PIM Kinases and Their Relevance to the PI3K/AKT/mTOR Pathway in the Regulation of Ovarian Cancer


Abstract
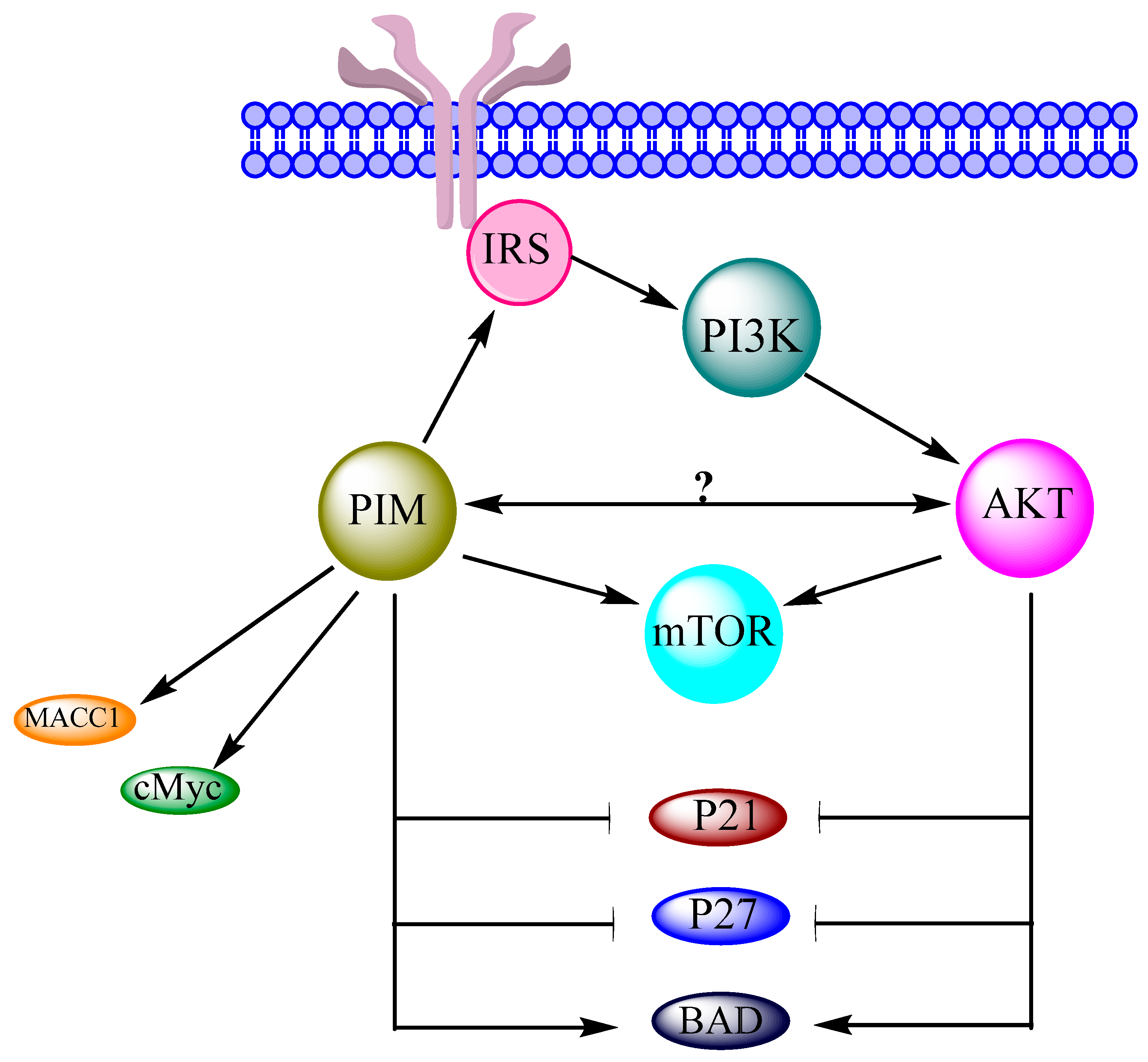
:1. Introduction
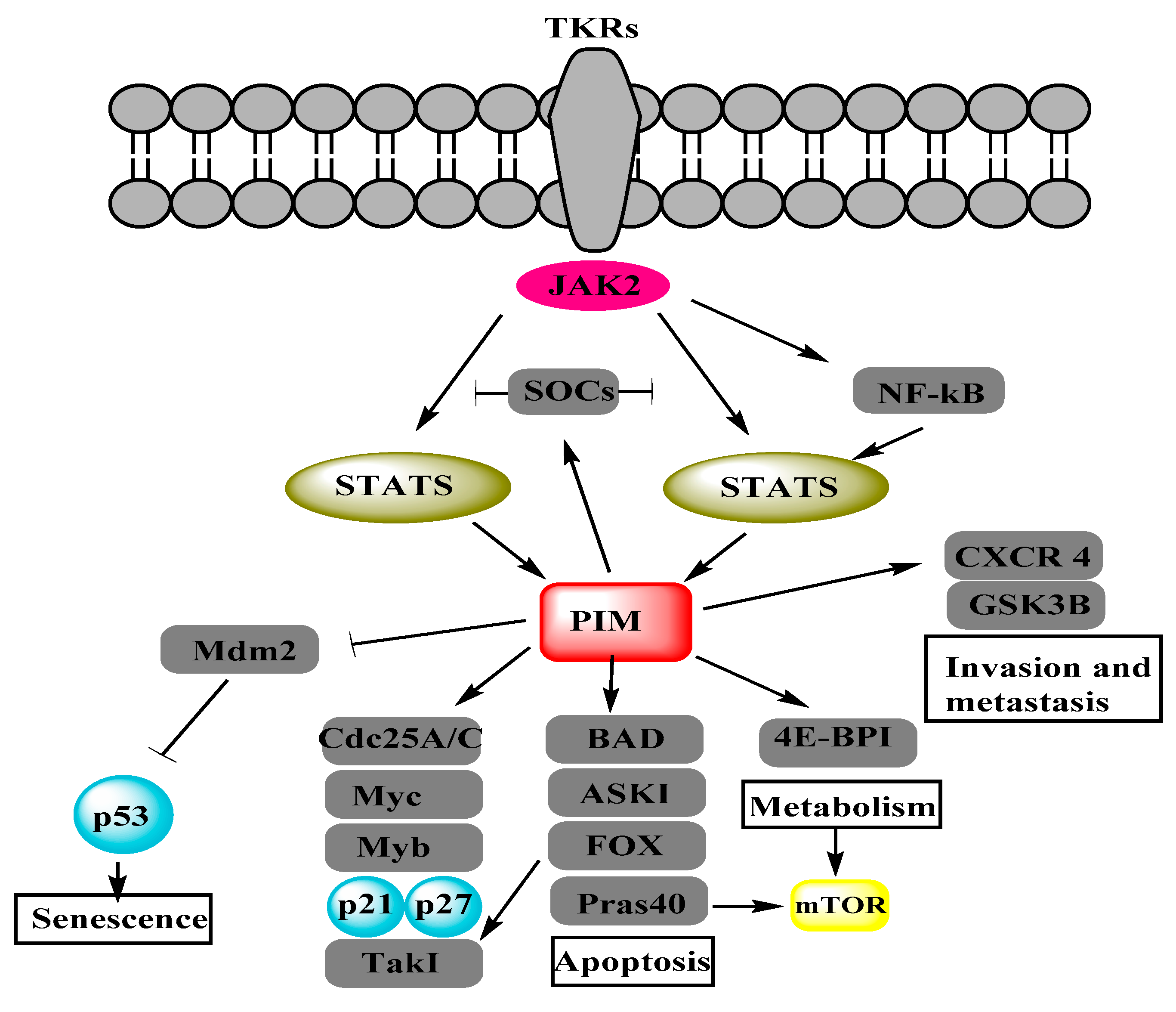
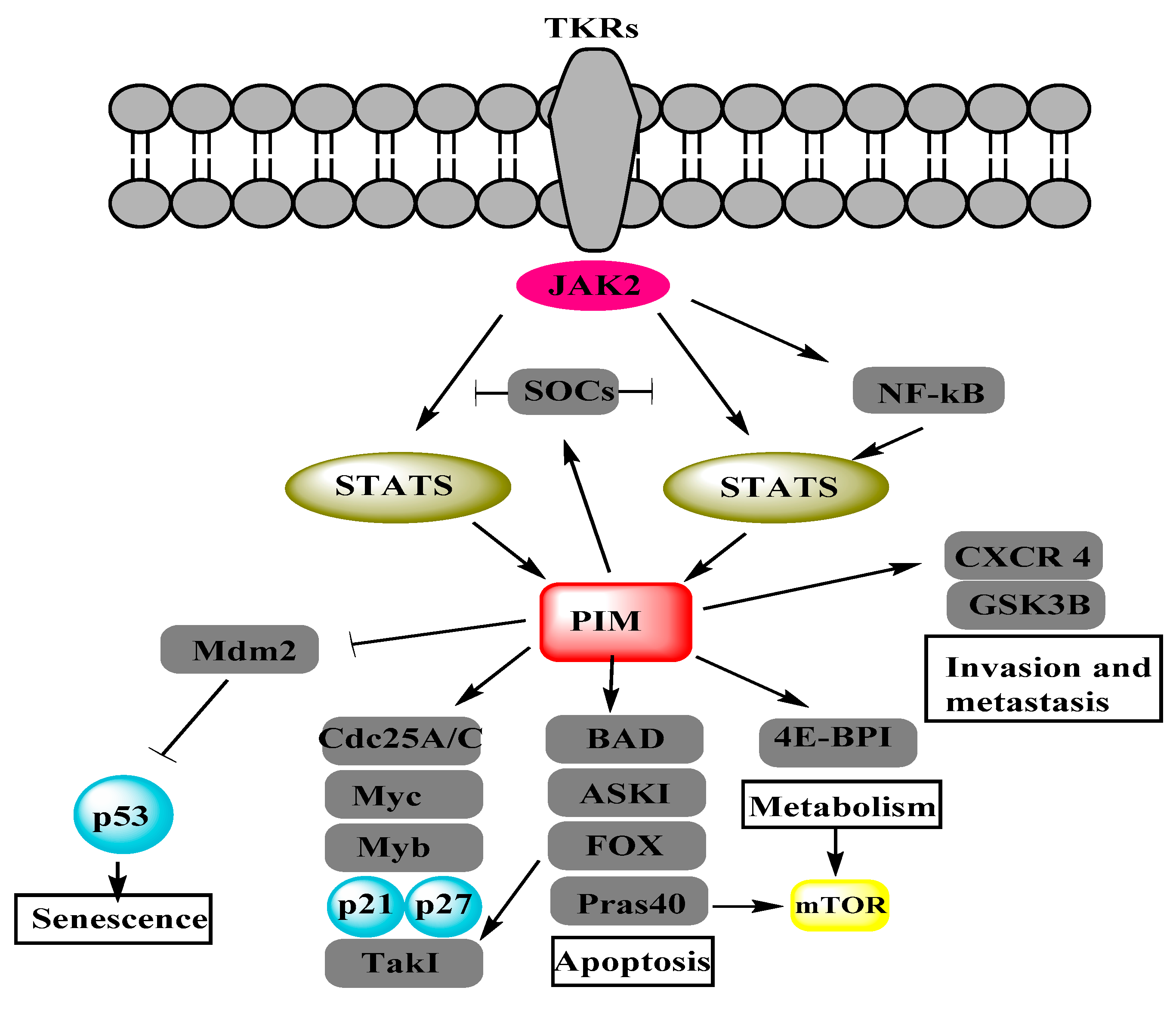
2. Interrelation of PI3K/AKT/mTOR and PIM Pathways in Other Cancers
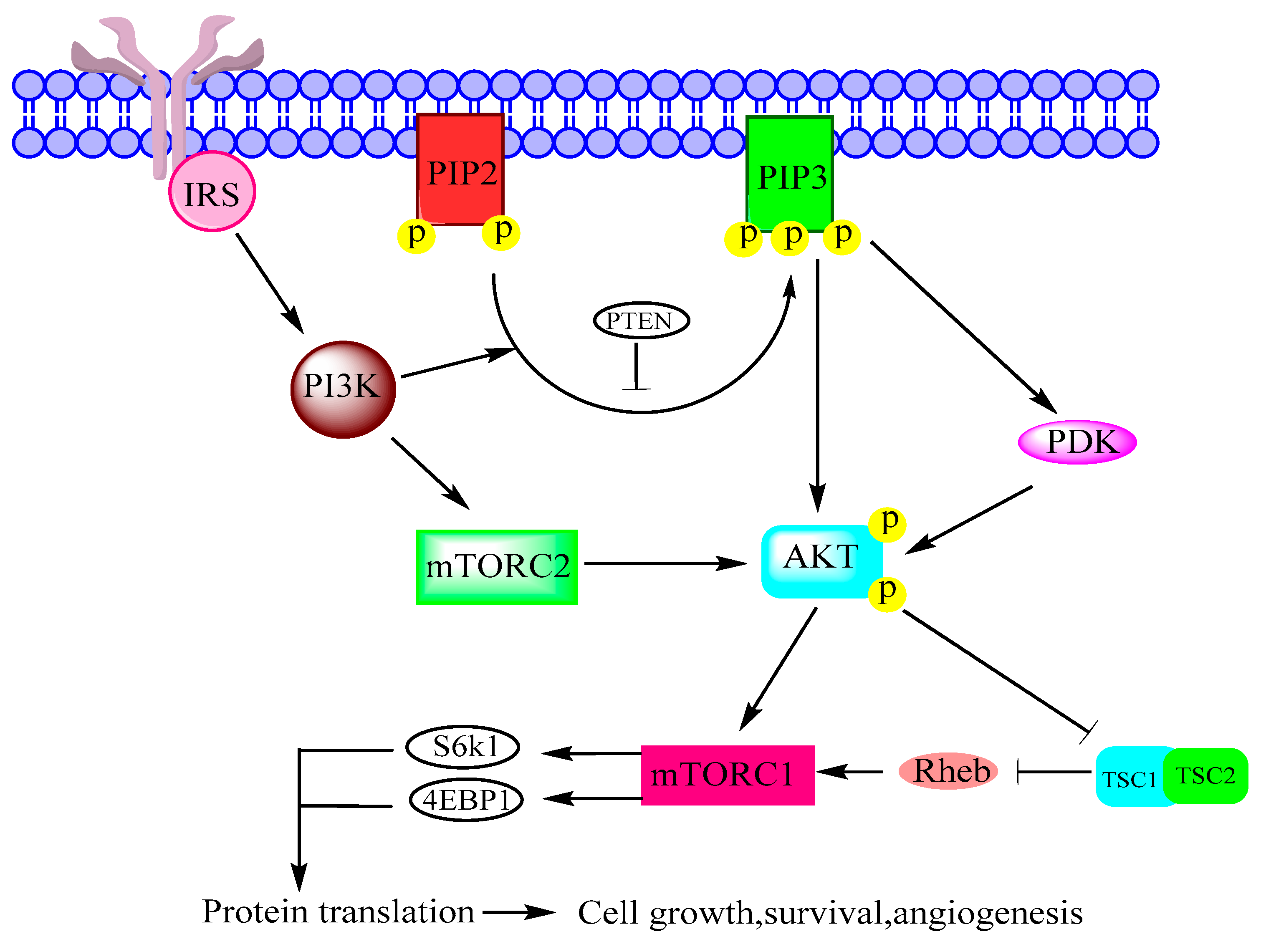
2.1. PI3K
2.2. AKT
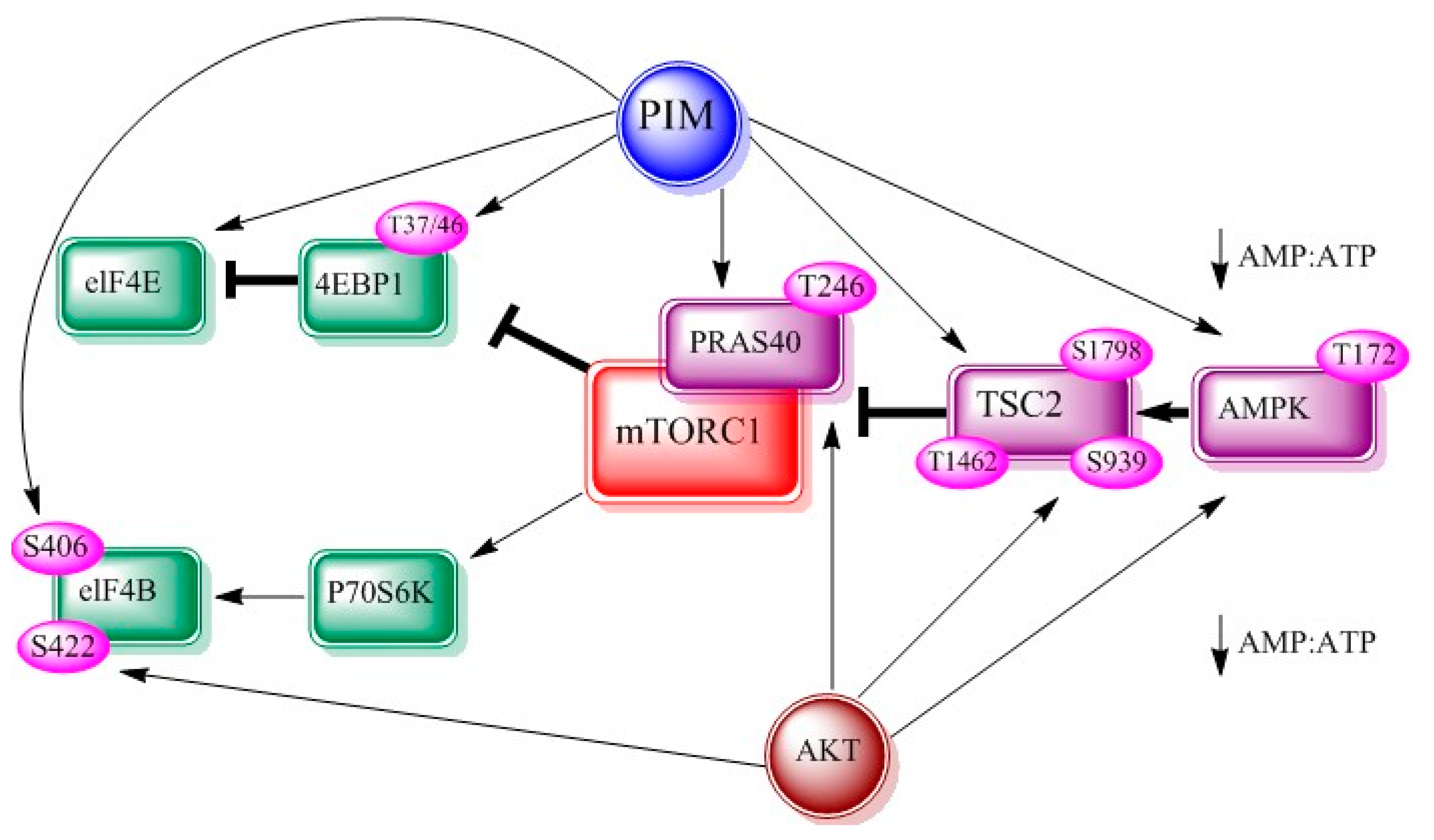
2.3. mTOR
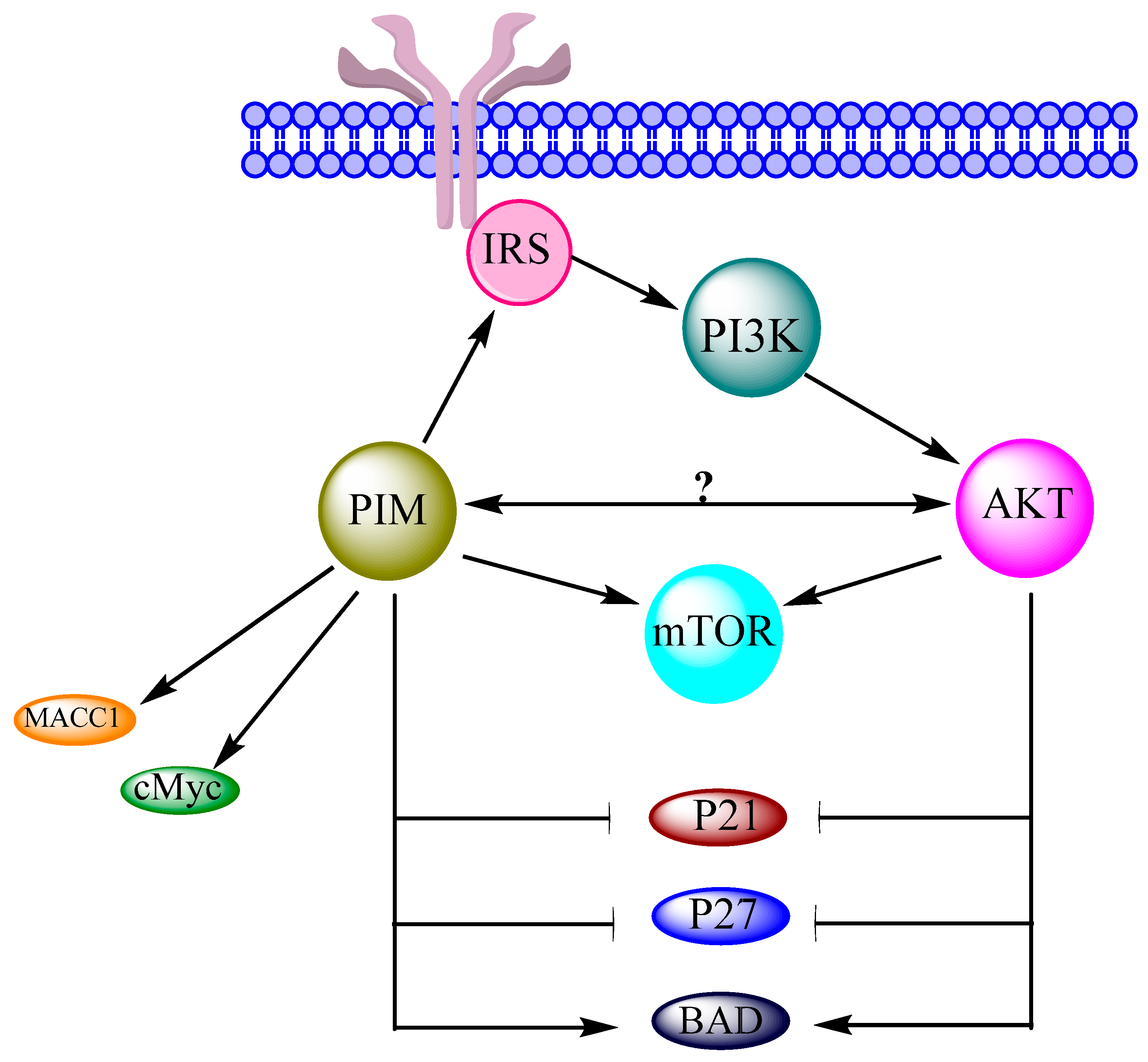
3. Interrelation of PI3K/AKT/mTOR and PIM Pathways in Ovarian Cancer
3.1. PI3K
3.2. AKT
3.3. mTOR
4. PIM Kinases and Proteins Involved in Cellular Functions of Ovarian Cancer Cells
4.1. cMyc
4.2. BAD
4.3. Cyclin-Dependent Kinases
4.4. Metastasis-Associated in Colon Cancer-1
5. Summary
6. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Yeung, T.L.; Leung, C.S.; Li, F.; Wong, S.S.; Mok, S.C. Targeting stromal-cancer cell crosstalk networks in ovarian cancer treatment. Biomolecules 2016, 6, 3. [Google Scholar] [CrossRef] [PubMed]
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2017. CA Cancer J. Clin. 2017, 67, 7–30. [Google Scholar] [CrossRef] [PubMed]
- Gizzo, S.; Noventa, M.; Quaranta, M.; Vitagliano, A.; Saccardi, C.; Litta, P.; Antona, D. A novel hysteroscopic approach for ovarian cancer screening/early diagnosis. Oncol. Lett. 2017, 13, 549–553. [Google Scholar] [CrossRef] [PubMed]
- Meinhold-Heerlein, I.; Hauptmann, S. The heterogeneity of ovarian cancer. Arch. Gynecol. Obstet. 2014, 289, 237–239. [Google Scholar] [CrossRef] [PubMed]
- Types and Stages of Ovarian Cancer. Available online: http://ovarian.org/about-ovarian-cancer/what-is-ovarian-cancer/types-a-stages (accessed on 14 January 2018).
- Mabuchi, S.; Kuroda, H.; Takahashi, R.; Sasano, T. The PI3K/AKT/mTOR pathway as a therapeutic target in ovarian cancer. Gynecol. Oncol. 2015, 137, 173–179. [Google Scholar] [CrossRef] [PubMed]
- Blanco-Aparicio, C.; Carnero, A. PIM kinases in cancer: Diagnostic, prognostic and treatment opportunities. Biochem. Pharmacol. 2013, 85, 629–643. [Google Scholar] [CrossRef] [PubMed]
- Warfel, N.A.; Kraft, A.S. PIM kinase (and AKT) biology and signaling in tumors. Pharmacol. Ther. 2015, 151, 41–49. [Google Scholar] [CrossRef] [PubMed]
- Tursynbay, Y.; Zhang, J.; Li, Z.; Tokay, T.; Zhumadilov, Z.; Wu, D.; Xie, Y. PIM-1 kinase as cancer drug target: An update. Biomed. Rep. 2016, 4, 140–146. [Google Scholar] [CrossRef] [PubMed]
- Xie, J.; Bai, J. SGI-1776, an imidazo pyridazine compound, inhibits the proliferation of ovarian cancer cells by inactivating PIM-1. Zhong Nan Da Xue Xue Bao Yi Xue Ban 2014, 39, 649–657. [Google Scholar] [PubMed]
- Zhuang, H.; Zhao, M.Y.; Hei, K.W.; Yang, B.C.; Sun, L.; Du, X.; Li, Y.M. Aberrant expression of PIM-3 promotes proliferation and migration of ovarian cancer cells. Asian Pac. J. Cancer Prev. 2015, 16, 3325–3331. [Google Scholar] [CrossRef] [PubMed]
- Domcke, S.; Sinha, R.; Levine, D.A.; Sander, C.; Schultz, N. Evaluating cell lines as tumour models by comparison of genomic profiles. Nat. Commun. 2013, 4. [Google Scholar] [CrossRef] [PubMed]
- Musiani, D.; Hammond, D.E.; Cirillo, L.; Erriquez, J.; Olivero, M.; Clague, M.J.; Di Renzo, M.F. PIM2 kinase is induced by cisplatin in ovarian cancer cells and limits drug efficacy. J. Proteome Res. 2014, 13, 4970–4982. [Google Scholar] [CrossRef] [PubMed]
