Structural Transition and Antibody Binding of EBOV GP and ZIKV E Proteins from Pre-Fusion to Fusion-Initiation State

 , , , , , ,
, , , , , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
- A trimer model of EBOV GP in the fusion-initiation state with the Niemann–Pick C1 (NPC1) receptor and neutralizing antibodies.
- A trimer model of ZIKV E in the fusion-initiation state with neutralizing antibodies and the surrounding 9-mer structure of ZIKV E in the pre-fusion state with neutralizing antibodies.
2. Results
2.1. Pre-Fusion and Fusion-Initiation State Structures of EBOV GP (“Spring-Loaded Model”)
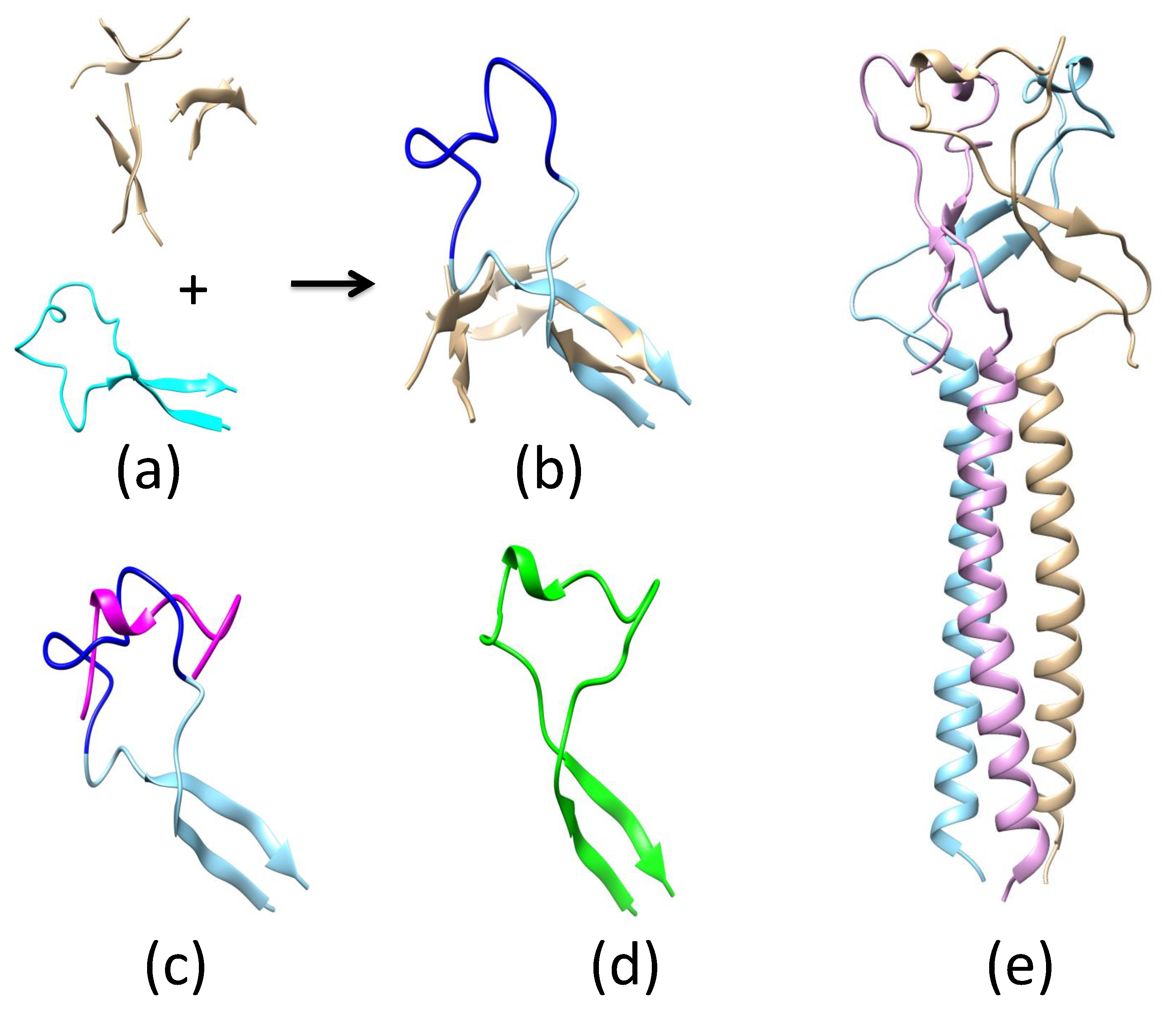
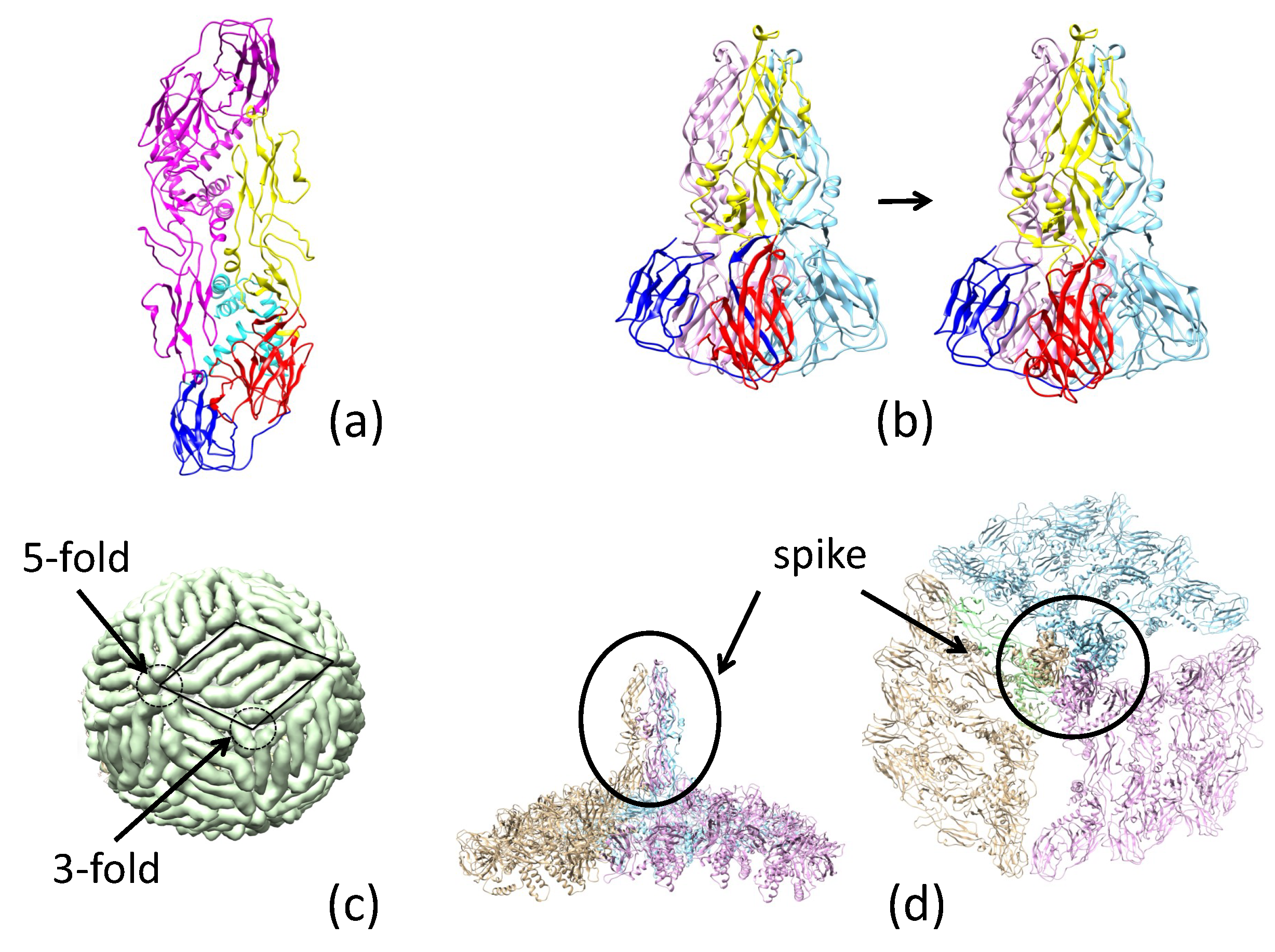
2.2. Pre-Fusion and Fusion Structure of ZIKV E
2.3. Neutralizing Antibody Blocks Viral Entry by Pre-Fusion and Fusion-Initiation-State Interactions
