Next‐Generation Sequencing‐Based RiboMethSeq Protocol for Analysis of tRNA 2′‐O‐Methylation


Abstract
:1. Introduction
1.1. Bacterial tRNA 2′-O-methylation
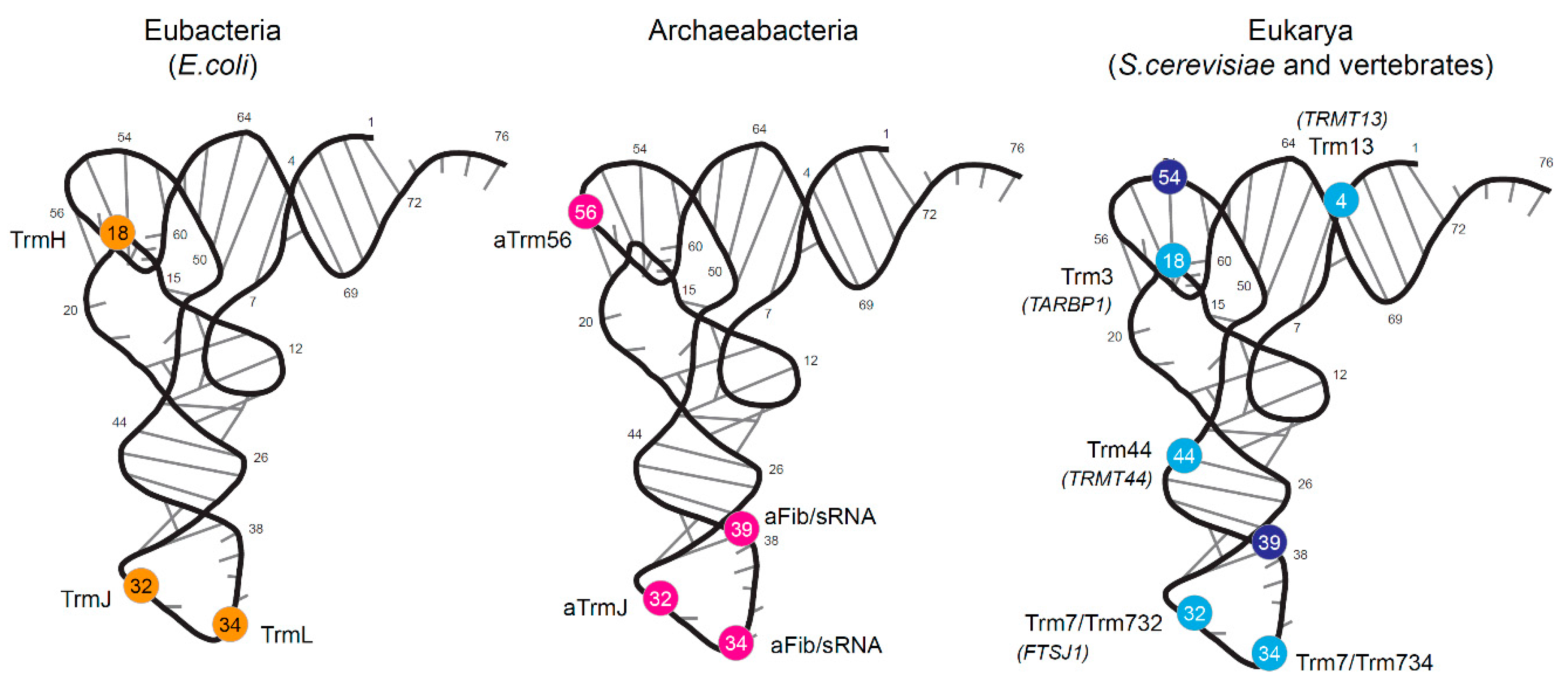
1.2. Archaeal tRNA 2′-O-Methylation
1.3. Eukaryotic tRNA 2′-O-Methylation
1.4. Functions of tRNA 2′-O-Methylation in tRNA Stability and Immunostimulation
1.5. Analytical Methods for Detection of 2′-O-Methylation in RNA
2. Results and Discussion
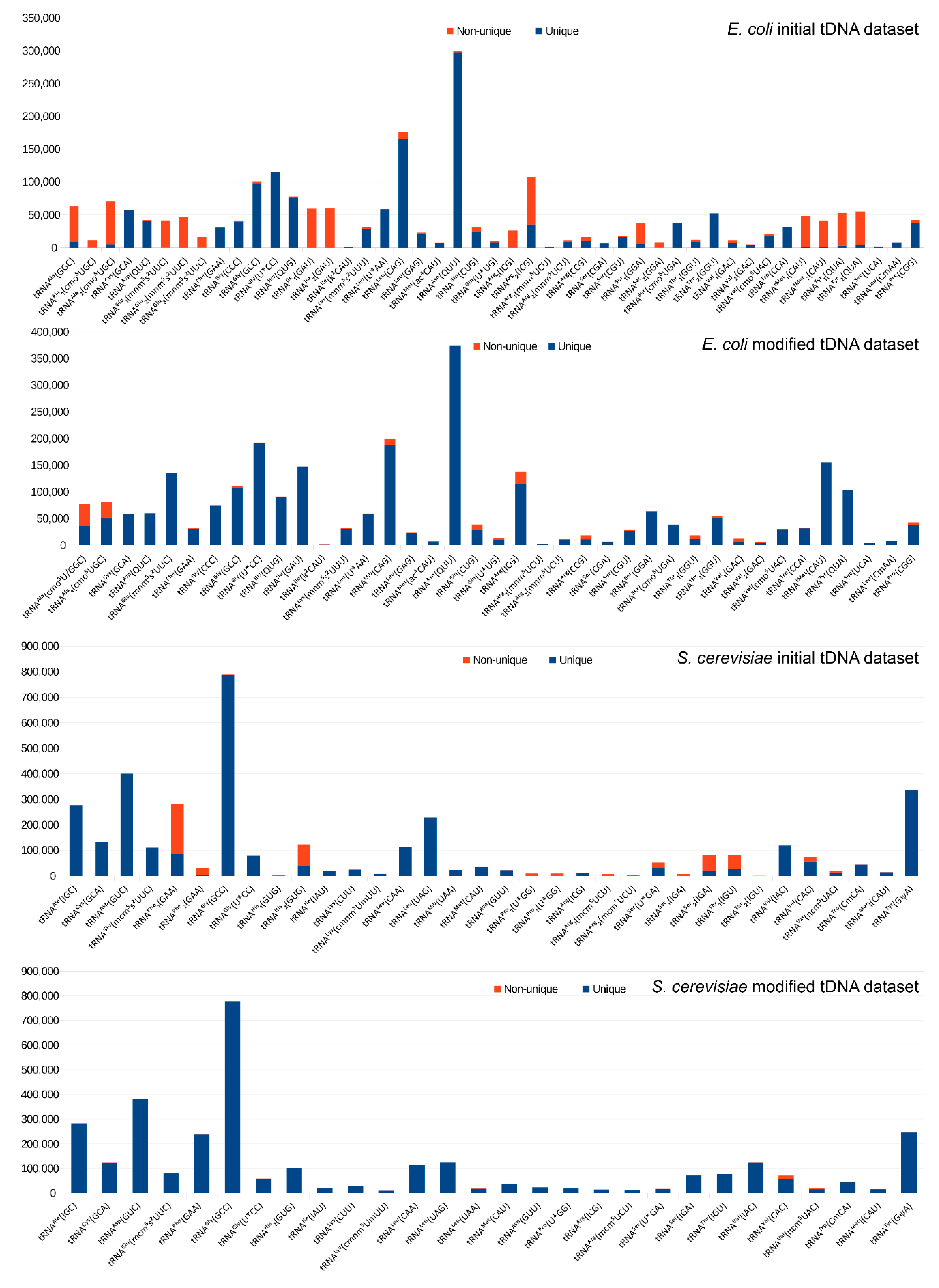
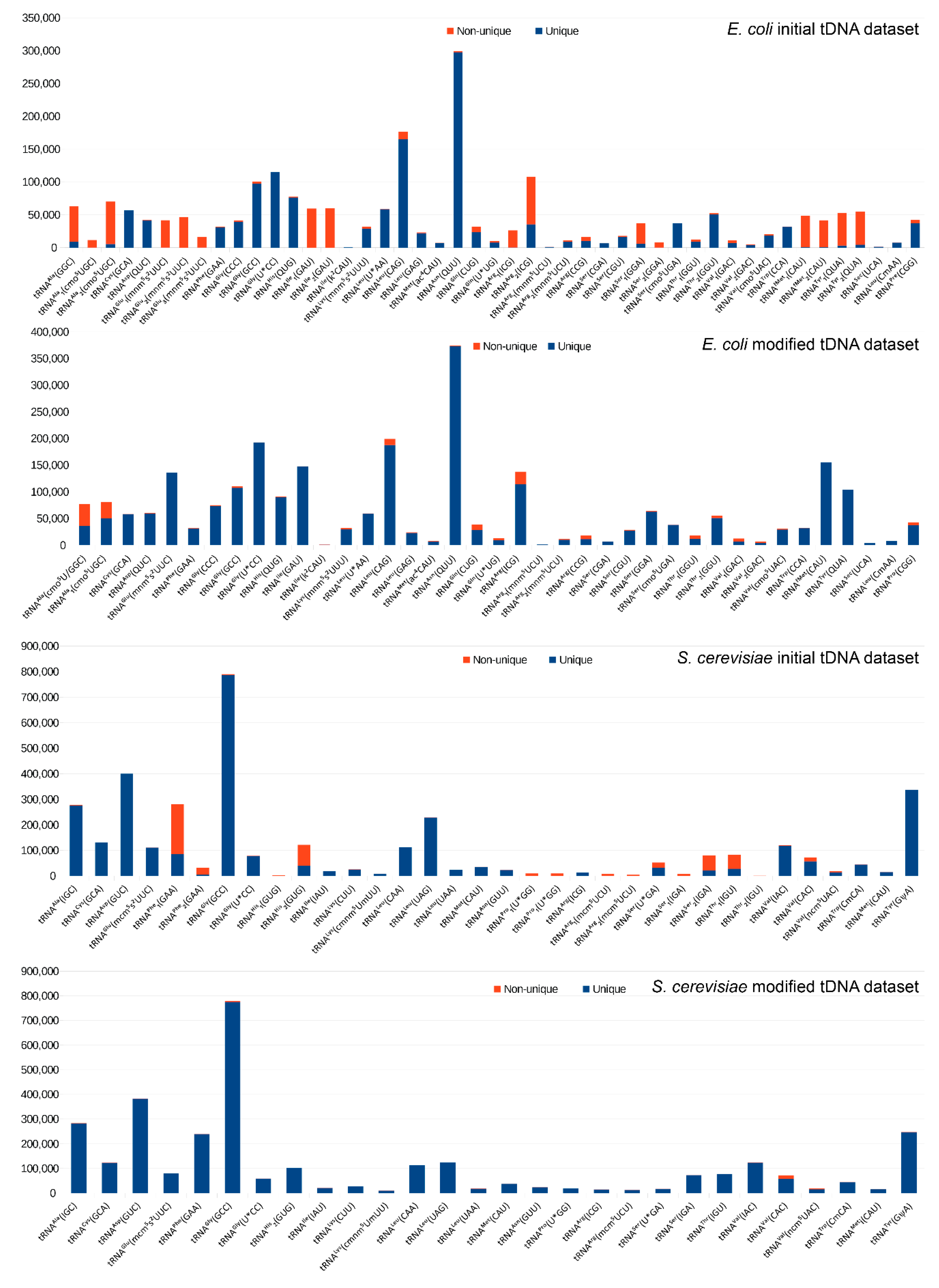
2.1. Construction of the Reference tDNA Dataset
2.2. Alignment and Analysis
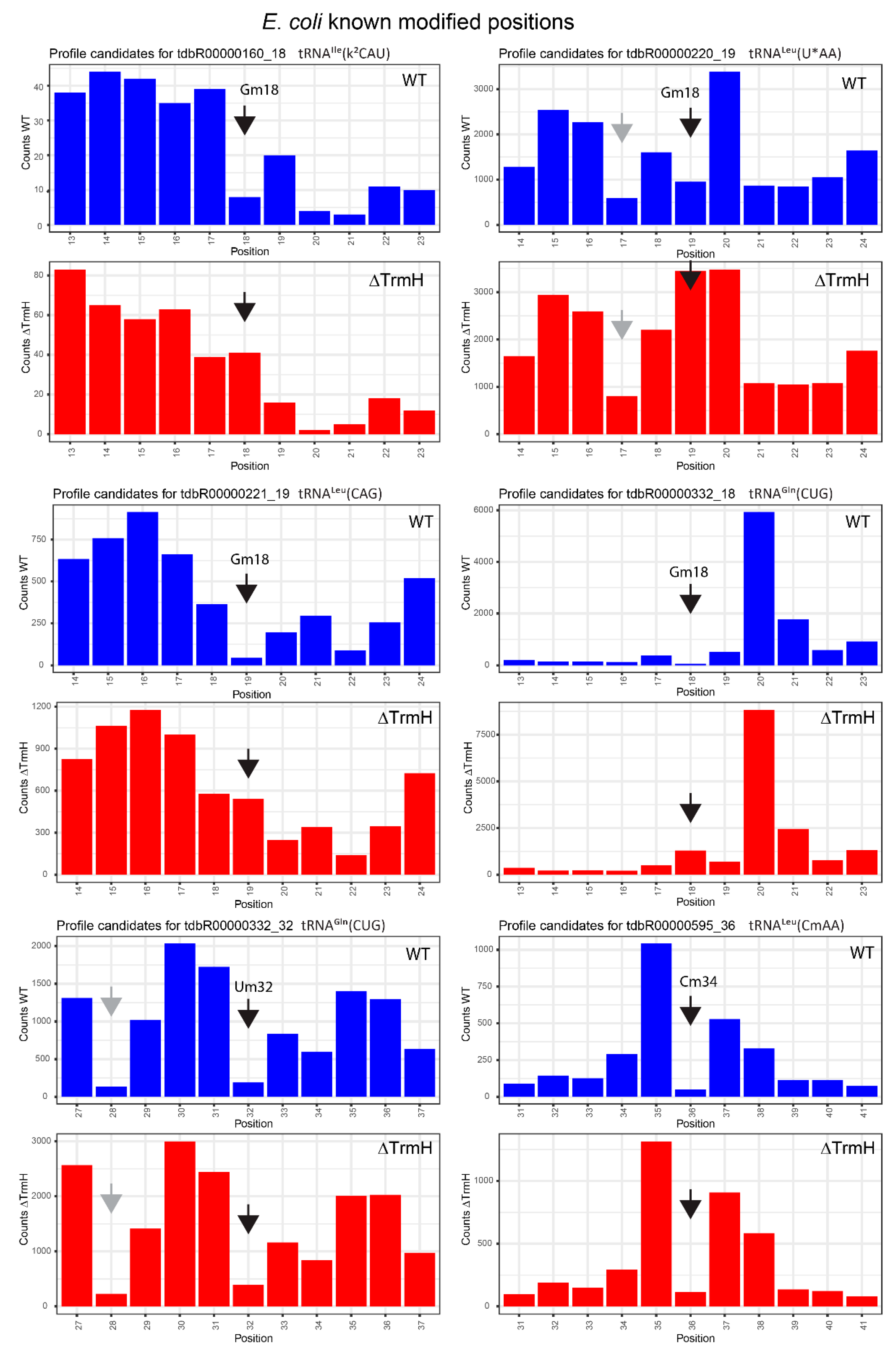
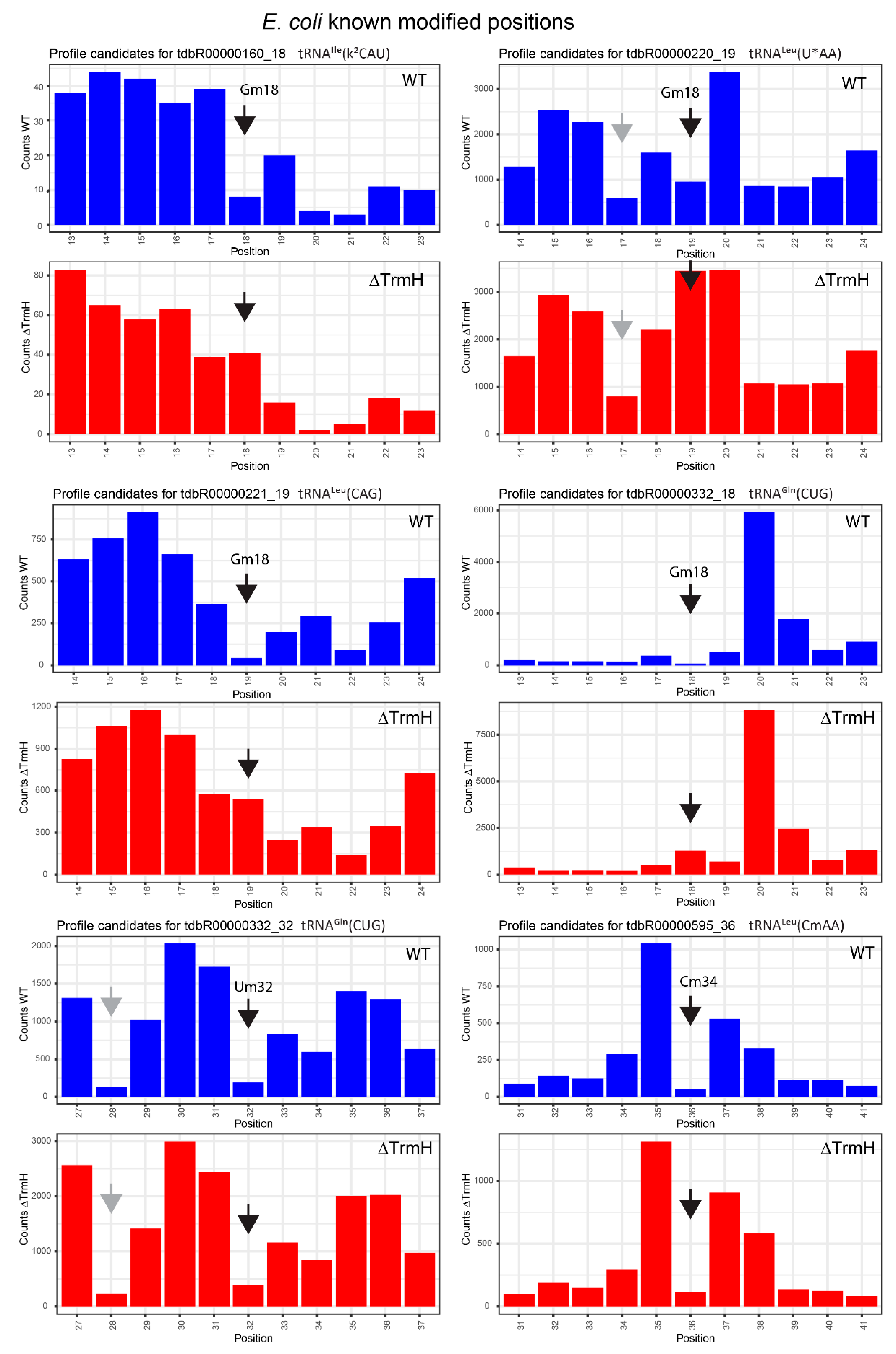
2.3. Profiles for tRNAs from WT and ΔTrmH E. coli Mutant Strains
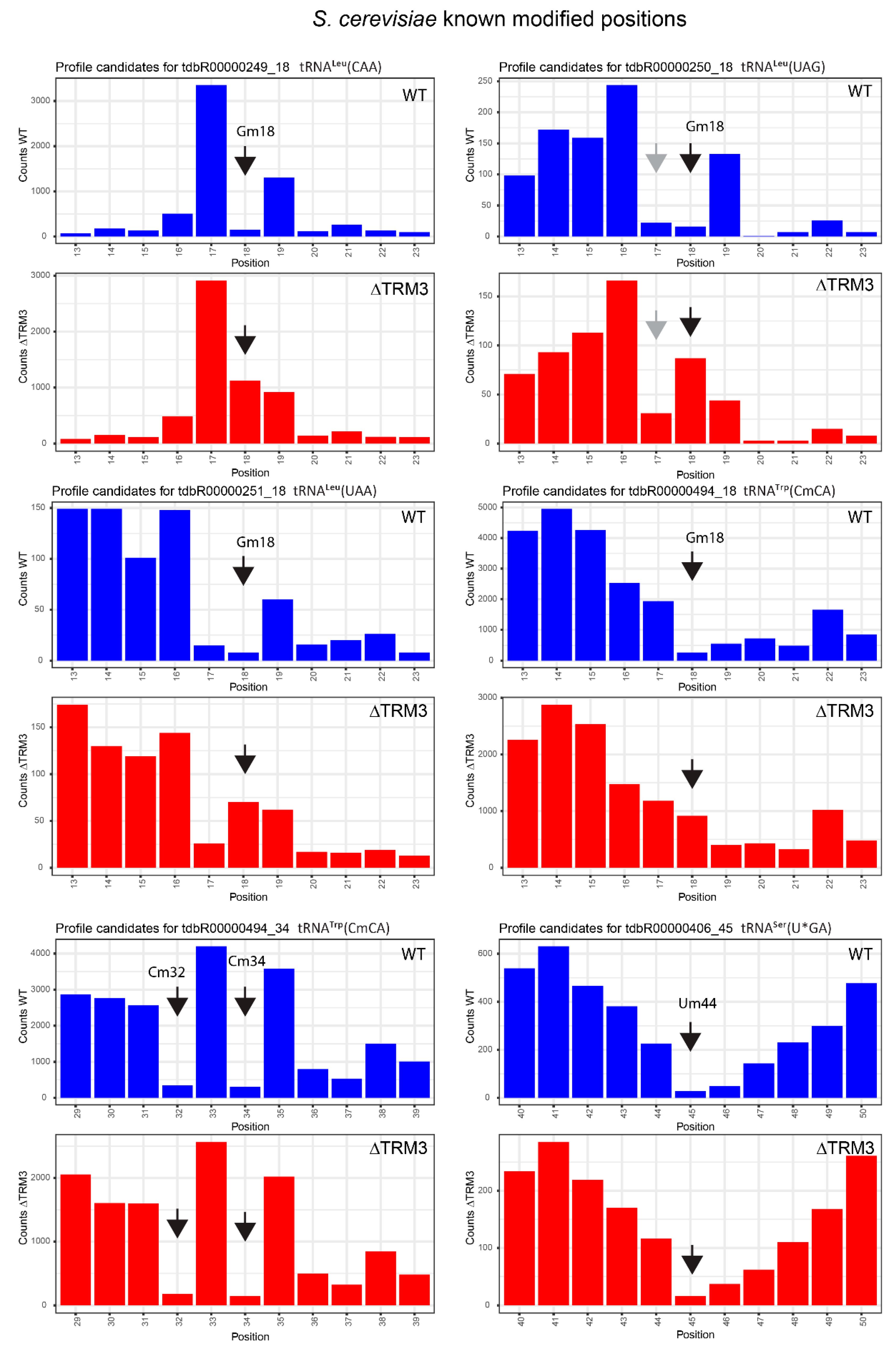
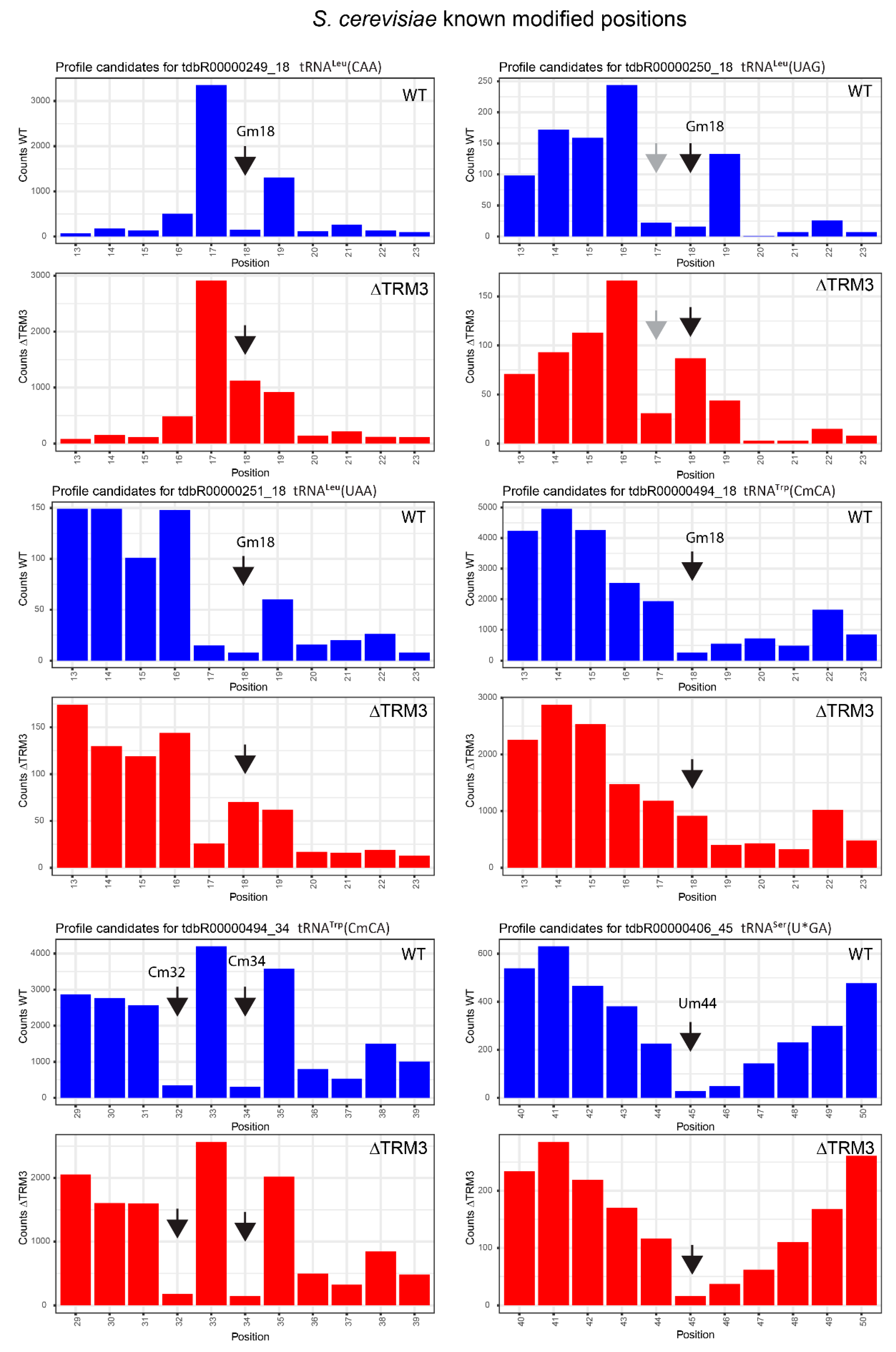
2.4. Analysis of 2′-O-Methylation in WT and ΔTRM3 Yeast Mutant Strains
2.5. Limitations
3. Materials and Methods
3.1. tRNA Sources
3.2. Isolation of tRNA
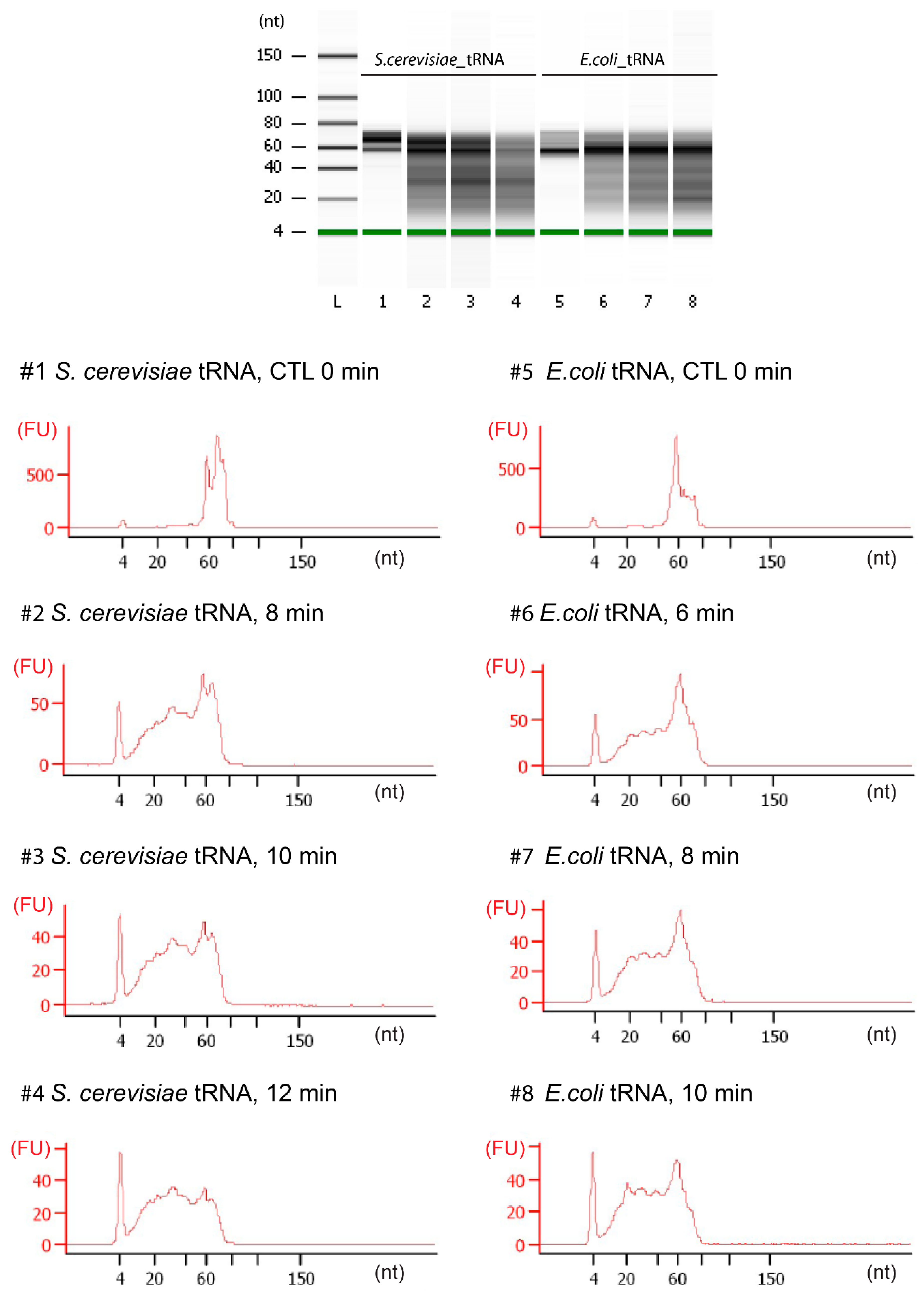
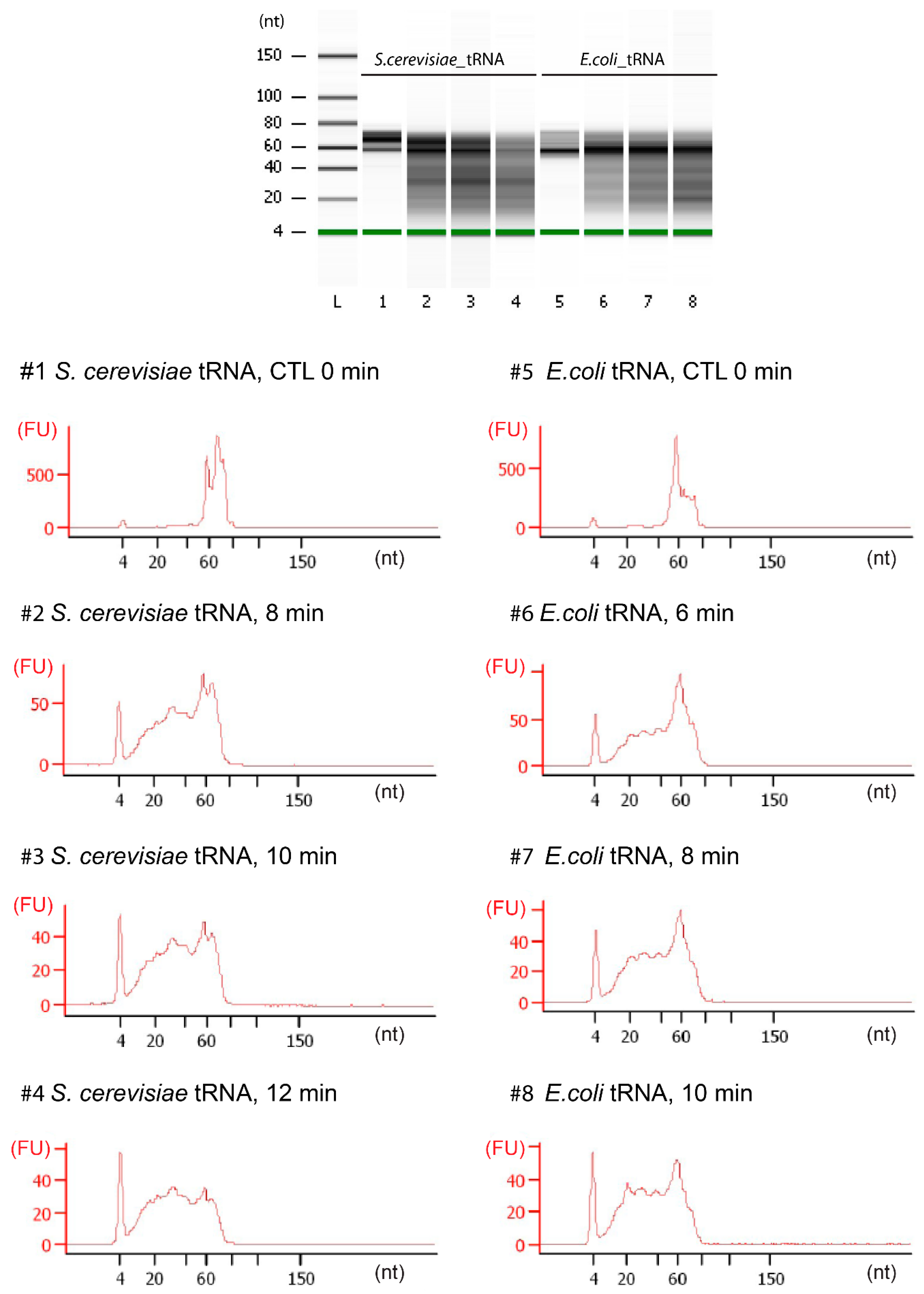
3.3. tRNA Fragmentation and Analysis
3.4. Library Preparation
3.5. Sequencing
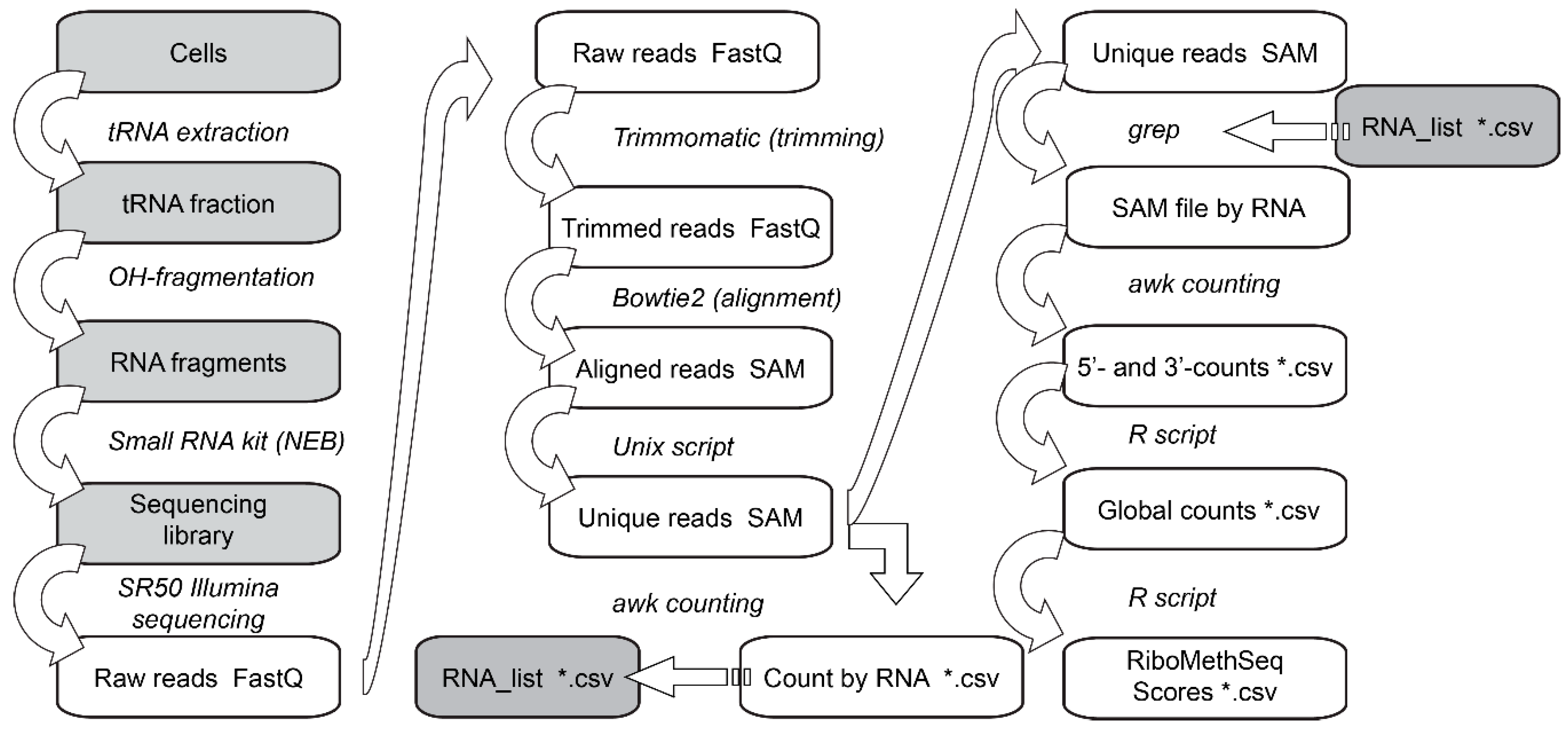
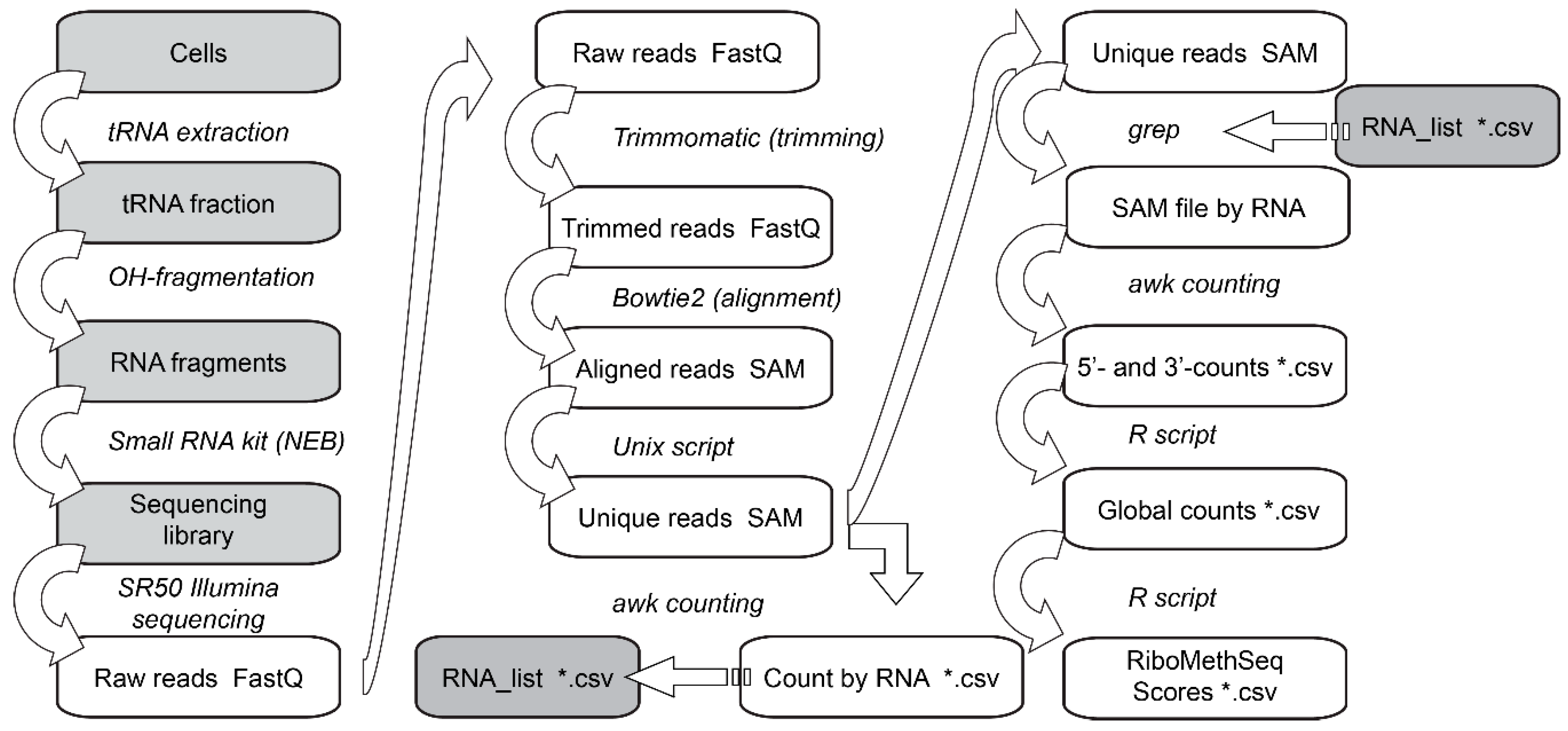
3.6. Data Analysis
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Cheng, X.; Roberts, R.J. AdoMet-dependent methylation, DNA methyltransferases and base flipping. Nucleic Acids Res. 2001, 29, 3784–3795. [Google Scholar] [CrossRef] [PubMed]
- Boschi-Muller, S.; Motorin, Y. Chemistry enters nucleic acids biology: Enzymatic mechanisms of RNA modification. Biochem. Biokhimiia 2013, 78, 1392–1404. [Google Scholar] [CrossRef] [PubMed]
- Motorin, Y.; Seidu-Larry, S.; Helm, M. DNA and RNA Pyrimidine Nucleobase Alkylation at the Carbon-5 Position. Adv. Exp. Med. Biol. 2016, 945, 19–33. [Google Scholar] [PubMed]
- Rana, A.K.; Ankri, S. Reviving the RNA World: An Insight into the Appearance of RNA Methyltransferases. Front. Genet. 2016, 7, 99. [Google Scholar] [CrossRef] [PubMed]
- Towns, W.L.; Begley, T.J. Transfer RNA methytransferases and their corresponding modifications in budding yeast and humans: Activities, predications, and potential roles in human health. DNA Cell Biol. 2012, 31, 434–454. [Google Scholar] [CrossRef] [PubMed]
- Motorin, Y.; Helm, M. RNA nucleotide methylation. Wiley Interdiscip. Rev. RNA 2011, 2, 611–631. [Google Scholar] [CrossRef] [PubMed]
- Gilbert, W.V.; Bell, T.A.; Schaening, C. Messenger RNA modifications: Form, distribution, and function. Science 2016, 352, 1408–1412. [Google Scholar] [CrossRef] [PubMed]
- Yue, Y.; Liu, J.; He, C. RNA N6-methyladenosine methylation in post-transcriptional gene expression regulation. Genes Dev. 2015, 29, 1343–1355. [Google Scholar] [CrossRef] [PubMed]
- Sarin, L.P.; Leidel, S.A. Modify or die?--RNA modification defects in metazoans. RNA Biol. 2014, 11, 1555–1567. [Google Scholar] [CrossRef] [PubMed]
- Machnicka, M.A.; Milanowska, K.; Osman Oglou, O.; Purta, E.; Kurkowska, M.; Olchowik, A.; Januszewski, W.; Kalinowski, S.; Dunin-Horkawicz, S.; Rother, K.M.; et al. MODOMICS: A database of RNA modification pathways--2013 update. Nucleic Acids Res. 2013, 41, D262–D267. [Google Scholar] [CrossRef] [PubMed]
