Antimicrobial Peptides: Diversity, Mechanism of Action and Strategies to Improve the Activity and Biocompatibility In Vivo


Abstract
1. Antimicrobial Peptides: History and Diversity
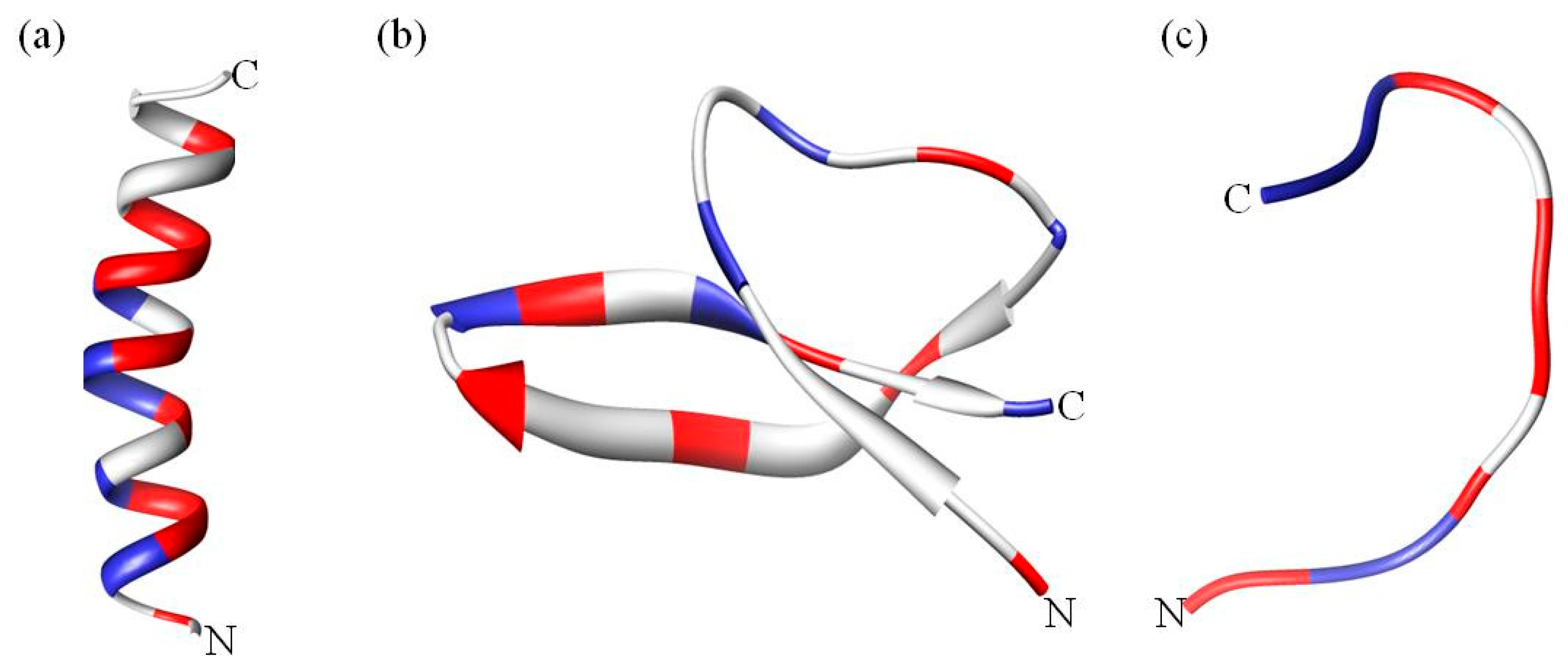
1.1. Categories of Antimicrobial Peptides
1.2. Common Properties of Antimicrobial Peptides
2. Mechanism of Antimicrobial Peptide Action
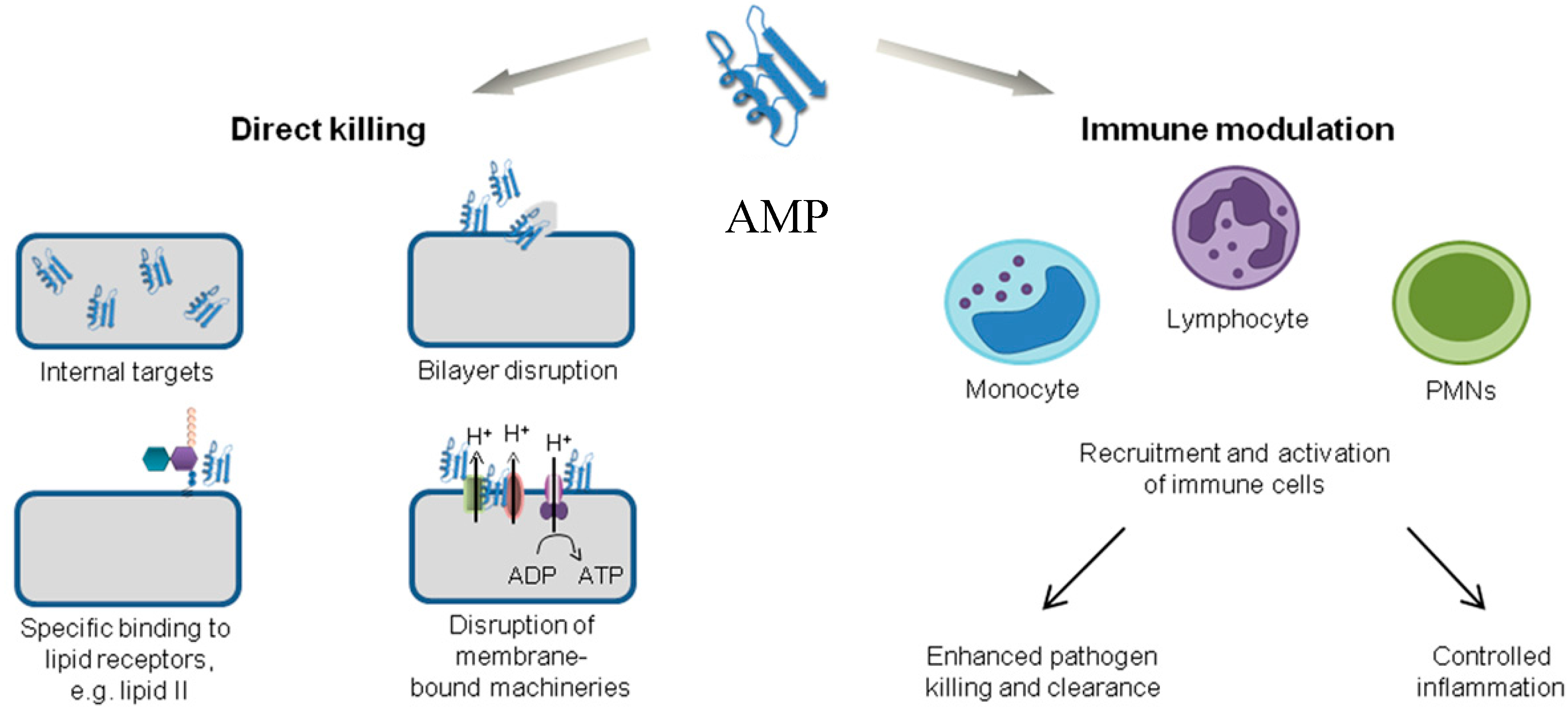
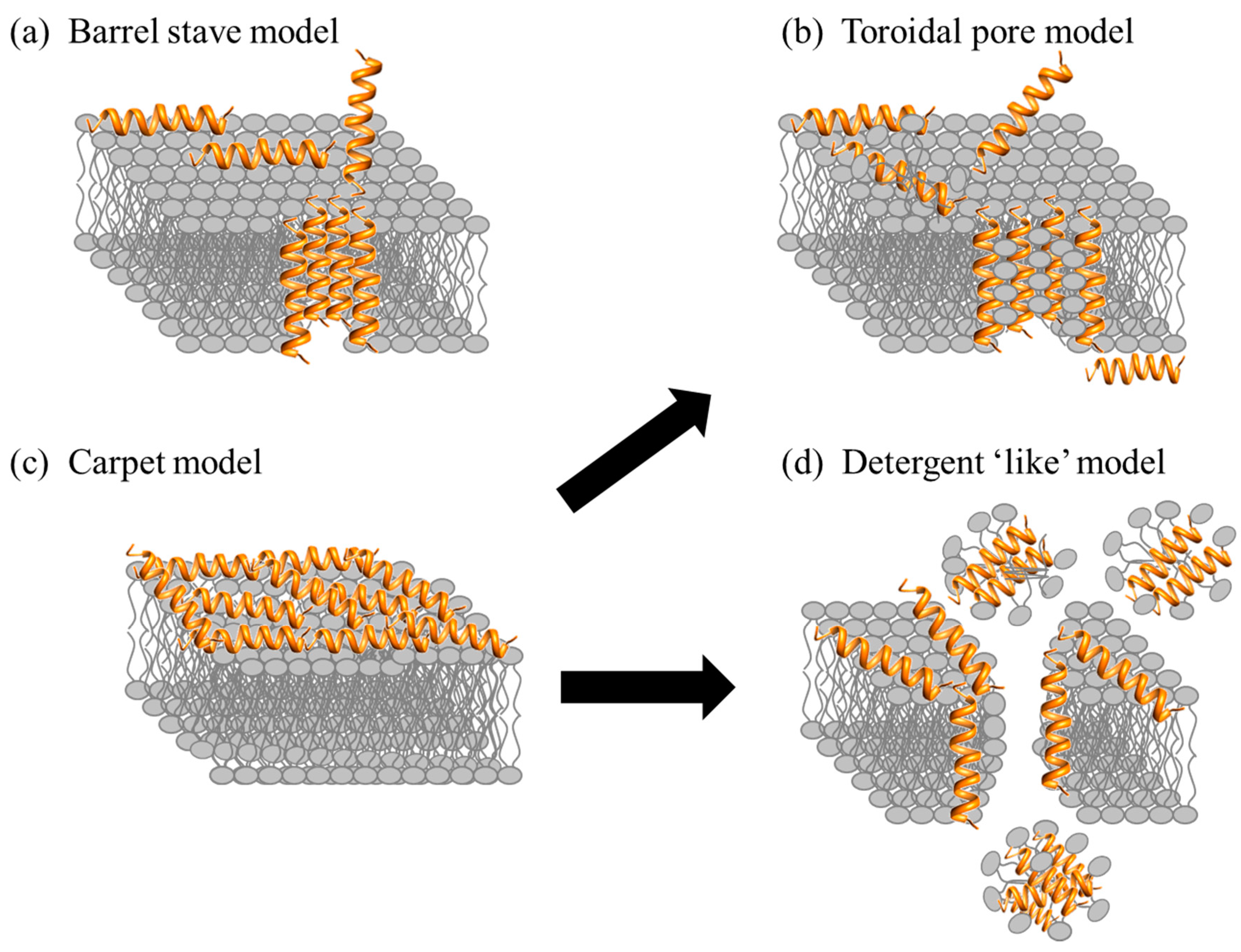
2.1. Direct Killing: Membrane Permeabilizing Mechanism of Action
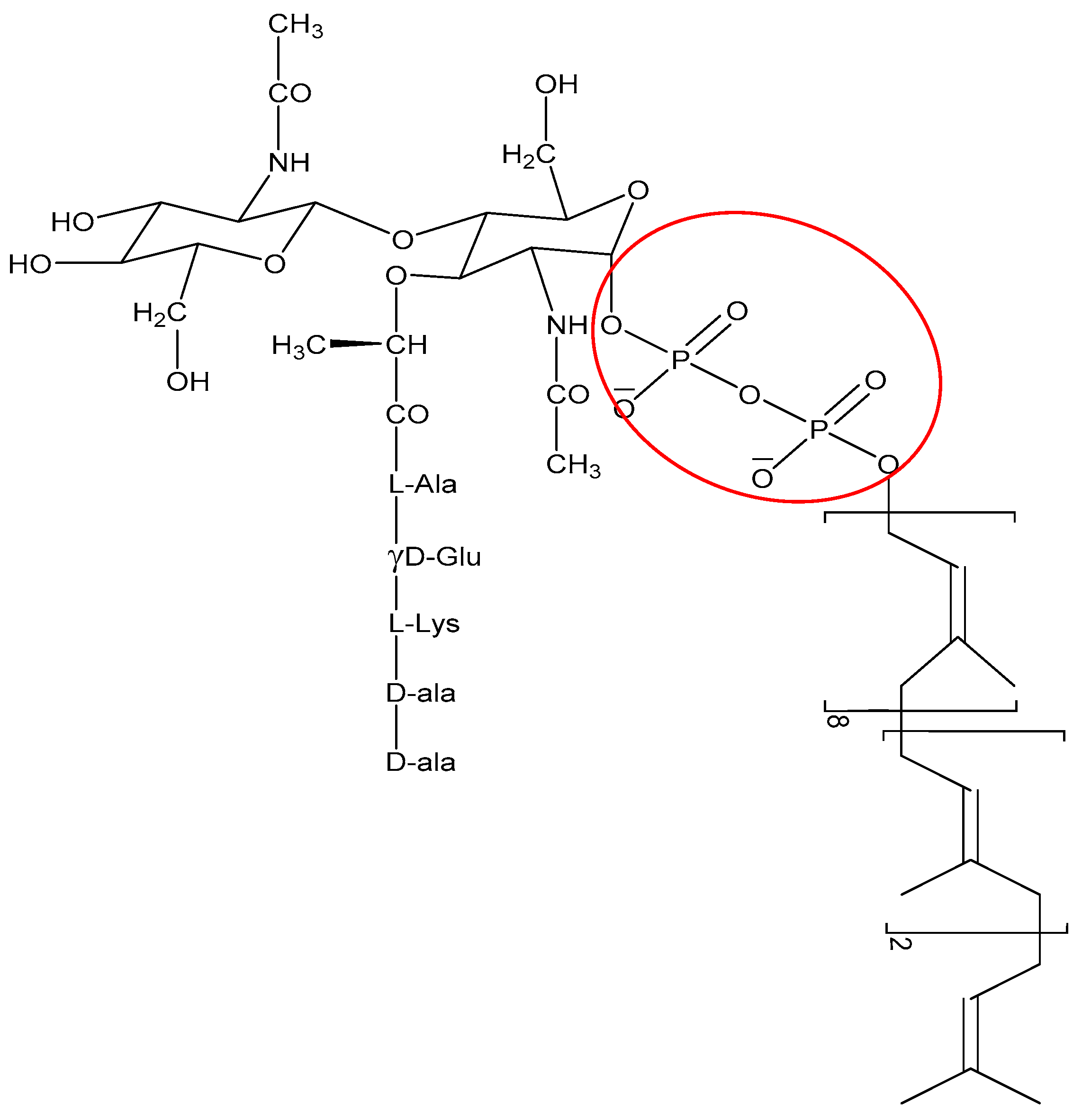
2.2. Direct Killing: Non Membrane Targeting Mechanisms of Action
2.3. Immune Modulation Mechanism of Action
3. Challenges with Antimicrobial Peptides
4. Strategies to Improve Antimicrobial Peptides
4.1. Chemical Modification of AMPs
4.2. Delivery Systems for AMPs
5. Conclusions
Acknowledgments
Conflicts of Interest
References
- Mattick, A.T.R.; Hirsch, A.; Berridge, N.J. Further observations on an inhibitory substance (nisin) from lactic streptococci. Lancet 1947, 250, 5–8. [Google Scholar] [CrossRef]
- Jenssen, H.; Hamill, P.; Hancock, R.E.W. Peptide Antimicrobial Agents. Clin. Microbiol. Rev. 2006, 19, 491–511. [Google Scholar] [CrossRef] [PubMed]
- Chatterjee, S.; Chatterjee, S.; Lad, S.J.; Phansalkar, M.S.; Rupp, R.H.; Ganguli, B.N.; Fehlhaber, H.W.; Kogler, H. Mersacidin, a new antibiotic from Bacillus. Fermentation, isolation, purification and chemical characterization. J. Antibiot. (Tokyo) 1992, 45, 832–838. [Google Scholar] [CrossRef] [PubMed]
- Skarnes, R.C.; Watson, D.W. Antimicrobial factors of normal tissues and fluids. Bacteriol. Rev. 1957, 21, 273–294. [Google Scholar] [PubMed]
- Steiner, H.; Hultmark, D.; Engström, Å.; Bennich, H.; Boman, H.G. Sequence and specificity of two antibacterial proteins involved in insect immunity. Nature 1981, 292, 246–248. [Google Scholar] [CrossRef] [PubMed]
