
Hsp90: A New Player in DNA Repair?




Abstract
1. Introduction
2. Hsp90 and Cancer
3. Structural Aspects of Hsp90 and Related Chaperones

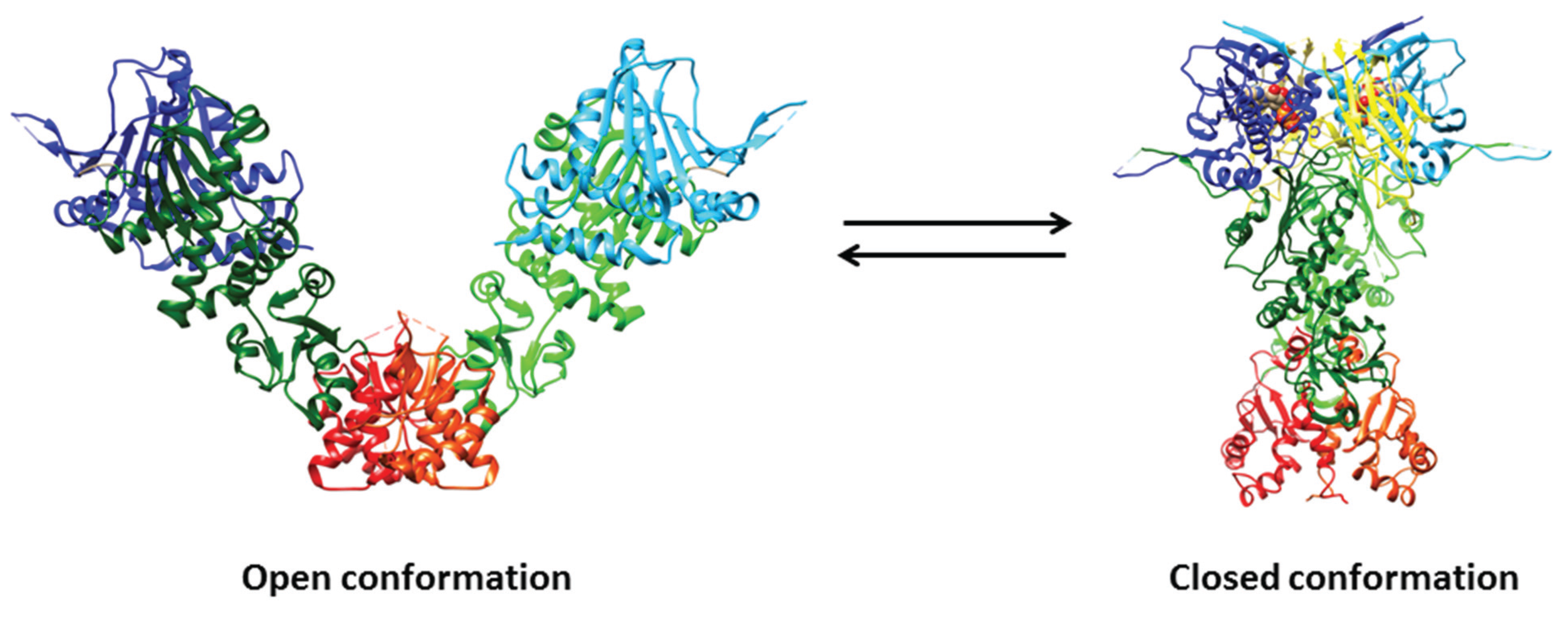{kind=link}
{kind=link}
{kind=link}
| Inhibitor | Derivatives | Structure | Pharmacokinetics (nM) | Clinical Study Stage | References |
|---|---|---|---|---|---|
| Geldanamycin (naturally derived from Streptomyces hygroscopicus) | 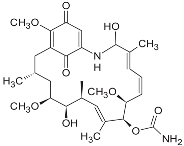 | GI50 = 1.0 × 10−1 nM; LC50 = 2.1 × 104 nM; Kd = 1.2 × 103 nM | Preclinical | [61,65,69]; www.medchemexpress.net | |
| 17-AAG (17-allylamino-17-desmethoxygeldanamycin; tanespimycin) |  | IC50 = 5.0 × 101–1.0 × 104 nM | Phase I/II/III | [70,71,72,73,74,75,76,77,78]; www.clinicaltrials.gov; www.medchemexpress.net | |
| 17-DMAG (17-dimethylaminoethylamino-17-demetoxygelanamycin; alvespimycin) | 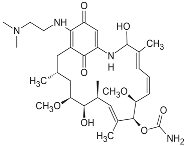 | IC50 = 6.0 × 101–3.0 × 103 nM; Kd = 3.5 × 102 nM | Phase I/II | [72,77,79,80,81]; www.clinicaltrials.gov | |
| PU-H71 | 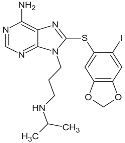 | IC50 = 5.0 × 101–3.0 × 102 nM | Phase I | [82,83,84]; www.clinicaltrials.gov | |
| NVP-AUY922 | 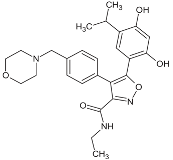 | IC50 = 1.3 × 101 nM (Hsp90α); IC50 = 2.1 × 101 nM (Hsp90β); GI50 = 2.0–4.0 × 101 nM; Kd = 1.7 nM | Phase I/II | [85,86,87]; www.medchemexpress.net; www.clinicaltrials.gov | |
| Radicicol (naturally derived from Diheterospora chlamydosporia) |  | Kd = 1.9 × 101 nM; IC50 = 2.0 × 101 nM | Preclinical | [65] | |
| STA9090 (Ganetespib) | 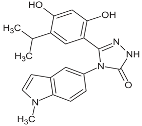 | IC50 = 1.0–5.0 × 101 nM | Phase I/II/III | [88,89,90,91]; www.clinicaltrials.gov; www.medchemexpress.net |
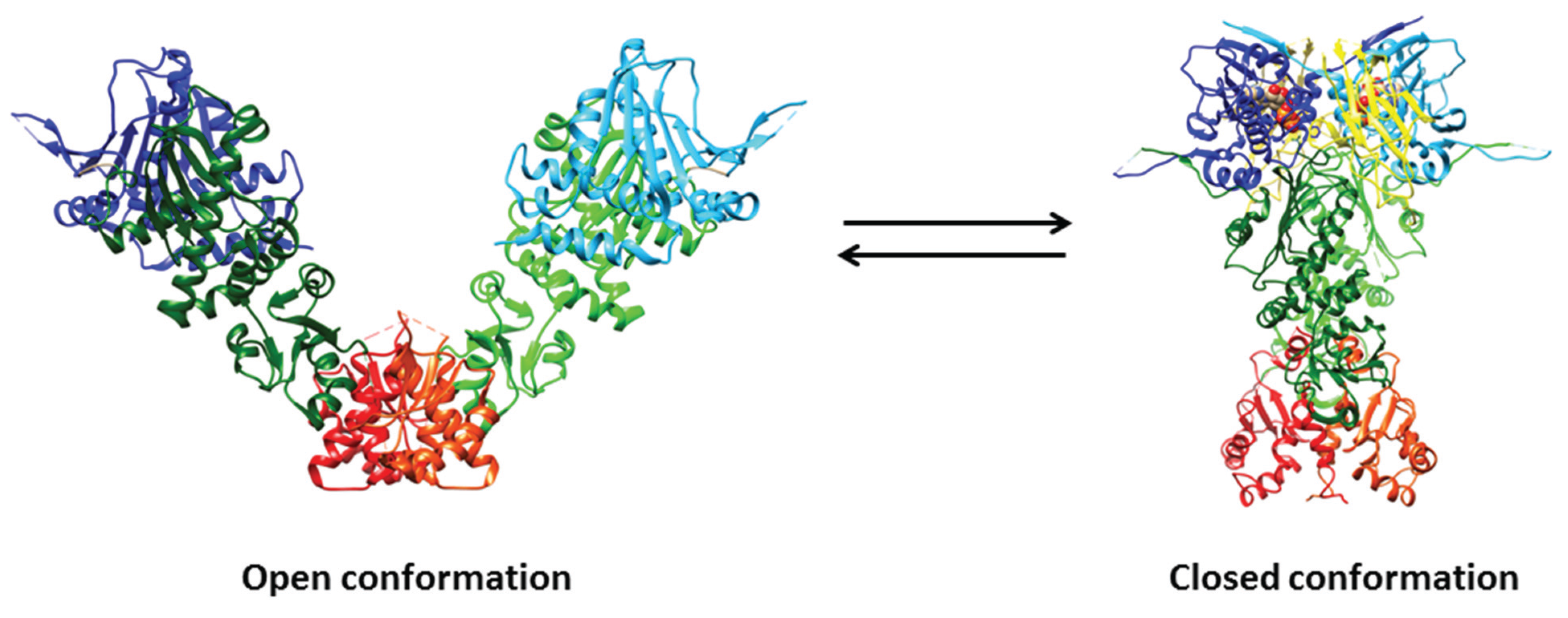
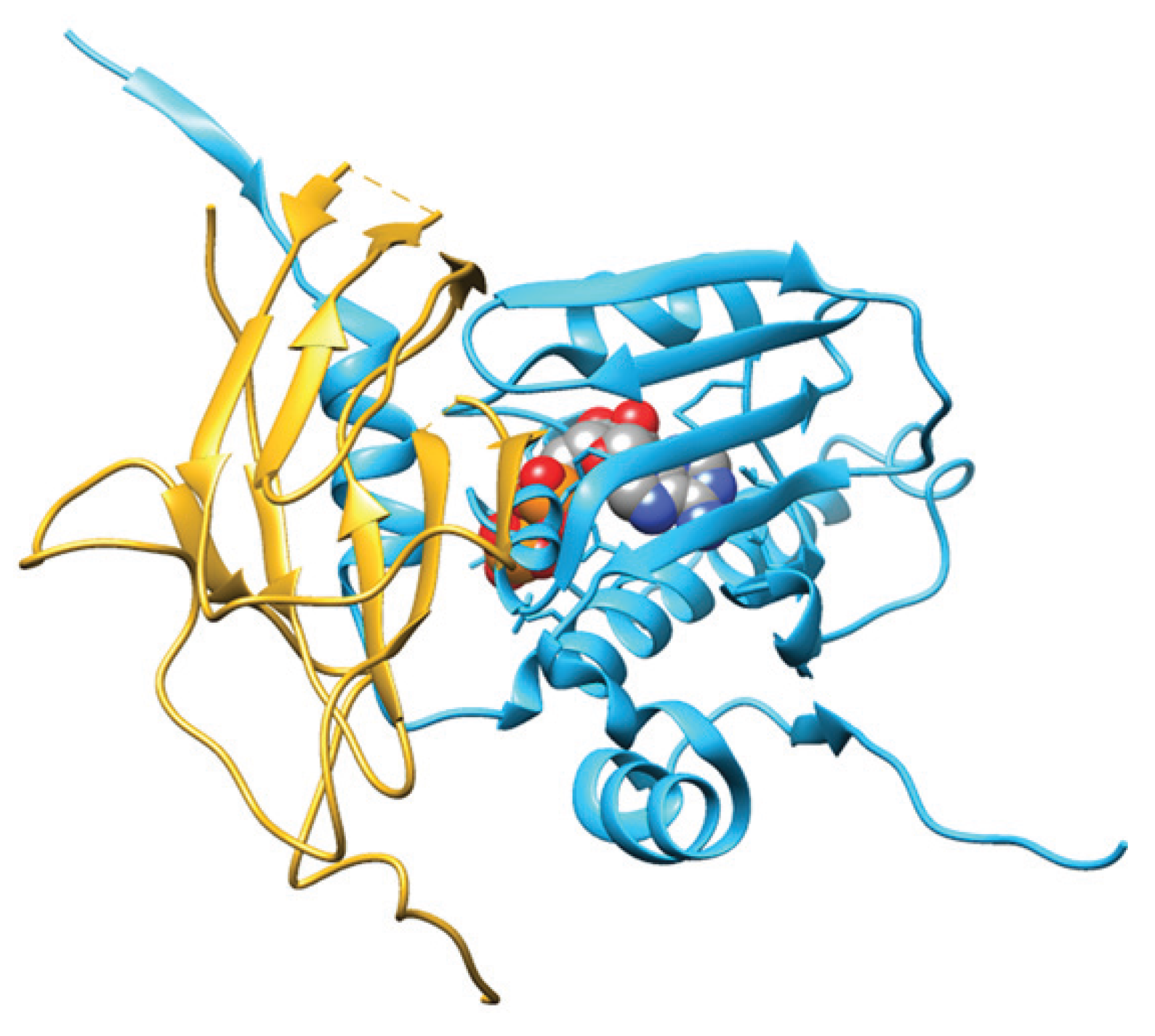
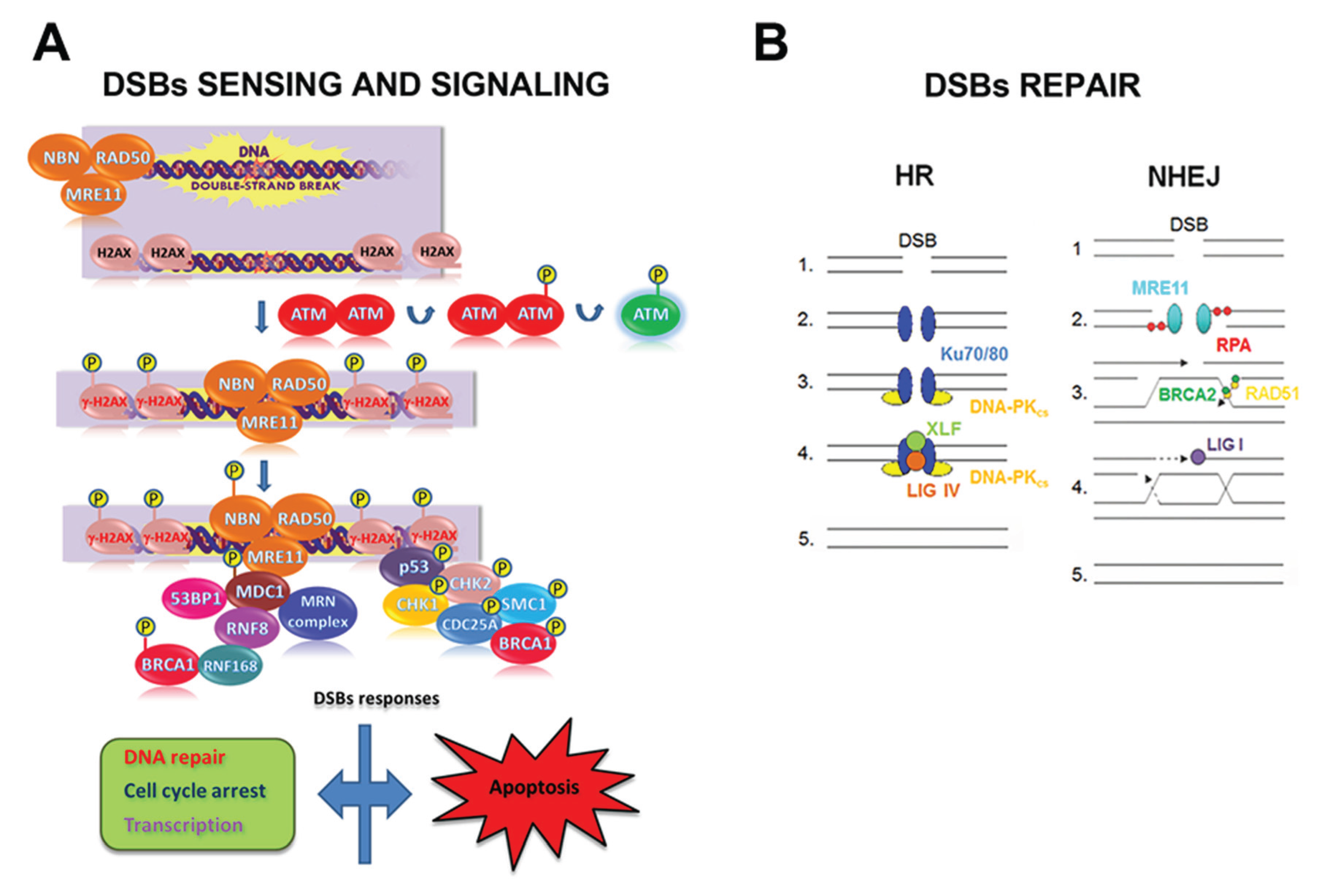
4. Role of Hsp90 in the Genome Stability Maintenance
4.1. The DNA Double-Strand Break Response
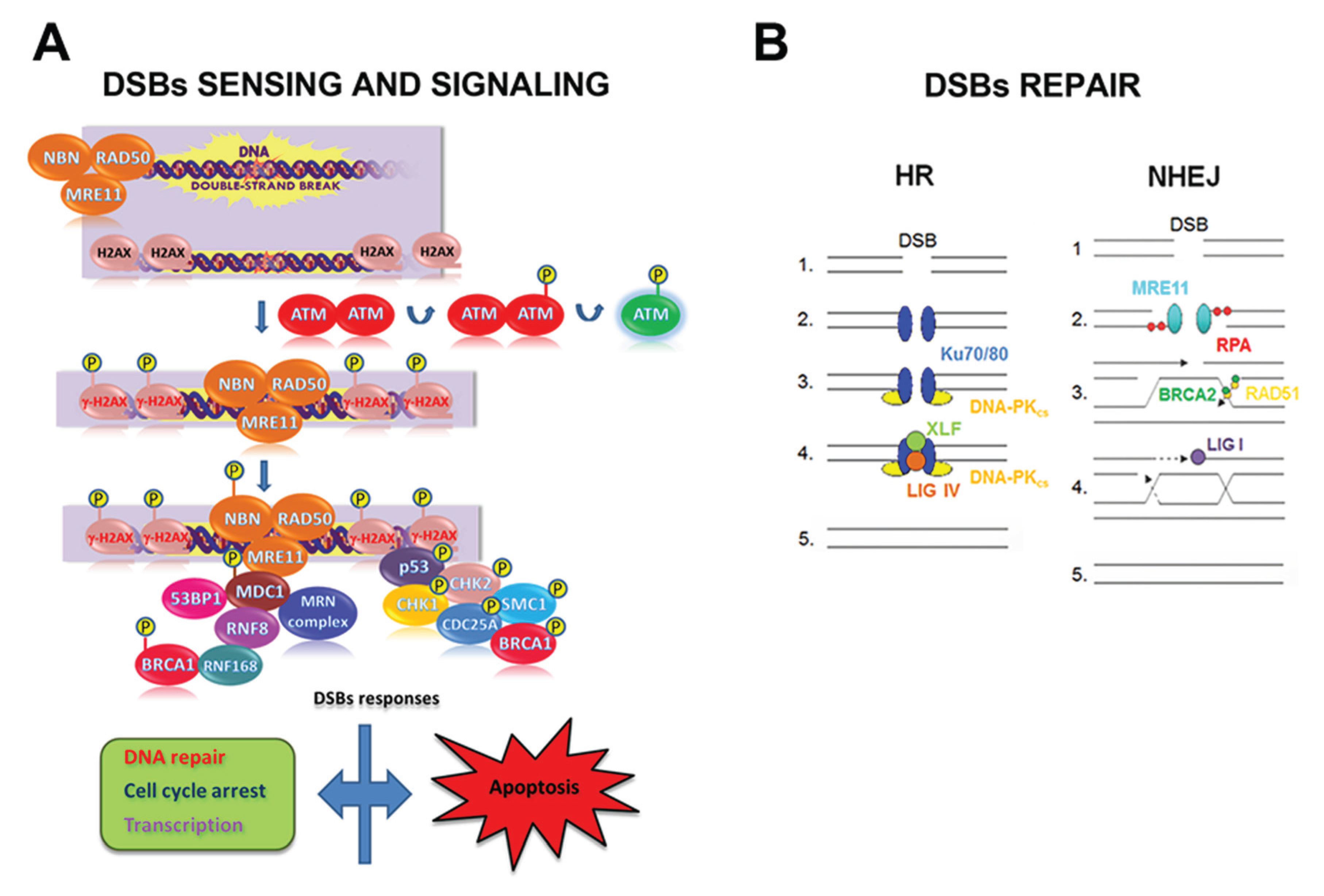
4.2. Hsp90 and the DNA Damage Response Clients
4.2.1. BRCA1, BRCA2, and RAD51
4.2.2. CHK1
4.2.3. DNA-PK
4.2.4. The FA Pathway
4.2.5. Histones
4.2.6. The MRE11/RAD50/NBN Complex
4.3. Other DNA Damage Response Clients
4.3.1. MSH2
4.3.2. PCNA and Polymerase η
4.3.3. XRCC1
5. Conclusions and Perspectives
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| 17-AAG | 17-(allylamino)-17-demethoxygeldanamycin |
| 17-DMAG | 17-dimethylaminoethylamino-17-demethoxygeldanamycin |
| BER | base excision repair |
| DDR | DNA damage response |
| DSB | DNA double-strand break |
| FA | Fanconi anemia |
| HR | homologous recombination repair |
| Hsp90 | heat shock protein 90 |
| IR | ionizing radiation |
| IRIF | IR-induced foci |
| MMR | DNA mismatch repair |
| NHEJ | non-homologous end-joining repair |
| NSCLC | non-small-cell lung cancer |
| Pol η | polymerase η |
| ssDNA | single-stranded regions of DNA |
| SSB | DNA single-strand break |
| TLS | translesion synthesis |
References
- Balch, W.E.; Morimoto, R.I.; Dillin, A.; Kelly, J.W. Adapting proteostasis for disease intervention. Science 2008, 319, 916–919. [Google Scholar] [CrossRef] [PubMed]
- Taipale, M.; Jarosz, D.F.; Lindquist, S. HSP90 at the hub of protein homeostasis: Emerging mechanistic insights. Nat. Rev. Mol. Cell Biol. 2010, 11, 515–528. [Google Scholar] [CrossRef] [PubMed]
- Makhnevych, T.; Houry, W.A. The role of Hsp90 in protein complex assembly. Biochim. Biophys. Acta 2012, 1823, 674–682. [Google Scholar] [CrossRef] [PubMed]
- Neckers, L. Heat shock protein 90: The cancer chaperone. J. Biosci. 2007, 32, 517–530. [Google Scholar] [CrossRef] [PubMed]
- Trepel, J.; Mollapour, M.; Giaccone, G.; Neckers, L. Targeting the dynamic HSP90 complex in cancer. Nat. Rev. Cancer 2010, 10, 537–549. [Google Scholar] [CrossRef] [PubMed]
- Wandinger, S.K.; Richter, K.; Buchner, J. The Hsp90 chaperone machinery. J. Biol. Chem. 2008, 283, 18473–18477. [Google Scholar] [CrossRef] [PubMed]
- Zhao, R.; Davey, M.; Hsu, Y.C.; Kaplanek, P.; Tong, A.; Parsons, A.B.; Krogan, N.; Cagney, G.; Mai, D.; Greenblatt, J.; et al. Navigating the chaperone network: An integrative map of physical and genetic interactions mediated by the Hsp90 chaperone. Cell 2005, 120, 715–727. [Google Scholar] [CrossRef] [PubMed]
- Csermely, P.; Schnaider, T.; Soti, C.; Prohaszka, Z.; Nardai, G. The 90-kDa molecular chaperone family: Structure, function, and clinical applications. A comprehensive review. Pharmacol. Ther. 1998, 79, 129–168. [Google Scholar] [CrossRef]
- Sreedhar, A.S.; Kalmar, E.; Csermely, P.; Shen, Y.F. Hsp90 isoforms: Functions, expression and clinical importance. FEBS Lett. 2004, 562, 11–15. [Google Scholar] [CrossRef]
- Echtenkamp, F.J.; Freeman, B.C. Expanding the cellular molecular chaperone network through the ubiquitous cochaperones. Biochim. Biophys. Acta 2012, 1823, 668–673. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Quadroni, M.; Potts, A.; Waridel, P. Hsp90 inhibition induces both protein-specific and global changes in the ubiquitinome. J. Proteom. 2015, 29, 120–215. [Google Scholar] [CrossRef] [PubMed]
- Young, J.C.; Moarefi, I.; Hartl, F.U. Hsp90: A specialized but essential protein-folding tool. J. Cell Biol. 2001, 154, 267–273. [Google Scholar] [CrossRef] [PubMed]
- Catlett, M.G.; Kaplan, K.B. Sgt1p is a unique co-chaperone that acts as a client adaptor to link Hsp90 to Skp1p. J. Biol. Chem. 2006, 281, 33739–33748. [Google Scholar] [CrossRef] [PubMed]
- Kimura, Y.; Rutherford, S.L.; Miyata, Y.; Yahara, I.; Freeman, B.C.; Yue, L.; Morimoto, R.I.; Lindquist, S. Cdc37 is a molecular chaperone with specific functions in signal transduction. Genes Dev. 1997, 11, 1775–1785. [Google Scholar] [CrossRef] [PubMed]
- Echtenkamp, F.J.; Zelin, E.; Oxelmark, E.; Woo, J.I.; Andrews, B.J.; Garabedian, M.; Freeman, B.C. Global functional map of the p23 molecular chaperone reveals an extensive cellular network. Mol. Cell 2011, 43, 229–241. [Google Scholar] [CrossRef] [PubMed]
- Soroka, J.; Wandinger, S.K.; Mausbacher, N.; Schreiber, T.; Richter, K.; Daub, H.; Buchner, J. Conformational switching of the molecular chaperone Hsp90 via regulated phosphorylation. Mol. Cell 2012, 45, 517–528. [Google Scholar] [CrossRef] [PubMed]
- Pratt, W.B.; Dittmar, K.D. Studies with Purified Chaperones Advance the Understanding of the Mechanism of Glucocorticoid Receptor-hsp90 Heterocomplex Assembly. Trends Endocrinol. Metab. 1998, 9, 244–252. [Google Scholar] [CrossRef]