- Hori, H. Methylated nucleosides in tRNA and tRNA methyltransferases. Front. Genet. 2014, 5, 144. [Google Scholar] [CrossRef] [PubMed]
- Gillingham, D.; Shahid, R. Catalysts for RNA and DNA modification. Curr. Opin. Chem. Biol. 2015, 25, 110–114. [Google Scholar] [CrossRef] [PubMed]
- El Yacoubi, B.; Bailly, M.; de Crécy-Lagard, V. Biosynthesis and function of posttranscriptional modifications of transfer RNAs. Annu. Rev. Genet. 2012, 46, 69–95. [Google Scholar] [CrossRef] [PubMed]
- Kawamura, T.; Anraku, R.; Hasegawa, T.; Tomikawa, C.; Hori, H. Transfer RNA methyltransferases from Thermoplasma acidophilum, a thermoacidophilic archaeon. Int. J. Mol. Sci. 2014, 16, 91–113. [Google Scholar] [CrossRef] [PubMed]
- Phillips, G.; de Crécy-Lagard, V. Biosynthesis and function of tRNA modifications in Archaea. Curr. Opin. Microbiol. 2011, 14, 335–341. [Google Scholar] [CrossRef] [PubMed]
- Gustafsson, C.; Reid, R.; Greene, P.J.; Santi, D.V. Identification of new RNA modifying enzymes by iterative genome search using known modifying enzymes as probes. Nucleic Acids Res. 1996, 24, 3756–3762. [Google Scholar] [CrossRef] [PubMed]
- Anantharaman, V.; Koonin, E.V.; Aravind, L. SPOUT: A class of methyltransferases that includes spoU and trmD RNA methylase superfamilies, and novel superfamilies of predicted prokaryotic RNA methylases. J. Mol. Microbiol. Biotechnol. 2002, 4, 71–75. [Google Scholar] [PubMed]
- Koonin, E.V.; Rudd, K.E. SpoU protein of Escherichia coli belongs to a new family of putative rRNA methylases. Nucleic Acids Res. 1993, 21, 5519. [Google Scholar] [CrossRef] [PubMed]
- Persson, B.C.; Jäger, G.; Gustafsson, C. The spoU gene of Escherichia coli, the fourth gene of the spoT operon, is essential for tRNA (Gm18) 2′-O-methyltransferase activity. Nucleic Acids Res. 1997, 25, 4093–4097. [Google Scholar] [CrossRef] [PubMed]
- Hori, H.; Suzuki, T.; Sugawara, K.; Inoue, Y.; Shibata, T.; Kuramitsu, S.; Yokoyama, S.; Oshima, T.; Watanabe, K. Identification and characterization of tRNA (Gm18) methyltransferase from Thermus thermophilus HB8: Domain structure and conserved amino acid sequence motifs. Genes Cells Devoted Mol. Cell. Mech. 2002, 7, 259–272. [Google Scholar] [CrossRef]
- Watanabe, K.; Nureki, O.; Fukai, S.; Ishii, R.; Okamoto, H.; Yokoyama, S.; Endo, Y.; Hori, H. Roles of conserved amino acid sequence motifs in the SpoU (TrmH) RNA methyltransferase family. J. Biol. Chem. 2005, 280, 10368–10377. [Google Scholar] [CrossRef] [PubMed]
- Pleshe, E.; Truesdell, J.; Batey, R.T. Structure of a class II TrmH tRNA-modifying enzyme from Aquifex aeolicus. Acta Crystallogr. Sect. F Struct. Biol. Cryst. Commun. 2005, 61, 722–728. [Google Scholar] [CrossRef] [PubMed]
- Purta, E.; van Vliet, F.; Tkaczuk, K.L.; Dunin-Horkawicz, S.; Mori, H.; Droogmans, L.; Bujnicki, J.M. The yfhQ gene of Escherichia coli encodes a tRNA:Cm32/Um32 methyltransferase. BMC Mol. Biol. 2006, 7, 23. [Google Scholar] [CrossRef] [PubMed]
- Somme, J.; Van Laer, B.; Roovers, M.; Steyaert, J.; Versées, W.; Droogmans, L. Characterization of two homologous 2′-O-methyltransferases showing different specificities for their tRNA substrates. RNA 2014, 20, 1257–1271. [Google Scholar] [CrossRef] [PubMed]
