Genome Editing in Model Strain Myxococcus xanthus DK1622 by a Site-Specific Cre/loxP Recombination System

 , ,
, , 
Abstract
1. Introduction
2. Results
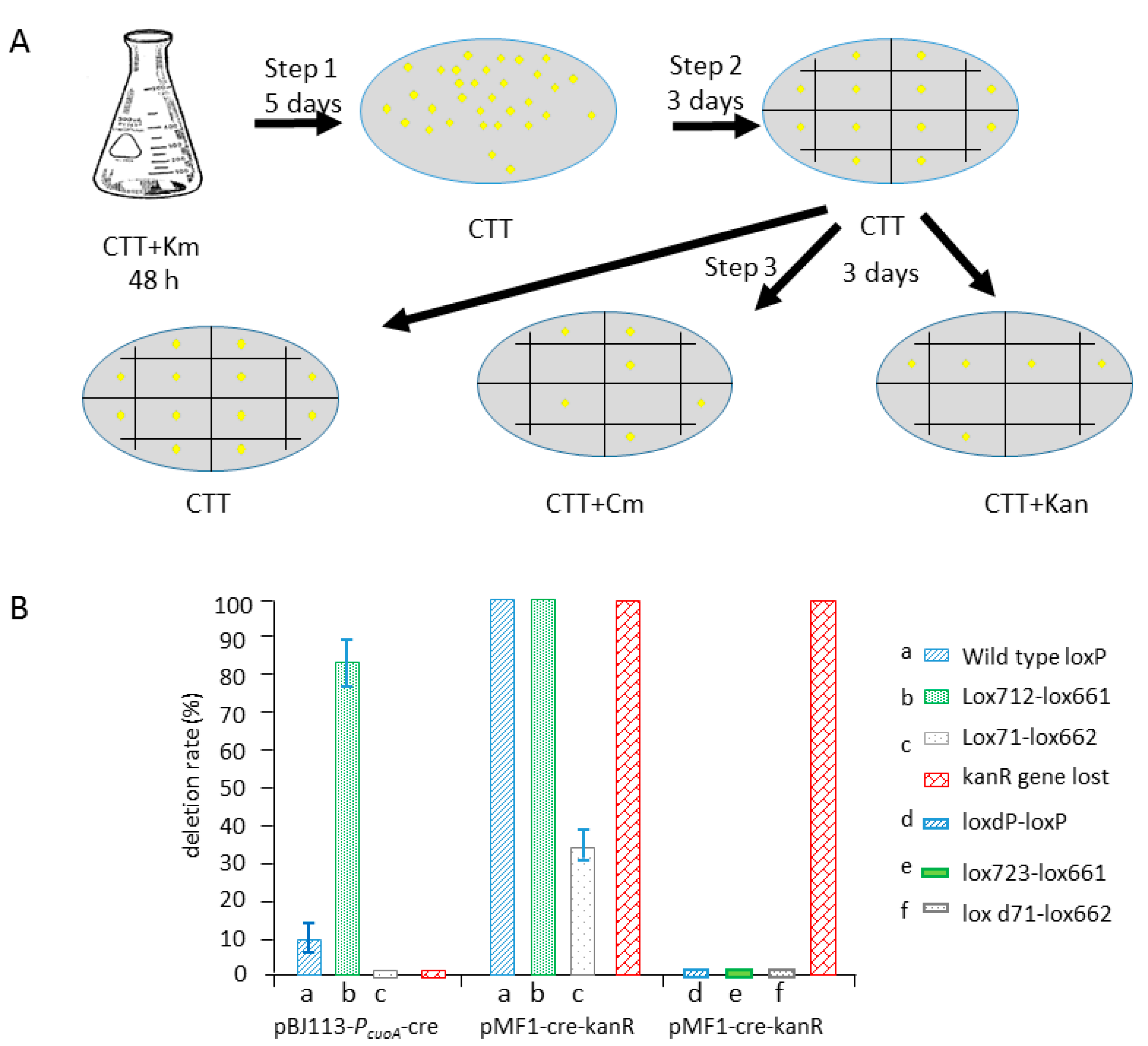
2.1. Efficient Deletion of Lox-Flanking Gene Fragment by Native Cre Recombinase in M. xanthus
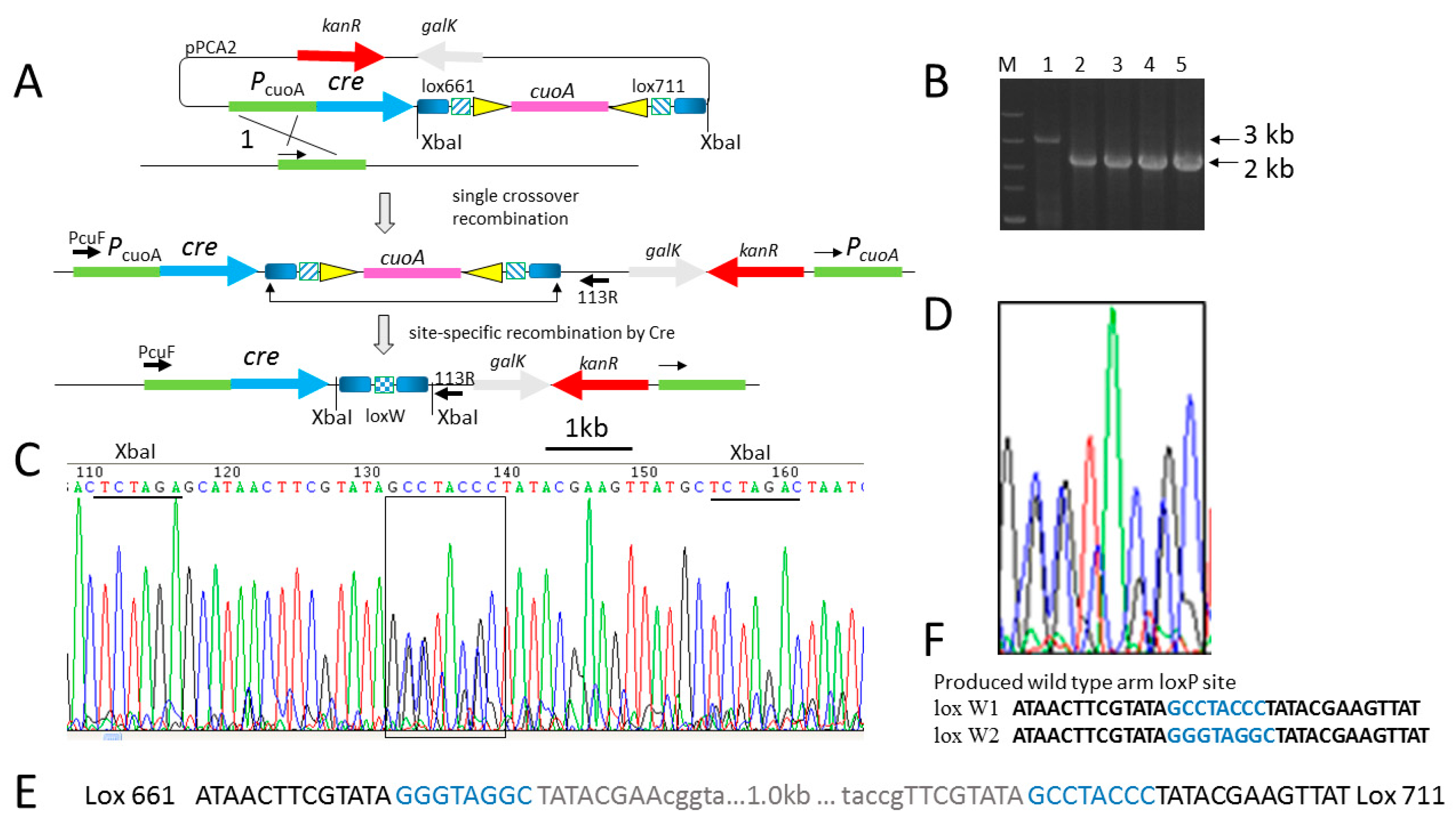
2.2. Cre Expression Driven by the Inducible Promoter PcuoA Was Found to Be Leaky during the Comparison of the Recombination Efficiencies of Different Lox Spacer Pairs by pBJ113-Cre Integration
2.3. Cre Expression from pZJY41-Self-Replicative Plasmid Has HigherEfficient Recombination of Different Lox Pairs
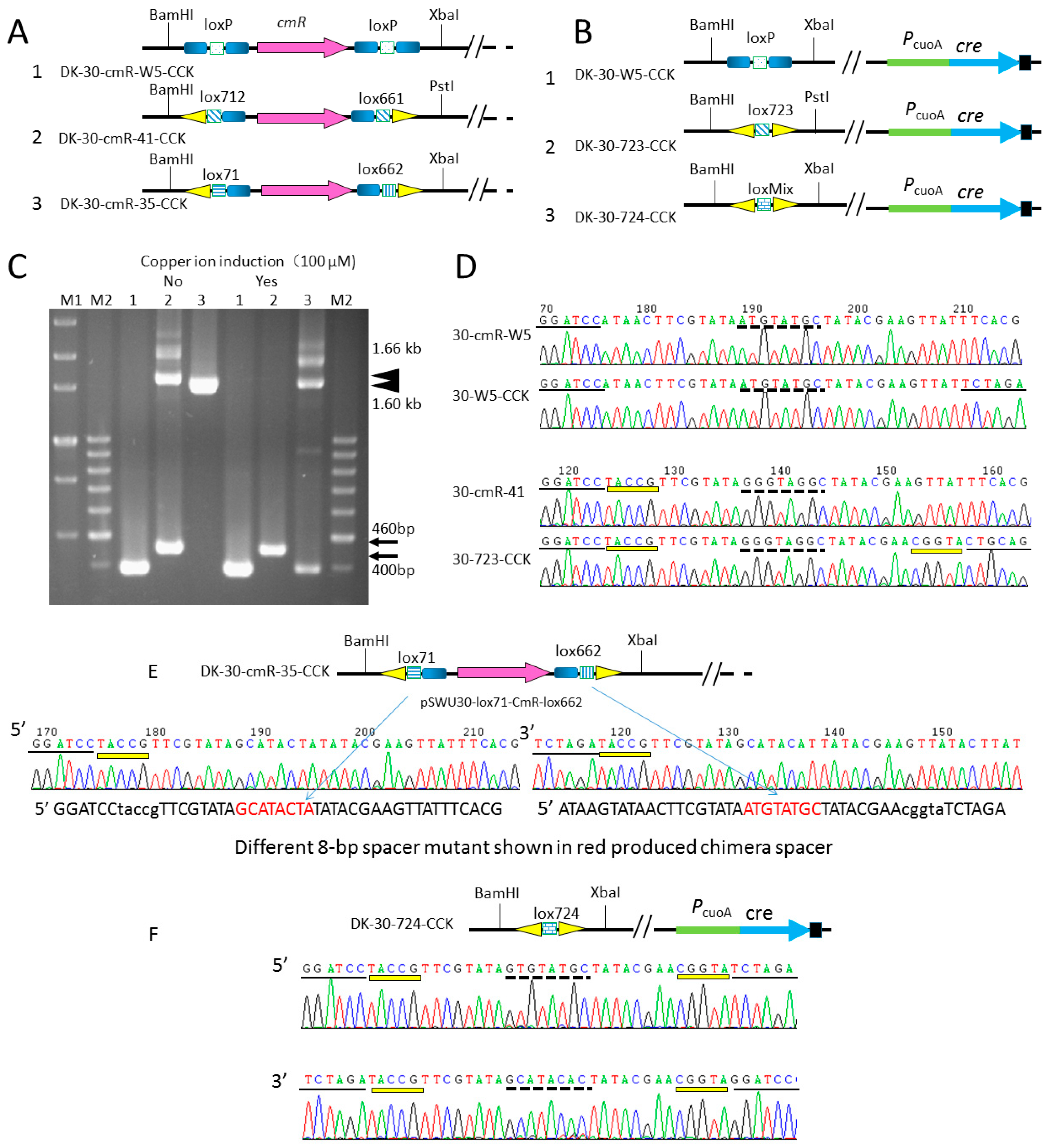
2.4. Comparison of the Recombination Efficiency in Different Expression Type (pBJ113 and pZJY41) with Different Lox Pairs
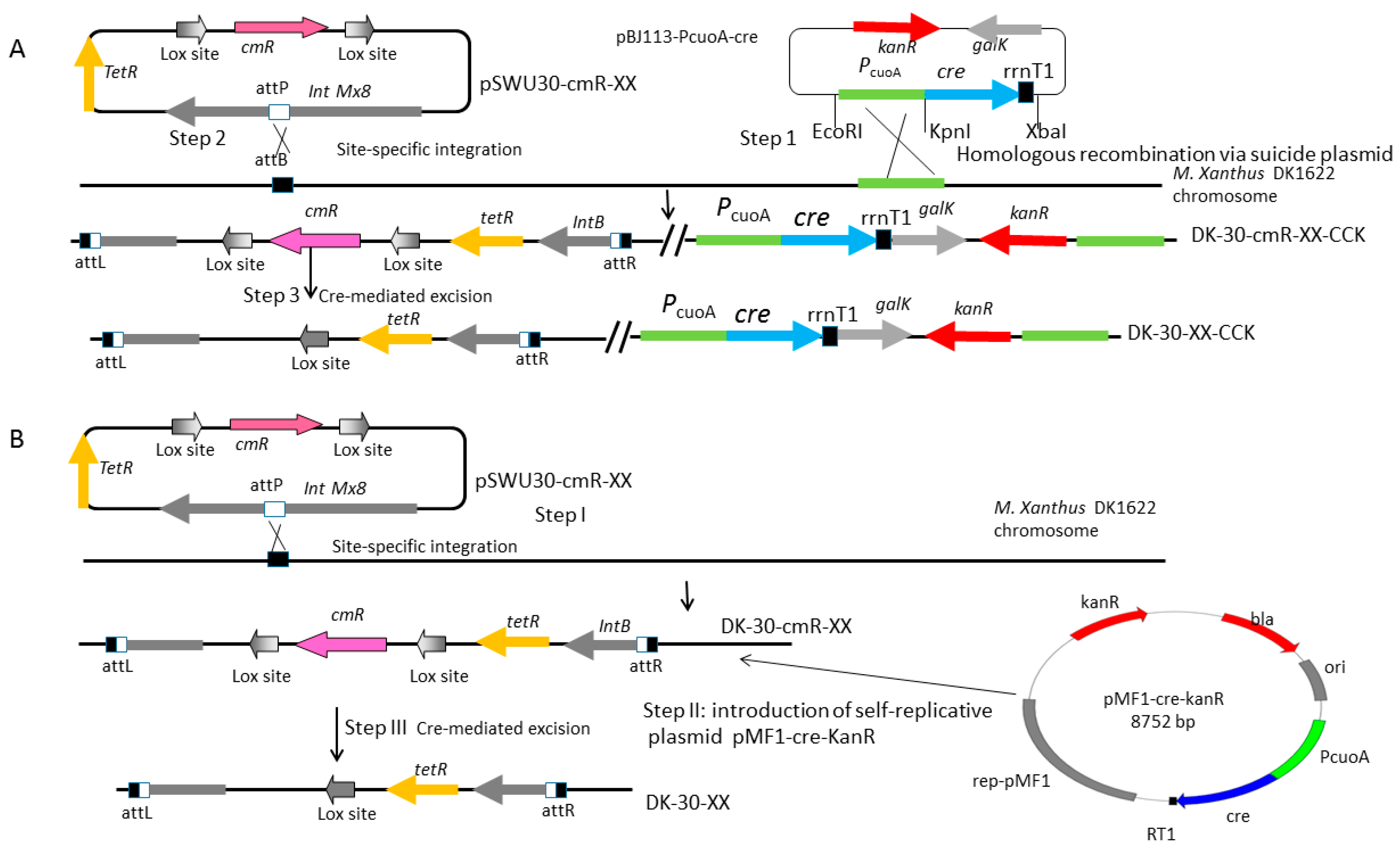
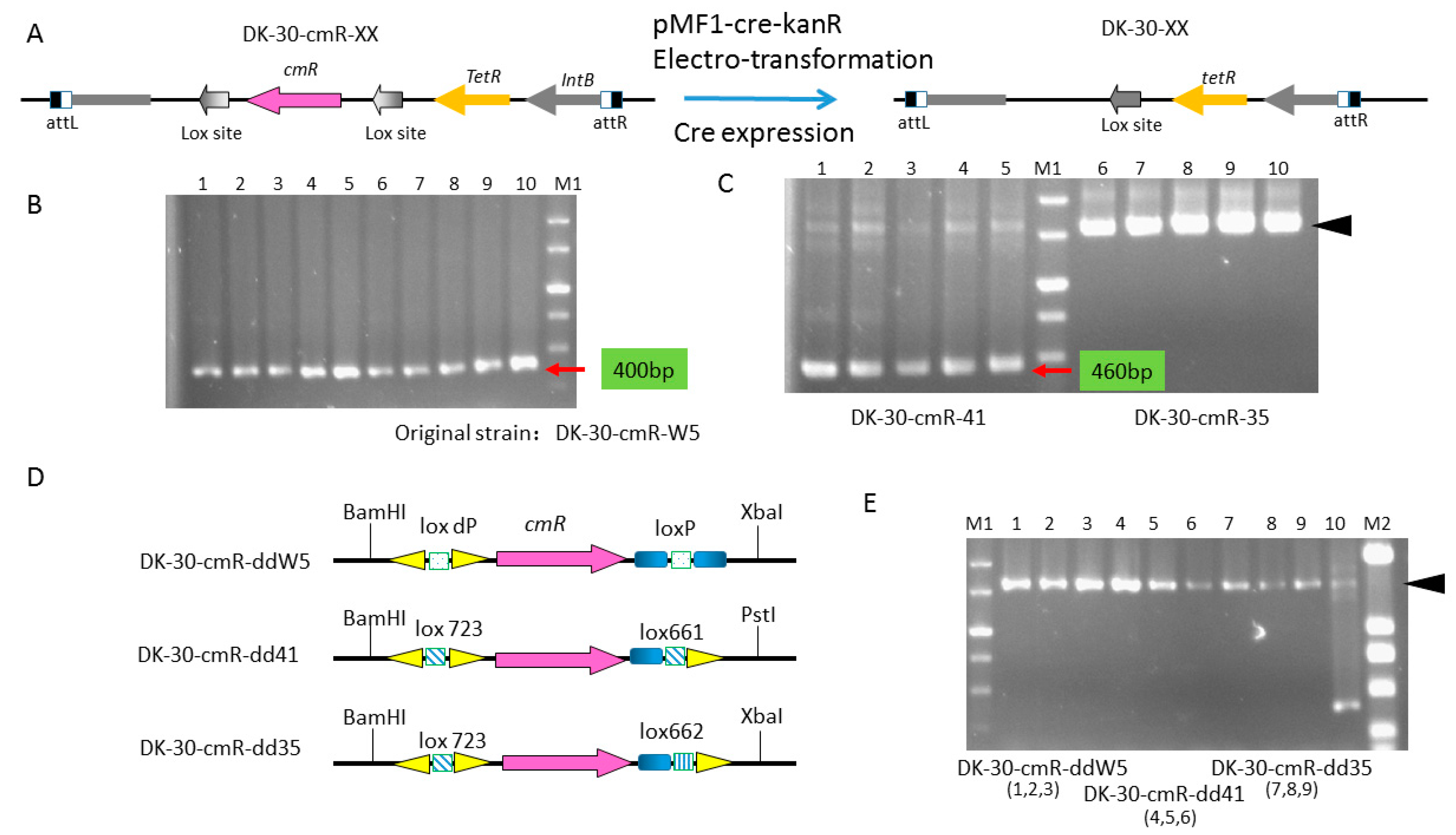
2.5. Construction of a Series of Self-Replicative Plasmids for the Expression of Native Cre Gene or Artificial Cre Gene in M. xanthus by Different Antibiotics Markers
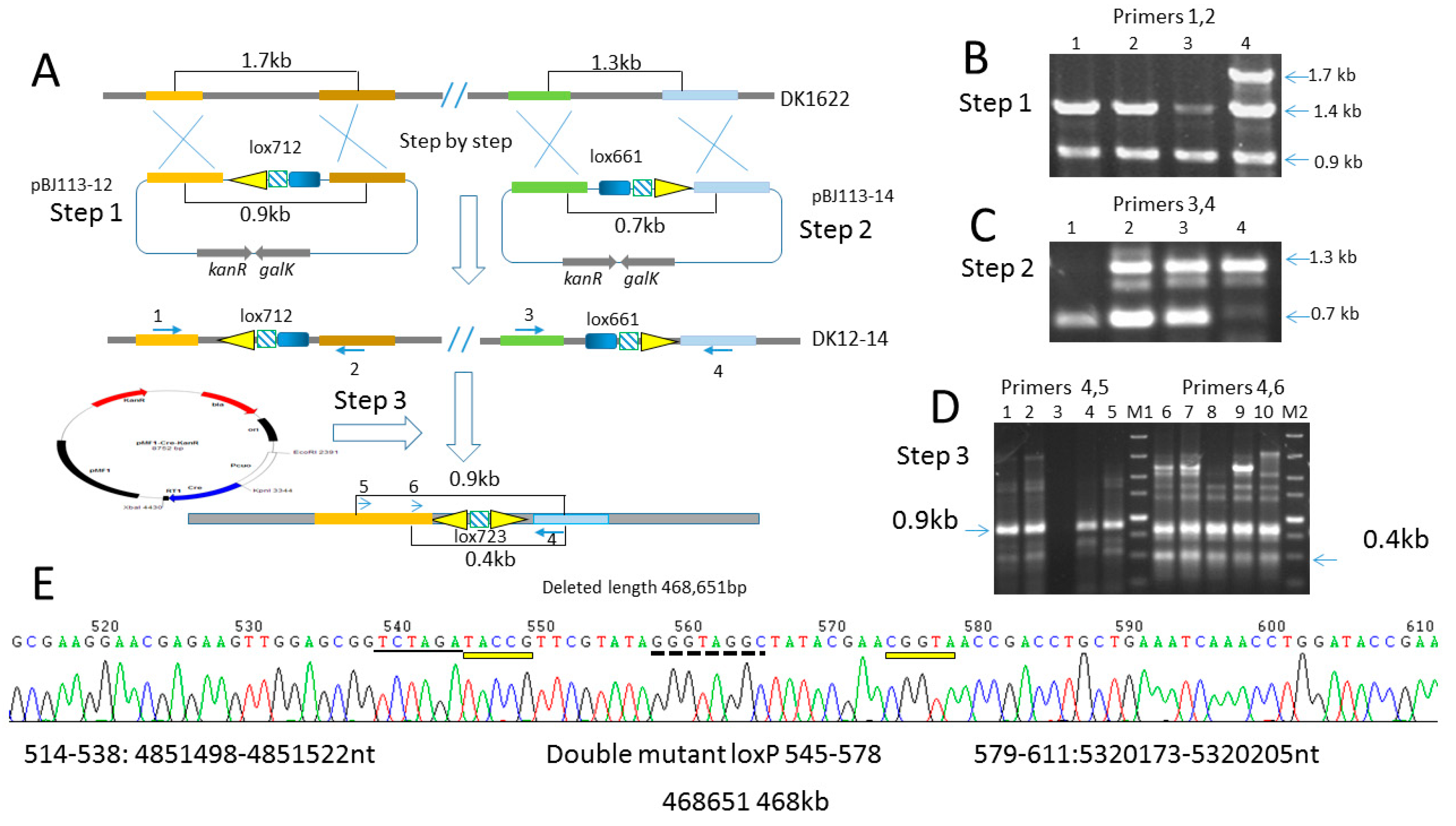
2.6. Deletion of 468 kb Large Fragment Covering Yellow Pigment Gene Cluster and Siderophore Gene Clusters
3. Discussion
4. Materials and Methods
4.1. Bacterial Strains, Growth Conditions, Plasmids, and Oligonucleotides
4.2. Construction of Lox-CmR-Lox-Containing Plasmids
4.3. Construction of the Series of Native Cre Expression Plasmids
4.4. Construction of the Series of Artificial Synthetic Cre Expression Plasmids
4.5. Construction of Homologous Arms Plasmids pBJ113-12 and pBJ113-14
4.6. Construction of the Mutant Strains: DK-30-cmR-XX-CCK Series Strains and DK-30-cmR-XX Series Strains
4.7. Construction of the Lox Site Containing Mutant Strain DK-12-14
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Reichenbach, H. Myxobacteria, producers of novel bioactive substances. J. Ind. Microbiol. Biotechnol. 2001, 27, 149–156. [Google Scholar] [CrossRef] [PubMed]