- Workman, P. Altered states: Selectively drugging the Hsp90 cancer chaperone. Trends Mol. Med. 2004, 10, 47–51. [Google Scholar] [CrossRef] [PubMed]
- Iannotti, A.M.; Rabideau, D.A.; Dougherty, J.J. Characterization of purified avian 90,000-Da heat shock protein. Arch. Biochem. Biophys. 1988, 264, 54–60. [Google Scholar] [CrossRef]
- Lees-Miller, S.P.; Anderson, C.W. The human double-stranded DNA-activated protein kinase phosphorylates the 90-kDa heat-shock protein, hsp90 alpha at two NH2-terminal threonine residues. J. Biol. Chem. 1989, 264, 17275–17280. [Google Scholar] [PubMed]
- Welch, W.J.; Feramisco, J.R. Purification of the major mammalian heat shock proteins. J. Biol. Chem. 1982, 257, 14949–14959. [Google Scholar] [PubMed]
- Welch, W.J. The role of heat-shock proteins as molecular chaperones. Curr. Opin. Cell Biol. 1991, 3, 1033–1038. [Google Scholar] [CrossRef]
- Jhaveri, K.; Ochiana, S.O.; Dunphy, M.P.; Gerecitano, J.F.; Corben, A.D.; Peter, R.I.; Janjigian, Y.Y.; Gomes-DaGama, E.M.; Koren, J., III; Modi, S.; et al. Heat shock protein 90 inhibitors in the treatment of cancer: Current status and future directions. Expert. Opin. Investig. Drugs 2014, 23, 611–628. [Google Scholar] [CrossRef] [PubMed]
- Proia, D.A.; Kaufmann, G.F. Targeting heat-shock protein 90 (Hsp90) as a complementary strategy to immune checkpoint blockade for cancer therapy. Cancer Immunol. Res. 2015, 3, 583–589. [Google Scholar] [CrossRef] [PubMed]
- Bhat, R.; Tummalapalli, S.R.; Rotella, D.P. Progress in the discovery and development of heat shock protein 90 (Hsp90) inhibitors. J. Med. Chem. 2014, 57, 8718–8728. [Google Scholar] [CrossRef] [PubMed]
- Jhaveri, K.; Taldone, T.; Modi, S.; Chiosis, G. Advances in the clinical development of heat shock protein 90 (Hsp90) inhibitors in cancers. Biochim. Biophys. Acta 2012, 1823, 742–755. [Google Scholar] [CrossRef] [PubMed]
- Hahn, J.S. The Hsp90 chaperone machinery: From structure to drug development. BMB. Rep. 2009, 42, 623–630. [Google Scholar] [CrossRef] [PubMed]
- Hong, D.S.; Banerji, U.; Tavana, B.; George, G.C.; Aaron, J.; Kurzrock, R. Targeting the molecular chaperone heat shock protein 90 (HSP90): Lessons learned and future directions. Cancer Treat. Rev. 2013, 39, 375–387. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Buchner, J. Structure, function and regulation of the Hsp90 machinery. Biomed. J. 2013, 36, 106–117. [Google Scholar] [PubMed]
- Lianos, G.D.; Alexiou, G.A.; Mangano, A.; Mangano, A.; Rausei, S.; Boni, L.; Dionigi, G.; Roukos, D.H. The role of heat shock proteins in cancer. Cancer Lett. 2015, 360, 114–118. [Google Scholar] [CrossRef] [PubMed]
- Luo, W.; Rodina, A.; Chiosis, G. Heat shock protein 90: Translation from cancer to Alzheimer’s disease treatment? BMC Neurosci. 2008, 9. [Google Scholar] [CrossRef] [PubMed]
- Paul, S.; Mahanta, S. Association of heat-shock proteins in various neurodegenerative disorders: Is it a master key to open the therapeutic door? Mol. Cell. Biochem. 2014, 386, 45–61. [Google Scholar] [CrossRef] [PubMed]
- Whitesell, L.; Lindquist, S.L. HSP90 and the chaperoning of cancer. Nat. Rev. Cancer 2005, 5, 761–772. [Google Scholar] [CrossRef] [PubMed]
- Yeyati, P.L.; van Heyningen, V. Incapacitating the evolutionary capacitor: Hsp90 modulation of disease. Curr. Opin. Genet. Dev. 2008, 18, 264–272. [Google Scholar] [CrossRef] [PubMed]
- Camphausen, K.; Tofilon, P.J. Inhibition of Hsp90: A multitarget approach to radiosensitization. Clin. Cancer Res. 2007, 13, 4326–4330. [Google Scholar] [CrossRef] [PubMed]
- Kamal, A.; Thao, L.; Sensintaffar, J.; Zhang, L.; Boehm, M.F.; Fritz, L.C.; Burrows, F.J. A high-affinity conformation of Hsp90 confers tumour selectivity on Hsp90 inhibitors. Nature 2003, 425, 407–410. [Google Scholar] [CrossRef] [PubMed]
- Kirschke, E.; Goswami, D.; Southworth, D.; Griffin, P.R.; Agard, D.A. Glucocorticoid receptor function regulated by coordinated action of the Hsp90 and Hsp70 chaperone cycles. Cell 2014, 157, 1685–1697. [Google Scholar] [CrossRef] [PubMed]
- Neckers, L.; Ivy, S.P. Heat shock protein 90. Curr. Opin. Oncol. 2003, 15, 419–424. [Google Scholar] [CrossRef] [PubMed]
- Sangster, T.A.; Lindquist, S.; Queitsch, C. Under cover: Causes, effects and implications of Hsp90-mediated genetic capacitance. Bioessays 2004, 26, 348–362. [Google Scholar] [CrossRef] [PubMed]
- Segnitz, B.; Gehring, U. The function of steroid hormone receptors is inhibited by the hsp90-specific compound geldanamycin. J. Biol. Chem. 1997, 272, 18694–18701. [Google Scholar] [CrossRef] [PubMed]
- Sharp, S.; Workman, P. Inhibitors of the HSP90 molecular chaperone: Current status. Adv. Cancer Res. 2006, 95, 323–348. [Google Scholar] [PubMed]
- Neckers, L.; Workman, P. Hsp90 molecular chaperone inhibitors: Are we there yet? Clin. Cancer Res. 2012, 18, 64–76. [Google Scholar] [CrossRef] [PubMed]
- Chiosis, G.; Neckers, L. Tumor selectivity of Hsp90 inhibitors: The explanation remains elusive. ACS Chem. Biol. 2006, 1, 279–284. [Google Scholar] [CrossRef] [PubMed]
- Ferrarini, M.; Heltai, S.; Zocchi, M.R.; Rugarli, C. Unusual expression and localization of heat-shock proteins in human tumor cells. Int. J. Cancer 1992, 51, 613–619. [Google Scholar] [CrossRef] [PubMed]
- Calderwood, S.K.; Khaleque, M.A.; Sawyer, D.B.; Ciocca, D.R. Heat shock proteins in cancer: Chaperones of tumorigenesis. Trends Biochem. Sci. 2006, 31, 164–172. [Google Scholar] [CrossRef] [PubMed]
- Chiosis, G.; Vilenchik, M.; Kim, J.; Solit, D. Hsp90: The vulnerable chaperone. Drug Discov. Today 2004, 9, 881–888. [Google Scholar] [CrossRef]
- Fionda, C.; Soriani, A.; Malgarini, G.; Iannitto, M.L.; Santoni, A.; Cippitelli, M. Heat shock protein-90 inhibitors increase MHC class I-related chain A and B ligand expression on multiple myeloma cells and their ability to trigger NK cell degranulation. J. Immunol. 2009, 183, 4385–4394. [Google Scholar] [CrossRef] [PubMed]
- Alexiou, G.A.; Vartholomatos, G.; Stefanaki, K.; Patereli, A.; Dova, L.; Karamoutsios, A.; Lallas, G.; Sfakianos, G.; Moschovi, M.; Prodromou, N. Expression of heat shock proteins in medulloblastoma. J. Neurosurg. Pediatr. 2013, 12, 452–457. [Google Scholar] [CrossRef] [PubMed]