- Armengod, M.-E.; Moukadiri, I.; Prado, S.; Ruiz-Partida, R.; Benítez-Páez, A.; Villarroya, M.; Lomas, R.; Garzón, M.J.; Martínez-Zamora, A.; Meseguer, S.; et al. Enzymology of tRNA modification in the bacterial MnmEG pathway. Biochimie 2012, 94, 1510–1520. [Google Scholar] [CrossRef] [PubMed]
- Benítez-Páez, A.; Villarroya, M.; Douthwaite, S.; Gabaldón, T.; Armengod, M.-E. YibK is the 2′-O-methyltransferase TrmL that modifies the wobble nucleotide in Escherichia coli tRNALeu isoacceptors. RNA 2010, 16, 2131–2143. [Google Scholar] [CrossRef] [PubMed]
- Jaroensuk, J.; Atichartpongkul, S.; Chionh, Y.H.; Wong, Y.H.; Liew, C.W.; McBee, M.E.; Thongdee, N.; Prestwich, E.G.; DeMott, M.S.; Mongkolsuk, S.; et al. Methylation at position 32 of tRNA catalyzed by TrmJ alters oxidative stress response in Pseudomonas aeruginosa. Nucleic Acids Res. 2016, 44, 10834–10848. [Google Scholar] [CrossRef] [PubMed]
- Grosjean, H.; Gaspin, C.; Marck, C.; Decatur, W.A.; de Crécy-Lagard, V. RNomics and Modomics in the halophilic archaea Haloferax volcanii: Identification of RNA modification genes. BMC Genom. 2008, 9, 470. [Google Scholar] [CrossRef] [PubMed]
- Joardar, A.; Malliahgari, S.R.; Skariah, G.; Gupta, R. 2′-O-methylation of the wobble residue of elongator pre-tRNAMet in Haloferax volcanii is guided by a box C/D RNA containing unique features. RNA Biol. 2011, 8, 782–791. [Google Scholar] [CrossRef] [PubMed]
- Singh, S.K.; Gurha, P.; Tran, E.J.; Maxwell, E.S.; Gupta, R. Sequential 2′-O-methylation of archaeal pre-tRNATrp nucleotides is guided by the intron-encoded but trans-acting box C/D ribonucleoprotein of pre-tRNA. J. Biol. Chem. 2004, 279, 47661–47671. [Google Scholar] [CrossRef] [PubMed]
- Clouet-d’Orval, B.; Gaspin, C.; Mougin, A. Two different mechanisms for tRNA ribose methylation in Archaea: A short survey. Biochimie 2005, 87, 889–895. [Google Scholar] [CrossRef] [PubMed]
- Renalier, M.-H.; Joseph, N.; Gaspin, C.; Thebault, P.; Mougin, A. The Cm56 tRNA modification in archaea is catalyzed either by a specific 2′-O-methylase, or a C/D sRNP. RNA 2005, 11, 1051–1063. [Google Scholar] [CrossRef] [PubMed]
- Kuratani, M.; Bessho, Y.; Nishimoto, M.; Grosjean, H.; Yokoyama, S. Crystal structure and mutational study of a unique SpoU family archaeal methylase that forms 2′-O-methylcytidine at position 56 of tRNA. J. Mol. Biol. 2008, 375, 1064–1075. [Google Scholar] [CrossRef] [PubMed]
- Cavaillé, J.; Chetouani, F.; Bachellerie, J.P. The yeast Saccharomyces cerevisiae YDL112w ORF encodes the putative 2′-O-ribose methyltransferase catalyzing the formation of Gm18 in tRNAs. RNA 1999, 5, 66–81. [Google Scholar] [CrossRef] [PubMed]
- Pintard, L.; Lecointe, F.; Bujnicki, J.M.; Bonnerot, C.; Grosjean, H.; Lapeyre, B. Trm7p catalyses the formation of two 2′-O-methylriboses in yeast tRNA anticodon loop. EMBO J. 2002, 21, 1811–1820. [Google Scholar] [CrossRef] [PubMed]
- Guy, M.P.; Podyma, B.M.; Preston, M.A.; Shaheen, H.H.; Krivos, K.L.; Limbach, P.A.; Hopper, A.K.; Phizicky, E.M. Yeast Trm7 interacts with distinct proteins for critical modifications of the tRNAPhe anticodon loop. RNA 2012, 18, 1921–1933. [Google Scholar] [CrossRef] [PubMed]
- Guy, M.P.; Phizicky, E.M. Conservation of an intricate circuit for crucial modifications of the tRNAPhe anticodon loop in eukaryotes. RNA 2015, 21, 61–74. [Google Scholar] [CrossRef] [PubMed]