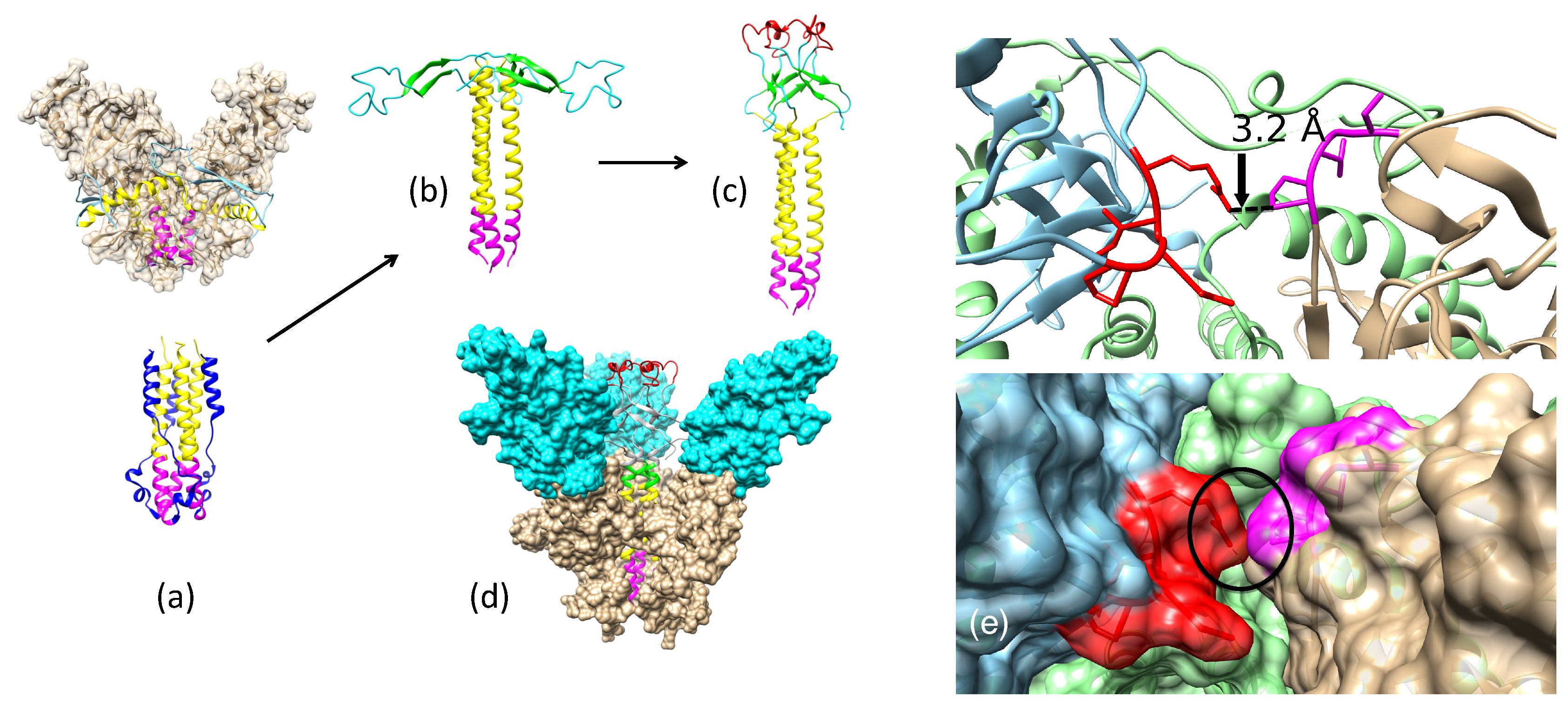
2.3.1. Antibody KZ52 Blocks EBOV Entry by Preventing GP Structural Transition from a Pre-Fusion to a Fusion-Initiation State
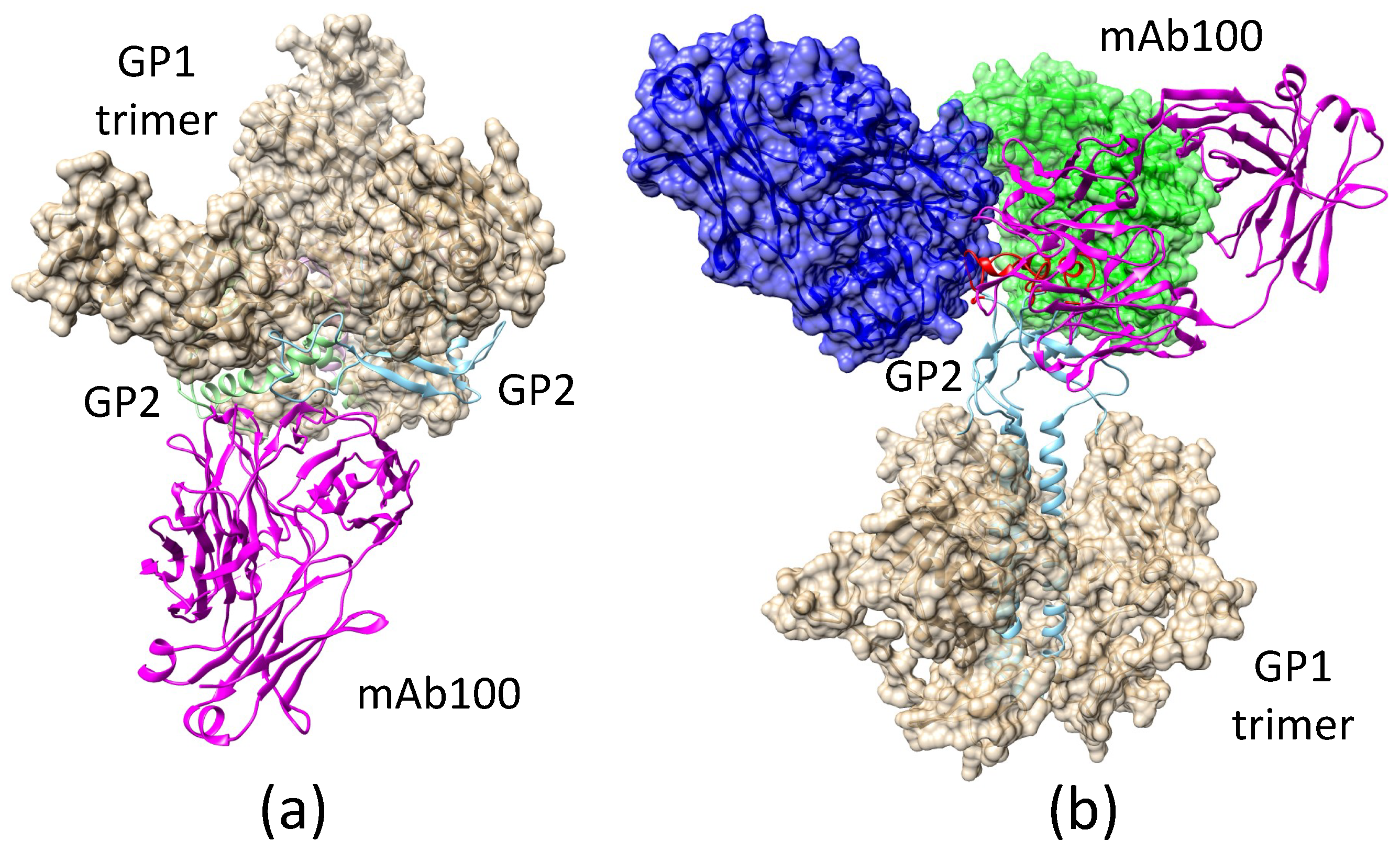
2.3.2. Antibody mAb100 Blocks EBOV Entry through Two Different Mechanisms
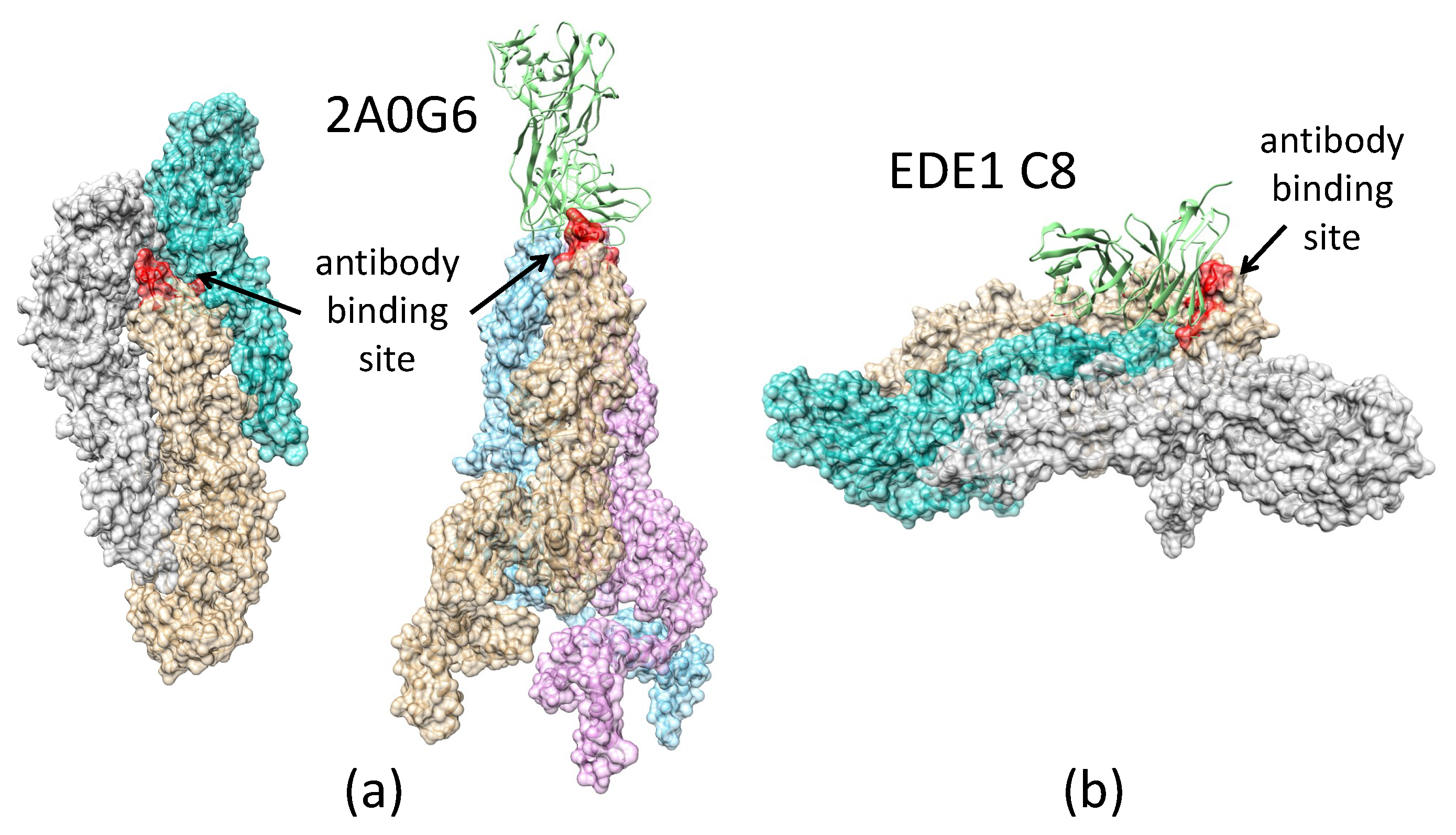
2.3.3. Antibody 2A0G6 Binds to ZIKV E in the Fusion-Initiation State and Blocks the Exposure of the Fusion Peptide
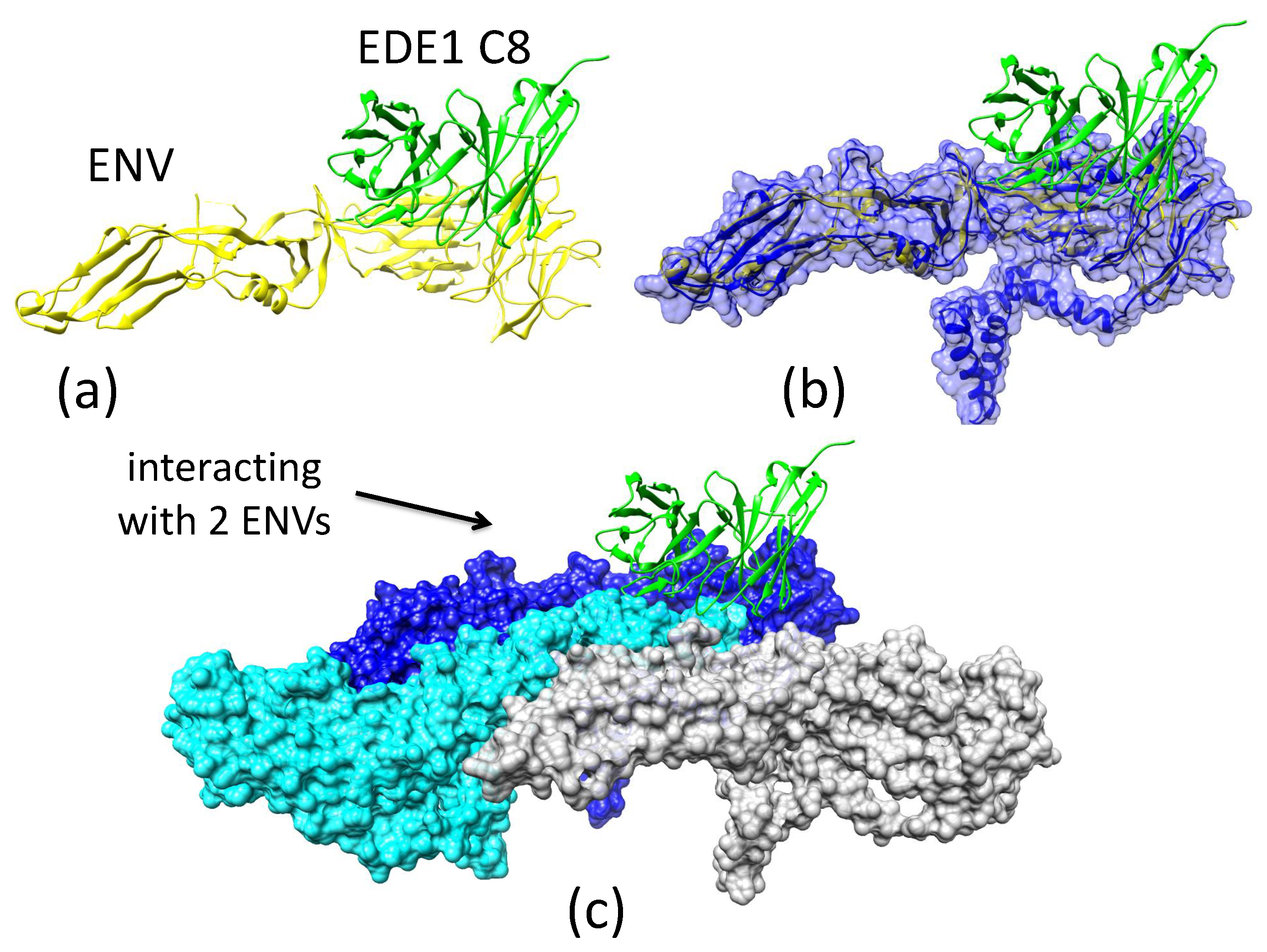
2.3.4. Antibody EDE1-C8 Blocks the Pre-Fusion to Fusion Transition of ZIKV E and Prevents Cell Entry
2.3.5. Videos Describing the Transitions from the Pre-Fusion to Fusion Structures
3. Discussion
4. Materials and Methods
4.1. Homology Modeling Using a Motif-Matching Fragment Assembly Method
4.2. Structure Alignment and Superposition
4.3. Structural Refinement