- Zagouri, F.; Sergentanis, T.N.; Nonni, A.; Papadimitriou, C.A.; Michalopoulos, N.V.; Domeyer, P.; Theodoropoulos, G.; Lazaris, A.; Patsouris, E.; Zogafos, E.; et al. Hsp90 in the continuum of breast ductal carcinogenesis: Evaluation in precursors, preinvasive and ductal carcinoma lesions. BMC Cancer 2010. [Google Scholar] [CrossRef] [PubMed]
- Nagaraju, G.P.; Long, T.E.; Park, W.; Landry, J.C.; Taliaferro-Smith, L.; Farris, A.B.; Diaz, R.; El Rayes, B.F. Heat shock protein 90 promotes epithelial to mesenchymal transition, invasion, and migration in colorectal cancer. Mol. Carcinog. 2014. [Google Scholar] [CrossRef] [PubMed]
- Annamalai, B.; Liu, X.; Gopal, U.; Isaacs, J.S. Hsp90 is an essential regulator of EphA2 receptor stability and signaling: Implications for cancer cell migration and metastasis. Mol. Cancer Res. 2009, 7, 1021–1032. [Google Scholar] [CrossRef] [PubMed]
- Kawabe, M.; Mandic, M.; Taylor, J.L.; Vasquez, C.A.; Wesa, A.K.; Neckers, L.M.; Storkus, W.J. Heat shock protein 90 inhibitor 17-dimethylaminoethylamino-17-demethoxygeldanamycin enhances EphA2+ tumor cell recognition by specific CD8+ T cells. Cancer Res. 2009, 69, 6995–7003. [Google Scholar] [CrossRef] [PubMed]
- Bisht, K.S.; Bradbury, C.M.; Mattson, D.; Kaushal, A.; Sowers, A.; Markovina, S.; Ortiz, K.L.; Sieck, L.K.; Isaacs, J.S.; Brechbiel, M.W.; et al. Geldanamycin and 17-allylamino-17-demethoxygeldanamycin potentiate the in vitro and in vivo radiation response of cervical tumor cells via the heat shock protein 90-mediated intracellular signaling and cytotoxicity. Cancer Res. 2003, 63, 8984–8995. [Google Scholar] [PubMed]
- Bull, E.E.; Dote, H.; Brady, K.J.; Burgan, W.E.; Carter, D.J.; Cerra, M.A.; Oswald, K.A.; Hollingshead, M.G.; Camphausen, K.; Tofilon, P.J. Enhanced tumor cell radiosensitivity and abrogation of G2 and S phase arrest by the Hsp90 inhibitor 17-(dimethylaminoethylamino)-17-demethoxygeldanamycin. Clin. Cancer Res. 2004, 10, 8077–8084. [Google Scholar] [CrossRef] [PubMed]
- Hirakawa, Y.; Suzuki, S.; Archibald, J.M.; Keeling, P.J.; Ishida, K. Overexpression of molecular chaperone genes in nucleomorph genomes. Mol. Biol. Evol. 2014, 31, 1437–1443. [Google Scholar] [CrossRef] [PubMed]
- Koll, T.T.; Feis, S.S.; Wright, M.H.; Teniola, M.M.; Richardson, M.M.; Robles, A.I.; Bradsher, J.; Capala, J.; Varticovski, L. HSP90 inhibitor, DMAG, synergizes with radiation of lung cancer cells by interfering with base excision and ATM-mediated DNA repair. Mol. Cancer Ther. 2008, 7, 1985–1992. [Google Scholar] [CrossRef] [PubMed]
- Noguchi, M.; Yu, D.; Hirayama, R.; Ninomiya, Y.; Sekine, E.; Kubota, N.; Ando, K.; Okayasu, R. Inhibition of homologous recombination repair in irradiated tumor cells pretreated with Hsp90 inhibitor 17-allylamino-17-demethoxygeldanamycin. Biochem. Biophys. Res. Commun. 2006, 351, 658–663. [Google Scholar] [CrossRef] [PubMed]
- Russell, J.S.; Burgan, W.; Oswald, K.A.; Camphausen, K.; Tofilon, P.J. Enhanced cell killing induced by the combination of radiation and the heat shock protein 90 inhibitor 17-allylamino-17- demethoxygeldanamycin: A multitarget approach to radiosensitization. Clin. Cancer Res. 2003, 9, 3749–3755. [Google Scholar] [PubMed]
- Workman, P.; Burrows, F.; Neckers, L.; Rosen, N. Drugging the cancer chaperone HSP90: Combinatorial therapeutic exploitation of oncogene addiction and tumor stress. Ann. NY Acad. Sci. 2007, 1113, 202–216. [Google Scholar] [CrossRef] [PubMed]
- Dote, H.; Burgan, W.E.; Camphausen, K.; Tofilon, P.J. Inhibition of hsp90 compromises the DNA damage response to radiation. Cancer Res. 2006, 66, 9211–9220. [Google Scholar] [CrossRef] [PubMed]
- Han, F.F.; Li, L.; Shang, B.Y.; Shao, R.G.; Zhen, Y.S. Hsp90 inhibitor geldanamycin enhances the antitumor efficacy of enediyne lidamycin in association with reduced DNA damage repair. Asian Pac. J. Cancer Prev. 2014, 15, 7043–7048. [Google Scholar] [CrossRef] [PubMed]
- Ri, M.; Iida, S.; Nakashima, T.; Miyazaki, H.; Mori, F.; Ito, A.; Inagaki, A.; Kusumoto, S.; Ishida, T.; Komatsu, H.; et al. Bortezomib-resistant myeloma cell lines: a Role for mutated PSMB5 in preventing the accumulation of unfolded proteins and fatal ER stress. Leukemia 2010, 24, 1506–1512. [Google Scholar] [CrossRef] [PubMed]
- Robles, A.I.; Wright, M.H.; Gandhi, B.; Feis, S.S.; Hanigan, C.L.; Wiestner, A.; Varticovski, L. Schedule-dependent synergy between the heat shock protein 90 inhibitor 17-(dimethylaminoethylamino)-17-demethoxygeldanamycin and doxorubicin restores apoptosis to p53-mutant lymphoma cell lines. Clin. Cancer Res. 2006, 12, 6547–6556. [Google Scholar] [CrossRef] [PubMed]
- Dehner, A.; Furrer, J.; Richter, K.; Schuster, I.; Buchner, J.; Kessler, H. NMR chemical shift perturbation study of the N-terminal domain of Hsp90 upon binding of ADP, AMP-PNP, geldanamycin, and radicicol. Chembiochem 2003, 4, 870–877. [Google Scholar] [CrossRef] [PubMed]
- Roe, S.M.; Prodromou, C.; O’Brien, R.; Ladbury, J.E.; Piper, P.W.; Pearl, L.H. Structural basis for inhibition of the Hsp90 molecular chaperone by the antitumor antibiotics radicicol and geldanamycin. J. Med. Chem. 1999, 42, 260–266. [Google Scholar] [CrossRef] [PubMed]
- Seo, Y.H. Small Molecule Inhibitors to Disrupt Protein-protein Interactions of Heat Shock Protein 90 Chaperone Machinery. J. Cancer Prev. 2015, 20, 5–11. [Google Scholar] [CrossRef] [PubMed]
- Stebbins, C.E.; Russo, A.A.; Schneider, C.; Rosen, N.; Hartl, F.U.; Pavletich, N.P. Crystal structure of an Hsp90-geldanamycin complex: Targeting of a protein chaperone by an antitumor agent. Cell 1997, 89, 239–250. [Google Scholar] [CrossRef]
- Jackson, S.E. Hsp90: Structure and function. Top. Curr. Chem. 2013, 328, 155–240. [Google Scholar] [PubMed]
- Supko, J.G.; Hickman, R.L.; Grever, M.R.; Malspeis, L. Preclinical pharmacologic evaluation of geldanamycin as an antitumor agent. Cancer Chemother. Pharmacol. 1995, 36, 305–315. [Google Scholar] [CrossRef] [PubMed]
- Banerji, U.; O’Donnell, A.; Scurr, M.; Pacey, S.; Stapleton, S.; Asad, Y.; Simmons, L.; Maloney, A.; Raynaud, F.; Campbell, M.; et al. Phase I pharmacokinetic and pharmacodynamic study of 17-allylamino, 17-demethoxygeldanamycin in patients with advanced malignancies. J. Clin. Oncol. 2005, 23, 4152–4161. [Google Scholar] [CrossRef] [PubMed]
- Ronnen, E.A.; Kondagunta, G.V.; Ishill, N.; Sweeney, S.M.; Deluca, J.K.; Schwartz, L.; Bacik, J.; Motzer, R.J. A phase II trial of 17-(Allylamino)-17-demethoxygeldanamycin in patients with papillary and clear cell renal cell carcinoma. Investig. New Drugs 2006, 24, 543–546. [Google Scholar] [CrossRef] [PubMed]