- Tipton, A.R.; Nyabuto, G.O.; Trendel, J.A.; Mazur, T.M.; Wilson, J.P.; Wadi, S.; Justinger, J.S.; Moore, G.L.; Nguyen, P.T.; Vestal, D.J. Guanylate-binding protein-1 protects ovarian cancer cell lines but not breast cancer cell lines from killing by paclitaxel. Biochem. Biophys. Res. Commun. 2016, 478, 1617–1623. [Google Scholar] [CrossRef] [PubMed]
- Dobbin, Z.C.; Landen, C.N. The importance of the PI3K/AKT/mTOR pathway in the progression of ovarian cancer. Int. J. Mol. Sci. 2013, 14, 8213–8227. [Google Scholar] [CrossRef] [PubMed]
- Cheaib, B.; Auguste, A.; Leary, A. The PI3K/AKT/mTOR pathway in ovarian cancer: Therapeutic opportunities and challenges. Chin. J. Cancer 2015, 34, 4–16. [Google Scholar] [CrossRef] [PubMed]
- Rebello, R.J.; Huglo, A.V.; Furic, L. PIM activity in tumours: A key node of therapy resistance. Adv. Biol. Regul. 2017. [Google Scholar] [CrossRef] [PubMed]
- Song, J.H.; Padi, S.K.; Luevano, L.A.; Minden, M.D.; DeAngelo, D.J.; Hardiman, G.; Ball, L.E.; Warfel, N.A.; Kraft, A.S. Insulin receptor substrate 1 is a substrate of the PIM protein kinases. Oncotarget 2016, 7, 20152–20165. [Google Scholar] [CrossRef] [PubMed]
- Brunen, D.; Garcia-Barchino, M.J.; Malani, D.; Basheer, N.J.; Lieftink, C.; Beijersbergen, R.L.; Murumagi, A.; Porkka, K.; Wolf, M.; Zwaan, C.M.; et al. Intrinsic resistance to PIM kinase inhibition in AML through p38a-mediated feedback activation of mTOR signaling. Oncotarget 2016, 7, 37407–37419. [Google Scholar] [CrossRef] [PubMed]
- Chen, K.; Li, J.N.; Chen, J.L. eIF4B is a key effector of oncogenic PIM and PI3K/AKT/mTOR signaling pathways during ABL-mediated cellular transformation. Cancer Res. 2016, 76. [Google Scholar] [CrossRef]
- Janus, J.M.; O’Shaughnessy, R.F.L.; Harwood, C.A.; Maffucci, T. Phosphoinositide 3-kinase-dependent signalling pathways in cutaneous squamous cell carcinomas. Cancers 2017, 9, 86. [Google Scholar] [CrossRef] [PubMed]
- Mantamadiotis, T. Towards targeting PI3K-dependent regulation of gene expression in brain cancer. Cancers 2017, 9, 60. [Google Scholar] [CrossRef] [PubMed]
- Crumbaker, M.; Khoja, L.; Joshua, A.M. AR signaling and the PI3K pathway in prostate cancer. Cancers 2017, 9, 34. [Google Scholar] [CrossRef] [PubMed]
- Piddock, R.E.; Bowles, K.M.; Rushworth, S.A. The role of PI3K isoforms in regulating bone marrow microenvironment signaling focusing on acute myeloid leukemia and multiple myeloma. Cancers 2017, 9, 29. [Google Scholar] [CrossRef] [PubMed]
- Freysoldt, B.; Schnaiter, A.; Fischer, L.; O’Neill, M.; Zimmermann, Y.; Hutter, G.; Hiddemann, W.; Dreyling, M.H. Cotargeting of PIM, PI3K and mTOR in mantle cell lymphoma (MCL). Blood 2015, 126, 5120. [Google Scholar]
- Reidy, M.; vanDijk, M.; Keane, N.; O’Neill, M.; O’Dwyer, M.E. Initial evaluation of novel dual PIM/PI3K and triple PIM/PI3K/mTOR inhibitors in multiple myeloma. Haematologica 2015, 100, 496–497. [Google Scholar]
- Wee, P.; Wang, Z.X. Epidermal growth factor receptor cell proliferation signaling pathways. Cancers 2017, 9, 52. [Google Scholar]
