
Molecular Mechanisms in the Pathogenesis of Alzheimer’s disease and Tauopathies-Prion-Like Seeded Aggregation and Phosphorylation

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Abnormal Tau in AD Brain
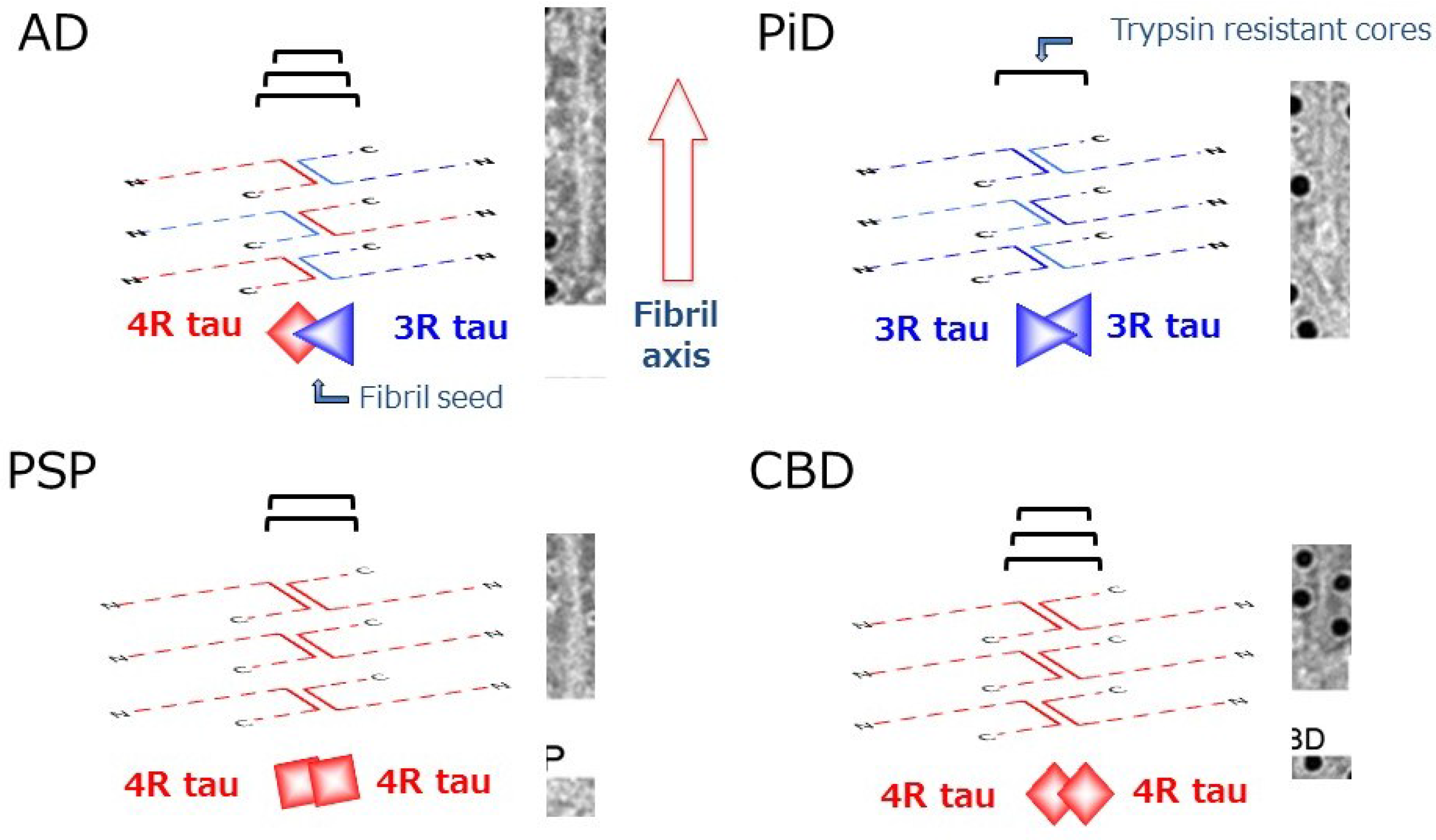
3. Tau-Related Neurodegeneration and Dementias
4. Relationship between Tau Aggregation and Phosphorylation
5. Conversion of Normal Tau into Amyloid-Like Fibrils
6. Molecular Mechanisms of Formation and Propagation of Pathological Tau Proteins
7. Conclusions
Acknowledgments
Conflicts of Interest
References
- Braak, H.; Braak, E. Neuropathological stageing of Alzheimer-related changes. Acta Neuropathol. 1991, 82, 239–259. [Google Scholar] [CrossRef] [PubMed]
- Goate, A.; Chartier-Harlin, M.C.; Mullan, M.; Brown, J.; Crawford, F.; Fidani, L.; Giuffra, L.; Haynes, A.; Irving, N.; James, L.; et al. Segregation of a missense mutation in the amyloid precursor protein gene with familial Alzheimer’s disease. Nature 1991, 349, 704–706. [Google Scholar] [CrossRef] [PubMed]
- Hardy, J.; Allsop, D. Amyloid deposition as the central event in the aetiology of Alzheimer’s disease. Trends Pharmacol. Sci. 1991, 12, 383–388. [Google Scholar] [CrossRef]
- Hardy, J.A.; Higgins, G.A. Alzheimer’s disease: The amyloid cascade hypothesis. Science 1992, 256, 184–185. [Google Scholar] [CrossRef] [PubMed]
- Hardy, J.; Selkoe, D.J. The amyloid hypothesis of Alzheimer’s disease: Progress and problems on the road to therapeutics. Science 2002, 297, 353–356. [Google Scholar] [CrossRef] [PubMed]
- Sherrington, R.; Rogaev, E.I.; Liang, Y.; Rogaeva, E.A.; Levesque, G.; Ikeda, M.; Chi, H.; Lin, C.; Li, G.; Holman, K.; et al. Cloning of a gene bearing missense mutations in early-onset familial Alzheimer’s disease. Nature 1995, 375, 754–760. [Google Scholar] [CrossRef] [PubMed]
- Levy-Lahad, E.; Wasco, W.; Poorkaj, P.; Romano, D.M.; Oshima, J.; Pettingell, W.H.; Yu, C.E.; Jondro, P.D.; Schmidt, S.D.; Wang, K.; et al. Candidate gene for the chromosome 1 familial Alzheimer’s disease locus. Science 1995, 269, 973–977. [Google Scholar] [CrossRef] [PubMed]
- Rogaev, E.I.; Sherrington, R.; Rogaeva, E.A.; Levesque, G.; Ikeda, M.; Liang, Y.; Chi, H.; Lin, C.; Holman, K.; Tsuda, T.; et al. Familial Alzheimer’s disease in kindreds with missense mutations in a gene on chromosome 1 related to the Alzheimer’s disease type 3 gene. Nature 1995, 376, 775–778. [Google Scholar] [CrossRef] [PubMed]
- De Strooper, B.; Saftig, P.; Craessaerts, K.; Vanderstichele, H.; Guhde, G.; Annaert, W.; von Figura, K.; van Leuven, F. Deficiency of presenilin-1 inhibits the normal cleavage of amyloid precursor protein. Nature 1998, 391, 387–390. [Google Scholar] [CrossRef] [PubMed]
- De Strooper, B.; Annaert, W.; Cupers, P.; Saftig, P.; Craessaerts, K.; Mumm, J.S.; Schroeter, E.H.; Schrijvers, V.; Wolfe, M.S.; Ray, W.J.; et al. A presenilin-1-dependent γ-secretase-like protease mediates release of notch intracellular domain. Nature 1999, 398, 518–522. [Google Scholar] [CrossRef] [PubMed]