- Guy, M.P.; Shaw, M.; Weiner, C.L.; Hobson, L.; Stark, Z.; Rose, K.; Kalscheuer, V.M.; Gecz, J.; Phizicky, E.M. Defects in tRNA Anticodon Loop 2′-O-Methylation Are Implicated in Nonsyndromic X-Linked Intellectual Disability due to Mutations in FTSJ1. Hum. Mutat. 2015, 36, 1176–1187. [Google Scholar] [CrossRef] [PubMed]
- Kotelawala, L.; Grayhack, E.J.; Phizicky, E.M. Identification of yeast tRNA Um44 2′-O-methyltransferase (Trm44) and demonstration of a Trm44 role in sustaining levels of specific tRNASer species. RNA 2008, 14, 158–169. [Google Scholar] [CrossRef] [PubMed]
- Wilkinson, M.L.; Crary, S.M.; Jackman, J.E.; Grayhack, E.J.; Phizicky, E.M. The 2′-O-methyltransferase responsible for modification of yeast tRNA at position 4. RNA 2007, 13, 404–413. [Google Scholar] [CrossRef] [PubMed]
- Gross, H.J.; Simsek, M.; Raba, M.; Limburg, K.; Heckman, J.; Raj Bhandary, U.L. 2′-O-methyl ribothymidine: a component of rabbit liver lysine transfer RNA. Nucleic Acids Res. 1974, 1, 35–43. [Google Scholar] [CrossRef] [PubMed]
- Païs de Barros, J.P.; Keith, G.; El Adlouni, C.; Glasser, A.L.; Mack, G.; Dirheimer, G.; Desgrès, J. 2′-O-methyl-5-formylcytidine (f5Cm), a new modified nucleotide at the “wobble” of two cytoplasmic tRNAsLeu(NAA) from bovine liver. Nucleic Acids Res. 1996, 24, 1489–1496. [Google Scholar] [CrossRef] [PubMed]
- Eichler, D.C. Characterization of a nucleolar 2′-O-methyltransferase and its involvement in the methylation of mouse precursor ribosomal RNA. Biochimie 1994, 76, 1115–1122. [Google Scholar] [CrossRef]
- Droogmans, L.; Haumont, E.; de Henau, S.; Grosjean, H. Enzymatic 2′-O-methylation of the wobble nucleoside of eukaryotic tRNAPhe: Specificity depends on structural elements outside the anticodon loop. EMBO J. 1986, 5, 1105–1109. [Google Scholar] [PubMed]
- Kawai, G.; Yamamoto, Y.; Kamimura, T.; Masegi, T.; Sekine, M.; Hata, T.; Iimori, T.; Watanabe, T.; Miyazawa, T.; Yokoyama, S. Conformational rigidity of specific pyrimidine residues in tRNA arises from posttranscriptional modifications that enhance steric interaction between the base and the 2′-hydroxyl group. Biochemistry 1992, 31, 1040–1046. [Google Scholar] [CrossRef] [PubMed]
- Kawai, G.; Ue, H.; Yasuda, M.; Sakamoto, K.; Hashizume, T.; McCloskey, J.A.; Miyazawa, T.; Yokoyama, S. Relation between functions and conformational characteristics of modified nucleosides found in tRNAs. Nucleic Acids Symp. Ser. 1991, 49–50. [Google Scholar]
- Kita, E.; Kashiba, S. Immunogenicity of transfer RNA isolated from a two-heptose rough mutant of Salmonella typhimurium LT2 in mouse typhoid infection. Immunology 1983, 50, 369–376. [Google Scholar] [PubMed]
- Gehrig, S.; Eberle, M.-E.; Botschen, F.; Rimbach, K.; Eberle, F.; Eigenbrod, T.; Kaiser, S.; Holmes, W.M.; Erdmann, V.A.; Sprinzl, M.; et al. Identification of modifications in microbial, native tRNA that suppress immunostimulatory activity. J. Exp. Med. 2012, 209, 225–233. [Google Scholar] [CrossRef] [PubMed]
- Jöckel, S.; Nees, G.; Sommer, R.; Zhao, Y.; Cherkasov, D.; Hori, H.; Ehm, G.; Schnare, M.; Nain, M.; Kaufmann, A.; Bauer, S. The 2′-O-methylation status of a single guanosine controls transfer RNA-mediated Toll-like receptor 7 activation or inhibition. J. Exp. Med. 2012, 209, 235–241. [Google Scholar] [CrossRef] [PubMed]