- Peltola, K.; Hollmen, M.; Maula, S.M.; Rainio, E.; Ristamaki, R.; Luukkaa, M.; Sandholm, J.; Sundvall, M.; Elenius, K.; Koskinen, P.J.; et al. PIM-1 kinase expression predicts radiation response in squamocellular carcinoma of head and neck and is under the control of epidermal growth factor receptor. Neoplasia 2009, 11, 629. [Google Scholar] [CrossRef] [PubMed]
- Chen, K.; Yang, J.L.; Li, J.N.; Wang, X.F.; Chen, Y.H.; Huang, S.L.; Chen, J.L. eIF4B is a convergent target and critical effector of oncogenic Pim and PI3K/AKT/mTOR signaling pathways in Abl transformants. Oncotarget 2016, 7, 10073–10089. [Google Scholar] [CrossRef] [PubMed]
- Iqbal, A.; Eckerdt, F.; Bell, J.; Nakano, I.; Giles, F.J.; Cheng, S.Y.; Lulla, R.R.; Goldman, S.; Platanias, L.C. Targeting of glioblastoma cell lines and glioma stem cells by combined PIM kinase and PI3K-P110 alpha inhibition. Oncotarget 2016, 7, 33192–33201. [Google Scholar] [CrossRef] [PubMed]
- Gyori, D.; Chessa, T.; Hawkins, P.T.; Stephens, L.R. Class (i) phosphoinositide 3-kinases in the tumor microenvironment. Cancers 2017, 9, 24. [Google Scholar] [CrossRef] [PubMed]
- Blanco-Aparicio, C.; Collazo, A.M.G.; Oyarzabal, J.; Leal, J.F.; Albaran, M.I.; Lima, F.R.; Pequeno, B.; Ajenjo, N.; Becerra, M.; Alfonso, P.; et al. PIM 1 kinase inhibitor ETP-45299 suppresses cellular proliferation and synergizes with PI3K inhibition. Cancer Lett. 2011, 300, 145–153. [Google Scholar] [CrossRef] [PubMed]
- Muraski, J.A.; Rota, M.; Misao, Y.; Fransioli, J.; Cottage, C.; Gude, N.; Esposito, G.; Delucchi, F.; Arcarese, M.; Alvarez, R.; et al. PIM-1 regulates cardiomyocyte survival downstream of AKT. Nat. Med. 2007, 13, 1467–1475. [Google Scholar] [CrossRef] [PubMed]
- Min, X.; Tang, J.; Wang, Y.; Yu, M.; Zhao, L.; Yang, H.; Zhang, P.; Ma, Y. PI3K-like kinases restrain PIM gene expression in endothelial cells. J. Huazhong Univ. Sci. Technol. Med. Sci. 2012, 32, 17–23. [Google Scholar] [CrossRef] [PubMed]
- Song, H.; Cui, J.; Simonyi, A.; Johnson, C.E.; Hubler, G.K.; DePalma, R.G.; Gu, Z. Linking blast physics to biological outcomes in mild traumatic brain injury: Narrative review and preliminary report of an open-field blast model. Behav. Brain Res. 2016, 340, 147–158. [Google Scholar] [CrossRef] [PubMed]
- Cen, B.; Xiong, Y.; Song, J.H.; Mahajan, S.; DuPont, R.; McEachern, K.; DeAngelo, D.J.; Cortes, J.E.; Minden, M.D.; Ebens, A.; et al. The PIM-1 protein kinase is an important regulator of MET receptor tyrosine kinase levels and signaling. Mol. Cell. Biol. 2014, 34, 2517–2532. [Google Scholar] [CrossRef] [PubMed]
- Guo, G.; Qiu, X.; Wang, S.; Chen, Y.; Rothman, P.B.; Wang, Z.; Chen, Y.; Wang, G.; Chen, J.L. Oncogenic E17K mutation in the pleckstrin homology domain of AKT1 promotes v-Abl-mediated pre-B-cell transformation and survival of PIM-deficient cells. Oncogene 2010, 29, 3845–3853. [Google Scholar] [CrossRef] [PubMed]
- Toren, P.; Zoubeidi, A. Rational cotargeting of PIM-1 and AKT in prostate cancer. Expert Rev. Anticancer 2013, 13, 937–939. [Google Scholar] [CrossRef] [PubMed]
- Viel, S.; Besson, L.; Marotel, M.; Walzer, T.; Marcais, A. Regulation of mTOR, metabolic fitness, and effector functions by cytokines in natural killer cells. Cancers 2017, 9, 132. [Google Scholar] [CrossRef] [PubMed]