- Wolfe, M.S.; Xia, W.; Ostaszewski, B.L.; Diehl, T.S.; Kimberly, W.T.; Selkoe, D.J. Two transmembrane aspartates in presenilin-1 required for presenilin endoproteolysis and γ-secretase activity. Nature 1999, 398, 513–517. [Google Scholar] [CrossRef] [PubMed]
- Chetelat, G. Alzheimer disease: Aβ-independent processes-rethinking preclinical AD. Nat. Rev. Neurol. 2013, 9, 123–124. [Google Scholar] [CrossRef] [PubMed]
- Giacobini, E.; Gold, G. Alzheimer disease therapy—moving from amyloid-β to tau. Nat. Rev. Neurol. 2013, 9, 677–686. [Google Scholar] [CrossRef] [PubMed]
- Herrup, K. The case for rejecting the amyloid cascade hypothesis. Nat. Neurosci. 2015, 18, 794–799. [Google Scholar] [CrossRef] [PubMed]
- Holmes, C.; Boche, D.; Wilkinson, D.; Yadegarfar, G.; Hopkins, V.; Bayer, A.; Jones, R.W.; Bullock, R.; Love, S.; Neal, J.W.; et al. Long-term effects of Aβ42 immunisation in Alzheimer’s disease: Follow-up of a randomised, placebo-controlled phase I trial. Lancet 2008, 372, 216–223. [Google Scholar] [CrossRef]
- Ostrowitzki, S.; Deptula, D.; Thurfjell, L.; Barkhof, F.; Bohrmann, B.; Brooks, D.J.; Klunk, W.E.; Ashford, E.; Yoo, K.; Xu, Z.X.; et al. Mechanism of amyloid removal in patients with Alzheimer disease treated with gantenerumab. Arch. Neurol. 2012, 69, 198–207. [Google Scholar] [CrossRef] [PubMed]
- Doody, R.S.; Raman, R.; Farlow, M.; Iwatsubo, T.; Vellas, B.; Joffe, S.; Kieburtz, K.; He, F.; Sun, X.; Thomas, R.G.; et al. A phase 3 trial of semagacestat for treatment of Alzheimer’s disease. N. Engl. J. Med. 2013, 369, 341–350. [Google Scholar] [CrossRef] [PubMed]
- Salloway, S.; Sperling, R.; Gilman, S.; Fox, N.C.; Blennow, K.; Raskind, M.; Sabbagh, M.; Honig, L.S.; Doody, R.; van Dyck, C.H.; et al. A phase 2 multiple ascending dose trial of bapineuzumab in mild to moderate Alzheimer disease. Neurology 2009, 73, 2061–2070. [Google Scholar] [CrossRef] [PubMed]
- Doody, R.S.; Thomas, R.G.; Farlow, M.; Iwatsubo, T.; Vellas, B.; Joffe, S.; Kieburtz, K.; Raman, R.; Sun, X.; Aisen, P.S.; et al. Phase 3 trials of solanezumab for mild-to-moderate Alzheimer’s disease. N. Engl. J. Med. 2014, 370, 311–321. [Google Scholar] [CrossRef] [PubMed]
- Goedert, M. Neurodegeneration. Alzheimer’s and Parkinson’s diseases: The prion concept in relation to assembled Aβ, tau, and α-synuclein. Science 2015. [Google Scholar] [CrossRef] [PubMed]
- Holmes, B.B.; Diamond, M.I. Prion-like properties of tau protein: The importance of extracellular tau as a therapeutic target. J. Biol. Chem. 2014, 289, 19855–19861. [Google Scholar] [CrossRef] [PubMed]
- Jucker, M.; Walker, L.C. Self-propagation of pathogenic protein aggregates in neurodegenerative diseases. Nature 2013, 501, 45–51. [Google Scholar] [CrossRef] [PubMed]
- Prusiner, S.B. Biology and genetics of prions causing neurodegeneration. Annu. Rev. Genet. 2013, 47, 601–623. [Google Scholar] [CrossRef] [PubMed]
- Frost, B.; Diamond, M.I. Prion-like mechanisms in neurodegenerative diseases. Nat. Rev. Neurosci. 2010, 11, 155–159. [Google Scholar] [CrossRef] [PubMed]