- Rimbach, K.; Kaiser, S.; Helm, M.; Dalpke, A.H.; Eigenbrod, T. 2′-O-Methylation within Bacterial RNA Acts as Suppressor of TLR7/TLR8 Activation in Human Innate Immune Cells. J. Innate Immun. 2015, 7, 482–493. [Google Scholar] [CrossRef] [PubMed]
- Oldenburg, M.; Krüger, A.; Ferstl, R.; Kaufmann, A.; Nees, G.; Sigmund, A.; Bathke, B.; Lauterbach, H.; Suter, M.; Dreher, S.; et al. TLR13 recognizes bacterial 23S rRNA devoid of erythromycin resistance-forming modification. Science 2012, 337, 1111–1115. [Google Scholar] [CrossRef] [PubMed]
- Maden, B.E.; Corbett, M.E.; Heeney, P.A.; Pugh, K.; Ajuh, P.M. Classical and novel approaches to the detection and localization of the numerous modified nucleotides in eukaryotic ribosomal RNA. Biochimie 1995, 77, 22–29. [Google Scholar] [CrossRef]
- Motorin, Y.; Muller, S.; Behm-Ansmant, I.; Branlant, C. Identification of modified residues in RNAs by reverse transcription-based methods. Methods Enzymol. 2007, 425, 21–53. [Google Scholar] [PubMed]
- Gehrke, C.W.; Kuo, K.C.; McCune, R.A.; Gerhardt, K.O.; Agris, P.F. Quantitative enzymatic hydrolysis of tRNAs: Reversed-phase high-performance liquid chromatography of tRNA nucleosides. J. Chromatogr. 1982, 230, 297–308. [Google Scholar] [CrossRef]
- Su, D.; Chan, C.T.Y.; Gu, C.; Lim, K.S.; Chionh, Y.H.; McBee, M.E.; Russell, B.S.; Babu, I.R.; Begley, T.J.; Dedon, P.C. Quantitative analysis of ribonucleoside modifications in tRNA by HPLC-coupled mass spectrometry. Nat. Protoc. 2014, 9, 828–841. [Google Scholar] [CrossRef] [PubMed]
- Cao, X.; Limbach, P.A. Enhanced detection of post-transcriptional modifications using a mass-exclusion list strategy for RNA modification mapping by LC-MS/MS. Anal. Chem. 2015, 87, 8433–8440. [Google Scholar] [CrossRef] [PubMed]
- Abbate, J.; Rottman, F. Gas chromatographic method for determination of 2′-O-methylation in RNA. Anal. Biochem. 1972, 47, 378–388. [Google Scholar] [CrossRef]
- Dominissini, D.; Moshitch-Moshkovitz, S.; Salmon-Divon, M.; Amariglio, N.; Rechavi, G. Transcriptome-wide mapping of N6-methyladenosine by m6A-seq based on immunocapturing and massively parallel sequencing. Nat. Protoc. 2013, 8, 176–189. [Google Scholar] [CrossRef] [PubMed]
- Dominissini, D.; Nachtergaele, S.; Moshitch-Moshkovitz, S.; Peer, E.; Kol, N.; Ben-Haim, M.S.; Dai, Q.; Di Segni, A.; Salmon-Divon, M.; Clark, W.C.; et al. The dynamic N1-methyladenosine methylome in eukaryotic messenger RNA. Nature 2016, 530, 441–446. [Google Scholar] [CrossRef] [PubMed]
- Schwartz, S.; Bernstein, D.A.; Mumbach, M.R.; Jovanovic, M.; Herbst, R.H.; León-Ricardo, B.X.; Engreitz, J.M.; Guttman, M.; Satija, R.; Lander, E.S.; et al. Transcriptome-wide mapping reveals widespread dynamic-regulated pseudouridylation of ncRNA and mRNA. Cell 2014, 159, 148–162. [Google Scholar] [CrossRef] [PubMed]
- Squires, J.E.; Patel, H.R.; Nousch, M.; Sibbritt, T.; Humphreys, D.T.; Parker, B.J.; Suter, C.M.; Preiss, T. Widespread occurrence of 5-methylcytosine in human coding and non-coding RNA. Nucleic Acids Res. 2012, 40, 5023–5033. [Google Scholar] [CrossRef] [PubMed]