4.4. Graphics
5. Conclusions
Supplementary Materials
Author Contributions
Acknowledgments
Conflicts of Interest
Appendix A
Appendix A.1. Modeling Fusion-State Structure of EBOV GP Using the Spring-Loaded Mechanism
Appendix A.2. Remodeling the Fusion Loop Structure
Appendix A.3. Determination of ZIKV E Domain III-1 Placement in the Fusion-State Structure
Appendix A.4. Binding of EDE1 C8 to ZIKV E’s on the Surface of the Virus
Appendix A.5. Animation
References
- Eckert, D.M.; Kim, P.S. Mechanisms of viral membrane fusion and its inhibition. Annu. Rev. Biochem. 2001, 70, 777–810. [Google Scholar] [CrossRef] [PubMed]
- Kielian, M.; Jungerwirth, S. Mechanisms of enveloped virus entry into cells. Mol. Biol. Med. 1990, 7, 17–31. [Google Scholar] [PubMed]
- Dimitrov, D.S. Virus entry: Molecular mechanisms and biomedical applications. Nat. Rev. Microbiol. 2004, 2, 109–122. [Google Scholar] [CrossRef] [PubMed]
- Más, V.; Melero, J.A. Entry of Enveloped Viruses into Host Cells: Membrane Fusion; Springer: Dordrecht, The Netherlands, 2013. [Google Scholar]
- Sullivan, N.; Sun, Y.; Sattentau, Q.; Thali, M.; Wu, D.; Denisova, G.; Gershoni, J.; Robinson, J.; Moore, J.; Sodroski, J. CD4-induced conformational changes in the human immunodeficiency virus Type 1 gp120 glycoprotein: Consequences for virus entry and neutralization. J. Virol. 1998, 72, 4694–4703. [Google Scholar] [PubMed]
- Riedel, C.; Vasishtan, D.; Siebert, C.A.; Whittle, C.; Lehmann, M.J.; Mothes, W.; Grünewald, K. Native structure of a retroviral envelope protein and its conformational change upon interaction with the target cell. J. Struct. Biol. 2017, 197, 172–180. [Google Scholar] [CrossRef] [PubMed]
- Tran, E.E.H.; Borgnia, M.J.; Kuybeda, O.; Schauder, D.M.; Bartesaghi, A.; Frank, G.A.; Sapiro, G.; Milne, J.L.S.; Subramaniam, S. Structural Mechanism of Trimeric HIV-1 Envelope Glycoprotein Activation. PLOS Pathog. 2012, 8, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Wilen, C.B.; Tilton, J.; Doms, R. HIV: Cell binding and entry. Cold Spring Harb. Perspect. Med. 2012, 2, a006866. [Google Scholar] [CrossRef] [PubMed]
- Berman, H.; Westbrook, J.; Feng, Z.; Gilliland, G.; Bhat, T.; Weissig, H.; Bourne, P. The Protein Data Bank. Nucleic Acids Res. 2000, 28, 235–242. [Google Scholar] [CrossRef] [PubMed]
- Ebola Data and Statistics. World Health Organization Situation Summary Data Published on 31 July 2015. Available online: http://apps.who.int/gho/data/view.ebola-sitrep.ebola-summary-20150731?lang=en (accessed on 17 July 2017).