- Goedert, M.; Spillantini, M.G.; Jakes, R.; Rutherford, D.; Crowther, R.A. Multiple isoforms of human microtubule-associated protein tau: Sequences and localization in neurofibrillary tangles of Alzheimer’s disease. Neuron 1989, 3, 519–526. [Google Scholar] [CrossRef]
- Weingarten, M.D.; Lockwood, A.H.; Hwo, S.Y.; Kirschner, M.W. A protein factor essential for microtubule assembly. Proc. Nat. Acad. Sci. USA 1975, 72, 1858–1862. [Google Scholar] [CrossRef] [PubMed]
- Goedert, M.; Jakes, R. Expression of separate isoforms of human tau protein: Correlation with the tau pattern in brain and effects on tubulin polymerization. EMBO J. 1990, 9, 4225–4230. [Google Scholar] [PubMed]
- Iqbal, K.; Liu, F.; Gong, C.X. Tau and neurodegenerative disease: The story so far. Nat. Rev. Neurol. 2016, 12, 15–27. [Google Scholar] [CrossRef] [PubMed]
- Goedert, M. Tau protein and the neurofibrillary pathology of Alzheimer’s disease. Trends Neurosci. 1993, 16, 460–465. [Google Scholar] [CrossRef]
- Goedert, M.; Spillantini, M.G.; Hasegawa, M.; Jakes, R.; Crowther, R.A.; Klug, A. Molecular dissection of the neurofibrillary lesions of Alzheimer’s disease. Cold Spring Harb. Symp. Quant. Biol. 1996, 61, 565–573. [Google Scholar] [PubMed]
- Crowther, R.A.; Wischik, C.M. Image reconstruction of the Alzheimer paired helical filament. EMBO J. 1985, 4, 3661–3665. [Google Scholar] [PubMed]
- Wischik, C.M.; Crowther, R.A. Subunit structure of the Alzheimer tangle. Br. Med. Bull. 1986, 42, 51–56. [Google Scholar] [PubMed]
- Wischik, C.M.; Novak, M.; Edwards, P.C.; Klug, A.; Tichelaar, W.; Crowther, R.A. Structural characterization of the core of the paired helical filament of Alzheimer disease. Proc. Nat. Acad. Sci. USA 1988, 85, 4884–4888. [Google Scholar] [CrossRef] [PubMed]
- Wischik, C.M.; Novak, M.; Thogersen, H.C.; Edwards, P.C.; Runswick, M.J.; Jakes, R.; Walker, J.E.; Milstein, C.; Roth, M.; Klug, A. Isolation of a fragment of tau derived from the core of the paired helical filament of Alzheimer disease. Proc. Nat. Acad. Sci. USA 1988, 85, 4506–4510. [Google Scholar] [CrossRef] [PubMed]
- Crowther, T.; Goedert, M.; Wischik, C.M. The repeat region of microtubule-associated protein tau forms part of the core of the paired helical filament of Alzheimer’s disease. Ann. Med. 1989, 21, 127–132. [Google Scholar] [CrossRef] [PubMed]
- Goedert, M.; Spillantini, M.G.; Potier, M.C.; Ulrich, J.; Crowther, R.A. Cloning and sequencing of the cdna encoding an isoform of microtubule-associated protein tau containing four tandem repeats: Differential expression of tau protein mrnas in human brain. EMBO J. 1989, 8, 393–399. [Google Scholar] [PubMed]
- Greenberg, S.G.; Davies, P. A preparation of Alzheimer paired helical filaments that displays distinct tau proteins by polyacrylamide gel electrophoresis. Proc. Nat. Acad. Sci. USA 1990, 87, 5827–5831. [Google Scholar] [CrossRef] [PubMed]
- Lee, V.M.; Balin, B.J.; Otvos, L., Jr.; Trojanowski, J.Q. A68: A major subunit of paired helical filaments and derivatized forms of normal tau. Science 1991, 251, 675–678. [Google Scholar] [CrossRef] [PubMed]