- Tserovski, L.; Marchand, V.; Hauenschild, R.; Blanloeil-Oillo, F.; Helm, M.; Motorin, Y. High-throughput sequencing for 1-methyladenosine (m1A) mapping in RNA. Methods 2016, 107, 110–121. [Google Scholar] [CrossRef] [PubMed]
- Birkedal, U.; Christensen-Dalsgaard, M.; Krogh, N.; Sabarinathan, R.; Gorodkin, J.; Nielsen, H. Profiling of ribose methylations in RNA by high-throughput sequencing. Angew. Chem. 2015, 54, 451–455. [Google Scholar] [CrossRef]
- Krogh, N.; Jansson, M.D.; Häfner, S.J.; Tehler, D.; Birkedal, U.; Christensen-Dalsgaard, M.; Lund, A.H.; Nielsen, H. Profiling of 2′-O-Me in human rRNA reveals a subset of fractionally modified positions and provides evidence for ribosome heterogeneity. Nucleic Acids Res. 2016, 44, 7884–7895. [Google Scholar] [CrossRef] [PubMed]
- Marchand, V.; Blanloeil-Oillo, F.; Helm, M.; Motorin, Y. Illumina-based RiboMethSeq approach for mapping of 2′-O-Me residues in RNA. Nucleic Acids Res. 2016, 44, e135. [Google Scholar] [CrossRef] [PubMed]
- Transfer RNA database (tRNAdb). Available online: http://trna.bioinf.uni-leipzig.de (accessed on 1 December 2016).
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [PubMed]
- Langmead, B.; Trapnell, C.; Pop, M.; Salzberg, S.L. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 2009, 10, R25. [Google Scholar] [CrossRef] [PubMed]
- Quinlan, A.R. BEDTools: The Swiss-Army Tool for Genome Feature Analysis. Curr. Protoc. Bioinform. 2014, 47, 1–34. [Google Scholar]
- The R Foundation. Available online: https://www.r-project.org/foundation/ (accessed on 1 December 2016).


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Organism | Enzyme | Enzyme Alt. Name | Position | Modification | tRNA | Anticodon | tRNAdb Reference |
|---|---|---|---|---|---|---|---|
| E. coli | TrmH | spoU | 18 | Gm | Ile | k2CAU | tdbR00000160 |
| 18 | Leu | cmnm5UmAA | tdbR00000220 | ||||
| 18 | Leu | CAG | tdbR00000221 | ||||
| 18 | Leu | GAG | tdbR00000222 | ||||
| 18 | Met | ac4CAU | tdbR00000276 | ||||
| 18 | Gln | CUG | tdbR00000332 | ||||
| 18 | Gln | U*UG | tdbR00000333 | ||||
| 18 | Ser | CGA | tdbR00000390 | ||||
| 18 | Ser | GGA | tdbR00000392-3 | ||||
| 18 | Ser | cmo5UGA | tdbR00000394 | ||||
| 18 | Tyr | QUA | tdbR00000545-46 | ||||
| 18 | Leu | CmAA | tdbR00000595 | ||||
| TrmJ | YfhQ, TrMet(Xm32) | 32 | Cm | Ser | cmo5UGA | tdbR00000394 | |
| 32 | Trp | CCA | tdbR00000484 | ||||
| 32 | Ini | CAU | tdbR00000510-11 | ||||
| 32 | Um | Gln | CUG | tdbR00000332 | |||
| 32 | Gln | U*UG | tdbR00000333 | ||||
| 32 | Pro | CGG | tdbR00000623 | ||||
| TrmL | yibK | 34 | Cm | Leu | CmAA | tdbR00000595 | |
| 34 | cmnm5Um | Leu | cmnm5UmAA | tdbR00000220 | |||
| H. volcanii | aTrmJ | HVO_2906 | 32 | Cm | Lys | U*UU | tdbR00000175 |
| Lys | ac4CUU | tdbR00000176 | |||||
| Trp | CmCA | tdbR00000478 | |||||
| Tyr | GUA | tdbR00000541 | |||||
| sRNA/aFib | 34 | Cm | Met | CmAU | tdbR00000271 | ||
| 34 | Cm | Trp | CmCA | tdbR00000478 | |||
| 39 | Um | Trp | CmCA | tdbR00000478 | |||
| aTrm56 | HVO_1173 | 56 | Cm | All known tRNAs | |||
| S. cerevisiae | Trm13 | YOL125w | 4 | Cm | Gly | GCC | tdbR00000129 |
| 4 | Cm | Pro | U*GG | tdbR00000323-4 | |||
| 4 | Am | His | GUG | tdbR00000145 | |||
| Trm3 | YDL112w | 18 | Gm | Leu | CAA | tdbR00000249 | |
| 18 | Leu | UAG | tdbR00000250 | ||||
| 18 | Leu | UAA | tdbR00000251 | ||||