- Goldman, B.S.; Nierman, W.C.; Kaiser, D.; Slater, S.C.; Durkin, A.S.; Eisen, J.A.; Ronning, C.M.; Barbazuk, W.B.; Blanchard, M.; Field, C. Evolution of sensory complexity recorded in a myxobacterial genome. Proc. Natl. Acad. Sci. USA 2006, 103, 15200–15205. [Google Scholar] [CrossRef] [PubMed]
- Kaiser, D.; Robinson, M.; Kroos, L. Myxobacteria, polarity, and multicellular morphogenesis. Cold Spring Harb. Perspect. Boil. 2010, 2, a000380. [Google Scholar] [CrossRef] [PubMed]
- Munoz-Dorado, J.; Marcos-Torres, F.J.; Garcia-Bravo, E.; Moraleda-Munoz, A.; Perez, J. Myxobacteria: Moving, Killing, Feeding, and Surviving Together. Front. Microbiol. 2016, 7, 781. [Google Scholar] [CrossRef] [PubMed]
- Gerth, K.; Pradella, S.; Perlova, O.; Beyer, S.; Müller, R. Myxobacteria: Proficient producers of novel natural products with various biological activities-past and future biotechnological aspects with the focus on the genus Sorangium. J. Biotechnol. 2003, 106, 233–253. [Google Scholar] [CrossRef] [PubMed]
- Kaiser, D. Social gliding is correlated with the presence of piliin Myxococcus xanthus. Proc. Natl. Acad. Sci. USA 1979, 76, 5952–5956. [Google Scholar] [CrossRef] [PubMed]
- Wenzel, S.C.; Müller, R. Recent developments towards the heterologous expression of complex bacterial natural product biosynthetic pathways. Curr. Opin. Biotechnol. 2005, 16, 594–606. [Google Scholar] [CrossRef] [PubMed]
- Wenzel, S.C.; Müller, R. Myxobacteria—Microbial factories for the production of bioactive secondary metabolites. Mol. Biosyst. 2009, 5, 567–574. [Google Scholar] [CrossRef] [PubMed]
- Wenzel, S.C.; Meiser, P.; Binz, T.M.; Mahmud, T.; Müller, R. Nonribosomal peptide biosynthesis: Point mutations and modules kipping lead to chemical diversity. Angew. Chem. 2006, 45, 2296–2301. [Google Scholar] [CrossRef] [PubMed]
- Bode, H.B.; Meiser, P.; Klefisch, T.; Cortina, N.S.; Krug, D.; Gohring, A.; Schwar, G.; Mahmud, T.; Elnakady, Y.A.; Müller, R. Mutasynthesis-derived myxalamids and origin of the isobutyryl-CoA starter unit of myxalamid B. Chembiochem 2007, 8, 2139–2144. [Google Scholar] [CrossRef] [PubMed]