- Smith, V.; Sausville, E.A.; Camalier, R.F.; Fiebig, H.H.; Burger, A.M. Comparison of 17-dimethylaminoethylamino-17-demethoxy-geldanamycin (17DMAG) and 17-allylamino-17-demethoxygeldanamycin (17AAG) in vitro: Effects on Hsp90 and client proteins in melanoma models. Cancer Chemother. Pharmacol. 2005, 56, 126–137. [Google Scholar] [CrossRef] [PubMed]
- Heath, E.I.; Gaskins, M.; Pitot, H.C.; Pili, R.; Tan, W.; Marschke, R.; Liu, G.; Hillman, D.; Sarkar, F.; Sheng, S.; et al. A phase II trial of 17-allylamino-17-demethoxygeldanamycin in patients with hormone-refractory metastatic prostate cancer. Clin. Prostate Cancer 2005, 4, 138–141. [Google Scholar] [CrossRef] [PubMed]
- Richardson, P.G.; Badros, A.Z.; Jagannath, S.; Tarantolo, S.; Wolf, J.L.; Albitar, M.; Berman, D.; Messina, M.; Anderson, K.C. Tanespimycin with bortezomib: Activity in relapsed/refractory patients with multiple myeloma. Br. J. Haematol. 2010, 150, 428–437. [Google Scholar] [CrossRef] [PubMed]
- Pacey, S.; Gore, M.; Chao, D.; Banerji, U.; Larkin, J.; Sarker, S.; Owen, K.; Asad, Y.; Raynaud, F.; Walton, M.; Judson, I.; Workman, P.; Eisen, T. A Phase II trial of 17-allylamino, 17-demethoxygeldanamycin (17-AAG, tanespimycin) in patients with metastatic melanoma. Investig. New Drugs 2012, 30, 341–349. [Google Scholar] [CrossRef] [PubMed]
- Oki, Y.; Copeland, A.; Romaguera, J.; Fayad, L.; Fanale, M.; Faria, S.C.; Medeiros, L.J.; Ivy, P.; Younes, A. Clinical experience with the heat shock protein-90 inhibitor, tanespimycin, in patients with relapsed lymphoma. Leuk. Lymphoma 2012, 53, 990–992. [Google Scholar] [CrossRef] [PubMed]
- Kitson, R.R.; Chang, C.H.; Xiong, R.; Williams, H.E.; Davis, A.L.; Lewis, W.; Dehn, D.L.; Siegel, D.; Roe, S.M.; Prodromou, C.; et al. Synthesis of 19-substituted geldanamycins with altered conformations and their binding to heat shock protein Hsp90. Nat. Chem. 2013, 5, 307–314. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Zheng, Z.; Zhao, Y.; Zhang, T.; Gu, X.; Yang, W. The heat shock protein 90 inhibitor 17-AAG suppresses growth and induces apoptosis in human cholangiocarcinoma cells. Clin. Exp. Med. 2013, 13, 323–328. [Google Scholar] [CrossRef] [PubMed]
- Hollingshead, M.; Alley, M.; Burger, A.M.; Borgel, S.; Pacula-Cox, C.; Fiebig, H.H.; Sausville, E.A. In vivo antitumor efficacy of 17-DMAG (17-dimethylaminoethylamino-17-demethoxygeldanamycin hydrochloride), a water-soluble geldanamycin derivative. Cancer Chemother. Pharmacol. 2005, 56, 115–125. [Google Scholar] [CrossRef] [PubMed]
- Onuoha, S.C.; Mukund, S.R.; Coulstock, E.T.; Sengerova, B.; Shaw, J.; McLaughlin, S.H.; Jackson, S.E. Mechanistic studies on Hsp90 inhibition by ansamycin derivatives. J. Mol. Biol. 2007, 372, 287–297. [Google Scholar] [CrossRef] [PubMed]
- Pacey, S.; Wilson, R.H.; Walton, M.; Eatock, M.M.; Hardcastle, A.; Zetterlund, A.; Arkenau, H.T.; Moreno-Farre, J.; Banerji, U.; Roels, B.; et al. A phase I study of the heat shock protein 90 inhibitor alvespimycin (17-DMAG) given intravenously to patients with advanced solid tumors. Clin. Cancer Res. 2011, 17, 1561–1570. [Google Scholar] [CrossRef] [PubMed]
- He, H.; Zatorska, D.; Kim, J.; Aguirre, J.; Llauger, L.; She, Y.; Wu, N.; Immormino, R.M.; Gewirth, D.T.; Chiosis, G. Identification of potent water soluble purine-scaffold inhibitors of the heat shock protein 90. J. Med. Chem. 2006, 49, 381–390. [Google Scholar] [CrossRef] [PubMed]
- Usmani, S.Z.; Bona, R.D.; Chiosis, G.; Li, Z. The anti-myeloma activity of a novel purine scaffold HSP90 inhibitor PU-H71 is via inhibition of both HSP90A and HSP90B1. J. Hematol. Oncol. 2010, 3. [Google Scholar] [CrossRef] [PubMed]
- Ambati, S.R.; Lopes, E.C.; Kosugi, K.; Mony, U.; Zehir, A.; Shah, S.K.; Taldone, T.; Moreira, A.L.; Meyers, P.A.; Chiosis, G.; et al. Pre-clinical efficacy of PU-H71, a novel HSP90 inhibitor, alone and in combination with bortezomib in Ewing sarcoma. Mol. Oncol. 2014, 8, 323–336. [Google Scholar] [CrossRef] [PubMed]
- Brough, P.A.; Aherne, W.; Barril, X.; Borgognoni, J.; Boxall, K.; Cansfield, J.E.; Cheung, K.M.; Collins, I.; Davies, N.G.; Drysdale, M.J.; et al. 4,5-diarylisoxazole Hsp90 chaperone inhibitors: Potential therapeutic agents for the treatment of cancer. J. Med. Chem. 2008, 51, 196–218. [Google Scholar] [CrossRef] [PubMed]
- Eccles, S.A.; Massey, A.; Raynaud, F.I.; Sharp, S.Y.; Box, G.; Valenti, M.; Patterson, L.; de Haven Brandon, A.; Gowan, S.; Boxall, F.; et al. NVP-AUY922: A novel heat shock protein 90 inhibitor active against xenograft tumor growth, angiogenesis, and metastasis. Cancer Res. 2008, 68, 2850–2860. [Google Scholar] [CrossRef] [PubMed]
- Ueno, T.; Tsukuda, K.; Toyooka, S.; Ando, M.; Takaoka, M.; Soh, J.; Asano, H.; Maki, Y.; Muraoka, T.; Tanaka, N.; et al. Strong anti-tumor effect of NVP-AUY922, a novel Hsp90 inhibitor, on non-small cell lung cancer. Lung Cancer 2012, 76, 26–31. [Google Scholar] [CrossRef] [PubMed]
- Ying, W.; Du, Z.; Sun, L.; Foley, K.P.; Proia, D.A.; Blackman, R.K.; Zhou, D.; Inoue, T.; Tatsuta, N.; Sang, J.; et al. Ganetespib, a unique triazolone-containing Hsp90 inhibitor, exhibits potent antitumor activity and a superior safety profile for cancer therapy. Mol. Cancer Ther. 2012, 11, 475–484. [Google Scholar] [CrossRef] [PubMed]
- Gomez-Casal, R.; Bhattacharya, C.; Epperly, M.W.; Basse, P.H.; Wang, H.; Wang, X.; Proia, D.A.; Greenberger, J.S.; Socinski, M.A.; Levina, V. The HSP90 Inhibitor Ganetespib Radiosensitizes Human Lung Adenocarcinoma Cells. Cancers 2015, 7, 876–907. [Google Scholar] [CrossRef] [PubMed]
- He, S.; Smith, D.L.; Sequeira, M.; Sang, J.; Bates, R.C.; Proia, D.A. The HSP90 inhibitor ganetespib has chemosensitizer and radiosensitizer activity in colorectal cancer. Investig. New Drugs. 2014, 32, 577–586. [Google Scholar] [CrossRef] [PubMed]
- Proia, D.A.; Bates, R.C. Ganetespib and HSP90: Translating preclinical hypotheses into clinical promise. Cancer. Res. 2014, 74, 1294–1300. [Google Scholar] [CrossRef] [PubMed]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera: A visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef] [PubMed]
- Shiau, A.K.; Harris, S.F.; Southworth, D.R.; Agard, D.A. Structural analysis of E. coli hsp90 reveals dramatic nucleotide-dependent conformational rearrangements. Cell 2006, 127, 329–340. [Google Scholar] [CrossRef] [PubMed]
