [CrossRef] [PubMed]
- Reetz, M.T.; Bocola, M.; Wang, L.W.; Sanchis, J.; Cronin, A.; Arand, M.; Zou, J.; Archelas, A.; Bottalla, A.L.; Naworyta, A.; et al. Directed evolution of an enantioselective epoxide hydrolase: Uncovering the source of enantioselectivity at each evolutionary stage. J. Am. Chem. Soc. 2009, 131, 7334–7343. [Google Scholar] [CrossRef] [PubMed]
- Gurell, A.; Widersten, M. Modification of substrate specificity resulting in an epoxide hydrolase with shifted enantiopreference for (2,3-Epoxypropyl)benzene. ChemBioChem 2010, 11, 1422–1429. [Google Scholar] [CrossRef] [PubMed]
- Carlsson, Å.J.; Bauer, P.; Ma, H.; Widersten, M. Obtaining optical purity for product diols in enzyme-catalyzed epoxide hydrolysis: Contributions from changes in both enantio- and regioselectivity. Biochemistry 2012, 51, 7627–7637. [Google Scholar] [CrossRef] [PubMed]
- Carlsson, Å.J.; Bauer, P.; Dobritzsch, D.; Kamerlin, S.C.L.; Widersten, M. Epoxide hydrolysis as a model system for understanding flux through a branched reaction scheme. IUCrJ 2018, 5, 269–282. [Google Scholar] [CrossRef] [PubMed]
- Hopmann, K.H.; Himo, F. Insights into the reaction mechanism of soluble epoxide hydrolase from theoretical active site mutants. J. Phys. Chem. B 2006, 110, 21299–21310. [Google Scholar] [CrossRef] [PubMed]
- Lonsdale, R.; Hoyle, S.; Grey, D.T.; Ridder, L.; Mulholland, A.J. Determinants of reactivity and selectivity in soluble epoxide hydrolase from quantum mechanics/molecular mechanics modeling. Biochemistry 2012, 51, 1774–1786. [Google Scholar] [CrossRef] [PubMed]
- Janfalk Carlsson, Å.; Bauer, P.; Dobritzsch, D.; Nilsson, M.; Kamerlin, S.C.L.; Widersten, M. Laboratory-Evolved Enzymes Provide Snapshots of the Development of Enantioconvergence in Enzyme-Catalyzed Epoxide Hydrolysis. ChemBioChem 2016, 17, 1693–1697. [Google Scholar] [CrossRef] [PubMed]
- Lind, M.E.S.; Himo, F. Quantum Chemical Modeling of Enantioconvergency in Soluble Epoxide Hydrolase. ACS Catal. 2016, 6, 8145–8155. [Google Scholar] [CrossRef]
- Lindberg, D.; Ahmad, S.; Widersten, M. Mutations in salt-bridging residues at the interface of the core and lid domains of epoxide hydrolase StEH1 affect regioselectivity, protein stability and hysteresis. Arch. Biochem. Biophys. 2010, 495, 165–173. [Google Scholar] [CrossRef] [PubMed]
- Bauer, P.; Carlsson, Å.J.; Amrein, B.A.; Dobritzsch, D.; Widersten, M.; Kamerlin, S.C.L. Conformational diversity and enantioconvergence in potato epoxide hydrolase 1. Org. Biomol. Chem. 2016, 1–48. [Google Scholar] [CrossRef] [PubMed]
- Amrein, B.A.; Bauer, P.; Duarte, F.; Janfalk Carlsson, Å.; Naworyta, A.; Mowbray, S.L.; Widersten, M.; Kamerlin, S.C.L. Expanding the Catalytic Triad in Epoxide Hydrolases and Related Enzymes. ACS Catal. 2015, 5, 5702–5713. [Google Scholar] [CrossRef] [PubMed]