- Bode, H.B.; Müller, R. The impact of bacterial genomicson natural product research. Angew. Chem. 2005, 44, 6828–6846. [Google Scholar] [CrossRef] [PubMed]
- ChristianSchley, M.O.A.; Remco, S.; Rolf, M.; Christian, G.H. Proteome Analysis of by Off-Line Two-Dimensional Chromatographic Separation Using Monolithic Poly-(styrene-divinylbenzene) Columns Combined with Ion-Trap Tandem Mass Spectrometry. J. Proteome Res. 2006, 5, 9. [Google Scholar]
- Bode, H.B.; Ring, M.W.; Schwar, G.; Altmeyer, M.O.; Kegler, C.; Jose, I.R.; Singer, M.; Müller, R. Identification of additional players in the alter native biosynthesis pathway to isovaleryl-CoA in the myxobacterium Myxococcus xanthus. Chembiochem 2009, 10, 128–140. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Feng, Z.; Tomura, T.; Suzuki, A.; Miyano, S.; Tsuge, T.; Mori, H.; Suh, J.W.; Iizuka, T.; Fudou, R. Heterologous production of the marine myxobacterial antibiotic haliangicin and its unnatural analogues gene rated by engineering of the biochemical pathway. Sci. Rep. 2016, 6, 22091. [Google Scholar] [CrossRef] [PubMed]
- Tu, Q.; Herrmann, J.; Hu, S.; Raju, R.; Bian, X.; Zhang, Y.; Müller, R. Genetic engineering and heterologous expression of the disorazol biosynthetic gene cluster via Red/ET recombineering. Sci. Rep. 2016, 6, 21066. [Google Scholar] [CrossRef] [PubMed]
- Fu, J.; Wenzel, S.C.; Perlova, O.; Wang, J.; Gross, F.; Tang, Z.; Yin, Y.; Stewart, A.F.; Muller, R.; Zhang, Y. Efficient transfer of two large secondary metabolite pathway gene clusters in toheterologous hosts by transposition. Nucleic Acids Res. 2008, 36, e113. [Google Scholar] [CrossRef] [PubMed]
- Zhu, L.P.; Yue, X.J.; Han, K.; Li, Z.F.; Zheng, L.S.; Yi, X.N.; Wang, H.L.; Zhang, Y.M.; Li, Y.Z. All opatric integrations selectively change host transcriptomes, leading to varied expression efficiencies of exotic genes in Myxococcus xanthus. Microb. Cell Factories 2015, 14, 105. [Google Scholar] [CrossRef] [PubMed]