- Ali, M.M.; Roe, S.M.; Vaughan, C.K.; Meyer, P.; Panaretou, B.; Piper, P.W.; Prodromou, C.; Pearl, L.H. Crystal structure of an Hsp90-nucleotide-p23/Sba1 closed chaperone complex. Nature 2006, 440, 1013–1017. [Google Scholar] [CrossRef] [PubMed]
- Bridge, G.; Rashid, S.; Martin, S.A. DNA mismatch repair and oxidative DNA damage: Implications for cancer biology and treatment. Cancers 2014, 6, 1597–1614. [Google Scholar] [CrossRef] [PubMed]
- Chistiakov, D.A.; Voronova, N.V.; Chistiakov, P.A. Genetic variations in DNA repair genes, radiosensitivity to cancer and susceptibility to acute tissue reactions in radiotherapy-treated cancer patients. Acta Oncol. 2008, 47, 809–824. [Google Scholar] [CrossRef] [PubMed]
- Hoeijmakers, J.H. Genome maintenance mechanisms for preventing cancer. Nature 2001, 411, 366–374. [Google Scholar] [CrossRef] [PubMed]
- Dezwaan, D.C.; Freeman, B.C. HSP90: The rosetta stone for cellular protein dynamics? Cell Cycle 2008, 7, 1006–1012. [Google Scholar] [CrossRef] [PubMed]
- Quanz, M.; Herbette, A.; Sayarath, M.; de Koning, L.; Dubois, T.; Sun, J.S.; Dutreix, M. Heat shock protein 90α (Hsp90α) is phosphorylated in response to DNA damage and accumulates in repair foci. J. Biol. Chem. 2012, 287, 8803–8815. [Google Scholar] [CrossRef] [PubMed]
- Solier, S.; Kohn, K.W.; Scroggins, B.; Xu, W.; Trepel, J.; Neckers, L.; Pommier, Y. Heat shock protein 90alpha (HSP90α), a substrate and chaperone of DNA-PK necessary for the apoptotic response. Proc. Natl. Acad. Sci. USA 2012, 109, 12866–12872. [Google Scholar] [CrossRef] [PubMed]
- Kumar, R.; Horikoshi, N.; Singh, M.; Gupta, A.; Misra, H.S.; Albuquerque, K.; Hunt, C.R.; Pandita, T.K. Chromatin modifications and the DNA damage response to ionizing radiation. Front. Oncol. 2012, 2. [Google Scholar] [CrossRef] [PubMed]
- Bassing, C.H.; Suh, H.; Ferguson, D.O.; Chua, K.F.; Manis, J.; Eckersdorff, M.; Gleason, M.; Bronson, R.; Lee, C.; Alt, F.W. Histone H2AX: A dosage-dependent suppressor of oncogenic translocations and tumors. Cell 2003, 114, 359–370. [Google Scholar] [CrossRef]
- Scully, R.; Xie, A. Double strand break repair functions of histone H2AX. Mutat. Res. 2013, 750, 5–14. [Google Scholar] [CrossRef] [PubMed]
- Kanaar, R.; Hoeijmakers, J.H.; van Gent, D.C. Molecular mechanisms of DNA double strand break repair. Trends Cell Biol. 1998, 8, 483–489. [Google Scholar] [CrossRef]
- Lieber, M.R.; Ma, Y.; Pannicke, U.; Schwarz, K. Mechanism and regulation of human non-homologous DNA end-joining. Nat. Rev. Mol. Cell Biol. 2003, 4, 712–720. [Google Scholar] [CrossRef] [PubMed]
- O’Driscoll, M.; Jeggo, P.A. The role of double-strand break repair—Insights from human genetics. Nat. Rev. Genet. 2006, 7, 45–54. [Google Scholar] [CrossRef] [PubMed]
- Van Gent, D.C.; Hoeijmakers, J.H.; Kanaar, R. Chromosomal stability and the DNA double-stranded break connection. Nat. Rev. Genet. 2001, 2, 196–206. [Google Scholar] [CrossRef] [PubMed]
- Lemaitre, C.; Soutoglou, E. Double strand break (DSB) repair in heterochromatin and heterochromatin proteins in DSB repair. DNA Repair 2014, 19, 163–168. [Google Scholar] [CrossRef] [PubMed]
- Pierce, A.J.; Jasin, M. NHEJ deficiency and disease. Mol. Cell 2001, 8, 1160–1161. [Google Scholar] [CrossRef]
- Rothkamm, K.; Kruger, I.; Thompson, L.H.; Lobrich, M. Pathways of DNA double-strand break repair during the mammalian cell cycle. Mol. Cell. Biol. 2003, 23, 5706–5715. [Google Scholar] [CrossRef] [PubMed]
- Chapman, J.R.; Taylor, M.R.; Boulton, S.J. Playing the end game: DNA double-strand break repair pathway choice. Mol. Cell 2012, 47, 497–510. [Google Scholar] [CrossRef] [PubMed]
- Lieber, M.R. The mechanism of double-strand DNA break repair by the nonhomologous DNA end-joining pathway. Annu. Rev. Biochem. 2010, 79, 181–211. [Google Scholar] [CrossRef] [PubMed]
- Goodarzi, A.A.; Jeggo, P.A. The repair and signaling responses to DNA double-strand breaks. Adv. Genet. 2013, 82, 1–45. [Google Scholar] [PubMed]
- Dynan, W.S.; Yoo, S. Interaction of Ku protein and DNA-dependent protein kinase catalytic subunit with nucleic acids. Nucleic Acids Res. 1998, 26, 1551–1559. [Google Scholar] [CrossRef] [PubMed]
- Ma, Y.; Pannicke, U.; Schwarz, K.; Lieber, M.R. Hairpin opening and overhang processing by an Artemis/DNA-dependent protein kinase complex in nonhomologous end joining and V(D)J recombination. Cell 2002, 108, 781–794. [Google Scholar] [CrossRef]
- Gupta, A.; Hunt, C.R.; Chakraborty, S.; Pandita, R.K.; Yordy, J.; Ramnarain, D.B.; Horikoshi, N.; Pandita, T.K. Role of 53BP1 in the regulation of DNA double-strand break repair pathway choice. Radiat. Res. 2014, 181, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Nimonkar, A.V.; Genschel, J.; Kinoshita, E.; Polaczek, P.; Campbell, J.L.; Wyman, C.; Modrich, P.; Kowalczykowski, S.C. BLM-DNA2-RPA-MRN and EXO1-BLM-RPA-MRN constitute two DNA end resection machineries for human DNA break repair. Genes Dev. 2011, 25, 350–362. [Google Scholar] [CrossRef] [PubMed]
- Di Masi, A.; Gullotta, F.; Cappadonna, V.; Leboffe, L.; Ascenzi, P. Cancer predisposing mutations in BRCT domains. IUBMB Life 2011, 63, 503–512. [Google Scholar] [CrossRef] [PubMed]
- Arlander, S.J.; Eapen, A.K.; Vroman, B.T.; McDonald, R.J.; Toft, D.O.; Karnitz, L.M. Hsp90 inhibition depletes Chk1 and sensitizes tumor cells to replication stress. J. Biol. Chem. 2003, 278, 52572–52577. [Google Scholar] [CrossRef] [PubMed]
- Fang, Q.; Inanc, B.; Schamus, S.; Wang, X.H.; Wei, L.; Brown, A.R.; Svilar, D.; Sugrue, K.F.; Goellner, E.M.; Zeng, X.; et al. HSP90 regulates DNA repair via the interaction between XRCC1 and DNA polymerase beta. Nat. Commun. 2014, 5. [Google Scholar] [CrossRef] [PubMed]
- Furuta, T.; Takemura, H.; Liao, Z.Y.; Aune, G.J.; Redon, C.; Sedelnikova, O.A.; Pilch, D.R.; Rogakou, E.P.; Celeste, A.; Chen, H.T.; et al. Phosphorylation of histone H2AX and activation of Mre11, Rad50, and Nbs1 in response to replication-dependent DNA double-strand breaks induced by mammalian DNA topoisomerase I cleavage complexes. J. Biol. Chem. 2003, 278, 20303–20312. [Google Scholar] [CrossRef] [PubMed]
- Schnaider, T.; Oikarinen, J.; Ishiwatari-Hayasaka, H.; Yahara, I.; Csermely, P. Interactions of Hsp90 with histones and related peptides. Life Sci. 1999, 65, 2417–2426. [Google Scholar] [CrossRef]