- Rose, P.W.; Prlić, A.; Altunkaya, A.; Bi, C.; Bradley, A.R.; Christie, C.H.; Di Costanzo, L.; Duarte, J.M.; Dutta, S.; Feng, Z.; et al. The RCSB protein data bank: Integrative view of protein, gene and 3D structural information. Nucleic Acids Res. 2017, 45. [Google Scholar] [CrossRef]
- Anandakrishnan, R.; Aguilar, B.; Onufriev, A.V. H++ 3.0: Automating pK prediction and the preparation of biomolecular structures for atomistic molecular modeling and simulations. Nucleic Acids Res. 2012, 40. [Google Scholar] [CrossRef] [PubMed]
- Kovalenko, A.; Luchko, T.; Gusarov, S.; Roe, D.R.; Simmerling, C.; Case, D.A.; Tuszynski, J. Three-dimensional molecular theory of solvation coupled with molecular dynamics in amber. J. Chem. Theory Comput. 2010, 6, 607–624. [Google Scholar] [CrossRef]
- Sindhikara, D.J.; Yoshida, N.; Hirata, F. Placevent: An algorithm for prediction of explicit solvent atom distribution-Application to HIV-1 protease and F-ATP synthase. J. Comput. Chem. 2012, 33, 1536–1543. [Google Scholar] [CrossRef] [PubMed]
- Case, D.A.; Babin, V.; Berryman, J.T.; Betz, R.M.; Cai, Q.; Cerutti, D.S.; Cheatham, T.E., III; Darden, T.A.; Duke, R.E.; Gohlke, H.; et al. Amber 14; University of San Francisco: San Francisco, CA, USA, 2014. [Google Scholar]
- Maier, J.A.; Martinez, C.; Kasavajhala, K.; Wickstrom, L.; Hauser, K.E.; Simmerling, C. ff14SB: Improving the Accuracy of Protein Side Chain and Backbone Parameters from ff99SB. J. Chem. Theory Comput. 2015, 11, 3696–3713. [Google Scholar] [CrossRef] [PubMed]
- Chovancova, E.; Pavelka, A.; Benes, P.; Strnad, O.; Brezovsky, J.; Kozlikova, B.; Gora, A.; Sustr, V.; Klvana, M.; Medek, P.; et al. CAVER 3.0: A Tool for the Analysis of Transport Pathways in Dynamic Protein Structures. PLoS Comput. Biol. 2012, 8. [Google Scholar] [CrossRef] [PubMed]
- Płuciennik, A.; Stolarczyk, M.; Bzówka, M.; Raczyńska, A.; Magdziarz, T.; Góra, A. BALCONY: An R package for MSA and functional compartments of protein variability analysis. BMC Bioinform. 2018. [Google Scholar] [CrossRef] [PubMed]
- Sander, C.; Schneider, R. Database of homology derived protein structures and the structural meaning of sequence alignment. Proteins Struct. Funct. Bioinform. 1991, 9, 56–68. [Google Scholar] [CrossRef] [PubMed]
- Berger, V.W.; Zhou, Y. Kolmogorov–Smirnov Test: Overview. Wiley StatsRef Stat. Ref. Online 2014. [Google Scholar] [CrossRef]
- Franzosa, E.A.; Xia, Y. Structural determinants of protein evolution are context-sensitive at the residue level. Mol. Biol. Evol. 2009, 26, 2387–2395. [Google Scholar] [CrossRef] [PubMed]
- Ramsey, D.C.; Scherrer, M.P.; Zhou, T.; Wilke, C.O. The relationship between relative solvent accessibility and evolutionary rate in protein evolution. Genetics 2011, 188, 479–488. [Google Scholar] [CrossRef] [PubMed]
- Horsman, G.P.; Liu, A.M.F.; Henke, E.; Bornscheuer, U.T.; Kazlauskas, R.J. Mutations in distant residues moderately increase the enantioselectivity of Pseudomonas fluorescens esterase towards methyl 3-bromo-2-methylpropanoate and ethyl 3-phenylbutyrate. Chem. Eur. J. 2003. [Google Scholar] [CrossRef] [PubMed]