- Ueki, T.; Inouye, S.; Inouye, M. Positive-negative KG cassettes for construction of multi-gene deletions using as ingle drug marker. Gene 1996, 183, 153–157. [Google Scholar] [CrossRef]
- Hoess, R.H.; Ziese, M.; Sternberg, N. P1 site-specific recombination: Nucleotide sequence of the recombining sites. Proc. Natl. Acad. Sci. USA 1982, 79, 3398–3402. [Google Scholar] [CrossRef] [PubMed]
- Kuhn, R.; Torres, R.M. Cre/loxP recombination system and gene targeting. Methods Mol. Boil. 2002, 180, 175–204. [Google Scholar]
- Gopaul, D.N.; Guo, F.; VanDuyne, G.D. Structure of the Hollidayjunction intermediate in Cre-loxP site-specific recombination. EMBO J. 1998, 17, 4175–4187. [Google Scholar] [CrossRef] [PubMed]
- Gomez-Santos, N.; Treuner-Lange, A.; Moraleda-Munoz, A.; Garcia-Bravo, E.; Garcia-Hernandez, R.; Martinez-Cayuela, M.; Perez, J.; Sogaard-Andersen, L.; Munoz-Dorado, J. Comprehensivese to fintegrative plasmid vectors for copper-inducible gene expression in Myxococcus xanthus. Appl. Environ. Microbiol. 2012, 78, 2515–2521. [Google Scholar] [CrossRef] [PubMed]
- Missirlis, P.I.; Smailus, D.E.; Holt, R.A. A high-through put screen identifying sequence and promiscuity characteristics of the loxP spacer region in Cre-mediated recombination. BMC Genom. 2006, 7, 73. [Google Scholar] [CrossRef] [PubMed]
- Albert, H.; Dale, E.C.; Lee, E.; Ow, D.W. Site-specific integration of DNA into wild-type and mutant lox sites placed in the plant genome. Plant J. 1995, 7, 649–659. [Google Scholar] [CrossRef] [PubMed]
- Fedoryshyn, M.; Welle, E.; Bechthold, A.; Luzhetskyy, A. Functional expression of the Cre recombinase in actinomycetes. Appl. Microbiol. Biotechnol. 2008, 78, 1065–1070. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.Y.; Zhong, L.; Shen, M.J.; Xia, Z.J.; Cheng, Q.X.; Sun, X.; Zhao, G.P.; Li, Y.Z.; Qin, Z.J. Discovery of the autonomously replicating plasmid pMF1 from Myxococcus fulvus and development of a gene cloning system in Myxococcus xanthus. Appl. Environ. Microbiol. 2008, 74, 1980–1987. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.J.; Wang, Y.; Li, Z.F.; Gong, Y.; Zhang, P.; Hu, W.C.; Sheng, D.H.; Li, Y.Z. Increasing on-target cleavage efficiency for CRISPR/Cas9-induced large fragment deletion in Myxococcus xanthus. Microb. Cell Factories 2017, 16, 142. [Google Scholar] [CrossRef] [PubMed]
- Fu, J.; Bian, X.; Hu, S.; Wang, H.; Huang, F.; Seibert, P.M.; Plaza, A.; Xia, L.; Muller, R.; Stewart, A.F. Full-length Rec Eenhances linear-linear homologous recombination and facilitates direct cloning for bioprospecting. Nat. Biotechnol. 2012, 30, 440–446. [Google Scholar] [CrossRef] [PubMed]
- Yanisch-Perron, C.; Vieira, J.; Messing, J. Improved M13 phage cloning vectors and hosts trains: Nucleotide sequences of the M13mp18 and pUC19 vectors. Gene 1985, 33, 103–119. [Google Scholar] [CrossRef]
- Julien, B.; Kaiser, A.D.; Garza, A. Spatial control of cell differentiation in Myxococcus xanthus. Proc. Natl. Acad. Sci. USA 2000, 97, 9098–9103. [Google Scholar] [CrossRef] [PubMed]
- Wu, S.S.; Wu, J.; Kaiser, D. The Myxococcus xanthus pilT locusis required for social gliding motility although pili are still produced. Mol. Microbiol. 1997, 23, 109–121. [Google Scholar] [CrossRef] [PubMed]
- Brosius, J. Plasmid vectors for the selection of promoters. Gene 1984, 27, 151–160. [Google Scholar] [CrossRef]
- Zhang, Y.; Buchholz, F.; Muyrers, J.P.; Stewart, A.F. A new logic for DNA engineering using recombination in Escherichia coli. Nat. Genet. 1998, 20, 123–128. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Name of LoxP | Left Arm | Spacer | Right Arm | Reference |
|---|---|---|---|---|
| Wild loxP | ATAACTTCGTATA | GCATACAT | TATACGAAGTTAT | [19] |
| lox 66 | ATAACTTCGTATA | GCATACAT | TATACGAAcggta | [24] |
| lox 71 | taccgTTCGTATA | GCATACAT | TATACGAAGTTAT | [24] |
| lox 661 | ATAACTTCGTATA | GGGTAGGC | TATACGAAcggta | This study |
| lox 711 | taccgTTCGTATA | GCCTACCC | TATACGAAGTTAT | This study |
| lox PC(W1) | ATAACTTCGTATA | GCCTACCC | TATACGAAGTTAT | This study |