- Patterson-Delafield, J.; Szklarek, D.; Martinez, R.J.; Lehrer, R.I. Microbicidal cationic proteins of rabbit alveolar macrophages: amino acid composition and functional attributes. Infect. Immun. 1981, 31, 723–731. [Google Scholar] [PubMed]
- Okada, M.; Natori, S. Purification and characterization of an antibacterial protein from haemolymph of Sarcophaga peregrina (flesh-fly) larvae. Biochem. J. 1983, 211, 727–734. [Google Scholar] [CrossRef] [PubMed]
- Zasloff, M. Magainins, a class of antimicrobial peptides from Xenopus skin: isolation, characterization of two active forms, and partial cDNA sequence of a precursor. Proc. Natl. Acad. Sci. USA 1987, 84, 5449–5453. [Google Scholar] [CrossRef] [PubMed]
- Tam, J.P.; Wang, S.; Wong, K.H.; Tan, W.L. Antimicrobial Peptides from Plants. Pharmaceuticals (Basel) 2015, 8, 711–757. [Google Scholar] [CrossRef] [PubMed]
- Nawrot, R.; Barylski, J.; Nowicki, G.; Broniarczyk, J.; Buchwald, W.; Goździcka-Józefiak, A. Plant antimicrobial peptides. Folia Microbiol. (Praha) 2014, 59, 181–196. [Google Scholar] [CrossRef] [PubMed]
- Stotz, H.U.; Thomson, J.G.; Wang, Y. Plant defensins: defense, development and application. Plant Signal. Behav. 2009, 4, 1010–1012. [Google Scholar] [CrossRef] [PubMed]
- Craik, D.J. Host-Defense Activities of Cyclotides. Toxins (Basel) 2012, 4, 139–156. [Google Scholar] [CrossRef] [PubMed]
- Hancock, R.E.W.; Brown, K.L.; Mookherjee, N. Host defence peptides from invertebrates—Emerging antimicrobial strategies. Immunobiology 2006, 211, 315–322. [Google Scholar] [CrossRef] [PubMed]
- Bachere, E.; Gueguen, Y.; Gonzalez, M.; de Lorgeril, J.; Garnier, J.; Romestand, B. Insights into the anti-microbial defense of marine invertebrates: the penaeid shrimps and the oyster Crassostrea gigas. Immunol. Rev. 2004, 198, 149–168. [Google Scholar] [CrossRef] [PubMed]
- Iwanaga, S.; Kawabata, S.-I. Evolution and phylogeny of defense molecules associated with innate immunity in horseshoe crab.