- Hasegawa, M.; Morishima-Kawashima, M.; Takio, K.; Suzuki, M.; Titani, K.; Ihara, Y. Protein sequence and mass spectrometric analyses of tau in the Alzheimer’s disease brain. J. Biol. Chem. 1992, 267, 17047–17054. [Google Scholar] [PubMed]
- Hasegawa, M.; Watanabe, A.; Takio, K.; Suzuki, M.; Arai, T.; Titani, K.; Ihara, Y. Characterization of two distinct monoclonal antibodies to paired helical filaments: Further evidence for fetal-type phosphorylation of the tau in paired helical filaments. J. Neurochem. 1993, 60, 2068–2077. [Google Scholar] [CrossRef] [PubMed]
- Morishima-Kawashima, M.; Hasegawa, M.; Takio, K.; Suzuki, M.; Titani, K.; Ihara, Y. Ubiquitin is conjugated with amino-terminally processed tau in paired helical filaments. Neuron 1993, 10, 1151–1160. [Google Scholar] [CrossRef]
- Morishima-Kawashima, M.; Hasegawa, M.; Takio, K.; Suzuki, M.; Yoshida, H.; Titani, K.; Ihara, Y. Proline-directed and non-proline-directed phosphorylation of PHF-tau. J. Biol. Chem. 1995, 270, 823–829. [Google Scholar] [CrossRef] [PubMed]
- Dan, A.; Takahashi, M.; Masuda-Suzukake, M.; Kametani, F.; Nonaka, T.; Kondo, H.; Akiyama, H.; Arai, T.; Mann, D.M.; Saito, Y.; et al. Extensive deamidation at asparagine residue 279 accounts for weak immunoreactivity of tau with RD4 antibody in Alzheimer’s disease brain. Acta Neuropathol. Commun. 2013. [Google Scholar] [CrossRef] [PubMed]
- Goedert, M.; Spillantini, M.G.; Cairns, N.J.; Crowther, R.A. Tau proteins of Alzheimer paired helical filaments: Abnormal phosphorylation of all six brain isoforms. Neuron 1992, 8, 159–168. [Google Scholar] [CrossRef]
- Biernat, J.; Mandelkow, E.M.; Schroter, C.; Lichtenberg-Kraag, B.; Steiner, B.; Berling, B.; Meyer, H.; Mercken, M.; Vandermeeren, A.; Goedert, M.; et al. The switch of tau protein to an Alzheimer-like state includes the phosphorylation of two serine-proline motifs upstream of the microtubule binding region. EMBO J. 1992, 11, 1593–1597. [Google Scholar] [PubMed]
- Bramblett, G.T.; Goedert, M.; Jakes, R.; Merrick, S.E.; Trojanowski, J.Q.; Lee, V.M. Abnormal tau phosphorylation at Ser396 in Alzheimer’s disease recapitulates development and contributes to reduced microtubule binding. Neuron 1993, 10, 1089–1099. [Google Scholar] [CrossRef]
- Goedert, M.; Jakes, R.; Crowther, R.A.; Six, J.; Lubke, U.; Vandermeeren, M.; Cras, P.; Trojanowski, J.Q.; Lee, V.M. The abnormal phosphorylation of tau protein at Ser-202 in Alzheimer disease recapitulates phosphorylation during development. Proc. Nat. Acad. Sci. USA 1993, 90, 5066–5070. [Google Scholar] [CrossRef] [PubMed]
- Morishima-Kawashima, M.; Hasegawa, M.; Takio, K.; Suzuki, M.; Yoshida, H.; Watanabe, A.; Titani, K.; Ihara, Y. Hyperphosphorylation of tau in PHF. Neurobiol. Aging 1995, 16, 365–371. [Google Scholar] [CrossRef]
- Iwatsubo, T.; Hasegawa, M.; Ihara, Y. Neuronal and glial tau-positive inclusions in diverse neurologic diseases share common phosphorylation characteristics. Acta Neuropathol. 1994, 88, 129–136. [Google Scholar] [CrossRef] [PubMed]
- Buee, L.; Delacourte, A. Comparative biochemistry of tau in progressive supranuclear palsy, corticobasal degeneration, FTDP-17 and Pick’s disease. Brain Pathol. 1999, 9, 681–693. [Google Scholar] [CrossRef] [PubMed]