- Malone, R.W.; Homan, J.; Callahan, M.V.; Glasspool-Malone, J.; Damodaran, L.; Schneider, A.D.B.; Zimler, R.; Talton, J.; Cobb, R.R.; Ruzic, I.; et al. Zika virus: Medical countermeasure development challenges. PLOS Negl. Trop. Dis. 2016, 10, e0004530. [Google Scholar] [CrossRef] [PubMed]
- Cunha, M.S.; Esposito, D.L.A.; Rocco, I.M.; Maeda, A.Y.; Vasami, F.G.S.; Nogueira, J.S.; de Souza, R.P.; Suzuki, A.; Addas-Carvalho, M.; Barjas-Castro, M.d.L.; et al. First complete genome sequence of Zika Virus (Flaviviridae, Flavivirus) from an autochthonous transmission in Brazil. Genome Announc. 2016, 4, e00032-16. [Google Scholar] [CrossRef] [PubMed]
- Dang, J.; Tiwari, S.K.; Lichinchi, G.; Qin, Y.; Patil, V.S.; Eroshkin, A.M.; Rana, T.M. Zika virus depletes neural progenitors in human cerebral organoids through activation of the innate immune receptor TLR3. Cell Stem Cell 2016, 19, 258–265. [Google Scholar] [CrossRef] [PubMed]
- Rasmussen, S.A.; Jamieson, D.J.; Honein, M.A.; Petersen, L.R. Zika virus and birth defects—Reviewing the evidence for causality. N. Engl. J. Med. 2016, 374, 1981–1987. [Google Scholar] [CrossRef] [PubMed]
- Huang, W.C.; Abraham, R.; Shim, B.S.; Choe, H.; Page, D.T. Zika virus infection during the period of maximal brain growth causes microcephaly and corticospinal neuron apoptosis in wild type mice. Sci. Rep. 2016, 6, 34793. [Google Scholar] [CrossRef] [PubMed]
- Govero, J.; Esakky, P.; Scheaffer, S.M.; Fernandez, E.; Drury, A.; Platt, D.J.; Gorman, M.J.; Richner, J.M.; Caine, E.A.; Salazar, V.; et al. Zika virus infection damages the testes in mice. Nature 2016, 540, 438–442. [Google Scholar] [CrossRef] [PubMed]
- Souza, B.S.F.; Sampaio, G.L.A.; Pereira, C.S.; Campos, G.S.; Sardi, S.I.; Freitas, L.A.R.; Figueira, C.P.; Paredes, B.D.; Nonaka, C.K.V.; Azevedo, C.M.; et al. Zika virus infection induces mitosis abnormalities and apoptotic cell death of human neural progenitor cells. Sci. Rep. 2016, 6, 39775. [Google Scholar] [CrossRef] [PubMed]
- Zika Data and Statistics. World Health Organization Situation Summary Data Published on 10 March 2017. Available online: http://www.who.int/emergencies/zika-virus/situation-report/10-march-2017/en/ (accessed on 17 July 2017).