- Tincu, J.A.; Taylor, S.W. Antimicrobial peptides from marine invertebrates. Antimicrob. Agents Chemother. 2004, 48, 3645–3654. [Google Scholar] [CrossRef] [PubMed]
- Masuda, M.; Nakashima, H.; Ueda, T.; Naba, H.; Ikoma, R.; Otaka, A.; Terakawa, Y.; Tamamura, H.; Ibuka, T.; Murakami, T.; et al. A novel anti-HIV synthetic peptide, T-22 ([Tyr5,12,Lys7]-polyphemusin II). Biochem. Biophys. Res. Commun. 1992, 189, 845–850. [Google Scholar] [CrossRef]
- Yang, D.; Biragyn, A.; Hoover, D.M.; Lubkowski, J.; Oppenheim, J.J. Multiple Roles of Antimicrobial Defensins, Cathelicidins, and Eosinophil-Derived Neurotoxin in Host Defense. Annu. Rev. Immunol. 2004, 22, 181–215. [Google Scholar] [CrossRef] [PubMed]
- Bowdish, D.M.E.; Davidson, D.J.; Lau, Y.E.; Lee, K.; Scott, M.G.; Hancock, R.E.W. Impact of LL-37 on anti-infective immunity. J. Leukoc. Biol. 2005, 77, 451–459. [Google Scholar] [CrossRef] [PubMed]
- Yang, D.; Biragyn, A.; Kwak, L.W.; Oppenheim, J.J. Mammalian defensins in immunity: More than just microbicidal. Trends Immunol. 2002, 23, 291–296. [Google Scholar] [CrossRef]
- Hilchie, A.L.; Wuerth, K.; Hancock, R.E.W. Immune modulation by multifaceted cationic host defense (antimicrobial) peptides. Nat. Chem. Biol. 2013, 9, 761–768. [Google Scholar] [CrossRef] [PubMed]
- Haney, E.F.; Hancock, R.E.W. Peptide design for antimicrobial and immunomodulatory applications. Biopolymers 2013, 100, 572–583. [Google Scholar] [CrossRef] [PubMed]
- Hancock, R.E.; Haney, E.F.; Gill, E.E. The immunology of host defence peptides: beyond antimicrobial activity. Nat. Publ. Gr. 2016, 16. [Google Scholar] [CrossRef] [PubMed]
- Bowdish, D.M.E.; Davidson, D.J.; Scott, M.G.; Hancock, R.E.W. Immunomodulatory activities of small host defense peptides. Antimicrob. Agents Chemother. 2005, 49, 1727–1732. [Google Scholar] [CrossRef] [PubMed]
- Nijnik, A.; Hancock, R. Host defence peptides: antimicrobial and immunomodulatory activity and potential applications for tackling antibiotic-resistant infections. Emerg. Health Threats J. 2009, 2, e1. [Google Scholar] [CrossRef] [PubMed]
- Veldhuizen, E.J.A.; Schneider, V.A.F.; Agustiandari, H.; van Dijk, A.; Tjeerdsma-van Bokhoven, J.L.M.; Bikker, F.J.; Haagsman, H.P. Antimicrobial and Immunomodulatory Activities of PR-39 Derived Peptides. PLoS ONE 2014, 9, e95939. [Google Scholar] [CrossRef] [PubMed]
- Felício, M.R.; Silva, O.N.; Gonçalves, S.; Santos, N.C.; Franco, O.L. Peptides with Dual Antimicrobial and Anticancer Activities. Front. Chem. 2017, 5, 5. [Google Scholar] [CrossRef] [PubMed]
- Gwyer Findlay, E.; Currie, S.M.; Davidson, D.J. Cationic Host Defence Peptides: Potential as Antiviral Therapeutics. BioDrugs 2013, 27, 479–493. [Google Scholar] [CrossRef] [PubMed]
- Blondelle, S.E.; Lohner, K.; Aguilar, M.-I. Lipid-induced conformation and lipid-binding properties of cytolytic and antimicrobial peptides: determination and biological specificity. Biochim. Biophys. Acta Biomembr. 1999, 1462, 89–108. [Google Scholar] [CrossRef]
- Brogden, K.A. Antimicrobial peptides: pore formers or metabolic inhibitors in bacteria? Nat. Rev. Microbiol. 2005, 3, 238–250. [Google Scholar] [CrossRef] [PubMed]
- Kościuczuk, E.M.; Lisowski, P.; Jarczak, J.; Strzałkowska, N.; Jóźwik, A.; Horbańczuk, J.; Krzyżewski, J.; Zwierzchowski, L.; Bagnicka, E. Cathelicidins: Family of antimicrobial peptides. A review. Mol. Biol. Rep. 2012, 39, 10957–10970. [Google Scholar] [CrossRef] [PubMed]
- Zairi, A.; Tangy, F.; Bouassida, K.; Hani, K. Dermaseptins and magainins: antimicrobial peptides from frogs’ skin-new sources for a promising spermicides microbicides-a mini review. J. Biomed. Biotechnol. 2009, 2009, 452567. [Google Scholar] [CrossRef] [PubMed]
- Lamb, H.M.; Wiseman, L.R. Pexiganan Acetate. Drugs 1998, 56, 1047–1052. [Google Scholar] [CrossRef] [PubMed]
- Ge, Y.; MacDonald, D.L.; Holroyd, K.J.; Thornsberry, C.; Wexler, H.; Zasloff, M. In vitro antibacterial properties of pexiganan, an analog of magainin. Antimicrob. Agents Chemother. 1999, 43, 782–788. [Google Scholar] [PubMed]
- Fox, J.L. Antimicrobial peptides stage a comeback. Nat. Biotechnol. 2013, 31, 379–382. [Google Scholar] [CrossRef] [PubMed]
- Rozek, T.; Bowie, J.H.; Wallace, J.C.; Tyler, M.J. The antibiotic and anticancer active aurein peptides from the Australian Bell FrogsLitoria aurea andLitoria raniformis. Part 2. Sequence determination using electrospray mass spectrometry. Rapid Commun. Mass Spectrom. 2000, 14, 2002–2011. [Google Scholar] [CrossRef]
- Rozek, T.; Wegener, K.L.; Bowie, J.H.; Olver, I.N.; Carver, J.A.; Wallace, J.C.; Tyler, M.J. The antibiotic and anticancer active aurein peptides from the Australian Bell Frogs Litoria aurea and Litoria raniformis. Eur. J. Biochem. 2000, 267, 5330–5341. [Google Scholar] [CrossRef] [PubMed]
- Pan, Y.L.; Cheng, J.T.; Hale, J.; Pan, J.; Hancock, R.E.; Straus, S.K. Characterization of the structure and membrane interaction of the antimicrobial peptides aurein 2.2 and 2.3 from Australian southern bell frogs. Biophys. J. 2007, 92, 2854–2864. [Google Scholar] [CrossRef] [PubMed]
- Cheng, J.T.J.; Hale, J.D.; Elliot, M.; Hancock, R.E.W.; Straus, S.K. Effect of membrane composition on antimicrobial peptides aurein 2.2 and 2.3 from Australian southern bell frogs. Biophys. J. 2009, 96, 552–565. [Google Scholar] [CrossRef] [PubMed]
- Cheng, J.T.J.; Hale, J.D.; Kindrachuk, J.; Jessen, H.; Elliott, M.; Hancock, R.E.W.; Straus, S.K. Importance of residue 13 and the C-terminus for the structure and activity of the antimicrobial peptide aurein 2.2. Biophys. J. 2010, 99, 2926–2935. [Google Scholar] [CrossRef] [PubMed]
- Cheng, J.T.J. Investigating the Structure-Function Relationship of Cationic Antimicrobial Peptides and Lipopeptides; University of British Columbia: Vancouver, BC, Canada, 2010. [Google Scholar] [CrossRef]
- Wenzel, M.; Senges, C.H.R.; Zhang, J.; Suleman, S.; Nguyen, M.; Kumar, P.; Chiriac, A.I.; Stepanek, J.J.; Raatschen, N.; May, C.; et al. Antimicrobial Peptides from the Aurein Family Form Ion-Selective Pores in Bacillus subtilis. ChemBioChem 2015, 16, 1101–1108. [Google Scholar] [CrossRef] [PubMed]
- Mura, M.; Wang, J.; Zhou, Y.; Pinna, M.; Zvelindovsky, A.V.; Dennison, S.R.; Phoenix, D.A. The effect of amidation on the behaviour of antimicrobial peptides. Eur. Biophys. J. 2016, 45, 195–207. [Google Scholar] [CrossRef] [PubMed]
- Dhople, V.; Krukemeyer, A.; Ramamoorthy, A. The human beta-defensin-3, an antibacterial peptide with multiple biological functions. Biochim. Biophys. Acta Biomembr. 2006, 1758, 1499–1512. [Google Scholar] [CrossRef] [PubMed]
- Lee, T.-H.; Hall, K.N.; Aguilar, M.-I. Antimicrobial Peptide Structure and Mechanism of Action: A Focus on the Role of Membrane Structure. Curr. Top. Med. Chem. 2016, 16, 25–39. [Google Scholar] [CrossRef] [PubMed]
- Wilson, C.L.; Ouellette, A.J.; Satchell, D.P.; Ayabe, T.; López-Boado, Y.S.; Stratman, J.L.; Hultgren, S.J.; Matrisian, L.M.; Parks, W.C. Regulation of intestinal alpha-defensin activation by the metalloproteinase matrilysin in innate host defense. Science 1999, 286, 113–117. [Google Scholar] [CrossRef] [PubMed]
- Salzman, N.H.; Ghosh, D.; Huttner, K.M.; Paterson, Y.; Bevins, C.L. Protection against enteric salmonellosis in transgenic mice expressing a human intestinal defensin. Nature 2003, 422, 522–526. [Google Scholar] [CrossRef] [PubMed]