- Smogorzewska, A.; Matsuoka, S.; Vinciguerra, P.; McDonald, E.R., III; Hurov, K.E.; Luo, J.; Ballif, B.A.; Gygi, S.P.; Hofmann, K.; D’Andrea, A.D.; et al. Identification of the FANCI protein, a monoubiquitinated FANCD2 paralog required for DNA repair. Cell 2007, 129, 289–301. [Google Scholar] [CrossRef] [PubMed]
- Stecklein, S.R.; Kumaraswamy, E.; Behbod, F.; Wang, W.; Chaguturu, V.; Harlan-Williams, L.M.; Jensen, R.A. BRCA1 and HSP90 cooperate in homologous and non-homologous DNA double-strand-break repair and G2/M checkpoint activation. Proc. Natl. Acad. Sci. USA 2012, 109, 13650–13655. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Zhang, X.; Wang, P.; Yu, X.; Essers, J.; Chen, D.; Kanaar, R.; Takeda, S.; Wang, Y. Characteristics of DNA-binding proteins determine the biological sensitivity to high-linear energy transfer radiation. Nucleic Acids Res. 2010, 38, 3245–3251. [Google Scholar] [CrossRef] [PubMed]
- Whitesell, L.; Mimnaugh, E.G.; de Costa, B.; Myers, C.E.; Neckers, L.M. Inhibition of heat shock protein HSP90-pp60v-src heteroprotein complex formation by benzoquinone ansamycins: Essential role for stress proteins in oncogenic transformation. Proc. Natl. Acad. Sci. USA 1994, 91, 8324–8328. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Zhang, T.; Schwartz, S.J.; Sun, D. New developments in Hsp90 inhibitors as anti-cancer therapeutics: Mechanisms, clinical perspective and more potential. Drug Resist. Updat. 2009, 12, 17–27. [Google Scholar] [CrossRef] [PubMed]
- Pacey, S.; Banerji, U.; Judson, I.; Workman, P. Hsp90 inhibitors in the clinic. Handb. Exp. Pharmacol. 2006, 331–358. [Google Scholar]
- Chiosis, G. Targeting chaperones in transformed systems—A focus on Hsp90 and cancer. Expert. Opin. Ther. Targets 2006, 10, 37–50. [Google Scholar] [CrossRef] [PubMed]
- Solit, D.B.; Chiosis, G. Development and application of Hsp90 inhibitors. Drug Discov. Today 2008, 13, 38–43. [Google Scholar] [CrossRef] [PubMed]
- Cheung, K.M.; Matthews, T.P.; James, K.; Rowlands, M.G.; Boxall, K.J.; Sharp, S.Y.; Maloney, A.; Roe, S.M.; Prodromou, C.; Pearl, L.H.; et al. The identification, synthesis, protein crystal structure and in vitro biochemical evaluation of a new 3,4-diarylpyrazole class of Hsp90 inhibitors. Bioorg. Med. Chem. Lett. 2005, 15, 3338–3343. [Google Scholar] [CrossRef] [PubMed]
- McDonald, E.; Jones, K.; Brough, P.A.; Drysdale, M.J.; Workman, P. Discovery and development of pyrazole-scaffold Hsp90 inhibitors. Curr. Top. Med. Chem. 2006, 6, 1193–1203. [Google Scholar] [CrossRef] [PubMed]
- Johnson, J.L. Evolution and function of diverse Hsp90 homologs and cochaperone proteins. Biochim. Biophys. Acta 2012, 1823, 607–613. [Google Scholar] [CrossRef] [PubMed]
- Ernst, A.; Anders, H.; Kapfhammer, H.; Orth, M.; Hennel, R.; Seidl, K.; Winssinger, N.; Belka, C.; Unkel, S.; Lauber, K. HSP90 inhibition as a means of radiosensitizing resistant, aggressive soft tissue sarcomas. Cancer Lett. 2015, 365, 211–222. [Google Scholar] [CrossRef] [PubMed]
- Falsone, S.F.; Gesslbauer, B.; Tirk, F.; Piccinini, A.M.; Kungl, A.J. A proteomic snapshot of the human heat shock protein 90 interactome. FEBS Lett. 2005, 579, 6350–6354. [Google Scholar] [CrossRef] [PubMed]
- Zaidi, S.; McLaughlin, M.; Bhide, S.A.; Eccles, S.A.; Workman, P.; Nutting, C.M.; Huddart, R.A.; Harrington, K.J. The HSP90 inhibitor NVP-AUY922 radiosensitizes by abrogation of homologous recombination resulting in mitotic entry with unresolved DNA damage. PLoS ONE 2012, 7, e35436. [Google Scholar] [CrossRef] [PubMed]
- Parameswaran, B.; Chiang, H.C.; Lu, Y.; Coates, J.; Deng, C.X.; Baer, R.; Lin, H.K.; Li, R.; Paull, T.T.; Hu, Y. Damage-induced BRCA1 phosphorylation by Chk2 contributes to the timing of end resection. Cell Cycle 2015, 14, 437–448. [Google Scholar] [CrossRef] [PubMed]
- Cortez, D.; Wang, Y.; Qin, J.; Elledge, S.J. Requirement of ATM-dependent phosphorylation of brca1 in the DNA damage response to double-strand breaks. Science 1999, 286, 1162–1166. [Google Scholar] [CrossRef] [PubMed]
- Yarden, R.I.; Pardo-Reoyo, S.; Sgagias, M.; Cowan, K.H.; Brody, L.C. BRCA1 regulates the G2/M checkpoint by activating Chk1 kinase upon DNA damage. Nat. Genet. 2002, 30, 285–289. [Google Scholar] [CrossRef] [PubMed]
- Bhattacharyya, A.; Ear, U.S.; Koller, B.H.; Weichselbaum, R.R.; Bishop, D.K. The breast cancer susceptibility gene BRCA1 is required for subnuclear assembly of Rad51 and survival following treatment with the DNA cross-linking agent cisplatin. J. Biol. Chem. 2000, 275, 23899–23903. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Nievera, C.J.; Lee, A.Y.; Wu, X. Cell cycle-dependent complex formation of BRCA1.CtIP.MRN is important for DNA double-strand break repair. J. Biol. Chem. 2008, 283, 7713–7720. [Google Scholar] [CrossRef] [PubMed]
- D’Andrea, A.D.; Grompe, M. The Fanconi anaemia/BRCA pathway. Nat .Rev. Cancer 2003, 3, 23–34. [Google Scholar] [CrossRef] [PubMed]
- Zhong, Q.; Chen, C.F.; Li, S.; Chen, Y.; Wang, C.C.; Xiao, J.; Chen, P.L.; Sharp, Z.D.; Lee, W.H. Association of BRCA1 with the hRad50-hMre11-p95 complex and the DNA damage response. Science 1999, 285, 747–750. [Google Scholar] [CrossRef] [PubMed]
- Scata, K.A.; El Deiry, W.S. p53, BRCA1 and breast Cancer chemoresistance. Adv. Exp. Med. Biol. 2007, 608, 70–86. [Google Scholar] [PubMed]
- Davies, A.A.; Masson, J.Y.; McIlwraith, M.J.; Stasiak, A.Z.; Stasiak, A.; Venkitaraman, A.R.; West, S.C. Role of BRCA2 in control of the RAD51 recombination and DNA repair protein. Mol. Cell 2001, 7, 273–282. [Google Scholar] [CrossRef]
- Segawa, T.; Fujii, Y.; Tanaka, A.; Bando, S.; Okayasu, R.; Ohnishi, K.; Kubota, N. Radiosensitization of human lung cancer cells by the novel purine-scaffold Hsp90 inhibitor, PU-H71. Int. J. Mol. Med. 2014, 33, 559–564. [Google Scholar] [PubMed]
- Dungey, F.A.; Caldecott, K.W.; Chalmers, A.J. Enhanced radiosensitization of human glioma cells by combining inhibition of poly(ADP-ribose) polymerase with inhibition of heat shock protein 90. Mol. Cancer Ther. 2009, 8, 2243–2254. [Google Scholar] [CrossRef] [PubMed]
- Yao, Q.; Weigel, B.; Kersey, J. Synergism between etoposide and 17-AAG in leukemia cells: Critical roles for Hsp90, FLT3, topoisomerase II, Chk1, and Rad51. Clin. Cancer Res. 2007, 13, 1591–1600. [Google Scholar] [CrossRef] [PubMed]
- Zhou, B.B.; Elledge, S.J. The DNA damage response: Putting checkpoints in perspective. Nature 2000, 408, 433–439. [Google Scholar] [PubMed]