- Faes, S.; Santoro, T.; Demartines, N.; Dormond, O. Evolving significance and future relevance of anti-angiogenic activity of mTOR inhibitors in cancer therapy. Cancers 2017, 9, 152. [Google Scholar] [CrossRef] [PubMed]
- Yao, Y.; Jones, E.; Inoki, K. Lysosomal regulation of mTORC1 by amino acids in mammalian cells. Biomolecules 2017, 7, 51. [Google Scholar] [CrossRef] [PubMed]
- Lu, J.; Zavorotinskaya, T.; Dai, Y.M.; Niu, X.H.; Castillo, J.; Sim, J.; Yu, J.J.; Wang, Y.Y.; Langowski, J.L.; Holash, J.; et al. PIM2 is required for maintaining multiple myeloma cell growth through modulating TSC2 phosphorylation. Blood 2013, 122, 1610–1620. [Google Scholar] [CrossRef] [PubMed]
- Beharry, Z.; Mahajan, S.; Zemskova, M.; Lin, Y.W.; Tholanikunnel, B.G.; Xia, Z.P.; Smith, C.D.; Kraft, A.S. The PIM protein kinases regulate energy metabolism and cell growth. Proc. Natl. Acad. Sci. USA 2011, 108, 528–533. [Google Scholar] [CrossRef] [PubMed]
- Bialopiotrowicz, E.; Gorniak, P.; Pula, B.; Noyszewska-Kania, M.; Makuch-Lasica, H.; Nowak, G.; Szydlowski, M.; Jablonska, E.; Sewastianik, T.; Polak, A.; et al. Microenvironment-induced expression of PIM kinases supports chronic lymphocytic leukemia cells survival and promotes CXCR4-mTOR pathway dependent migration. Blood 2016, 128, 3239. [Google Scholar]
- Chen, L.S.; Yang, J.Y.; Liang, H.; Cortes, J.E.; Gandhi, V. Protein profiling identifies mTOR pathway modulation and cytostatic effects of PIM kinase inhibitor, AZD1208, in acute myeloid leukemia. Leuk. Lymphoma 2016, 57, 2863–2873. [Google Scholar] [CrossRef] [PubMed]
- Levine, D.A.; Bogomolniy, F.; Yee, C.J.; Lash, A.; Barakat, R.R.; Borgen, P.I.; Boyd, J. Frequent mutation of the PIK3CA gene in ovarian and breast cancers. Clin. Cancer Res. 2005, 11, 2875–2878. [Google Scholar] [CrossRef] [PubMed]
- Philp, A.J.; Campbell, I.G.; Leet, C.; Vincan, E.; Rockman, S.P.; Whitehead, R.H.; Thomas, R.J.; Phillips, W.A. The phosphatidylinositol 3′-kinase p85α gene is an oncogene in human ovarian and colon tumors. Cancer Res. 2001, 61, 7426–7429. [Google Scholar] [PubMed]
- Romero, I.; Bast, R.C., Jr. Minireview: Human ovarian cancer: Biology, current management, and paths to personalizing therapy. Endocrinology 2012, 153, 1593–1602. [Google Scholar] [CrossRef] [PubMed]
- Kinross, K.M.; Montgomery, K.G.; Kleinschmidt, M.; Waring, P.; Ivetac, I.; Tikoo, A.; Saad, M.; Hare, L.; Roh, V.; Mantamadiotis, T.; et al. An activating PIK3CA mutation coupled with pten loss is sufficient to initiate ovarian tumorigenesis in mice. J. Clin. Investig. 2012, 122, 553–557. [Google Scholar] [CrossRef] [PubMed]
- Fruman, D.A.; Rommel, C. PI3K and cancer: Lessons, challenges and opportunities. Nat. Rev. Drug Discov. 2014, 13, 140–156. [Google Scholar] [CrossRef] [PubMed]
- Vanhaesebroeck, B.; Khwaja, A. PI3K delta inhibition hits a sensitive spot in B cell malignancies. Cancer Cell 2014, 25, 269–271. [Google Scholar] [CrossRef] [PubMed]
- Liefers-Visser, J.A.L.; Meijering, R.A.M.; Reyners, A.K.L.; van der Zee, A.G.J.; de Jong, S. Igf system targeted therapy: Therapeutic opportunities for ovarian cancer. Cancer Treat. Rev. 2017, 60, 90–99. [Google Scholar] [CrossRef] [PubMed]
- Faes, S.; Dormond, O. PI3K and AKT: Unfaithful partners in cancer. Int. J. Mol. Sci. 2015, 16, 21138–21152. [Google Scholar] [CrossRef] [PubMed]