- Da Mata, É.C.G.; Mourão, C.B.F.; Rangel, M.; Schwartz, E.F. Antiviral activity of animal venom peptides and related compounds. J. Venom. Anim. Toxins Incl. Trop. Dis. 2017, 23, 3. [Google Scholar] [CrossRef] [PubMed]
- Miyata, T.; Tokunaga, F.; Yoneya, T.; Yoshikawa, K.; Iwanaga, S.; Niwa, M.; Takao, T.; Shimonishi, Y. Antimicrobial peptides, isolated from horseshoe crab hemocytes, tachyplesin II, and polyphemusins I and II: chemical structures and biological activity. J. Biochem. 1989, 106, 663–668. [Google Scholar] [CrossRef] [PubMed]
- Niidome, T.; Kobayashi, K.; Arakawa, H.; Hatakeyama, T.; Aoyagi, H. Structure–activity relationship of an antibacterial peptide, maculatin 1.1, from the skin glands of the tree frog, Litoria genimaculata. J. Pept. Sci. 2004, 10, 414–422. [Google Scholar] [CrossRef] [PubMed]
- Sikorska, E.; Greber, K.; Rodziewicz-Motowidło, S.; Szultka, Ł.; Łukasiak, J.; Kamysz, W. Synthesis and antimicrobial activity of truncated fragments and analogs of citropin 1.1: The solution structure of the SDS micelle-bound citropin-like peptides. J. Struct. Biol. 2009, 168, 250–258. [Google Scholar] [CrossRef] [PubMed]
- Park, C.B.; Kim, H.S.; Kim, S.C. Mechanism of Action of the Antimicrobial Peptide Buforin II: Buforin II Kills Microorganisms by Penetrating the Cell Membrane and Inhibiting Cellular Functions. Biochem. Biophys. Res. Commun. 1998, 244, 253–257. [Google Scholar] [CrossRef] [PubMed]
- Ganz, T. Defensins: Antimicrobial peptides of innate immunity. Nat. Rev. Immunol. 2003, 3, 710–720. [Google Scholar] [CrossRef] [PubMed]
- Ulm, H.; Wilmes, M.; Shai, Y.; Sahl, H.-G. Antimicrobial Host Defensins—Specific Antibiotic Activities and Innate Defense Modulation. Front. Immunol. 2012, 3, 249. [Google Scholar] [CrossRef] [PubMed]
- Lehrer, R.I.; Barton, A.; Daher, K.A.; Harwig, S.S.; Ganz, T.; Selsted, M.E. Interaction of human defensins with Escherichia coli. Mechanism of bactericidal activity. J. Clin. Investig. 1989, 84, 553–561. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, D.; Shukla, S.K.; Prakash, O.; Zhang, G. Structural determinants of host defense peptides for antimicrobial activity and target cell selectivity. Biochimie 2010, 92, 1236–1241. [Google Scholar] [CrossRef] [PubMed]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera?A visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.; Lasker, K.; Schneidman-Duhovny, D.; Webb, B.; Huang, C.C.; Pettersen, E.F.; Goddard, T.D.; Meng, E.C.; Sali, A.; Ferrin, T.E. UCSF Chimera, MODELLER, and IMP: An integrated modeling system. J. Struct. Biol. 2012, 179, 269–278. [Google Scholar] [CrossRef] [PubMed]
- Huang, C.C.; Meng, E.C.; Morris, J.H.; Pettersen, E.F.; Ferrin, T.E. Enhancing UCSF Chimera through web services. Nucleic Acids Res. 2014, 42, W478–W484. [Google Scholar] [CrossRef] [PubMed]
- Nelson, D.L.; David, L.; Cox, M.M.; Lehninger, A.L. Lehninger Principles of Biochemistry; W.H. Freeman and Company: New York, NY, USA, 2013; ISBN 1429234148. [Google Scholar]
- Falla, T.J.; Karunaratne, D.N.; Hancock, R.E. Mode of action of the antimicrobial peptide indolicidin. J. Biol. Chem. 1996, 271, 19298–19303. [Google Scholar] [CrossRef] [PubMed]
- Rozek, A.; Friedrich, C.L.; Hancock, R.E. Structure of the bovine antimicrobial peptide indolicidin bound to dodecylphosphocholine and sodium dodecyl sulfate micelles. Biochemistry 2000, 39, 15765–15774. [Google Scholar] [CrossRef] [PubMed]
- Falcao, C.B.; Pérez-Peinado, C.; de la Torre, B.G.; Mayol, X.; Zamora-Carreras, H.; Jiménez, M.Á.; Rádis-Baptista, G.; Andreu, D. Structural Dissection of Crotalicidin, a Rattlesnake Venom Cathelicidin, Retrieves a Fragment with Antimicrobial and Antitumor Activity. J. Med. Chem. 2015, 58, 8553–8563. [Google Scholar] [CrossRef] [PubMed]
- Gagnon, M.-C.; Strandberg, E.; Grau-Campistany, A.; Wadhwani, P.; Reichert, J.; Bürck, J.; Rabanal, F.; Auger, M.; Paquin, J.-F.; Ulrich, A.S. Influence of the Length and Charge on the Activity of α-Helical Amphipathic Antimicrobial Peptides. Biochemistry 2017, 56, 1680–1695. [Google Scholar] [CrossRef] [PubMed]
- Dathe, M.; Nikolenko, H.; Meyer, J.; Beyermann, M.; Bienert, M. Optimization of the antimicrobial activity of magainin peptides by modification of charge. FEBS Lett. 2001, 501, 146–150. [Google Scholar] [CrossRef]
- Lyu, Y.; Yang, Y.; Lyu, X.; Dong, N.; Shan, A. Antimicrobial activity, improved cell selectivity and mode of action of short PMAP-36-derived peptides against bacteria and Candida. Sci. Rep. 2016, 6, 27258. [Google Scholar] [CrossRef] [PubMed]
- Hong, S.Y.; Park, T.G.; Lee, K.H. The effect of charge increase on the specificity and activity of a short antimicrobial peptide. Peptides 2001, 22, 1669–1674. [Google Scholar] [CrossRef]
- Jiang, Z.; Vasil, A.I.; Hale, J.D.; Hancock, R.E.W.; Vasil, M.L.; Hodges, R.S. Effects of net charge and the number of positively charged residues on the biological activity of amphipathic alpha-helical cationic antimicrobial peptides. Biopolymers 2008, 90, 369–383. [Google Scholar] [CrossRef] [PubMed]
- Yeaman, M.R.; Yount, N.Y. Mechanisms of Antimicrobial Peptide Action and Resistance. Pharmacol. Rev. 2003, 55, 27–55. [Google Scholar] [CrossRef] [PubMed]
- Yin, L.M.; Edwards, M.A.; Li, J.; Yip, C.M.; Deber, C.M. Roles of hydrophobicity and charge distribution of cationic antimicrobial peptides in peptide-membrane interactions. J. Biol. Chem. 2012, 287, 7738–7745. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Guarnieri, M.T.; Vasil, A.I.; Vasil, M.L.; Mant, C.T.; Hodges, R.S. Role of peptide hydrophobicity in the mechanism of action of alpha-helical antimicrobial peptides. Antimicrob. Agents Chemother. 2007, 51, 1398–1406. [Google Scholar] [CrossRef] [PubMed]
- Mihajlovic, M.; Lazaridis, T. Charge distribution and imperfect amphipathicity affect pore formation by antimicrobial peptides. Biochim. Biophys. Acta Biomembr. 2012, 1818, 1274–1283. [Google Scholar] [CrossRef] [PubMed]
- Hawrani, A.; Howe, R.A.; Walsh, T.R.; Dempsey, C.E. Origin of Low Mammalian Cell Toxicity in a Class of Highly Active Antimicrobial Amphipathic Helical Peptides. J. Biol. Chem. 2008, 283, 18636–18645. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Mant, C.T.; Farmer, S.W.; Hancock, R.E.W.; Vasil, M.L.; Hodges, R.S. Rational Design of α-Helical Antimicrobial Peptides with Enhanced Activities and Specificity/Therapeutic Index. J. Biol. Chem. 2005, 280, 12316–12329. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Z.; Vasil, A.I.; Gera, L.; Vasil, M.L.; Hodges, R.S. Rational Design of α-Helical Antimicrobial Peptides to Target Gram-negative Pathogens, Acinetobacter baumannii and Pseudomonas aeruginosa: Utilization of Charge, “Specificity Determinants,” Total Hydrophobicity, Hydrophobe Type and Location as Design Para. Chem. Biol. Drug Des. 2011, 77, 225–240. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.-K.; Song, J.; Gong, F.; Li, S.-B.; Chang, H.-Y.; Xie, H.-M.; Gao, H.-W.; Tan, Y.-X.; Ji, S.-P. Design of an α-helical antimicrobial peptide with improved cell-selective and potent anti-biofilm activity. Sci. Rep. 2016, 6, 27394. [Google Scholar] [CrossRef] [PubMed]
- Edwards, I.A.; Elliott, A.G.; Kavanagh, A.M.; Zuegg, J.; Blaskovich, M.A.T.; Cooper, M.A. Contribution of Amphipathicity and Hydrophobicity to the Antimicrobial Activity and Cytotoxicity of β-Hairpin Peptides. ACS Infect. Dis. 2016, 2, 442–450. [Google Scholar] [CrossRef] [PubMed]