- Quick, J.; Loman, N.J.; Duraffour, S.; Simpson, J.T.; Severi, E.; Cowley, L.; Bore, J.A.; Koundouno, R.; Dudas, G.; Mikhail, A.; et al. Real-time, portable genome sequencing for Ebola surveillance. Nature 2016, 530, 228–232. [Google Scholar] [CrossRef] [PubMed]
- Nanbo, A.; Imai, M.; Watanabe, S.; Noda, T.; Takahashi, K.; Neumann, G.; Halfmann, P.; Kawaoka, Y. Ebolavirus is internalized into host cells via macropinocytosis in a viral glycoprotein-dependent manner. PLOS Pathog. 2010, 6, 1–20. [Google Scholar] [CrossRef] [PubMed]
- Aleksandrowicz, P.; Marzi, A.; Biedenkopf, N.; Beimforde, N.; Becker, S.; Hoenen, T.; Feldmann, H.; Schnittler, H.J. Ebola virus enters host cells by macropinocytosis and clathrin-mediated endocytosis. J. Infect. Dis. 2011, 204, S957–S967. [Google Scholar] [CrossRef] [PubMed]
- Volchkov, V.E.; Feldmann, H.; Volchkova, V.A.; Klenk, H.D. Processing of the Ebola virus glycoprotein by the proprotein convertase furin. Proc. Natl. Acad. Sci. USA 1998, 95, 5762–5767. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Manicassamy, B.; Caffrey, M.; Rong, L. Characterization of the receptor-binding domain of Ebola glycoprotein in viral entry. Virol. Sin. 2011, 26, 156–170. [Google Scholar] [CrossRef] [PubMed]
- Manicassamy, B.; Wang, J.; Jiang, H.; Rong, L. Comprehensive analysis of Ebola Virus GP1 in viral entry. J. Virol. 2005, 79, 4793–4805. [Google Scholar] [CrossRef] [PubMed]
- Weissenhorn, W.; Carfí, A.; Lee, K.H.; Skehel, J.J.; Wiley, D.C. Crystal structure of the Ebola Virus Membrane Fusion Subunit, GP2, from the Envelope Glycoprotein ectodomain. Mol. Cell 1998, 2, 605–616. [Google Scholar] [CrossRef]
- Lee, J.E.; Fusco, M.L.; Hessell, A.J.; Oswald, W.B.; Burton, D.R.; Saphire, E.O. Structure of the Ebola virus glycoprotein bound to an antibody from a human survivor. Nature 2008, 454, 177–182. [Google Scholar] [CrossRef] [PubMed]
- Brister, J.R.; Bao, Y.; Zhdanov, S.A.; Ostapchuck, Y.; Chetvernin, V.; Kiryutin, B.; Zaslavsky, L.; Kimelman, M.; Tatusova, T.A. Virus variation resource—Recent updates and future directions. Nucleic Acids Res. 2014, 42, D660–D665. [Google Scholar] [CrossRef] [PubMed]
- Malashkevich, V.N.; Schneider, B.J.; McNally, M.L.; Milhollen, M.A.; Pang, J.X.; Kim, P.S. Core structure of the envelope glycoprotein GP2 from Ebola virus at 1.9-Å resolution. Proc. Natl. Acad. Sci. USA 1999, 96, 2662–2667. [Google Scholar] [CrossRef] [PubMed]
- Dias, J.M.; Kuehne, A.I.; Abelson, D.M.; Bale, S.; Wong, A.C.; Halfmann, P.; Muhammad, M.A.; Fusco, M.L.; Zak, S.E.; Kang, E.; et al. A shared structural solution for neutralizing Ebolaviruses. Nat. Struct. Mol. Biol. 2011, 18, 1424–1427. [Google Scholar] [CrossRef] [PubMed]
- Bale, S.; Dias, J.M.; Fusco, M.L.; Hashiguchi, T.; Wong, A.C.; Liu, T.; Keuhne, A.I.; Li, S.; Woods, V.L.; Chandran, K.; et al. Structural basis for differential neutralization of Ebolaviruses. Viruses 2012, 4, 447–470. [Google Scholar] [CrossRef] [PubMed]
- Freitas, M.S.; Gaspar, L.P.; Lorenzoni, M.; Almeida, F.C.L.; Tinoco, L.W.; Almeida, M.S.; Maia, L.F.; Degrève, L.; Valente, A.P.; Silva, J.L. Structure of the Ebola Fusion Peptide in a membrane-mimetic environment and the interaction with lipid rafts. J. Biol. Chem. 2007, 282, 27306–27314. [Google Scholar] [CrossRef] [PubMed]
- Gregory, S.M.; Harada, E.; Liang, B.; Delos, S.E.; White, J.M.; Tamm, L.K. Structure and function of the complete internal fusion loop from Ebolavirus glycoprotein 2. Proc. Natl. Acad. Sci. USA 2011, 108, 11211–11216. [Google Scholar] [CrossRef] [PubMed]
- Murin, C.D.; Fusco, M.L.; Bornholdt, Z.A.; Qiu, X.; Olinger, G.G.; Zeitlin, L.; Kobinger, G.P.; Ward, A.B.; Saphire, E.O. Structures of protective antibodies reveal sites of vulnerability on Ebola virus. Proc. Natl. Acad. Sci. USA 2014, 111, 17182–17187. [Google Scholar] [CrossRef] [PubMed]
- Tran, E.E.H.; Simmons, J.A.; Bartesaghi, A.; Shoemaker, C.J.; Nelson, E.; White, J.M.; Subramaniam, S. Spatial localization of the Ebola Virus Glycoprotein Mucin-Like Domain determined by Cryo-Electron Tomography. J. Virol. 2014, 88, 10958–10962. [Google Scholar] [CrossRef] [PubMed]
- Lindenbach, B.D.; Rice, C.M. Molecular biology of flaviviruses. Adv. Virus Res. 2003, 59, 23–61. [Google Scholar] [PubMed]
- Modis, Y.; Ogata, S.; Clements, D.; Harrison, S.C. Structure of the dengue virus envelope protein after membrane fusion. Nature 2004, 427, 313–319. [Google Scholar] [CrossRef] [PubMed]