- Brown, J.S.; Banerji, U. Maximising the potential of AKT inhibitors as anti-cancer treatments. Pharmacol. Ther. 2017, 172, 101–115. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.C.; Chang, Y.C.; Lan, M.S.; Breslin, M. Leptin stimulates ovarian cancer cell growth and inhibits apoptosis by increasing cyclin D1 and MCL-1 expression via the activation of the MEK/ERK1/2 and PI3K/AKT signaling pathways. Int. J. Oncol. 2016, 49, 847. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Bai, M.; Ning, C.; Xie, B.; Zhang, J.; Liao, H.; Xiong, J.; Tao, X.; Yan, D.; Xi, X.; et al. Gankyrin facilitates follicle-stimulating hormone-driven ovarian cancer cell proliferation through the PI3K/AKT/HIF-1 α/cyclin D1 pathway. Oncogene 2016, 35, 2506–2517. [Google Scholar] [CrossRef] [PubMed]
- Jin, Y.F.; Huang, M.H.; Wang, Y.Y.; Yi, C.Y.; Deng, Y.; Chen, Y.N.; Jiang, L.F.; Wang, J.; Shen, Q.; Liu, R.; et al. C-yes enhances tumor migration and invasion via PI3K/AKT pathway in epithelial ovarian cancer. Exp. Mol. Pathol. 2016, 101, 50–57. [Google Scholar] [CrossRef] [PubMed]
- Choi, H.S.; Kim, M.K.; Choi, Y.K.; Shin, Y.C.; Cho, S.G.; Ko, S.G. Rhus verniciflua stokes (RVS) and butein induce apoptosis of paclitaxel-resistant SKOV-3/PAX ovarian cancer cells through inhibition of AKT phosphorylation. BMC Complement. Altern. Med. 2016, 16, 122. [Google Scholar] [CrossRef] [PubMed]
- Ding, F.; Liu, T.F.; Yu, N.N.; Li, S.H.; Zhang, X.F.; Zheng, G.H.; Lv, C.M.; Mou, K.; Xu, J.; Li, B.; et al. Nitidine chloride inhibits proliferation, induces apoptosis via the AKT pathway and exhibits a synergistic effect with doxorubicin in ovarian cancer cells. Mol. Med. Rep. 2016, 14, 2853–2859. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.; Piao, H.L.; Li, Y.H.; Qiu, Q.; Li, D.J.; Du, M.R.; Tsang, B.K. Inhibition of AKT sensitizes chemoresistant ovarian cancer cells to cisplatin by abrogating S and G2/M arrest. Exp. Mol. Pathol. 2016, 100, 506–513. [Google Scholar] [CrossRef] [PubMed]
- Li, X.Y.; Lu, W.M.; Shen, W.F.; Wu, Y.; Liu, Y.P.; Tuo, Y.; Liu, Y.L. Growth inhibitory effect of stevioside on ovarian cancer through AKT/ERK pathway. Biomed. Res. India 2017, 28, 1820–1827. [Google Scholar]
- Lv, Y.; Li, F.L.; Liu, P.S. miR-151 promotes ovarian cancer through activation of AKT/mTOR signaling pathway by decreasing rhogdia. Int. J. Clin. Exp. Pathol. 2016, 9, 11222–11229. [Google Scholar]
- Hayakawa, J.; Ohmichi, M.; Kurachi, H.; Kanda, Y.; Hisamoto, K.; Nishio, Y.; Adachi, K.; Tasaka, K.; Kanzaki, T.; Murata, Y. Inhibition of bad phosphorylation either at serine 112 via extracellullar signal-regulated protein kinase cascade or at serine 136 via AKT cascade sensitizes human ovarian cancer cells to cisplatin. Cancer Res. 2000, 60, 5988–5994. [Google Scholar] [PubMed]
- Mabuchi, S.; Ohmichi, M.; Kimura, A.; Hisamoto, K.; Hayakawa, J.; Nishio, Y.; Adachi, K.; Takahashi, K.; Arimoto-Ishida, E.; Nakatsuji, Y.; et al. Inhibition of phosphorylation of bad and RAF-1 by AKT sensitizes human ovarian cancer cells to paclitaxel. J. Biol. Chem. 2002, 277, 33490–33500. [Google Scholar] [CrossRef] [PubMed]