| 18 | Trp | CmCA | tdbR00000494 | ||||
| 18 | Ser | IGA | tdbR00000407-8 | ||||
| 18 | Ser | U*GA | tdbR00000406 | ||||
| 18 | His | GUG | tdbR00000145 | ||||
| 18 | Tyr | GψA | tdbR00000555 | ||||
| Trm7/Trm732 | YBR061C/YMR259c | 32 | Cm | Trp | CmCA | tdbR00000494 | |
| 32 | Phe | GmAA | tdbR00000083-4 | ||||
| 32 | Leu | UAA | tdbR00000251 | ||||
| Trm7/Trm734 | YBR061C/Rtt10 | 34 | Cm | Trp | CmCA | tdbR00000494 | |
| 34 | Gm | Phe | GmAA | tdbR00000083-4 | |||
| 34 | cmnm5Um | Lys | cmnm5UmUU | tdbR00000193 | |||
| Trm44 | YPL030w | 44 | Um | Ser | IGA | tdbR00000407-8 | |
| 44 | Ser | U*GA | tdbR00000406 | ||||
| H. sapiens | hTrm13 | CCDC76 | 4 | Cm | Gly | CCC | tdbR00000135 |
| 4 | Um | Gly | GCC | tdbR00000136 | |||
| hTrm3 | TARBP1 | 18 | Gm | Asn | NUG | tdbR00000306 | |
| 18 | Gln | U*UG | tdbR00000345 | ||||
| 18 | Gln | U*UG | tdbR00000346 | ||||
| 18 | Ser | UGA | tdbR00000428 | ||||
| hTrm7 | FTSJ1 | 32 | Cm | Phe | GmAA | tdbR00000103 | |
| 32 | Asn | NUG | tdbR00000306 | ||||
| 32 | Gln | U*UG | tdbR00000345 | ||||
| 32 | Gln | U*UG | tdbR00000346 | ||||
| 32 | Um | Ala | IGC | tdbR00000016 | |||
| 32 | Ala | IGC | tdbR00000017 | ||||
| 34 | Cm | Met | CmAU | tdbR00000289 | |||
| 34 | Gm | Phe | GmAA | tdbR00000103 | |||
| unknown | 39 | ψm | Gly | CCC | tdbR00000135 | ||
| hTrm44 | METTL19 | 44 | Um | Leu | NAA | tdbR00000269 | |
| 44 | Ser | UGA | tdbR00000428 | ||||
| unknown | 54 | m5Um | Glu | CUC | tdbR00000062 | ||
| Escherichia coli | |||||||||
|---|---|---|---|---|---|---|---|---|---|
| WT E. coli | ΔTrmH E. coli | Difference | |||||||
| tdbR Number | tRNA | Modification | Position * | ScoreMean | MethScore | ScoreMean | MethScore | ScoreMean | MethScore |
| tdbR00000160 | tRNAIle(k2CAU) | Gm18 | 18 | 0.92 | 0.68 | 0.29 | −0.37 | 0.63 | 1.05 |
| tdbR00000332 | tRNAGln(CUG) | Gm18 | 18 | 0.97 | 0.85 | 0.94 | 0.79 | 0.03 | 0.06 |
| tdbR00000333 | tRNAGln(U*UG) | Gm18 | 18 | 0.81 | 0.7 | 0.76 | 0.67 | 0.05 | 0.04 |
| tdbR00000390 | tRNASer(CGA) | Gm18 | 18 | 0.6 | 0.25 | 0.24 | −0.3 | 0.35 | 0.55 |
| tdbR00000392-393 | tRNASer(GGA) | Gm18 | 18 | 0.86 | 0.76 | 0.26 | 0.32 | 0.6 | 0.44 |
| tdbR00000394 | tRNASer(cmo5UGA) | Gm18 | 18 | 0.98 | 0.88 | 0.94 | 0.8 | 0.04 | 0.08 |
| tdbR00000545-546 | tRNATyr(QUA) | Gm18 | 18 | 0.93 | 0.84 | 0.35 | 0.17 | 0.58 | 0.67 |
| tdbR00000220 | tRNALeu(cmnm5UmAA) | Gm18 | 19 | 0.87 | 0.42 | 0.46 | −0.78 | 0.42 | 1.21 |
| tdbR00000221 | tRNALeu(CAG) | Gm18 | 19 | 0.97 | 0.89 | 0.35 | −0.02 | 0.62 | 0.9 |
| tdbR00000222 | tRNALeu(GAG) | Gm18 | 19 | 0.99 | 0.95 | 0.25 | −0.48 | 0.74 | 1.44 |
| tdbR00000276 | tRNAMet(ac4CAU) | Gm18 | 19 | 0.58 | 0.42 | 0.75 | 0.44 | −0.17 | −0.02 |
| tdbR00000595 | tRNALeu(CmAA) | Gm18 | 19 | 0.93 | 0.9 | 0.73 | 0.67 | 0.19 | 0.22 |
| tdbR00000332 | tRNAGln(CUG) | Um32 | 32 | 0.97 | 0.85 | 0.94 | 0.79 | 0.03 | 0.06 |
| tdbR00000333 | tRNAGln(U*UG) | Um32 | 32 | 0.81 | 0.7 | 0.76 | 0.67 | 0.05 | 0.04 |
| tdbR00000484 | tRNATrp(CCA) | Cm32 | 33 | 0.99 | 0.92 | 0.99 | 0.91 | 0 | 0.01 |
| tdbR00000394 | tRNASer(cmo5UGA) | Cm32 | 34 | 0.98 | 0.88 | 0.94 | 0.8 | 0.04 | 0.08 |
| tdbR00000510-511 | tRNAMeti(CAU) | Cm32 | 34 | 0.97 | 0.84 | 0.97 | 0.85 | 0 | −0.02 |