| lox PG(W2) | ATAACTTCGTATA | GGGTAGGC | TATACGAAGTTAT | This study |
| lox 712 | taccgTTCGTATA | GGGTAGGC | TATACGAAGTTAT | This study |
| lox 662 | ATAACTTCGTATA | TAGTATGC | TATACGAAcggta | This study |
| lox 723 | taccgTTCGTATA | GGGTAGGC | TATACGAAcggta | This study |
| lox dp | taccgTTCGTATA | GCATACAT | TATACGAAcggta | This study |
| Strains | Genotype | Reference or Source |
|---|---|---|
| E. coli | ||
| E. coli TOP10 | F–mcrA Δ(mrr-hsdRMS-mcrBC) Φ80lacZΔM15 ΔlacX74 recA1 araD139 Δ(araleu) 7697 galUgalKrpsL (StrR) endA1 nupG | Thermo Fisher Scientific (MA, USA) |
| E. coli DH5α | F– Φ80lacZΔM15 Δ(lacZYA-argF) U169 recA1 endA1 hsdR17 (rK–, mK+) phoAsupE44 λ– thi-1 gyrA96 relA1 | Thermo Ffisher Scientific (MA, USA) |
| E. coli GB05 | F-mcrA ∆(mrr-hsdRMS-mcrBC) φ80lacZ∆M15 ∆lacX74 recA1 endA1 araD139 ∆(ara, leu)7697 galUgalK λ rpsLnupGfhuA::IS2 recET redα, phage T1-resisten | [16] |
| E. coli GB05-dir | GB2005, araC-BAD-ETγA | [28] |
| M. xanthus | ||
| DK1622 | Wild type | [6] |
| DK-WGC | DK1622::pBJ113-PcuoA-cre-lox661-cuoA-lox711, Kan | this study |
| DK-WG | DK1622::pBJ113-PcuoA-cre-loxW1, Kan | this study |
| DK-WC | DK1622::pBJ113-PcuoA-cre-loxW2, Kan | this study |
| DK-30-cmR-W5 | DK1622::pSWU30-loxP-cmR-loxP, TetR, CmR | this study |
| DK-30-cmR-41 | DK1622::pSWU30-lox712-cmR-lox661, TetR, CmR | this study |
| DK-30-cmR-35 | DK1622::pSWU30-lox71-cmR-lox662, TetR, CmR | this study |
| DK-30-cmR-ddW5 | DK1622::pSWU30-loxdP-cmR-lox661, TetR, CmR | this study |
| DK-30-cmR-dd41 | DK1622::pSWU30-lox723-cmR-lox661, TetR, CmR | this study |
| DK-30-cmR-dd35 | DK1622::pSWU30-lox d71-cmR-lox661, TetR, CmR | this study |
| DK-30-cmR-W5-CCK | DK-30-cmR-W5::pBJ113Cre, KanR, TetR, CmR | this study |
| DK-30-cmR-41-CCK | DK-30-cmR-41::pBJ113Cre, KanR, TetR, CmR | this study |
| DK-30-cmR-35-CCK | DK-30-cmR-35::pBJ113Cre, KanR, TetR, CmR | this study |
| DK-30-W5-CCK | DK-30-W5::pBJ113-cre, KanR, TetR | this study |
| DK-30-41-CCK | DK-30-41::pBJ113-cre, KanR, TetR | this study |