- Macdonald, A.; Campbell, D.G.; Toth, R.; McLauchlan, H.; Hastie, C.J.; Arthur, J.S. PIM kinases phosphorylate multiple sites on bad and promote 14-3-3 binding and dissociation from BCL-Xl. BMC Cell Biol. 2006, 7, 1. [Google Scholar] [CrossRef] [PubMed]
- Yan, B.; Zemskova, M.; Holder, S.; Chin, V.; Kraft, A.; Koskinen, P.J.; Lilly, M. The PIM-2 kinase phosphorylates bad on serine 112 and reverses bad-induced cell death. J. Biol. Chem. 2003, 278, 45358–45367. [Google Scholar] [CrossRef] [PubMed]
- Liu, A.L.; Liao, H.Q.; Li, Z.L.; Liu, J.; Zhou, C.L.; Guo, Z.F.; Xie, H.Y.; Peng, C.Y. New insights into mTOR signal pathways in ovarian-related diseases: Polycystic ovary syndrome and ovarian cancer. Asian Pac. J. Cancer Prev. 2016, 17, 5087–5094. [Google Scholar] [PubMed]
- Shao, W.Y.; Yang, Y.L.; Yan, H.; Huang, Q.; Liu, K.J.; Zhang, S. Phenethyl isothiocyanate suppresses the metastasis of ovarian cancer associated with the inhibition of CRM1-mediated nuclear export and mTOR-STAT3 pathway. Cancer Biol. Ther. 2017, 18, 26–35. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Tang, Y.; Yu, H.J.; Yin, Q.Y.; Li, M.D.; Shi, L.J.; Zhang, W.; Li, D.R.; Li, L. CCL18 from tumor-cells promotes epithelial ovarian cancer metastasis via mTOR signaling pathway. Mol. Carcinog. 2016, 55, 1688–1699. [Google Scholar] [CrossRef] [PubMed]
- Xia, R.; Chen, S.X.; Qin, Q.; Chen, Y.; Zhang, W.W.; Zhu, R.R.; Deng, A.M. Oridonin suppresses proliferation of human ovarian cancer cells via blockage of mTOR signaling. Asian Pac. J. Cancer Prev. 2016, 17, 667–671. [Google Scholar] [CrossRef] [PubMed]
- Feng, S.J.; Jin, Y.; Cui, M.J.; Zheng, J.H. Lysine-specific demethylase 1 (LSD1) inhibitor s2101 induces autophagy via the AKT/mTOR pathway in SKOV3 ovarian cancer cells. Med. Sci. Monit. 2016, 22, 4742–4748. [Google Scholar] [CrossRef] [PubMed]
- Xu, S.; Fu, G.B.; Tao, Z.; OuYang, J.; Kong, F.; Jiang, B.H.; Wan, X.; Chen, K. miR-497 decreases cisplatin resistance in ovarian cancer cells by targeting mTOR/P70S6K1. Oncotarget 2015, 6, 26457–26471. [Google Scholar] [CrossRef] [PubMed]
- Casey, S.C.; Tong, L.; Li, Y.L.; Do, R.; Walz, S.; Fitzgerald, K.N.; Gouw, A.M.; Baylot, V.; Gutgemann, I.; Eilers, M.; et al. Myc regulates the antitumor immune response through CD47 and PD-L1. Science 2016, 352, 227–231. [Google Scholar] [CrossRef] [PubMed]
- Cohen, J.; Bowman, S.; Szabo, G.; Umetsu, S.E.; Krings, G.; Laszik, Z. Discordant expression of CMYC and PD-L1 in various human tumors. Mod. Pathol. 2017, 30, 455. [Google Scholar]
- Xu, B.; Lefringhouse, J.; Liu, Z.; West, D.; Baldwin, L.A.; Ou, C.; Chen, L.; Napier, D.; Chaiswing, L.; Brewer, L.D.; et al. Inhibition of the integrin/FAK signaling axis and c-Myc synergistically disrupts ovarian cancer malignancy. Oncogenesis 2017, 6, e295. [Google Scholar] [CrossRef] [PubMed]
- Yuan, J.L.; Wang, K.N.; Xi, M.R. miR-494 inhibits epithelial ovarian cancer growth by targeting c-Myc. Med. Sci. Monit. 2016, 22, 617–624. [Google Scholar] [CrossRef] [PubMed]
- Vivas-Mejia, P.E.; Reyes, J.; Sood, A.K. C-MYC is a potential therapeutic target for cisplatin-resistant ovarian cancer. Mol. Cancer Res. 2015, 13. [Google Scholar] [CrossRef]