- Dai, Y.; Grant, S. New insights into checkpoint kinase 1 in the DNA damage response signaling network. Clin. Cancer Res. 2010, 16, 376–383. [Google Scholar] [CrossRef] [PubMed]
- Oakley, G.G.; Patrick, S.M. Replication protein A: Directing traffic at the intersection of replication and repair. Fron. Biosci. 2010, 15, 883–900. [Google Scholar] [CrossRef]
- Gonzalez Besteiro, M.A.; Gottifredi, V. The fork and the kinase: A DNA replication tale from a CHK1 perspective. Mutat. Res. Rev. Mutat. Res. 2015, 763, 168–180. [Google Scholar] [CrossRef] [PubMed]
- Roos-Mattjus, P.; Hopkins, K.M.; Oestreich, A.J.; Vroman, B.T.; Johnson, K.L.; Naylor, S.; Lieberman, H.B.; Karnitz, L.M. Phosphorylation of human Rad9 is required for genotoxin-activated checkpoint signaling. J. Biol. Chem. 2003, 278, 24428–24437. [Google Scholar] [CrossRef] [PubMed]
- Zachos, G.; Rainey, M.D.; Gillespie, D.A. Chk1-deficient tumour cells are viable but exhibit multiple checkpoint and survival defects. EMBO J. 2003, 22, 713–723. [Google Scholar] [CrossRef] [PubMed]
- Dittmann, K.; Mayer, C.; Fehrenbacher, B.; Schaller, M.; Raju, U.; Milas, L.; Chen, D.J.; Kehlbach, R.; Rodemann, H.P. Radiation-induced epidermal growth factor receptor nuclear import is linked to activation of DNA-dependent protein kinase. J. Biol. Chem. 2005, 280, 31182–31189. [Google Scholar] [CrossRef] [PubMed]
- Bertrand, R.; Solary, E.; O’Connor, P.; Kohn, K.W.; Pommier, Y. Induction of a common pathway of apoptosis by staurosporine. Exp. Cell Res. 1994, 211, 314–321. [Google Scholar] [CrossRef] [PubMed]
- Strasser, A.; Jost, P.J.; Nagata, S. The many roles of FAS receptor signaling in the immune system. Immunity 2009, 30, 180–192. [Google Scholar] [CrossRef] [PubMed]
- Solier, S.; Pommier, Y. The apoptotic ring: A novel entity with phosphorylated histones H2AX and H2B and activated DNA damage response kinases. Cell Cycle 2009, 8, 1853–1859. [Google Scholar] [CrossRef] [PubMed]
- Solier, S.; Sordet, O.; Kohn, K.W.; Pommier, Y. Death receptor-induced activation of the Chk2- and histone H2AX-associated DNA damage response pathways. Mol. Cell. Biol. 2009, 29, 68–82. [Google Scholar] [CrossRef] [PubMed]
- Vasilevskaya, I.A.; O’Dwyer, P.J. 17-Allylamino-17-demethoxygeldanamycin overcomes TRAIL resistance in colon cancer cell lines. Biochem. Pharmacol. 2005, 70, 580–589. [Google Scholar] [CrossRef] [PubMed]
- Solier, S.; Pommier, Y. MDC1 cleavage by caspase-3: A novel mechanism for inactivating the DNA damage response during apoptosis. Cancer Res. 2011, 71, 906–913. [Google Scholar] [CrossRef] [PubMed]
- Nakanishi, K.; Yang, Y.G.; Pierce, A.J.; Taniguchi, T.; Digweed, M.; D’Andrea, A.D.; Wang, Z.Q.; Jasin, M. Human Fanconi anemia monoubiquitination pathway promotes homologous DNA repair. Proc. Natl. Acad. Sci. USA 2005, 102, 1110–1115. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, K.; Ishiai, M.; Matsushita, N.; Arakawa, H.; Lamerdin, J.E.; Buerstedde, J.M.; Tanimoto, M.; Harada, M.; Thompson, L.H.; Takata, M. Fanconi anemia FANCG protein in mitigating radiation- and enzyme-induced DNA double-strand breaks by homologous recombination in vertebrate cells. Mol. Cell. Biol. 2003, 23, 5421–5430. [Google Scholar] [CrossRef] [PubMed]
- Oda, T.; Hayano, T.; Miyaso, H.; Takahashi, N.; Yamashita, T. Hsp90 regulates the Fanconi anemia DNA damage response pathway. Blood 2007, 109, 5016–5026. [Google Scholar] [CrossRef] [PubMed]
- Yamashita, T.; Oda, T.; Sekimoto, T. Hsp90 and the Fanconi anemia pathway: A molecular link between protein quality control and the DNA damage response. Cell Cycle 2007, 6, 2232–2235. [Google Scholar] [CrossRef] [PubMed]
- Gospodinov, A.; Herceg, Z. Chromatin structure in double strand break repair. DNA Repair 2013, 12, 800–810. [Google Scholar] [CrossRef] [PubMed]
- Jeggo, P.A.; Downs, J.A. Roles of chromatin remodellers in DNA double strand break repair. Exp. Cell Res. 2014, 329, 69–77. [Google Scholar] [CrossRef] [PubMed]
- Csordas, A. On the biological role of histone acetylation. Biochem. J. 1990, 265, 23–38. [Google Scholar] [CrossRef] [PubMed]
- Hill, C.S.; Rimmer, J.M.; Green, B.N.; Finch, J.T.; Thomas, J.O. Histone-DNA interactions and their modulation by phosphorylation of -Ser-Pro-X-Lys/Arg- motifs. EMBO J. 1991, 10, 1939–1948. [Google Scholar] [PubMed]
- Khadake, J.R.; Rao, M.R. Condensation of DNA and chromatin by an SPKK-containing octapeptide repeat motif present in the C-terminus of histone H1. Biochemistry 1997, 36, 1041–1051. [Google Scholar] [CrossRef] [PubMed]
- Tarkka, T.; Oikarinen, J.; Grundstrom, T. Nucleotide and calcium-induced conformational changes in histone H1. FEBS Lett. 1997, 406, 56–60. [Google Scholar] [CrossRef]
- Jiricny, J. The multifaceted mismatch-repair system. Nat. Rev. Mol. Cell Biol. 2006, 7, 335–346. [Google Scholar] [CrossRef] [PubMed]
- Mo, C.; Dai, Y.; Kang, N.; Cui, L.; He, W. Ectopic expression of human MutS homologue 2 on renal carcinoma cells is induced by oxidative stress with interleukin-18 promotion via p38 mitogen-activated protein kinase (MAPK) and c-Jun N-terminal kinase (JNK) signaling pathways. J. Biol. Chem. 2012, 287, 19242–19254. [Google Scholar] [CrossRef] [PubMed]
- Tung, H.Y.; Plunkett, B.; Huang, S.K.; Zhou, Y. Murine mast cells secrete and respond to interleukin-33. J. Interferon Cytokine Res. 2014, 34, 141–147. [Google Scholar] [CrossRef] [PubMed]
- Curtin, N.J.; Harris, A.L.; Aherne, G.W. Mechanism of cell death following thymidylate synthase inhibition: 2′-deoxyuridine-5′-triphosphate accumulation, DNA damage, and growth inhibition following exposure to CB3717 and dipyridamole. Cancer Res. 1991, 51, 2346–2352. [Google Scholar] [PubMed]
- Van der Wilt, C.L.; Kuiper, C.M.; Peters, G.J. Combination studies of antifolates with 5-fluorouracil in colon cancer cell lines. Oncol. Res. 1999, 11, 383–391. [Google Scholar] [PubMed]
- White, I.N. Tamoxifen: Is it safe? Comparison of activation and detoxication mechanisms in rodents and in humans. Curr. Drug Metab. 2003, 4, 223–239. [Google Scholar] [CrossRef] [PubMed]
- Olivo-Marston, S.E.; Mechanic, L.E.; Mollerup, S.; Bowman, E.D.; Remaley, A.T.; Forman, M.R.; Skaug, V.; Zheng, Y.L.; Haugen, A.; Harris, C.C. Serum estrogen and tumor-positive estrogen receptor-alpha are strong prognostic classifiers of non-small-cell lung cancer survival in both men and women. Carcinogenesis 2010, 31, 1778–1786. [Google Scholar] [CrossRef] [PubMed]