| tdbR00000623 | tRNAPro(CGG) | Um32 | 34 | 0.98 | 0.93 | 0.97 | 0.92 | 0 | 0.01 |
| tdbR00000595 | tRNALeu(CmAA) | Cm34 | 36 | 1 | 0.94 | 0.99 | 0.88 | 0.01 | 0.05 |
| tdbR00000220 | tRNALeu(cmnm5UmAA) | cmnm5Um34 | 36 | 1 | 0.94 | 0.98 | 0.81 | 0.02 | 0.13 |
| Saccharomyces cerevisae | |||||||||
| WT S. cerevisiae | ΔTRM3 S. cerevisiae | Difference | |||||||
| tdbR Number | tRNA | Modification | Position * | ScoreMean | MethScore | ScoreMean | MethScore | ScoreMean | MethScore |
| tdbR00000129 | tRNAGly(GCC) | Cm4 | 5 | 0.89 | 0.63 | 0.93 | 0.76 | −0.04 | −0.13 |
| tdbR00000323-4 | tRNAPro(U*GG) | Cm4 | 5 | 0.95 | 0.83 | 0.95 | 0.81 | 0.01 | 0.02 |
| tdbR00000145 | tRNAHis(GUG) | Am5 | 6 | 0.98 | 0.89 | 0.96 | 0.85 | 0.01 | 0.03 |
| tdbR00000249 | tRNALeu(CAA) | Gm18 | 18 | 0.99 | 0.89 | 0.84 | 0.03 | 0.15 | 0.87 |
| tdbR00000250 | tRNALeu(UAG) | Gm18 | 18 | 0.93 | 0.84 | −0.35 | −0.46 | 1.28 | 1.29 |
| tdbR00000251 | tRNALeu(UAA) | Gm18 | 18 | 0.93 | 0.86 | −0.16 | −0.14 | 1.10 | 1.01 |
| tdbR00000406 | tRNASer(U*GA) | Gm18 | 18 | 0.91 | 0.85 | 0.75 | 0.57 | 0.15 | 0.29 |
| tdbR00000407-8 | tRNASer(IGA) | Gm18 | 18 | 0.95 | 0.81 | −0.35 | −0.80 | 1.30 | 1.61 |
| tdbR00000494 | tRNATrp(CmCA) | Gm18 | 18 | 0.93 | 0.81 | 0.26 | −0.05 | 0.67 | 0.87 |
| tdbR00000145 | tRNAHis(GUG) | Gm18 | 19 | 0.91 | 0.80 | −0.98 | −0.84 | 1.89 | 1.64 |
| tdbR00000555 | tRNATyr(GψA) | Gm18 | 19 | 0.96 | 0.92 | 0.09 | 0.13 | 0.87 | 0.79 |
| tdbR00000494 | tRNATrp(CmCA) | Cm32 | 32 | 0.99 | 0.86 | 0.99 | 0.88 | 0.00 | −0.02 |
| tdbR00000083-4 | tRNAPhe(GmAA) | Cm32 | 33 | 0.98 | 0.79 | 0.98 | 0.81 | 0.00 | −0.03 |
| tdbR00000251 | tRNALeu(UAA) | Cm32 | 34 | 0.92 | 0.55 | 0.95 | 0.69 | −0.03 | −0.14 |
| tdbR00000494 | tRNATrp(CmCA) | Cm34 | 34 | 0.99 | 0.87 | 0.99 | 0.89 | 0.00 | −0.03 |
| tdbR00000083-4 | tRNAPhe(GmAA) | Gm34 | 35 | 0.96 | 0.74 | 0.93 | 0.63 | 0.03 | 0.10 |
| tdbR00000193 | tRNALys(cmnm5UmUU) | cmnm5Um34 | 35 | 0.64 | 0.30 | 0.58 | 0.26 | 0.06 | 0.04 |
| tdbR00000406 | tRNASer(U*GA) | Um44 | 45 | 0.93 | 0.86 | 0.93 | 0.83 | −0.01 | 0.03 |
| tdbR00000407-8 | tRNASer(IGA) | Um44 | 45 | 0.90 | 0.78 | 0.86 | 0.71 | 0.04 | 0.07 |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Marchand, V.; Pichot, F.; Thüring, K.; Ayadi, L.; Freund, I.; Dalpke, A.; Helm, M.; Motorin, Y. Next‐Generation Sequencing‐Based RiboMethSeq Protocol for Analysis of tRNA 2′‐O‐Methylation. Biomolecules 2017, 7, 13. https://doi.org/10.3390/biom7010013
Marchand V, Pichot F, Thüring K, Ayadi L, Freund I, Dalpke A, Helm M, Motorin Y. Next‐Generation Sequencing‐Based RiboMethSeq Protocol for Analysis of tRNA 2′‐O‐Methylation. Biomolecules. 2017; 7(1):13. https://doi.org/10.3390/biom7010013
Chicago/Turabian StyleMarchand, Virginie, Florian Pichot, Kathrin Thüring, Lilia Ayadi, Isabel Freund, Alexander Dalpke, Mark Helm, and Yuri Motorin. 2017. "Next‐Generation Sequencing‐Based RiboMethSeq Protocol for Analysis of tRNA 2′‐O‐Methylation" Biomolecules 7, no. 1: 13. https://doi.org/10.3390/biom7010013
APA StyleMarchand, V., Pichot, F., Thüring, K., Ayadi, L., Freund, I., Dalpke, A., Helm, M., & Motorin, Y. (2017). Next‐Generation Sequencing‐Based RiboMethSeq Protocol for Analysis of tRNA 2′‐O‐Methylation. Biomolecules, 7(1), 13. https://doi.org/10.3390/biom7010013