- Lee, V.M.; Goedert, M.; Trojanowski, J.Q. Neurodegenerative tauopathies. Annu. Rev. Neurosci. 2001, 24, 1121–1159. [Google Scholar] [CrossRef] [PubMed]
- Goedert, M.; Hasegawa, M. The tauopathies: Toward an experimental animal model. Am. J. Pathol. 1999, 154, 1–6. [Google Scholar] [CrossRef]
- Kovacs, G.G. Invited review: Neuropathology of tauopathies: Principles and practice. Neuropathol. Appl. Neurobiol. 2015, 41, 3–23. [Google Scholar] [CrossRef] [PubMed]
- Umeda, Y.; Taniguchi, S.; Arima, K.; Piao, Y.S.; Takahashi, H.; Iwatsubo, T.; Mann, D.; Hasegawa, M. Alterations in human tau transcripts correlate with those of neurofilament in sporadic tauopathies. Neurosci. Lett. 2004, 359, 151–154. [Google Scholar] [CrossRef] [PubMed]
- Poorkaj, P.; Bird, T.D.; Wijsman, E.; Nemens, E.; Garruto, R.M.; Anderson, L.; Andreadis, A.; Wiederholt, W.C.; Raskind, M.; Schellenberg, G.D. Tau is a candidate gene for chromosome 17 frontotemporal dementia. Ann. Neurol. 1998, 43, 815–825. [Google Scholar] [CrossRef] [PubMed]
- Spillantini, M.G.; Goedert, M.; Crowther, R.A.; Murrell, J.R.; Farlow, M.R.; Ghetti, B. Familial multiple system tauopathy with presenile dementia: A disease with abundant neuronal and glial tau filaments. Proc. Nat. Acad. Sci. USA 1997, 94, 4113–4118. [Google Scholar] [CrossRef] [PubMed]
- Hutton, M.; Lendon, C.L.; Rizzu, P.; Baker, M.; Froelich, S.; Houlden, H.; Pickering-Brown, S.; Chakraverty, S.; Isaacs, A.; Grover, A.; et al. Association of missense and 5'-splice-site mutations in tau with the inherited dementia FTDP-17. Nature 1998, 393, 702–705. [Google Scholar] [CrossRef] [PubMed]
- Spillantini, M.G.; Murrell, J.R.; Goedert, M.; Farlow, M.R.; Klug, A.; Ghetti, B. Mutation in the tau gene in familial multiple system tauopathy with presenile dementia. Proc. Nat. Acad. Sci. USA 1998, 95, 7737–7741. [Google Scholar] [CrossRef] [PubMed]
- Goedert, M.; Crowther, R.A.; Spillantini, M.G. Tau mutations cause frontotemporal dementias. Neuron 1998, 21, 955–958. [Google Scholar] [CrossRef]
- Goedert, M.; Ghetti, B.; Spillantini, M.G. Frontotemporal dementia: Implications for understanding Alzheimer disease. Cold Spring Harb. Perspect. Med. 2012. [Google Scholar] [CrossRef] [PubMed]
- Taniguchi-Watanabe, S.; Arai, T.; Kametani, F.; Nonaka, T.; Masuda-Suzukake, M.; Tarutani, A.; Murayama, S.; Saito, Y.; Arima, K.; Yoshida, M.; et al. Biochemical classification of tauopathies by immunoblot, protein sequence and mass spectrometric analyses of sarkosyl-insoluble and trypsin-resistant tau. Acta Neuropathol. 2016, 131, 267–280. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, A.; Hasegawa, M.; Suzuki, M.; Takio, K.; Morishima-Kawashima, M.; Titani, K.; Arai, T.; Kosik, K.S.; Ihara, Y. In vivo phosphorylation sites in fetal and adult rat tau. J. Biol. Chem. 1993, 268, 25712–25717. [Google Scholar] [PubMed]
- Matsuo, E.S.; Shin, R.W.; Billingsley, M.L.; van deVoorde, A.; O’Connor, M.; Trojanowski, J.Q.; Lee, V.M. Biopsy-derived adult human brain tau is phosphorylated at many of the same sites as Alzheimer’s disease paired helical filament tau. Neuron 1994, 13, 989–1002. [Google Scholar] [CrossRef]