- Tokuriki, N.; Tawfik, D.S. Stability effects of mutations and protein evolvability. Curr. Opin. Struct. Biol. 2009, 19, 596–604. [Google Scholar] [CrossRef] [PubMed]
- Bednar, D.; Beerens, K.; Sebestova, E.; Bendl, J.; Khare, S.; Chaloupkova, R.; Prokop, Z.; Brezovsky, J.; Baker, D.; Damborsky, J. FireProt: Energy- and Evolution-Based Computational Design of Thermostable Multiple-Point Mutants. PLoS Comput. Biol. 2015. [Google Scholar] [CrossRef] [PubMed]
- Oda, K.; Kinoshita, M. Physicochemical origin of high correlation between thermal stability of a protein and its packing efficiency: A theoretical study for staphylococcal nuclease mutants. Biophys. Physicobiol. 2015, 12, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Koudelakova, T.; Chaloupkova, R.; Brezovsky, J.; Prokop, Z.; Sebestova, E.; Hesseler, M.; Khabiri, M.; Plevaka, M.; Kulik, D.; Kuta Smatanova, I.; et al. Engineering enzyme stability and resistance to an organic cosolvent by modification of residues in the access tunnel. Angew. Chem. Int. Ed. 2013, 52, 1959–1963. [Google Scholar] [CrossRef] [PubMed]
- Wijma, H.J.; Floor, R.J.; Jekel, P.A.; Baker, D.; Marrink, S.J.; Janssen, D.B. Computationally designed libraries for rapid enzyme stabilization. Protein Eng. Des. Sel. 2014, 27, 49–58. [Google Scholar] [CrossRef] [PubMed]
- Brezovsky, J.; Babkova, P.; Degtjarik, O.; Fortova, A.; Gora, A.; Iermak, I.; Rezacova, P.; Dvorak, P.; Kuta Smatanova, I.; Prokop, Z.; et al. Engineering a de Novo Transport Tunnel. ACS Catal. 2016, 6, 7597–7610. [Google Scholar] [CrossRef]
- Pavlova, M.; Klvana, M.; Prokop, Z.; Chaloupkova, R.; Banas, P.; Otyepka, M.; Wade, R.C.; Tsuda, M.; Nagata, Y.; Damborsky, J. Redesigning dehalogenase access tunnels as a strategy for degrading an anthropogenic substrate. Nat. Chem. Biol. 2009, 5, 727–733. [Google Scholar] [CrossRef] [PubMed]
- Hendil-Forssell, P.; Martinelle, M.; Syré, P.-O. As featured in: Exploring water as building bricks in enzyme engineering. Chem. Commun. 2015, 51, 17221–17224. [Google Scholar] [CrossRef] [PubMed]
- Fink, M.J.; Syrén, P.O. Redesign of water networks for efficient biocatalysis. Curr. Opin. Chem. Biol. 2017, 37, 107–114. [Google Scholar] [CrossRef] [PubMed]
- Subramanian, K.; Góra, A.; Spruijt, R.; Mitusińska, K.; Suarez-Diez, M.; Martins dos Santos, V.; Schaap, P.J. Modulating D-amino acid oxidase (DAAO) substrate specificity through facilitated solvent access. PLoS ONE 2018, 13, e0198990. [Google Scholar] [CrossRef] [PubMed]
- Fox, J.M.; Kang, K.; Sastry, M.; Sherman, W.; Sankaran, B.; Zwart, P.H.; Whitesides, G.M. Water-Restructuring Mutations Can Reverse the Thermodynamic Signature of Ligand Binding to Human Carbonic Anhydrase. Angew. Chem. Int. Ed. 2017. [Google Scholar] [CrossRef]