- Falla, T.J.; Hancock, R.E. Improved activity of a synthetic indolicidin analog. Antimicrob. Agents Chemother. 1997, 41, 771–775. [Google Scholar] [PubMed]
- Smirnova, M.P.; Afonin, V.G.; Shpen’, V.M.; Tyagotin, Y.V.; Kolodkin, N.I. Structure–Function Relationship between Analogues of the Antibacterial Peptide Indolicidin. I. Synthesis and Biological Activity of Analogues with Increased Amphipathicity and Elevated Net Positive Charge of the Molecule. Russ. J. Bioorg. Chem. 2004, 30, 409–416. [Google Scholar] [CrossRef]
- Breukink, E.; Wiedemann, I.; van Kraaij, C.; Kuipers, O.P.; Sahl, H.G.; de Kruijff, B. Use of the cell wall precursor lipid II by a pore-forming peptide antibiotic. Science 1999, 286, 2361–2364. [Google Scholar] [CrossRef] [PubMed]
- Fleury, Y.; Dayem, M.A.; Montagne, J.J.; Chaboisseau, E.; Le Caer, J.P.; Nicolas, P.; Delfour, A. Covalent structure, synthesis, and structure-function studies of mesentericin Y 105(37), a defensive peptide from gram-positive bacteria Leuconostoc mesenteroides. J. Biol. Chem. 1996, 271, 14421–14429. [Google Scholar] [CrossRef] [PubMed]
- Shai, Y. Mode of action of membrane active antimicrobial peptides. Biopolymers 2002, 66, 236–248. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Rozek, A.; Hancock, R.E. Interaction of cationic antimicrobial peptides with model membranes. J. Biol. Chem. 2001, 276, 35714–35722. [Google Scholar] [CrossRef] [PubMed]
- Guilhelmelli, F.; Vilela, N.; Albuquerque, P.; Derengowski Lda, S.; Silva-Pereira, I.; Kyaw, C.M. Antibiotic development challenges: the various mechanisms of action of antimicrobial peptides and of bacterial resistance. Front. Microbiol. 2013, 4, 353. [Google Scholar] [CrossRef] [PubMed]
- Zasloff, M. Antimicrobial peptides of multicellular organisms. Nature 2002, 415, 389–395. [Google Scholar] [CrossRef] [PubMed]
- Bevers, E.; Comfurius, P.; Zwaal, R. Regulatory Mechanisms in Maintenance and Modulation of Transmembrane Lipid Asymmetry: Pathophysiological Implications. Lupus 1996, 5, 480–487. [Google Scholar] [CrossRef] [PubMed]
- Matsuzaki, K.; Sugishita, K.; Ishibe, N.; Ueha, M.; Nakata, S.; Miyajima, K.; Epand, R.M. Relationship of Membrane Curvature to the Formation of Pores by Magainin 2. Biochemistry 1998, 37, 11856–11863. [Google Scholar] [CrossRef] [PubMed]
- Epand, R.M.; Vogel, H.J. Diversity of antimicrobial peptides and their mechanisms of action. Biochim. Biophys. Acta Biomembr. 1999, 1462, 11–28. [Google Scholar] [CrossRef]
- Jouhet, J. Importance of the hexagonal lipid phase in biological membrane organization. Front. Plant Sci. 2013, 4, 494. [Google Scholar] [CrossRef] [PubMed]
- Alvares, D.S.; Ruggiero Neto, J.; Ambroggio, E.E. Phosphatidylserine lipids and membrane order precisely regulate the activity of Polybia-MP1 peptide. Biochim. Biophys. Acta Biomembr. 2017, 1859, 1067–1074. [Google Scholar] [CrossRef] [PubMed]
- Strömstedt, A.A.; Kristiansen, P.E.; Gunasekera, S.; Grob, N.; Skjeldal, L.; Göransson, U. Selective membrane disruption by the cyclotide kalata B7: Complex ions and essential functional groups in the phosphatidylethanolamine binding pocket. Biochim. Biophys. Acta Biomembr. 2016, 1858, 1317–1327. [Google Scholar] [CrossRef] [PubMed]
- Phoenix, D.A.; Harris, F.; Mura, M.; Dennison, S.R. The increasing role of phosphatidylethanolamine as a lipid receptor in the action of host defence peptides. Prog. Lipid Res. 2015, 59, 26–37. [Google Scholar] [CrossRef] [PubMed]
- Drin, G.; Antonny, B. Amphipathic helices and membrane curvature. FEBS Lett. 2010, 584, 1840–1847. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, N.W.; Wong, G.C.L. Antimicrobial peptides and induced membrane curvature: Geometry, coordination chemistry, and molecular engineering. Curr. Opin. Solid State Mater. Sci. 2013, 17, 151–163. [Google Scholar] [CrossRef] [PubMed]
- Epand, R.M.; Walker, C.; Epand, R.F.; Magarvey, N.A. Molecular mechanisms of membrane targeting antibiotics. Biochim. Biophys. Acta Biomembr. 2016, 1858, 980–987. [Google Scholar] [CrossRef] [PubMed]
- Andersson, D.I.; Hughes, D.; Kubicek-Sutherland, J.Z. Mechanisms and consequences of bacterial resistance to antimicrobial peptides. Drug Resist. Updat. 2016, 26, 43–57. [Google Scholar] [CrossRef] [PubMed]
- Ehrenstein, G.; Lecar, H. Electrically gated ionic channels in lipid bilayers. Q. Rev. Biophys. 1977, 10, 1–34. [Google Scholar] [CrossRef] [PubMed]
- Breukink, E.; de Kruijff, B. The lantibiotic nisin, a special case or not? Biochim. Biophys. Acta 1999, 1462, 223–234. [Google Scholar] [CrossRef]
- Wimley, W.C. Describing the Mechanism of Antimicrobial Peptide Action with the Interfacial Activity Model. ACS Chem. Biol. 2010, 5, 905–917. [Google Scholar] [CrossRef] [PubMed]
- Rapaport, D.; Shai, Y. Interaction of fluorescently labeled pardaxin and its analogues with lipid bilayers. J. Biol. Chem. 1991, 266, 23769–23775. [Google Scholar] [PubMed]
- Shai, Y.; Bach, D.; Yanovsky, A. Channel formation properties of synthetic pardaxin and analogues. J. Biol. Chem. 1990, 265, 20202–20209. [Google Scholar] [PubMed]
- Uematsu, N.; Matsuzaki, K. Polar Angle as a Determinant of Amphipathic α-Helix-Lipid Interactions: A Model Peptide Study. Biophys. J. 2000, 79, 2075–2083. [Google Scholar] [CrossRef]
- Sparr, E.; Ash, W.L.; Nazarov, P.V.; Rijkers, D.T.S.; Hemminga, M.A.; Tieleman, D.P.; Killian, J.A. Self-association of Transmembrane α-Helices in Model Membranes. J. Biol. Chem. 2005, 280, 39324–39331. [Google Scholar] [CrossRef] [PubMed]
- Cheng, J.T.J.; Hale, J.D.; Elliott, M.; Hancock, R.E.W.; Straus, S.K. The importance of bacterial membrane composition in the structure and function of aurein 2.2 and selected variants. Biochim. Biophys. Acta Biomembr. 2011, 1808, 622–633. [Google Scholar] [CrossRef] [PubMed]
- Sitaram, N.; Nagaraj, R. Interaction of antimicrobial peptides with biological and model membranes: Structural and charge requirements for activity. Biochim. Biophys. Acta 1999, 1462, 29–54. [Google Scholar] [CrossRef]
- Fernandez, D.I.; Le Brun, A.P.; Whitwell, T.C.; Sani, M.-A.; James, M.; Separovic, F. The antimicrobial peptide aurein 1.2 disrupts model membranes via the carpet mechanism. Phys. Chem. Chem. Phys. 2012, 14, 15739. [Google Scholar] [CrossRef] [PubMed]
- Gee, M.L.; Burton, M.; Grevis-James, A.; Hossain, M.A.; McArthur, S.; Palombo, E.A.; Wade, J.D.; Clayton, A.H.A. Imaging the action of antimicrobial peptides on living bacterial cells. Sci. Rep. 2013, 3, 1557. [Google Scholar] [CrossRef] [PubMed]
- Choi, H.; Rangarajan, N.; Weisshaar, J.C. Lights, Camera, Action! Antimicrobial Peptide Mechanisms Imaged in Space and Time. Trends Microbiol. 2016, 24, 111–122. [Google Scholar] [CrossRef] [PubMed]
- Manzini, M.C.; Perez, K.R.; Riske, K.A.; Bozelli, J.C.; Santos, T.L.; da Silva, M.A.; Saraiva, G.K.V.; Politi, M.J.; Valente, A.P.; Almeida, F.C.L.; et al. Peptide: Lipid ratio and membrane surface charge determine the mechanism of action of the antimicrobial peptide BP100. Conformational and functional studies. Biochim. Biophys. Acta Biomembr. 2014, 1838, 1985–1999. [Google Scholar] [CrossRef] [PubMed]
- Malanovic, N.; Lohner, K. Antimicrobial Peptides Targeting Gram-Positive Bacteria. Pharmaceuticals (Basel) 2016, 9. [Google Scholar] [CrossRef] [PubMed]