- Yamamoto, H.; Hasegawa, M.; Ono, T.; Tashima, K.; Ihara, Y.; Miyamoto, E. Dephosphorylation of fetal-tau and paired helical filaments-tau by protein phosphatases 1 and 2A and calcineurin. J. Biochem. 1995, 118, 1224–1231. [Google Scholar] [PubMed]
- Von Bergen, M.; Friedhoff, P.; Biernat, J.; Heberle, J.; Mandelkow, E.M.; Mandelkow, E. Assembly of tau protein into Alzheimer paired helical filaments depends on a local sequence motif 306VQIVYK311 forming β structure. Proc. Nat. Acad. Sci. USA 2000, 97, 5129–5134. [Google Scholar] [CrossRef] [PubMed]
- Arai, T.; Ikeda, K.; Akiyama, H.; Nonaka, T.; Hasegawa, M.; Ishiguro, K.; Iritani, S.; Tsuchiya, K.; Iseki, E.; Yagishita, S.; et al. Identification of amino-terminally cleaved tau fragments that distinguish progressive supranuclear palsy from corticobasal degeneration. Ann. Neurol. 2004, 55, 72–79. [Google Scholar] [CrossRef] [PubMed]
- Ksiezak-Reding, H.; Yen, S.H. Structural stability of paired helical filaments requires microtubule-binding domains of tau: A model for self-association. Neuron 1991, 6, 717–728. [Google Scholar] [CrossRef]
- Hasegawa, M.; Watanabe, S.; Kondo, H.; Akiyama, H.; Mann, D.M.; Saito, Y.; Murayama, S. 3r and 4r tau isoforms in paired helical filaments in Alzheimer’s disease. Acta Neuropathol. 2014, 127, 303–305. [Google Scholar] [CrossRef] [PubMed]
- Probst, A.; Tolnay, M.; Langui, D.; Goedert, M.; Spillantini, M.G. Pick’s disease: Hyperphosphorylated tau protein segregates to the somatoaxonal compartment. Acta Neuropathol. 1996, 92, 588–596. [Google Scholar] [CrossRef] [PubMed]
- Arai, T.; Ikeda, K.; Akiyama, H.; Tsuchiya, K.; Iritani, S.; Ishiguro, K.; Yagishita, S.; Oda, T.; Odawara, T.; Iseki, E. Different immunoreactivities of the microtubule-binding region of tau and its molecular basis in brains from patients with Alzheimer’s disease, Pick’s disease, progressive supranuclear palsy and corticobasal degeneration. Acta Neuropathol. 2003, 105, 489–498. [Google Scholar] [PubMed]
- Aoyagi, H.; Hasegawa, M.; Tamaoka, A. Fibrillogenic nuclei composed of P301L mutant tau induce elongation of P301L tau but not wild-type tau. J. Biol. Chem. 2007, 282, 20309–20318. [Google Scholar] [CrossRef] [PubMed]
- Frost, B.; Jacks, R.L.; Diamond, M.I. Propagation of tau misfolding from the outside to the inside of a cell. J. Biol. Chem. 2009, 284, 12845–12852. [Google Scholar] [CrossRef] [PubMed]
- Nonaka, T.; Watanabe, S.T.; Iwatsubo, T.; Hasegawa, M. Seeded aggregation and toxicity of α-synuclein and tau: Cellular models of neurodegenerative diseases. J. Biol. Chem. 2010, 285, 34885–34898. [Google Scholar] [CrossRef] [PubMed]
- Clavaguera, F.; Bolmont, T.; Crowther, R.A.; Abramowski, D.; Frank, S.; Probst, A.; Fraser, G.; Stalder, A.K.; Beibel, M.; Staufenbiel, M.; et al. Transmission and spreading of tauopathy in transgenic mouse brain. Nat. Cell Biol. 2009, 11, 909–913. [Google Scholar] [CrossRef] [PubMed]