- Kirschner, A.N.; Wang, J.; Van der Meer, R.; Anderson, P.D.; Franco-Coronel, O.E.; Kushner, M.H.; Everett, J.H.; Hameed, O.; Keeton, E.K.; Ahdesmaki, M.; Grosskurth, S.E.; Huszar, D.; Abdulkadir, S. PIM kinase inhibitor AZD1208 for treatment of MYC-driven prostate cancer. J. Natl. Cancer Inst. 2015, 107. [Google Scholar] [CrossRef] [PubMed]
- Hao, H.X.; Wang, D.; Vanasse, G.; Caponigro, G. The PAN-PIM inhibitor PIM447 enhances the antitumor activity of lenalidomide in multiple myeloma cells via synergistic inhibition of c-MYC. Cancer Res. 2016, 76, 4630. [Google Scholar] [CrossRef]
- Emmanouilidi, A.; Falasca, M. Targeting PDK1 for chemosensitization of cancer cells. Cancers 2017, 9, 140. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Lee, W.C.; Huang, B.M.; Chia, Y.C.; Chen, Y.C. 16-hydroxycleroda-3, 13-dien-15, 16-olide inhibits the proliferation and induces mitochondrial-dependent apoptosis through AKT, mTOR, and MEK-ERK pathways in human renal carcinoma cells. Phytomedicine 2017, 36, 95–107. [Google Scholar] [CrossRef] [PubMed]
- Ning, Y.X.; Luo, X.; Xu, M.; Feng, X.; Wang, J. LET-7d increases ovarian cancer cell sensitivity to a genistein analog by targeting c-MYC. Oncotarget 2017, 8, 74836–74845. [Google Scholar] [CrossRef] [PubMed]
- Kaur, M.; Cole, M.D. MYC acts via the pten tumor suppressor to elicit autoregulation and genome-wide gene repression by activation of the EZH2 methyltransferase. Cancer Res. 2013, 73, 695–705. [Google Scholar] [CrossRef] [PubMed]
- Bansal, N.; Marchion, D.; Bicaku, E.; Cottrill, H.; Xiong, Y.; Chen, N.; Kamath, S.; Apte, S.; Wenham, R.; Lancaster, J. BCL2 antagonist of cell death kinases, phosphatases, and ovarian cancer sensitivity to cisplatin. J. Gynecol. Oncol. 2012, 23, 35–42. [Google Scholar] [CrossRef] [PubMed]
- Marchion, D.C.; Cottrill, H.M.; Xiong, Y.; Chen, N.; Bicaku, E.; Fulp, W.J.; Bansal, N.; Chon, H.S.; Stickles, X.B.; Kamath, S.G.; et al. Bad phosphorylation determines ovarian cancer chemosensitivity and patient survival. Clin. Cancer Res. 2011, 17, 6356–6366. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Xu, R.C.; Zhang, X.L.; Niu, X.L.; Qu, Y.; Li, L.Z.; Meng, X.Y. Interleukin-8 secretion by ovarian cancer cells increases anchorage-independent growth, proliferation, angiogenic potential, adhesion and invasion. Cytokine 2012, 59, 145–155. [Google Scholar] [CrossRef] [PubMed]
- Cicenas, J.; Kalyan, K.; Sorokinas, A.; Jatulyte, A.; Valiunas, D.; Kaupinis, A.; Valius, M. Highlights of the latest advances in research on CDK inhibitors. Cancers 2014, 6, 2224–2242. [Google Scholar] [CrossRef] [PubMed]
- Peyressatre, M.; Prevel, C.; Pellerano, M.; Morris, M.C. Targeting cyclin-dependent kinases in human cancers: From small molecules to peptide inhibitors. Cancers 2015, 7, 179–237. [Google Scholar] [CrossRef] [PubMed]
- Kanwal, A.; Kaur, M.; Singh, A.; Gupta, S.; Sachan, M. Hypo/unmethylated promoter status of CDK2 gene correlates with its over-expression in ovarian cancer in North Indian population. Cell. Mol. Biol. 2016, 62, 67–72. [Google Scholar] [PubMed]
- Etemadmoghadam, D.; Au-Yeung, G.; Wall, M.; Mitchell, C.; Kansara, M.; Loehrer, E.; Batzios, C.; George, J.; Ftouni, S.; Weir, B.A.; et al. Resistance to CDK2 inhibitors is associated with selection of polyploid cells in CCNE1-amplified ovarian cancer. Clin. Cancer Res. 2013, 19, 5960–5971. [Google Scholar] [CrossRef] [PubMed]