| DK-30-35-CCK | DK-30-35::pBJ11-cre, KanR, TetR | this study |
| DK-30-cmR-W5-Pcre | DK-30-cmR-W5,pZJY41-cre, KanR, TetR, CmR | this study |
| DK-30-cmR-41-Pcre | DK-30-cmR-41,pZJY41Cre, KanR, TetR, CmR | this study |
| DK-30-cmR-35-Pcre | DK-30-cmR-35,pZJY41Cre, KanR, TetR, CmR | this study |
| DK-W5 | DK-30-W5, TetR | this study |
| DK-41 | DK-30-41, TetR | this study |
| DK-35-2 | DK-30-35-2, TetR | this study |
| DK-p12lox712 | DK1622::pBJ113-12lox712, KanR | this study |
| DK-12lox712 | DK1622::lox712 | this study |
| DK-12lox712-p14lox661 | DK1622::lox712::pBJ113-14lox661, KanR | this study |
| DK-12lox712-14lox661 | DK1622::lox712::lox661 | this study |
| Name | Description | Reference or Source |
|---|---|---|
| pUC19 | pBR322 ori, AmpR | [29] |
| pNG10A | PcuoATetR from pUC19 | [22] |
| pMAT3 | PcuoA Mx8 attPintegraseTetR from pSWU30 | [22] |
| pBJ113 | pBR322 ori,galK; KanR | [30] |
| pSWU30 | pBR322 ori, Site-specific integration vector with Mx8 attBintegration site, TetR | [31] |
| pZJY41 | pMF1 ori, pBR322 ori, KanR | [26] |
| pUC19-cre | pBR322 ori, native cre geneAmpR | this study |
| pUC57-cre(a) | pBR322 ori, codon-optimized artificial cregene, AmpR | this study |
| pUC19-PcuoA-cre | native cre gene promoted by PcuoA, AmpR | this study |
| pBJ113-PcuoA-cre | this study | |
| pPCA2 | pBJ113-PcuoA-cre-lox-Frag-lox | this study |
| pSWU30-CmR-W5 | pSWU30, contained the cassette of loxP-cmR-loxP, TetR, CmR | this study |
| pSWU30-cmR-41 | pSWU30, contained the cassette of lox712-cmR-lox661, TetR, CmR | this study |
| pSWU30-cmR-35 | pSWU30, contained the cassette of lox71-CmR-lox662, TetR, CmR | this study |
| pSWU30-cmR-ddW5 | pSWU30, contained the cassette of loxdp-cmR-lox66, TetR, CmR | this study |
| pSWU30-cmR-dd41 | pSWU30, contained the cassette of lox723-cmR-lox661, TetR, CmR | this study |
| pSWU30-cmR-dd35 | pSWU30, contained the cassette of lox723-cmR-lox662, TetR, CmR | this study |
| pMF1-cre-kanR | pMF1ori, pBR322 ori, native cre gene driven by PcuoA, KanR | this study |