- Clavaguera, F.; Akatsu, H.; Fraser, G.; Crowther, R.A.; Frank, S.; Hench, J.; Probst, A.; Winkler, D.T.; Reichwald, J.; Staufenbiel, M.; et al. Brain homogenates from human tauopathies induce tau inclusions in mouse brain. Proc. Nat. Acad. Sci. USA 2013, 110, 9535–9540. [Google Scholar] [CrossRef] [PubMed]
- Sanders, D.W.; Kaufman, S.K.; DeVos, S.L.; Sharma, A.M.; Mirbaha, H.; Li, A.; Barker, S.J.; Foley, A.C.; Thorpe, J.R.; Serpell, L.C.; et al. Distinct tau prion strains propagate in cells and mice and define different tauopathies. Neuron 2014, 82, 1271–1288. [Google Scholar] [CrossRef] [PubMed]
- Goedert, M.; Clavaguera, F.; Tolnay, M. The propagation of prion-like protein inclusions in neurodegenerative diseases. Trends Neurosci. 2010, 33, 317–325. [Google Scholar] [CrossRef] [PubMed]
- Aguzzi, A.; Rajendran, L. The transcellular spread of cytosolic amyloids, prions, and prionoids. Neuron 2009, 64, 783–790. [Google Scholar] [CrossRef] [PubMed]
- Brundin, P.; Melki, R.; Kopito, R. Prion-like transmission of protein aggregates in neurodegenerative diseases. Nat. Rev. Mol. Cell Biol. 2010, 11, 301–307. [Google Scholar] [CrossRef] [PubMed]
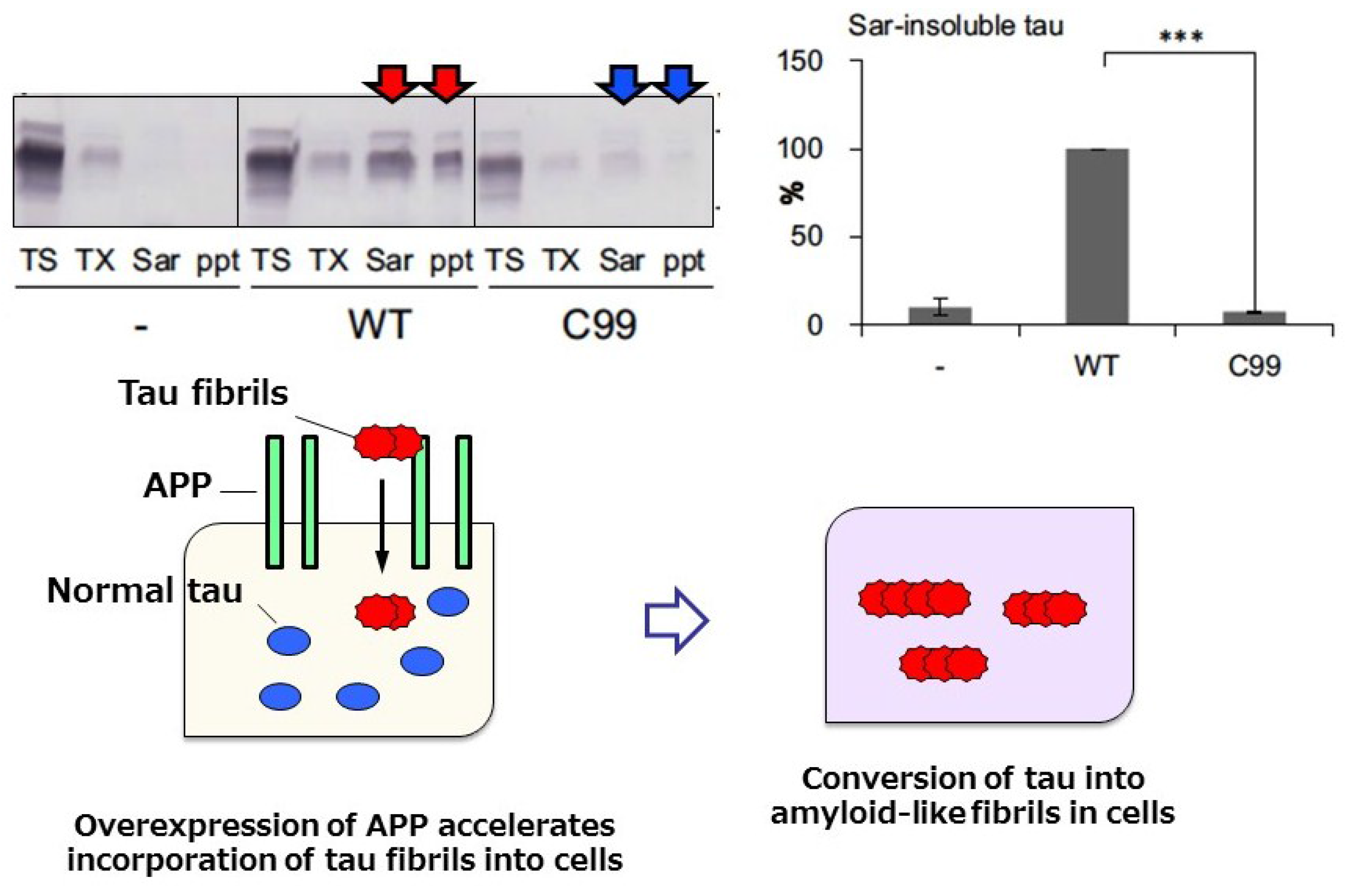
- Takahashi, M.; Miyata, H.; Kametani, F.; Nonaka, T.; Akiyama, H.; Hisanaga, S.; Hasegawa, M. Extracellular association of app and tau fibrils induces intracellular aggregate formation of tau. Acta Neuropathol. 2015, 129, 895–907. [Google Scholar] [CrossRef] [PubMed]
- Braak, H.; Del Tredici, K. The pathological process underlying Alzheimer’s disease in individuals under thirty. Acta Neuropathol. 2011, 121, 171–181. [Google Scholar] [CrossRef] [PubMed]
- Mann, D.M.; Hardy, J. Amyloid or tau: The chicken or the egg? Acta Neuropathol. 2013, 126, 609–613. [Google Scholar] [CrossRef] [PubMed]
- Braak, H.; Del Tredici, K. Reply: The early pathological process in sporadic Alzheimer’s disease. Acta Neuropathol. 2013, 126, 615–618. [Google Scholar] [CrossRef] [PubMed]
- Braak, H.; Zetterberg, H.; Del Tredici, K.; Blennow, K. Intraneuronal tau aggregation precedes diffuse plaque deposition, but amyloid-β changes occur before increases of tau in cerebrospinal fluid. Acta Neuropathol. 2013, 126, 631–641. [Google Scholar] [CrossRef] [PubMed]
- Jellinger, K.A.; Alafuzoff, I.; Attems, J.; Beach, T.G.; Cairns, N.J.; Crary, J.F.; Dickson, D.W.; Hof, P.R.; Hyman, B.T.; Jack, C.R., Jr.; et al. PART, a distinct tauopathy, different from classical sporadic Alzheimer disease. Acta Neuropathol. 2015, 129, 757–762. [Google Scholar] [CrossRef] [PubMed]
- Duyckaerts, C.; Braak, H.; Brion, J.P.; Buee, L.; Del Tredici, K.; Goedert, M.; Halliday, G.; Neumann, M.; Spillantini, M.G.; Tolnay, M.; et al. PART is part of Alzheimer disease. Acta Neuropathol. 2015, 129, 749–756. [Google Scholar] [CrossRef] [PubMed]
- Braak, H.; Del Tredici, K. Amyloid-β may be released from non-junctional varicosities of axons generated from abnormal tau-containing brainstem nuclei in sporadic Alzheimer’s disease: A hypothesis. Acta Neuropathol. 2013, 126, 303–306. [Google Scholar] [CrossRef] [PubMed]