- Syrén, P.O.; Hammer, S.C.; Claasen, B.; Hauer, B. Entropy is key to the formation of pentacyclic terpenoids by enzyme-catalyzed polycyclization. Angew. Chem. Int. Ed. 2014. [Google Scholar] [CrossRef]
- Breiten, B.; Lockett, M.R.; Sherman, W.; Fujita, S.; Al-Sayah, M.; Lange, H.; Bowers, C.M.; Heroux, A.; Krilov, G.; Whitesides, G.M. Water networks contribute to enthalpy/entropy compensation in protein-ligand binding. J. Am. Chem. Soc. 2013. [Google Scholar] [CrossRef] [PubMed]
- Kong, X.-D.; Yuan, S.; Li, L.; Chen, S.; Xu, J.-H.; Zhou, J. Engineering of an epoxide hydrolase for efficient bioresolution of bulky pharmaco substrates. Proc. Natl. Acad. Sci. USA 2014, 111, 15717–15722. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cavity I | Cavity II | Cavity III | |
|---|---|---|---|
| MD1 | I180, T182, Y183, R184, F189, D265*, L266, A299, H300*, F301 | H31, G32, F33, E35 *, S39, W40, H104 *, D105 *, G107, H300 *, F301 | F33, P34, F158, F168, V176, L177, I180, F191, F223, G227, F228, T229, G230, A231, V232 |
| MD2 | I180, T182, Y183, R184, F189, W210, L266, A299, H300*, F301, E305 | G32, F33, P34, E35*, H104*, D105*, H300*, F301 | F33, Y154*, F158, F168, I180, F189, F191, I200 |
| MD3 | I180, L181, T182, Y183, R184, D185, P186, A187, F189, L207, W210, F264, D265*, L266, A299, H300*, F301, E305 | - | F33, P34, Y154*, F158, F168, V176, K179, I180, F189, L197, I200, F228 |
| MD4 | I180, T182, Y183, R184, D185, F189, A203, L207, S208, W210, F264, D265*, L266, A299, H300*, F301, E305 | H31, G32, F33, E35*, S39, W40, H104*, D105*, L128, H300 *, F301, V302, S303 | F33, P34, Y154*, F158, F168, V176, L177, K179, I180, F189, F191, L197, I200, F228 |
| MD5 | K179, I180, L181, T182, Y183, R184, D185, P186, A187, A203, P204, L207, S208, W210, F264, D265 *, L266, A298, A299, H300 *, F301, V302, Q304, E305 | G32, F33, P34, E35*, H104*, D105*, H300*, F301, | F33, Y154*, F158, F168, V176, K179, I180, F189, F191, L197, I200, F228, V232 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mitusińska, K.; Magdziarz, T.; Bzówka, M.; Stańczak, A.; Góra, A. Exploring Solanum tuberosum Epoxide Hydrolase Internal Architecture by Water Molecules Tracking. Biomolecules 2018, 8, 143. https://doi.org/10.3390/biom8040143
Mitusińska K, Magdziarz T, Bzówka M, Stańczak A, Góra A. Exploring Solanum tuberosum Epoxide Hydrolase Internal Architecture by Water Molecules Tracking. Biomolecules. 2018; 8(4):143. https://doi.org/10.3390/biom8040143
Chicago/Turabian StyleMitusińska, Karolina, Tomasz Magdziarz, Maria Bzówka, Agnieszka Stańczak, and Artur Góra. 2018. "Exploring Solanum tuberosum Epoxide Hydrolase Internal Architecture by Water Molecules Tracking" Biomolecules 8, no. 4: 143. https://doi.org/10.3390/biom8040143
APA StyleMitusińska, K., Magdziarz, T., Bzówka, M., Stańczak, A., & Góra, A. (2018). Exploring Solanum tuberosum Epoxide Hydrolase Internal Architecture by Water Molecules Tracking. Biomolecules, 8(4), 143. https://doi.org/10.3390/biom8040143