- Ko, J.C.; Chiu, H.C.; Syu, J.J.; Jian, Y.J.; Chen, C.Y.; Jian, Y.T.; Huang, Y.J.; Wo, T.Y.; Lin, Y.W. Tamoxifen enhances erlotinib-induced cytotoxicity through down-regulating AKT-mediated thymidine phosphorylase expression in human non-small-cell lung cancer cells. Biochem. Pharmacol. 2014, 88, 119–127. [Google Scholar] [CrossRef] [PubMed]
- Ko, J.C.; Chiu, H.C.; Syu, J.J.; Chen, C.Y.; Jian, Y.T.; Huang, Y.J.; Wo, T.Y.; Jian, Y.J.; Chang, P.Y.; Wang, T.J.; Lin, Y.W. Down-regulation of MSH2 expression by Hsp90 inhibition enhances cytotoxicity affected by tamoxifen in human lung cancer cells. Biochem. Biophys. Res. Commun. 2015, 456, 506–512. [Google Scholar] [CrossRef] [PubMed]
- Friedberg, E.C.; Lehmann, A.R.; Fuchs, R.P. Trading places: How do DNA polymerases switch during translesion DNA synthesis? Mol. Cell 2005, 18, 499–505. [Google Scholar] [CrossRef] [PubMed]
- Lehmann, A.R.; Niimi, A.; Ogi, T.; Brown, S.; Sabbioneda, S.; Wing, J.F.; Kannouche, P.L.; Green, C.M. Translesion synthesis: Y-family polymerases and the polymerase switch. DNA Repair 2007, 6, 891–899. [Google Scholar] [CrossRef] [PubMed]
- Yang, W.; Woodgate, R. What a difference a decade makes: Insights into translesion DNA synthesis. Proc. Natl. Acad. Sci. USA 2007, 104, 15591–15598. [Google Scholar] [CrossRef] [PubMed]
- Masutani, C.; Kusumoto, R.; Iwai, S.; Hanaoka, F. Mechanisms of accurate translesion synthesis by human DNA polymerase eta. EMBO J. 2000, 19, 3100–3109. [Google Scholar] [CrossRef] [PubMed]
- McCulloch, S.D.; Kokoska, R.J.; Kunkel, T.A. Efficiency, fidelity and enzymatic switching during translesion DNA synthesis. Cell Cycle 2004, 3, 580–583. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Kannouche, P.L.; Wing, J.; Lehmann, A.R. Interaction of human DNA polymerase eta with monoubiquitinated PCNA: A possible mechanism for the polymerase switch in response to DNA damage. Mol. Cell 2004, 14, 491–500. [Google Scholar] [CrossRef]
- Watanabe, K.; Tateishi, S.; Kawasuji, M.; Tsurimoto, T.; Inoue, H.; Yamaizumi, M. Rad18 guides poleta to replication stalling sites through physical interaction and PCNA monoubiquitination. EMBO J. 2004, 23, 3886–3896. [Google Scholar] [CrossRef] [PubMed]
- Sekimoto, T.; Oda, T.; Pozo, F.M.; Murakumo, Y.; Masutani, C.; Hanaoka, F.; Yamashita, T. The molecular chaperone Hsp90 regulates accumulation of DNA polymerase eta at replication stalling sites in UV-irradiated cells. Mol. Cell 2010, 37, 79–89. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Heuvelman, D.M.; Carroll, J.A.; Dufield, D.R.; Masferrer, J.L. Geldanamycin-induced PCNA degradation in isolated Hsp90 complex from cancer cells. Cancer Investig. 2010, 28, 635–641. [Google Scholar] [CrossRef] [PubMed]
- Lindahl, T.; Prigent, C.; Barnes, D.E.; Lehmann, A.R.; Satoh, M.S.; Roberts, E.; Nash, R.A.; Robins, P.; Daly, G. DNA joining in mammalian cells. Cold Spring Harb. Symp. Quant. Biol. 1993, 58, 619–624. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.J.; Wilson, D.M., III. Overview of base excision repair biochemistry. Curr. Mol. Pharmacol. 2012, 5, 3–13. [Google Scholar] [CrossRef] [PubMed]
- Svilar, D.; Goellner, E.M.; Almeida, K.H.; Sobol, R.W. Base excision repair and lesion-dependent subpathways for repair of oxidative DNA damage. Antioxid. Redox Signal. 2011, 14, 2491–2507. [Google Scholar] [CrossRef] [PubMed]
- Braithwaite, E.K.; Kedar, P.S.; Stumpo, D.J.; Bertocci, B.; Freedman, J.H.; Samson, L.D.; Wilson, S.H. DNA polymerases beta and lambda mediate overlapping and independent roles in base excision repair in mouse embryonic fibroblasts. PLoS ONE 2010, 5, e12229. [Google Scholar] [CrossRef] [PubMed]
- Caldecott, K.W. XRCC1 and DNA strand break repair. DNA Repair 2003, 2, 955–969. [Google Scholar] [CrossRef]
- Shim, H.J.; Yun, J.Y.; Hwang, J.E.; Bae, W.K.; Cho, S.H.; Lee, J.H.; Kim, H.N.; Shin, M.H.; Kweon, S.S.; Lee, J.H.; et al. BRCA1 and XRCC1 polymorphisms associated with survival in advanced gastric cancer treated with taxane and cisplatin. Cancer Sci. 2010, 101, 1247–1254. [Google Scholar] [CrossRef] [PubMed]
- Caldecott, K.W. Single-strand break repair and genetic disease. Nat. Rev. Genet. 2008, 9, 619–631. [Google Scholar] [PubMed]
- Brem, R.; Hall, J. XRCC1 is required for DNA single-strand break repair in human cells. Nucleic Acids Res. 2005, 33, 2512–2520. [Google Scholar] [CrossRef] [PubMed]
- Toulany, M.; Dittmann, K.; Fehrenbacher, B.; Schaller, M.; Baumann, M.; Rodemann, H.P. PI3K-Akt signaling regulates basal, but MAP-kinase signaling regulates radiation-induced XRCC1 expression in human tumor cells in vitro. DNA Repair 2008, 7, 1746–1756. [Google Scholar] [CrossRef] [PubMed]
- Della-Maria, J.; Zhou, Y.; Tsai, M.S.; Kuhnlein, J.; Carney, J.P.; Paull, T.T.; Tomkinson, A.E. Human Mre11/human Rad50/Nbs1 and DNA ligase IIIalpha/XRCC1 protein complexes act together in an alternative nonhomologous end joining pathway. J. Biol. Chem. 2011, 286, 33845–33853. [Google Scholar] [CrossRef] [PubMed]
- Saribasak, H.; Maul, R.W.; Cao, Z.; McClure, R.L.; Yang, W.; McNeill, D.R.; Wilson, D.M., III; Gearhart, P.J. XRCC1 suppresses somatic hypermutation and promotes alternative nonhomologous end joining in Igh genes. J. Exp. Med. 2011, 208, 2209–2216. [Google Scholar] [CrossRef] [PubMed]
- Tung, C.L.; Jian, Y.J.; Syu, J.J.; Wang, T.J.; Chang, P.Y.; Chen, C.Y.; Jian, Y.T.; Lin, Y.W. Down-regulation of ERK1/2 and AKT-mediated X-ray repair cross-complement group 1 protein (XRCC1) expression by Hsp90 inhibition enhances the gefitinib-induced cytotoxicity in human lung cancer cells. Exp. Cell. Res. 2015, 334, 126–135. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pennisi, R.; Ascenzi, P.; Di Masi, A. Hsp90: A New Player in DNA Repair? Biomolecules 2015, 5, 2589-2618. https://doi.org/10.3390/biom5042589
Pennisi R, Ascenzi P, Di Masi A. Hsp90: A New Player in DNA Repair? Biomolecules. 2015; 5(4):2589-2618. https://doi.org/10.3390/biom5042589
Chicago/Turabian StylePennisi, Rosa, Paolo Ascenzi, and Alessandra Di Masi. 2015. "Hsp90: A New Player in DNA Repair?" Biomolecules 5, no. 4: 2589-2618. https://doi.org/10.3390/biom5042589
APA StylePennisi, R., Ascenzi, P., & Di Masi, A. (2015). Hsp90: A New Player in DNA Repair? Biomolecules, 5(4), 2589-2618. https://doi.org/10.3390/biom5042589