| pMF1-cre-tetR | pMF1 ori, pBR322 ori, native cre gene driven by PcuoA, TetR | this study |
| pMF1-cre-cmR | pMF1 ori, pBR322 ori, native cre gene driven by PcuoA, CmR | this study |
| pMF1-cre(a)-kanR | pMF1ori, pBR322 ori, artificial cre gene driven by PcuoA, KanR | this study |
| pMF1-cre(a)-tetR | pMF1 ori, pBR322 ori, artificial cre gene driven by PcuoA, TetR | this study |
| pMF1-cre(a)-cmR | pMF1 ori, pBR322 ori, artificial cre gene driven by PcuoA, CmR | this study |
| pBJ113-12 | Two fragments amplified by two primer pairs contained one lox712 (S12uu-130F, S12uu-1638R2; S12uF2, S12uR2) were cloned into EcoRI, XbaI, HindIII sites of pBJ113, respectively | this study |
| pBJ113-14 | Two fragments amplified by two primer pairs contained one lox661 (S14E-451F, S14E-1457R; S14E-2057F, S14E-3649R) were recombined with linearized pBJ113 by Red/ET | this study |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yang, Y.-J.; Singh, R.P.; Lan, X.; Zhang, C.-S.; Li, Y.-Z.; Li, Y.-Q.; Sheng, D.-H. Genome Editing in Model Strain Myxococcus xanthus DK1622 by a Site-Specific Cre/loxP Recombination System. Biomolecules 2018, 8, 137. https://doi.org/10.3390/biom8040137
Yang Y-J, Singh RP, Lan X, Zhang C-S, Li Y-Z, Li Y-Q, Sheng D-H. Genome Editing in Model Strain Myxococcus xanthus DK1622 by a Site-Specific Cre/loxP Recombination System. Biomolecules. 2018; 8(4):137. https://doi.org/10.3390/biom8040137
Chicago/Turabian StyleYang, Ying-Jie, Raghvendra Pratap Singh, Xin Lan, Cheng-Sheng Zhang, Yue-Zhong Li, Yi-Qiang Li, and Duo-Hong Sheng. 2018. "Genome Editing in Model Strain Myxococcus xanthus DK1622 by a Site-Specific Cre/loxP Recombination System" Biomolecules 8, no. 4: 137. https://doi.org/10.3390/biom8040137
APA StyleYang, Y.-J., Singh, R. P., Lan, X., Zhang, C.-S., Li, Y.-Z., Li, Y.-Q., & Sheng, D.-H. (2018). Genome Editing in Model Strain Myxococcus xanthus DK1622 by a Site-Specific Cre/loxP Recombination System. Biomolecules, 8(4), 137. https://doi.org/10.3390/biom8040137