- Taniguchi, S.; Suzuki, N.; Masuda, M.; Hisanaga, S.; Iwatsubo, T.; Goedert, M.; Hasegawa, M. Inhibition of heparin-induced tau filament formation by phenothiazines, polyphenols, and porphyrins. J. Biol. Chem. 2005, 280, 7614–7623. [Google Scholar] [CrossRef] [PubMed]
- Masuda, M.; Suzuki, N.; Taniguchi, S.; Oikawa, T.; Nonaka, T.; Iwatsubo, T.; Hisanaga, S.; Goedert, M.; Hasegawa, M. Small molecule inhibitors of α-synuclein filament assembly. Biochemistry 2006, 45, 6085–6094. [Google Scholar] [CrossRef] [PubMed]
- Harrington, C.R.; Storey, J.M.; Clunas, S.; Harrington, K.A.; Horsley, D.; Ishaq, A.; Kemp, S.J.; Larch, C.P.; Marshall, C.; Nicoll, S.L.; et al. Cellular models of aggregation-dependent template-directed proteolysis to characterize tau aggregation inhibitors for treatment of Alzheimer disease. J. Biol. Chem. 2015, 290, 10862–10875. [Google Scholar] [CrossRef] [PubMed]
- Wischik, C.M.; Harrington, C.R.; Storey, J.M. Tau-aggregation inhibitor therapy for Alzheimer’s disease. Biochem. Pharmacol. 2014, 88, 529–539. [Google Scholar] [CrossRef] [PubMed]
- Asuni, A.A.; Boutajangout, A.; Quartermain, D.; Sigurdsson, E.M. Immunotherapy targeting pathological tau conformers in a tangle mouse model reduces brain pathology with associated functional improvements. J. Neurosci. 2007, 27, 9115–9129. [Google Scholar] [CrossRef] [PubMed]
- Umeda, T.; Eguchi, H.; Kunori, Y.; Matsumoto, Y.; Taniguchi, T.; Mori, H.; Tomiyama, T. Passive immunotherapy of tauopathy targeting pSer413-tau: A pilot study in mice. Ann. Clin. Transl. Neurol. 2015, 2, 241–255. [Google Scholar] [CrossRef] [PubMed]
- Flight, M.H. Neurodegenerative disease: Tau immunotherapy targets transcellular propagation. Nat. Rev. Drug Discov. 2013. [Google Scholar] [CrossRef] [PubMed]
- Wischik, C.M.; Staff, R.T.; Wischik, D.J.; Bentham, P.; Murray, A.D.; Storey, J.M.; Kook, K.A.; Harrington, C.R. Tau aggregation inhibitor therapy: An exploratory phase 2 study in mild or moderate Alzheimer’s disease. J. Alzheimers Dis. 2015, 44, 705–720. [Google Scholar] [PubMed]
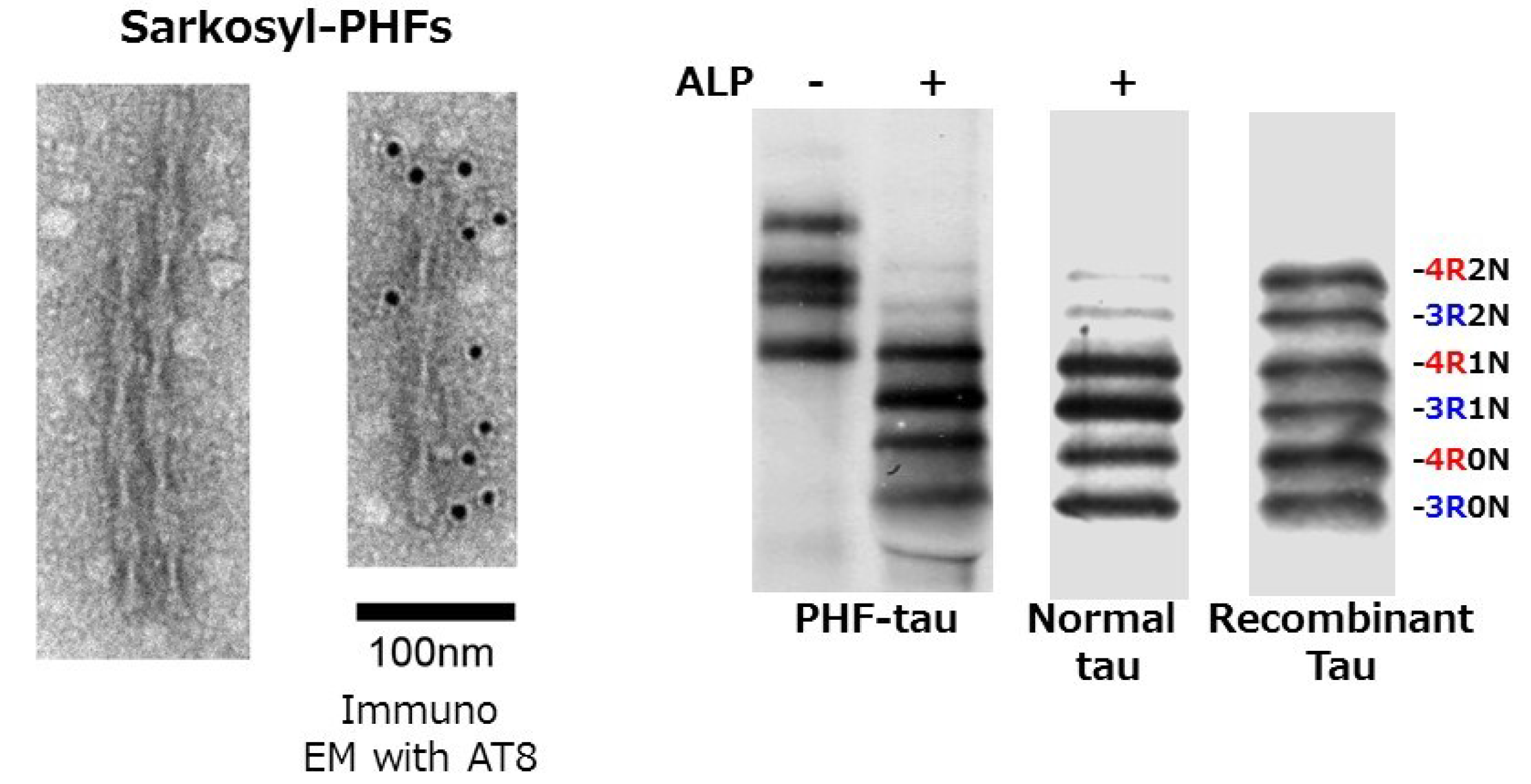
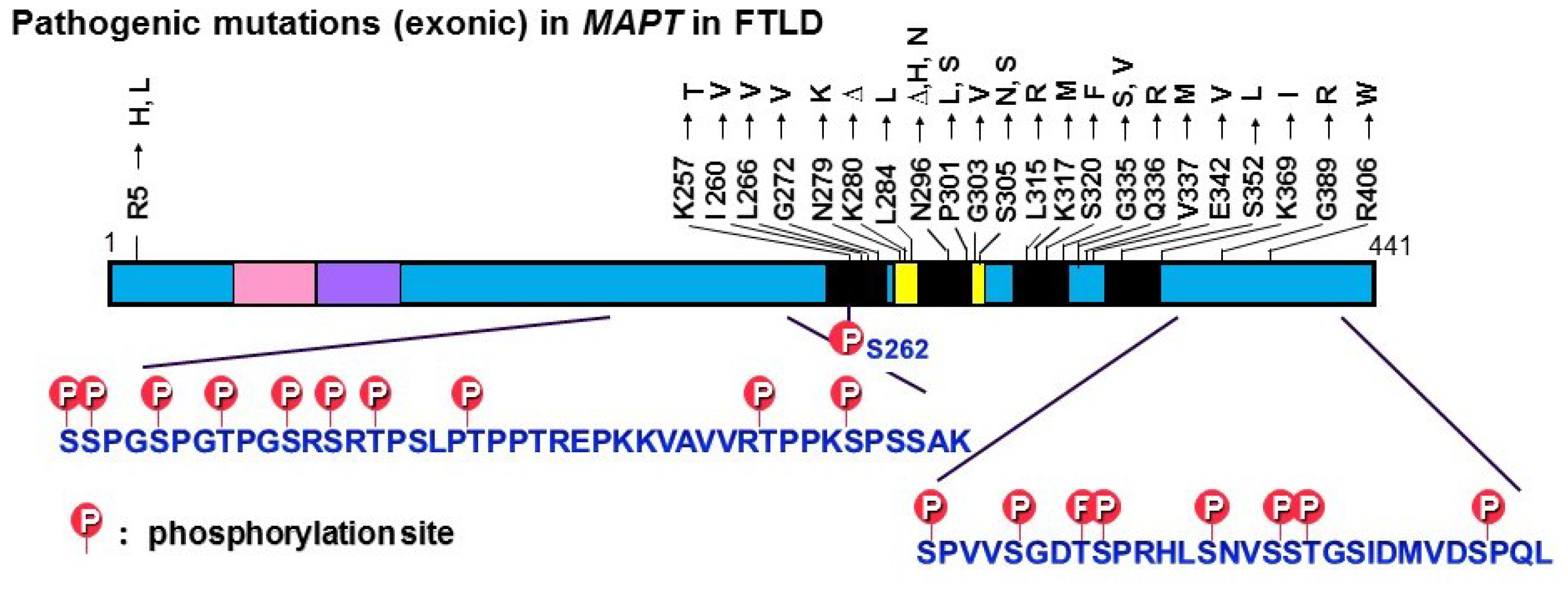
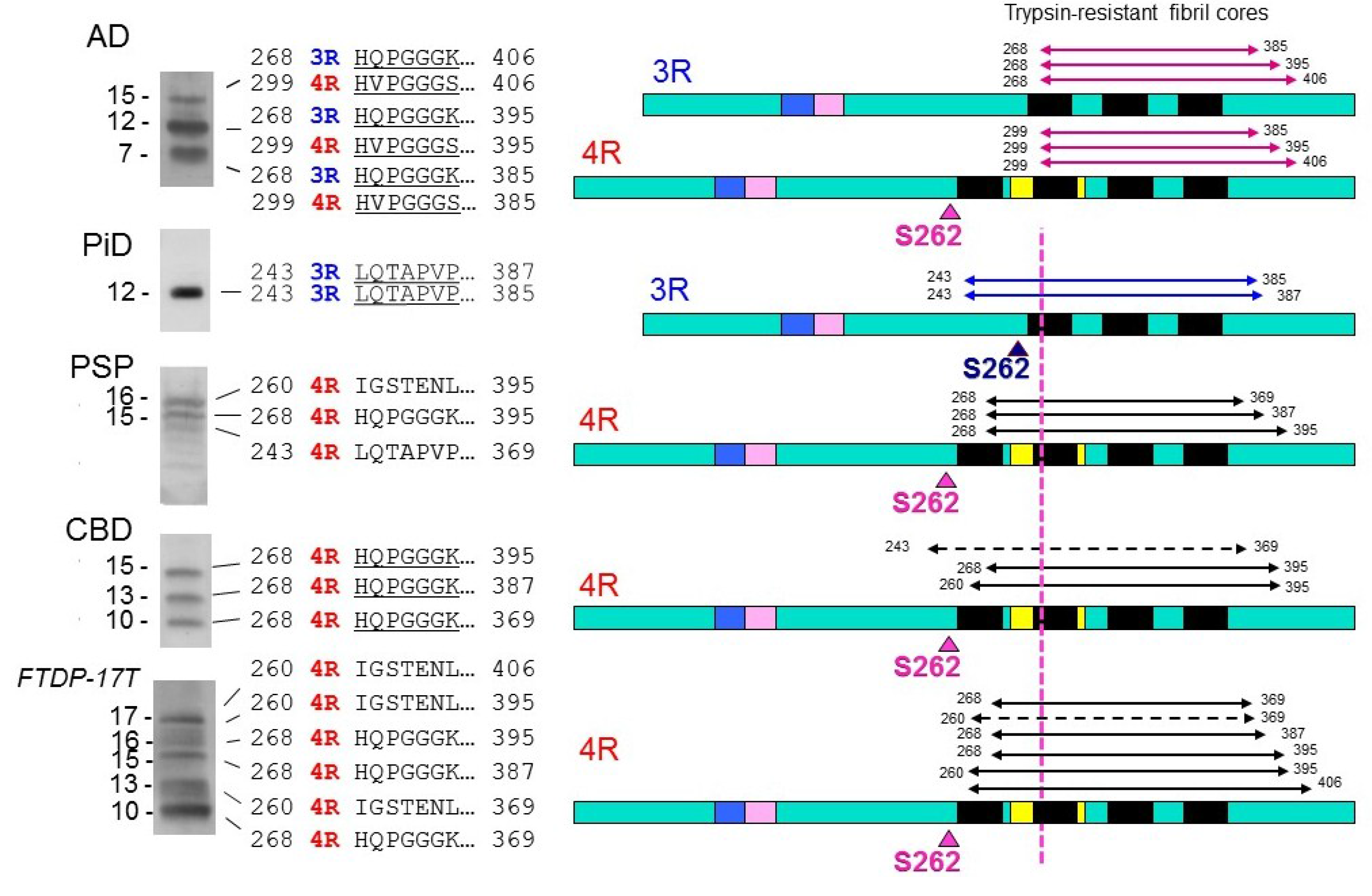
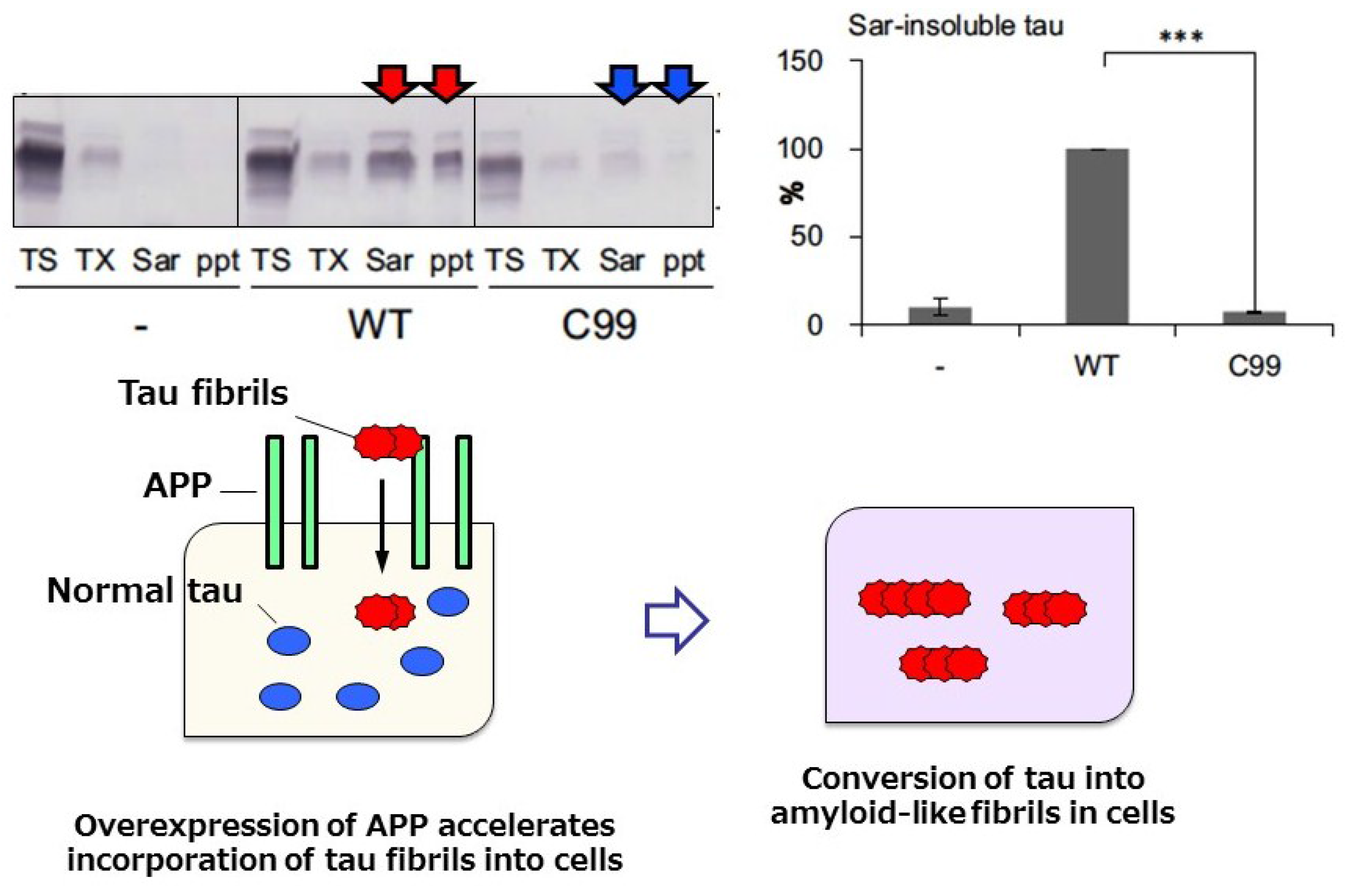
© 2016 by the author; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hasegawa, M. Molecular Mechanisms in the Pathogenesis of Alzheimer’s disease and Tauopathies-Prion-Like Seeded Aggregation and Phosphorylation. Biomolecules 2016, 6, 24. https://doi.org/10.3390/biom6020024
Hasegawa M. Molecular Mechanisms in the Pathogenesis of Alzheimer’s disease and Tauopathies-Prion-Like Seeded Aggregation and Phosphorylation. Biomolecules. 2016; 6(2):24. https://doi.org/10.3390/biom6020024
Chicago/Turabian StyleHasegawa, Masato. 2016. "Molecular Mechanisms in the Pathogenesis of Alzheimer’s disease and Tauopathies-Prion-Like Seeded Aggregation and Phosphorylation" Biomolecules 6, no. 2: 24. https://doi.org/10.3390/biom6020024
APA StyleHasegawa, M. (2016). Molecular Mechanisms in the Pathogenesis of Alzheimer’s disease and Tauopathies-Prion-Like Seeded Aggregation and Phosphorylation. Biomolecules, 6(2), 24. https://doi.org/10.3390/biom6020024