- Münch, D. Structural variations of the cell wall precursor lipid II in Gram-positive bacteria—Impact on binding and efficacy of antimicrobial peptides. Biochim. Biophys. Acta Biomembr. 2015, 1848, 3062–3071. [Google Scholar] [CrossRef] [PubMed]
- De Leeuw, E.; Li, C.; Zeng, P.; Li, C.; Buin, M.D.; Lu, W.-Y.; Breukink, E.; Lu, W. Functional interaction of human neutrophil peptide-1 with the cell wall precursor lipid II. FEBS Lett. 2010, 584, 1543–1548. [Google Scholar] [CrossRef] [PubMed]
- Wade, D.; Boman, A.; Wåhlin, B.; Drain, C.M.; Andreu, D.; Boman, H.G.; Merrifield, R.B. All-D amino acid-containing channel-forming antibiotic peptides. Proc. Natl. Acad. Sci. USA 1990, 87, 4761–4765. [Google Scholar] [CrossRef] [PubMed]
- Vunnam, S.; Juvvadi, P.; Merrifield, R.B. Synthesis and antibacterial action of cecropin and proline-arginine-rich peptides from pig intestine. J. Pept. Res. 1997, 49, 59–66. [Google Scholar] [CrossRef] [PubMed]
- Subbalakshmi, C.; Sitaram, N. Mechanism of antimicrobial action of indolicidin. FEMS Microbiol. Lett. 1998, 160, 91–96. [Google Scholar] [CrossRef] [PubMed]
- Sharma, H.; Nagaraj, R.; Rodrigues, D.; de Sousa, D.; da Silva, E.; de Moraes, L. Human β-Defensin 4 with Non-Native Disulfide Bridges Exhibit Antimicrobial Activity. PLoS ONE 2015, 10, e0119525. [Google Scholar] [CrossRef] [PubMed]
- Boman, H.G.; Agerberth, B.; Boman, A. Mechanisms of action on Escherichia coli of cecropin P1 and PR-39, two antibacterial peptides from pig intestine. Infect. Immun. 1993, 61, 2978–2984. [Google Scholar] [PubMed]
- Afacan, N.J.; Yeung, A.T.Y.; Pena, O.M.; Hancock, R.E.W. Therapeutic potential of host defense peptides in antibiotic-resistant infections. Curr. Pharm. Des. 2012, 18, 807–819. [Google Scholar] [CrossRef] [PubMed]
- Mader, J.S.; Hoskin, D.W. Cationic antimicrobial peptides as novel cytotoxic agents for cancer treatment. Expert Opin. Investig. Drugs 2006, 15, 933–946. [Google Scholar] [CrossRef] [PubMed]
- Lai, Y.; Gallo, R.L. AMPed up immunity: how antimicrobial peptides have multiple roles in immune defense. Trends Immunol. 2009, 30, 131–141. [Google Scholar] [CrossRef] [PubMed]
- Hancock, R.E.W.; Nijnik, A.; Philpott, D.J. Modulating immunity as a therapy for bacterial infections. Nat. Rev. Microbiol. 2012, 10, 243–254. [Google Scholar] [CrossRef] [PubMed]
- Niyonsaba, F.; Iwabuchi, K.; Someya, A.; Hirata, M.; Matsuda, H.; Ogawa, H.; Nagaoka, I. A cathelicidin family of human antibacterial peptide LL-37 induces mast cell chemotaxis. Immunology 2002, 106, 20–26. [Google Scholar] [CrossRef] [PubMed]
- García, J.-R.; Jaumann, F.; Schulz, S.; Krause, A.; Rodríguez-Jiménez, J.; Forssmann, U.; Adermann, K.; Klüver, E.; Vogelmeier, C.; Becker, D.; et al. Identification of a novel, multifunctional *-defensin (human *-defensin 3) with specific antimicrobial activity. Cell Tissue Res. 2001, 306, 257–264. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.J. Dendritic cell subsets and lineages, and their functions in innate and adaptive immunity. Cell 2001, 106, 259–262. [Google Scholar] [CrossRef]
- Nijnik, A.; Madera, L.; Ma, S.; Waldbrook, M.; Elliott, M.R.; Easton, D.M.; Mayer, M.L.; Mullaly, S.C.; Kindrachuk, J.; Jenssen, H.; et al. Synthetic Cationic Peptide IDR-1002 Provides Protection against Bacterial Infections through Chemokine Induction and Enhanced Leukocyte Recruitment. J. Immunol. 2010, 184, 2539–2550. [Google Scholar] [CrossRef] [PubMed]
- Scott, M.G.; Dullaghan, E.; Mookherjee, N.; Glavas, N.; Waldbrook, M.; Thompson, A.; Wang, A.; Lee, K.; Doria, S.; Hamill, P.; et al. An anti-infective peptide that selectively modulates the innate immune response. Nat. Biotechnol. 2007, 25, 465–472. [Google Scholar] [CrossRef] [PubMed]
- Nicholls, E.F.; Madera, L.; Hancock, R.E.W. Immunomodulators as adjuvants for vaccines and antimicrobial therapy. Ann. N. Y. Acad. Sci. 2010, 1213, 46–61. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Dhillon, P.; Yan, H.; Farmer, S.; Hancock, R.E. Interactions of bacterial cationic peptide antibiotics with outer and cytoplasmic membranes of Pseudomonas aeruginosa. Antimicrob. Agents Chemother. 2000, 44, 3317–3321. [Google Scholar] [CrossRef] [PubMed]
- Vaara, M. New approaches in peptide antibiotics. Curr. Opin. Pharmacol. 2009, 9, 571–576. [Google Scholar] [CrossRef] [PubMed]
- Gentilucci, L.; De Marco, R.; Cerisoli, L. Chemical modifications designed to improve peptide stability: incorporation of non-natural amino acids, pseudo-peptide bonds, and cyclization. Curr. Pharm. Des. 2010, 16, 3185–3203. [Google Scholar] [CrossRef] [PubMed]
- Nordström, R.; Malmsten, M. Delivery systems for antimicrobial peptides. Adv. Colloid Interface Sci. 2017, 242, 17–34. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Zhang, M.; Qiu, S.; Wang, J.; Peng, J.; Zhao, P.; Zhu, R.; Wang, H.; Li, Y.; Wang, K.; et al. Antimicrobial activity and stability of the D-amino acid substituted derivatives of antimicrobial peptide polybia-MPI. AMB Express 2016, 6, 122. [Google Scholar] [CrossRef] [PubMed]
- Kindrachuk, J.; Scruten, E.; Attah-Poku, S.; Bell, K.; Potter, A.; Babiuk, L.A.; Griebel, P.J.; Napper, S. Stability, toxicity, and biological activity of host defense peptide BMAP28 and its inversed and retro-inversed isomers. Biopolymers 2011, 96, 14–24. [Google Scholar] [CrossRef] [PubMed]
- Falciani, C.; Lozzi, L.; Pollini, S.; Luca, V.; Carnicelli, V.; Brunetti, J.; Lelli, B.; Bindi, S.; Scali, S.; Di Giulio, A.; et al. Isomerization of an antimicrobial peptide broadens antimicrobial spectrum to gram-positive bacterial pathogens. PLoS ONE 2012, 7, e46259. [Google Scholar] [CrossRef] [PubMed]
- Davies, J.S.; Elmore, D.T. Amino Acids, Peptides and Proteins. Volume 36, A Review of the Literature Published during 2003-2004; RSC Pub: Cambridge, UK, 2007; ISBN 9781847558459. [Google Scholar]
- Fjell, C.D.; Hiss, J.A.; Hancock, R.E.W.; Schneider, G. Designing antimicrobial peptides: form follows function. Nat. Rev. Drug Discov. 2011, 11, 37. [Google Scholar] [CrossRef] [PubMed]
- Berthold, N.; Czihal, P.; Fritsche, S.; Sauer, U.; Schiffer, G.; Knappe, D.; Alber, G.; Hoffmann, R. Novel Apidaecin 1b Analogs with Superior Serum Stabilities for Treatment of Infections by Gram-Negative Pathogens. Antimicrob. Agents Chemother. 2013, 57, 402–409. [Google Scholar] [CrossRef] [PubMed]
- Papanastasiou, E.A.; Hua, Q.; Sandouk, A.; Son, U.H.; Christenson, A.J.; Van Hoek, M.L.; Bishop, B.M. Role of acetylation and charge in antimicrobial peptides based on human β-defensin-3. APMIS 2009, 117, 492–499. [Google Scholar] [CrossRef] [PubMed]
- Jayawardene, D.S.; Dass, C. The effect of N-terminal acetylation and the inhibition activity of acetylated enkephalins on the aminopeptidase M-catalyzed hydrolysis of enkephalins. Peptides 1999, 20, 963–970. [Google Scholar] [CrossRef]
- Nguyen, L.T.; Chau, J.K.; Perry, N.A.; de Boer, L.; Zaat, S.A.J.; Vogel, H.J. Serum Stabilities of Short Tryptophan- and Arginine-Rich Antimicrobial Peptide Analogs. PLoS ONE 2010, 5, e12684. [Google Scholar] [CrossRef] [PubMed]
- Liu, B.; Zhang, W.; Gou, S.; Huang, H.; Yao, J.; Yang, Z.; Liu, H.; Zhong, C.; Liu, B.; Ni, J.; et al. Intramolecular cyclization of the antimicrobial peptide Polybia-MPI with triazole stapling: influence on stability and bioactivity. J. Pept. Sci. 2017. [Google Scholar] [CrossRef] [PubMed]