- Carr, C.M.; Kim, P.S. A spring-loaded mechanism for the conformational change of influenza hemagglutinin. Cell 1993, 73, 823–832. [Google Scholar] [CrossRef]
- Carr, C.M.; Chaudhry, C.; Kim, P.S. Influenza hemagluttinin is spring-loaded by a metastable native conformation. Proc. Natl. Acad. Sci. USA 1997, 94, 14306–14313. [Google Scholar] [CrossRef] [PubMed]
- White, J.M.; Delos, S.E.; Brecher, M.; Schornberg, K. Structures and mechanisms of viral membrane fusion proteins. Crit. Rev. Biochem. Mol. Biol. 2008, 43, 189–219. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Shi, Y.; Song, J.; Qi, J.; Lu, G.; Yan, J.; Gao, G. Ebola viral Glycoprotein bound to its endosomal receptor Niemann-Pick {C1}. Cell 2016, 164, 258–268. [Google Scholar] [CrossRef] [PubMed]
- Dube, D.; Schornberg, K.L.; Shoemaker, C.J.; Delos, S.E.; Stantchev, T.S.; Clouse, K.A.; Broder, C.C.; White, J.M. Cell adhesion-dependent membrane trafficking of a binding partner for the ebolavirus glycoprotein is a determinant of viral entry. Proc. Natl. Acad. Sci. USA 2010, 107, 16637–16642. [Google Scholar] [CrossRef] [PubMed]
- Stiasny, K.; Heinz, F.X. Flavivirus membrane fusion. J. Gen. Virol. 2006, 87, 2755–2766. [Google Scholar] [CrossRef] [PubMed]
- Stiasny, K.; Fritz, R.; Pangerl, K.; Heinz, F.X. Molecular mechanisms of flavivirus membrane fusion. Amino Acids 2011, 41, 1159–1163. [Google Scholar] [CrossRef] [PubMed]
- Moller-Tank, S.; Maury, W. Ebola Virus entry: A curious and complex series of events. PLOS Pathog. 2015, 11, e1004731. [Google Scholar] [CrossRef] [PubMed]
- Misasi, J.; Gilman, M.S.A.; Kanekiyo, M.; Gui, M.; Cagigi, A.; Mulangu, S.; Corti, D.; Ledgerwood, J.E.; Lanzavecchia, A.; Cunningham, J.; et al. Structural and molecular basis for Ebola virus neutralization by protective human antibodies. Science 2016, 351, 1343–1346. [Google Scholar] [CrossRef] [PubMed]
- Dai, L.; Song, J.; Lu, X.; Deng, Y.Q.; Musyoki, A.M.; Cheng, H.; Zhang, Y.; Yuan, Y.; Song, H.; Haywood, J.; et al. Structures of the Zika Virus Envelope Protein and its complex with a flavivirus broadly protective antibody. Cell Host Microbe 2016, 19, 696–704. [Google Scholar] [CrossRef] [PubMed]
- Tung, C.S.; McMahon, B.H. A structural model of the E. coli PhoB Dimer in the transcription initiation complex. BMC Struct. Biol. 2012, 12, 3. [Google Scholar] [CrossRef] [PubMed]
- Meng, E.; Pattersen, E.; Souch, G.; Huang, C.; Ferrin, T. Tools for integrated sequence-structure analysis with UCSF Chimera. BMC Bioinform. 2006, 7. [Google Scholar]
- Hess, B.; Kutzner, C.; van der Spoel, D.; Lindahl, E. GROMACS 4: Algorithms for highly efficient, load-balanced, and scalable molecular simulation. J. Chem. Theory Comput. 2008, 4, 435–447. [Google Scholar] [CrossRef] [PubMed]
- Pattersen, E.; Goddard, T.; Huang, C.; Couch, G.; Greenblatt, D.; Meng, E.; Ferrin, T. UCSF Chimera—A visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef] [PubMed]
- Sikic, K.; Tomic, S.; Carugo, O. Systematic comparison of crystal and NMR protein structures deposited in the Protein Data Bank. Open Biochem. J. 2010, 4, 83–95. [Google Scholar] [CrossRef] [PubMed]
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lappala, A.; Nishima, W.; Miner, J.; Fenimore, P.; Fischer, W.; Hraber, P.; Zhang, M.; McMahon, B.; Tung, C.-S. Structural Transition and Antibody Binding of EBOV GP and ZIKV E Proteins from Pre-Fusion to Fusion-Initiation State. Biomolecules 2018, 8, 25. https://doi.org/10.3390/biom8020025
Lappala A, Nishima W, Miner J, Fenimore P, Fischer W, Hraber P, Zhang M, McMahon B, Tung C-S. Structural Transition and Antibody Binding of EBOV GP and ZIKV E Proteins from Pre-Fusion to Fusion-Initiation State. Biomolecules. 2018; 8(2):25. https://doi.org/10.3390/biom8020025
Chicago/Turabian StyleLappala, Anna, Wataru Nishima, Jacob Miner, Paul Fenimore, Will Fischer, Peter Hraber, Ming Zhang, Benjamin McMahon, and Chang-Shung Tung. 2018. "Structural Transition and Antibody Binding of EBOV GP and ZIKV E Proteins from Pre-Fusion to Fusion-Initiation State" Biomolecules 8, no. 2: 25. https://doi.org/10.3390/biom8020025
APA StyleLappala, A., Nishima, W., Miner, J., Fenimore, P., Fischer, W., Hraber, P., Zhang, M., McMahon, B., & Tung, C.-S. (2018). Structural Transition and Antibody Binding of EBOV GP and ZIKV E Proteins from Pre-Fusion to Fusion-Initiation State. Biomolecules, 8(2), 25. https://doi.org/10.3390/biom8020025