- Dall’Acqua, A.; Monica, S.; Baldassarre, G. CDK6 controls platinum sensitivity via the regulation of FOXO3a/ATR: A new actionable pathway for ovarian cancer patients. Clin. Cancer Res. 2016, 22. [Google Scholar] [CrossRef]
- Xia, B.R.; Yang, S.S.; Liu, T.B.; Lou, G. miR-211 suppresses epithelial ovarian cancer proliferation and cell-cycle progression by targeting cyclin D1 and CDK6. Mol. Cancer 2015, 14, 57. [Google Scholar] [CrossRef] [PubMed]
- Iyengar, M.; Coffman, L.; Buckanovich, R. CDK4/6 inhibition as maintenance therapy in ovarian cancer. Clin. Cancer Res. 2016, 22. [Google Scholar] [CrossRef]
- Meng, Q.; Xia, C.; Fang, J.; Rojanasakul, Y.; Jiang, B.H. Role of PI3K and AKT specific isoforms in ovarian cancer cell migration, invasion and proliferation through the P70S6K1 pathway. Cell. Signal. 2006, 18, 2262–2271. [Google Scholar] [CrossRef] [PubMed]
- Zhang, R.T.; Shi, H.R.; Ren, F.; Li, X.; Zhang, M.H.; Feng, W.; Jia, Y.Y. Knockdown of MACC1 expression increases cisplatin sensitivity in cisplatin-resistant epithelial ovarian cancer cells. Oncol. Rep. 2016, 35, 2466–2472. [Google Scholar] [CrossRef] [PubMed]
- Guo, B.Q.; Yao, N.; Gan, H.Y.; Yu, L. TGF β1 upregulates the expression of MACC1 to promote invasion and metastasis of ovarian cancer. Int. J. Clin. Exp. Med. 2016, 9, 12629–12638. [Google Scholar]
- Li, H.Y.; Zhang, H.; Zhao, S.J.; Shi, Y.; Yao, J.G.; Zhang, Y.Y.; Guo, H.H.; Liu, X.S. Overexpression of MACC1 and the association with hepatocyte growth factor/c-MET in epithelial ovarian cancer. Oncol. Lett. 2015, 9, 1989–1996. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Pan, C.Q.; Guo, L.H.; Wu, M.W.; Guo, J.; Peng, S.; Wu, Q.Y.; Zuo, Q. A new mechanism of trastuzumab resistance in gastric cancer: MACC1 promotes the warburg effect via activation of the PI3K/AKT signaling pathway. J. Hematol. Oncol. 2016, 9, 76. [Google Scholar] [CrossRef] [PubMed]
- Stein, U.; Smith, J.; Walther, W.; Arlt, F. MACC1 controls met what a difference an Sp1 site makes. Cell Cycle 2009, 8, 2467–2469. [Google Scholar] [CrossRef] [PubMed]
- Zhang, R.T.; Shi, H.R.; Chen, Z.M.; Wu, Q.H.; Ren, F.; Huang, H.L. Effects of metastasis-associated in colon cancer 1 inhibition by small hairpin RNA on ovarian carcinoma ovcar-3 cells. J. Exp. Clin. Cancer Res. 2011, 30, 83. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Aziz, A.U.R.; Farid, S.; Qin, K.; Wang, H.; Liu, B. PIM Kinases and Their Relevance to the PI3K/AKT/mTOR Pathway in the Regulation of Ovarian Cancer. Biomolecules 2018, 8, 7. https://doi.org/10.3390/biom8010007
Aziz AUR, Farid S, Qin K, Wang H, Liu B. PIM Kinases and Their Relevance to the PI3K/AKT/mTOR Pathway in the Regulation of Ovarian Cancer. Biomolecules. 2018; 8(1):7. https://doi.org/10.3390/biom8010007
Chicago/Turabian StyleAziz, Aziz Ur Rehman, Sumbal Farid, Kairong Qin, Hanqin Wang, and Bo Liu. 2018. "PIM Kinases and Their Relevance to the PI3K/AKT/mTOR Pathway in the Regulation of Ovarian Cancer" Biomolecules 8, no. 1: 7. https://doi.org/10.3390/biom8010007
APA StyleAziz, A. U. R., Farid, S., Qin, K., Wang, H., & Liu, B. (2018). PIM Kinases and Their Relevance to the PI3K/AKT/mTOR Pathway in the Regulation of Ovarian Cancer. Biomolecules, 8(1), 7. https://doi.org/10.3390/biom8010007