- Som, A.; Vemparala, S.; Ivanov, I.; Tew, G.N. Synthetic mimics of antimicrobial peptides. Biopolymers 2008, 90, 83–93. [Google Scholar] [CrossRef] [PubMed]
- Rotem, S.; Mor, A. Antimicrobial peptide mimics for improved therapeutic properties. Biochim. Biophys. Acta Biomembr. 2009, 1788, 1582–1592. [Google Scholar] [CrossRef] [PubMed]
- Giuliani, A.; Rinaldi, A.C. Beyond natural antimicrobial peptides: multimeric peptides and other peptidomimetic approaches. Cell. Mol. Life Sci. 2011, 68, 2255–2266. [Google Scholar] [CrossRef] [PubMed]
- Kapoor, R.; Wadman, M.W.; Dohm, M.T.; Czyzewski, A.M.; Spormann, A.M.; Barron, A.E. Antimicrobial peptoids are effective against Pseudomonas aeruginosa biofilms. Antimicrob. Agents Chemother. 2011, 55, 3054–3057. [Google Scholar] [CrossRef] [PubMed]
- Chongsiriwatana, N.P.; Patch, J.A.; Czyzewski, A.M.; Dohm, M.T.; Ivankin, A.; Gidalevitz, D.; Zuckermann, R.N.; Barron, A.E. Peptoids that mimic the structure, function, and mechanism of helical antimicrobial peptides. Proc. Natl. Acad. Sci. USA 2008, 105, 2794–2799. [Google Scholar] [CrossRef] [PubMed]
- Andreev, K.; Martynowycz, M.W.; Ivankin, A.; Huang, M.L.; Kuzmenko, I.; Meron, M.; Lin, B.; Kirshenbaum, K.; Gidalevitz, D. Cyclization Improves Membrane Permeation by Antimicrobial Peptoids. Langmuir 2016, 32, 12905–12913. [Google Scholar] [CrossRef] [PubMed]
- Reinhardt, A.; Neundorf, I. Design and Application of Antimicrobial Peptide Conjugates. Int. J. Mol. Sci. 2016, 17, 701. [Google Scholar] [CrossRef] [PubMed]
- Syryamina, V.N.; Samoilova, R.I.; Tsvetkov, Y.D.; Ischenko, A.V.; De Zotti, M.; Gobbo, M.; Toniolo, C.; Formaggio, F.; Dzuba, S.A. Peptides on the Surface: Spin-Label EPR and PELDOR Study of Adsorption of the Antimicrobial Peptides Trichogin GA IV and Ampullosporin A on the Silica Nanoparticles. Appl. Magn. Reson. 2016, 47, 309–320. [Google Scholar] [CrossRef]
- Godoy-Gallardo, M.; Mas-Moruno, C.; Yu, K.; Manero, J.M.; Gil, F.J.; Kizhakkedathu, J.N.; Rodriguez, D. Antibacterial Properties of hLf1–11 Peptide onto Titanium Surfaces: A Comparison Study Between Silanization and Surface Initiated Polymerization. Biomacromolecules 2015, 16, 483–496. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.-Y.; Chang, H.-Y.; Lu, J.-K.; Huang, Y.-C.; Harroun, S.G.; Tseng, Y.-T.; Li, Y.-J.; Huang, C.-C.; Chang, H.-T. Self-Assembly of Antimicrobial Peptides on Gold Nanodots: Against Multidrug-Resistant Bacteria and Wound-Healing Application. Adv. Funct. Mater. 2015, 25, 7189–7199. [Google Scholar] [CrossRef]
- Chaudhari, A.A.; Ashmore, D.; deb Nath, S.; Kate, K.; Dennis, V.; Singh, S.R.; Owen, D.R.; Palazzo, C.; Arnold, R.D.; Miller, M.E.; Pillai, S.R. A novel covalent approach to bio-conjugate silver coated single walled carbon nanotubes with antimicrobial peptide. J. Nanobiotechnology 2016, 14, 58. [Google Scholar] [CrossRef] [PubMed]
- Galdiero, E.; Siciliano, A.; Maselli, V.; Gesuele, R.; Guida, M.; Fulgione, D.; Galdiero, S.; Lombardi, L.; Falanga, A. An integrated study on antimicrobial activity and ecotoxicity of quantum dots and quantum dots coated with the antimicrobial peptide indolicidin. Int. J. Nanomed. 2016, 11, 4199–4211. [Google Scholar] [CrossRef] [PubMed]
- Kanchanapally, R.; Viraka Nellore, B.P.; Sinha, S.S.; Pedraza, F.; Jones, S.J.; Pramanik, A.; Chavva, S.R.; Tchounwou, C.; Shi, Y.; Vangara, A.; et al. Antimicrobial peptide-conjugated graphene oxide membrane for efficient removal and effective killing of multiple drug resistant bacteria. RSC Adv. 2015, 5, 18881–18887. [Google Scholar] [CrossRef] [PubMed]
- Dostalova, S.; Moulick, A.; Milosavljevic, V.; Guran, R.; Kominkova, M.; Cihalova, K.; Heger, Z.; Blazkova, L.; Kopel, P.; Hynek, D.; et al. Antiviral activity of fullerene C60 nanocrystals modified with derivatives of anionic antimicrobial peptide maximin H5. Monatshefte für Chem. Chem. Mon. 2016, 147, 905–918. [Google Scholar] [CrossRef]
- Vivero-Escoto, J.L.; Slowing, I.I.; Trewyn, B.G.; Lin, V.S.-Y. Mesoporous Silica Nanoparticles for Intracellular Controlled Drug Delivery. Small 2010, 6, 1952–1967. [Google Scholar] [CrossRef] [PubMed]
- Urbán, P.; Valle-Delgado, J.J.; Moles, E.; Marques, J.; Díez, C.; Fernàndez-Busquets, X. Nanotools for the delivery of antimicrobial peptides. Curr. Drug Targets 2012, 13, 1158–1172. [Google Scholar] [CrossRef] [PubMed]
- Malmsten, M. Soft drug delivery systems. Soft Matter 2006, 2, 760. [Google Scholar] [CrossRef]
- Li, P.; Nielsen, H.M.; Müllertz, A. Oral delivery of peptides and proteins using lipid-based drug delivery systems. Expert Opin. Drug Deliv. 2012, 9, 1289–1304. [Google Scholar] [CrossRef] [PubMed]
- Ron-Doitch, S.; Sawodny, B.; Kühbacher, A.; David, M.M.N.; Samanta, A.; Phopase, J.; Burger-Kentischer, A.; Griffith, M.; Golomb, G.; Rupp, S. Reduced cytotoxicity and enhanced bioactivity of cationic antimicrobial peptides liposomes in cell cultures and 3D epidermis model against HSV. J. Control. Release 2016, 229, 163–171. [Google Scholar] [CrossRef] [PubMed]
- Taylor, T.M.; Gaysinsky, S.; Davidson, P.M.; Bruce, B.D.; Weiss, J. Characterization of Antimicrobial-bearing Liposomes by ζ-Potential, Vesicle Size, and Encapsulation Efficiency. Food Biophys. 2007, 2, 1–9. [Google Scholar] [CrossRef]
- Sadiq, S.; Imran, M.; Habib, H.; Shabbir, S.; Ihsan, A.; Zafar, Y.; Hafeez, F.Y. Potential of monolaurin based food-grade nano-micelles loaded with nisin Z for synergistic antimicrobial action against Staphylococcus aureus. LWT Food Sci. Technol. 2016, 71, 227–233. [Google Scholar] [CrossRef]
- D’Angelo, I.; Casciaro, B.; Miro, A.; Quaglia, F.; Mangoni, M.L.; Ungaro, F. Overcoming barriers in Pseudomonas aeruginosa lung infections: Engineered nanoparticles for local delivery of a cationic antimicrobial peptide. Colloids Surfaces B Biointerfaces 2015, 135, 717–725. [Google Scholar] [CrossRef] [PubMed]
- Yüksel, E.; Karakeçili, A.; Demirtaş, T.T.; Gümüşderelioğlu, M. Preparation of bioactive and antimicrobial PLGA membranes by magainin II/EGF functionalization. Int. J. Biol. Macromol. 2016, 86, 162–168. [Google Scholar] [CrossRef] [PubMed]
- Dong, P.; Zhou, Y.; He, W.; Hua, D. A strategy for enhanced antibacterial activity against Staphylococcus aureus by the assembly of alamethicin with a thermo-sensitive polymeric carrier. Chem. Commun. 2016, 52, 896–899. [Google Scholar] [CrossRef] [PubMed]
- Davis, F.F. The origin of pegnology. Adv. Drug Deliv. Rev. 2002, 54, 457–458. [Google Scholar] [CrossRef]
- Veronese, F.M.; Mero, A. The Impact of PEGylation on Biological Therapies. BioDrugs 2008, 22, 315–329. [Google Scholar] [CrossRef] [PubMed]
- Imura, Y.; Nishida, M.; Ogawa, Y.; Takakura, Y.; Matsuzaki, K. Action mechanism of tachyplesin I and effects of PEGylation. Biochim. Biophys. Acta Biomembr. 2007, 1768, 1160–1169. [Google Scholar] [CrossRef] [PubMed]
- Imura, Y.; Nishida, M.; Matsuzaki, K. Action mechanism of PEGylated magainin 2 analogue peptide. Biochim. Biophys. Acta Biomembr. 2007, 1768, 2578–2585. [Google Scholar] [CrossRef] [PubMed]
- Guiotto, A.; Pozzobon, M.; Canevari, M.; Manganelli, R.; Scarin, M.; Veronese, F.M. PEGylation of the antimicrobial peptide nisin A: problems and perspectives. Farmaco 2003, 58, 45–50. [Google Scholar] [CrossRef]
- Singh, S.; Papareddy, P.; Mörgelin, M.; Schmidtchen, A.; Malmsten, M. Effects of PEGylation on Membrane and Lipopolysaccharide Interactions of Host Defense Peptides. Biomacromolecules 2014, 15, 1337–1345. [Google Scholar] [CrossRef] [PubMed]
- Imran Ul-Haq, M.; Lai, B.F.L.; Chapanian, R.; Kizhakkedathu, J.N. Influence of architecture of high molecular weight linear and branched polyglycerols on their biocompatibility and biodistribution. Biomaterials 2012, 33, 9135–9147. [Google Scholar] [CrossRef] [PubMed]
- Kong, M.; Chen, X.G.; Xing, K.; Park, H.J. Antimicrobial properties of chitosan and mode of action: A state of the art review. Int. J. Food Microbiol. 2010, 144, 51–63. [Google Scholar] [CrossRef] [PubMed]
- Sahariah, P.; Sørensen, K.K.; Hjálmarsdóttir, M.Á.; Sigurjónsson, Ó.E.; Jensen, K.J.; Másson, M.; Thygesen, M.B. Antimicrobial peptide shows enhanced activity and reduced toxicity upon grafting to chitosan polymers. Chem. Commun. 2015, 51, 11611–11614. [Google Scholar] [CrossRef] [PubMed]
- Barbosa, M.; Vale, N.; Costa, F.M.T.A.; Martins, M.C.L.; Gomes, P. Tethering antimicrobial peptides onto chitosan: Optimization of azide-alkyne “click” reaction conditions. Carbohydr. Polym. 2017, 165, 384–393. [Google Scholar] [CrossRef] [PubMed]
- Lequeux, I.; Ducasse, E.; Jouenne, T.; Thebault, P. Addition of antimicrobial properties to hyaluronic acid by grafting of antimicrobial peptide. Eur. Polym. J. 2014, 51, 182–190. [Google Scholar] [CrossRef]
- Shenoi, R.A.; Kalathottukaren, M.T.; Travers, R.J.; Lai, B.F.L.; Creagh, A.L.; Lange, D.; Yu, K.; Weinhart, M.; Chew, B.H.; Du, C.; et al. Affinity-based design of a synthetic universal reversal agent for heparin anticoagulants. Sci. Transl. Med. 2014, 6. [Google Scholar] [CrossRef] [PubMed]
- Imran ul-haq, M.; Hamilton, J.L.; Lai, B.F.L.; Shenoi, R.A.; Horte, S.; Constantinescu, I.; Leitch, H.A.; Kizhakkedathu, J.N. Design of long circulating nontoxic dendritic polymers for the removal of iron in vivo. ACS Nano 2013, 7, 10704–10716. [Google Scholar] [CrossRef] [PubMed]
- Kainthan, R.K.; Janzen, J.; Kizhakkedathu, J.N.; Devine, D.V.; Brooks, D.E. Hydrophobically derivatized hyperbranched polyglycerol as a human serum albumin substitute. Biomaterials 2008, 29, 1693–1704. [Google Scholar] [CrossRef] [PubMed]
- Hamilton, J.L.; Imran Ul-Haq, M.; Abbina, S.; Kalathottukaren, M.T.; Lai, B.F.L.; Hatef, A.; Unniappan, S.; Kizhakkedathu, J.N. In vivo efficacy, toxicity and biodistribution of ultra-long circulating desferrioxamine based polymeric iron chelator. Biomaterials 2016, 102, 58–71. [Google Scholar] [CrossRef] [PubMed]
- Kalathottukaren, M.T.; Abraham, L.; Kapopara, P.R.; Lai, B.F.L.; Shenoi, R.A.; Rosell, F.I.; Conway, E.M.; Pryzdial, E.L.G.; Morrissey, J.H.; Haynes, C.A.; et al. Alteration of blood clotting and lung damage by protamine are avoided using the heparin and polyphosphate inhibitor UHRA. Blood 2017, 129, 1368–1379. [Google Scholar] [CrossRef] [PubMed]
- Chapanian, R.; Kwan, D.H.; Constantinescu, I.; Shaikh, F.A.; Rossi, N.A.A.; Withers, S.G.; Kizhakkedathu, J.N. Enhancement of biological reactions on cell surfaces via macromolecular crowding. Nat. Commun. 2014, 5, 4683. [Google Scholar] [CrossRef] [PubMed]
- Du, C.; Mendelson, A.A.; Guan, Q.; Chapanian, R.; Chafeeva, I.; da Roza, G.; Kizhakkedathu, J.N. The size-dependent efficacy and biocompatibility of hyperbranched polyglycerol in peritoneal dialysis. Biomaterials 2014, 35, 1378–1389. [Google Scholar] [CrossRef] [PubMed]
- Gao, S.; Guan, Q.; Chafeeva, I.; Brooks, D.E.; Nguan, C.Y.C.; Kizhakkedathu, J.N.; Du, C. Hyperbranched polyglycerol as a colloid in cold organ preservation solutions. PLoS ONE 2015, 10, 1–22. [Google Scholar] [CrossRef] [PubMed]
- Kumar, P.; Shenoi, R.A.; Lai, B.F.L.; Nguyen, M.; Kizhakkedathu, J.N.; Straus, S.K. Conjugation of Aurein 2.2 to HPG Yields an Antimicrobial with Better Properties. Biomacromolecules 2015, 16, 913–923. [Google Scholar] [CrossRef] [PubMed]
- Kumar, P.; Takayesu, A.; Abbasi, U.; Kalathottukaren, M.T.; Abbina, S.; Kizhakkedathu, J.N.; Straus, S.K. Antimicrobial Peptide–Polymer Conjugates with High Activity: Influence of Polymer Molecular Weight and Peptide Sequence on Antimicrobial Activity, Proteolysis, and Biocompatibility. ACS Appl. Mater. Interfaces 2017. [Google Scholar] [CrossRef] [PubMed]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Category a | Peptides | Unique Structural/Sequence Feature | Source |
|---|---|---|---|
| α helical peptides | Aurein 1–2 [36,37] | Amidated C-terminus | Frogs |
| Mellitin [23] | Amidated C-terminus | Bees | |
| Brevinin 1 [48] | - | Frogs | |
| Maculatins [50] | Amidated C-terminus | Frogs | |
| Citropin [51] | Amidated C-terminus | Frogs | |
| Buforin II [52] | - | Toad | |
| Cathelicidins [31] | |||
| • LL-37 b | Amidated C-terminus | Humans | |
| • BMAP-27,28,34 b | - | Bovine | |
| • Magainins | - | Frogs | |
| • Cecropin | Amidated C-terminus | Insect | |
| β sheet peptides | Cathelicidins [31] | ||
| • Protegrins | Cysteine rich | Pigs | |
| • Bactenecin | Disulfide forming loop/Arginine rich | Bovine | |
| Defensins c [20,53,54,55] | |||
| • α defensins | Three disulfide bonds | Mammals | |
| • β defensins | Three disulfide bonds | Mammals | |
| • θ defensins | Three disulfide bonds and cyclic | Gorilla | |
| Tachyplesins [16] and Polyphemusin [49] | Cysteine/arginine rich and amidated C-terminus | Horse Crab | |
| Extended/flexible | Cathelicidins [31] | ||
| • PR-39 b | Proline and arginine rich | Pigs | |
| • Tritrpticin | Tryptophan and arginine rich | Pigs | |
| • Indolicidin | Tryptophan and amidated C-terminus | Bovine | |
| • Crotalicidin 15–34 | Lysine rich | Snakes | |
| Histatins [56] | Histidine rich and amidated C-terminus | Humans |
| Peptide | Progress | Application | AMP Analogue (Host) |
|---|---|---|---|
| Pexiganan | Phase III | Topical application for diabetic foot ulcers | Magainin (frogs) |
| OP145 | Phase I/II | Bacterial ear infection | LL-37 (humans) |
| Omiganan | Phase III | Topical cream for prevention of catheter infection, severe acne, rosacea, atopic dermatitis | Indolicidin (bovine) |
| PAC 113 | Phase II | Mouth wash for fungal/yeast infection | Histatin (humans) |
| Iseganan | Phase III | Treatment of inflammation and ulceration of digestive system mucous membrane | Protegrin-1 (pigs) |
| IMX942 | Phase II | Intravenous administration against hospital-acquired bacterial infections | Synthetic analogue of IDR-1 a |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kumar, P.; Kizhakkedathu, J.N.; Straus, S.K. Antimicrobial Peptides: Diversity, Mechanism of Action and Strategies to Improve the Activity and Biocompatibility In Vivo. Biomolecules 2018, 8, 4. https://doi.org/10.3390/biom8010004
Kumar P, Kizhakkedathu JN, Straus SK. Antimicrobial Peptides: Diversity, Mechanism of Action and Strategies to Improve the Activity and Biocompatibility In Vivo. Biomolecules. 2018; 8(1):4. https://doi.org/10.3390/biom8010004
Chicago/Turabian StyleKumar, Prashant, Jayachandran N. Kizhakkedathu, and Suzana K. Straus. 2018. "Antimicrobial Peptides: Diversity, Mechanism of Action and Strategies to Improve the Activity and Biocompatibility In Vivo" Biomolecules 8, no. 1: 4. https://doi.org/10.3390/biom8010004
APA StyleKumar, P., Kizhakkedathu, J. N., & Straus, S. K. (2018). Antimicrobial Peptides: Diversity, Mechanism of Action and Strategies to Improve the Activity and Biocompatibility In Vivo. Biomolecules, 8(1), 4. https://doi.org/10.3390/biom8010004