
Targeting the Checkpoint to Kill Cancer Cells

Abstract
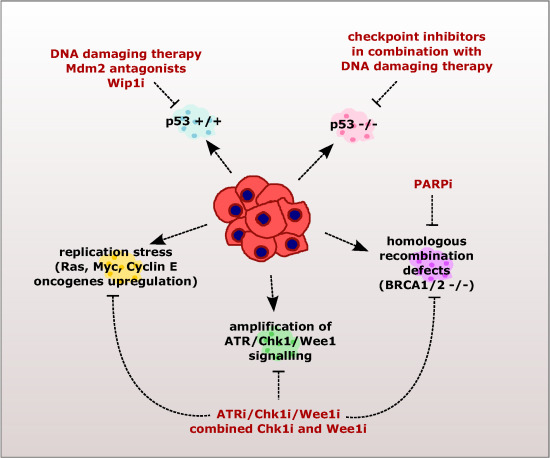
:
1. Introduction
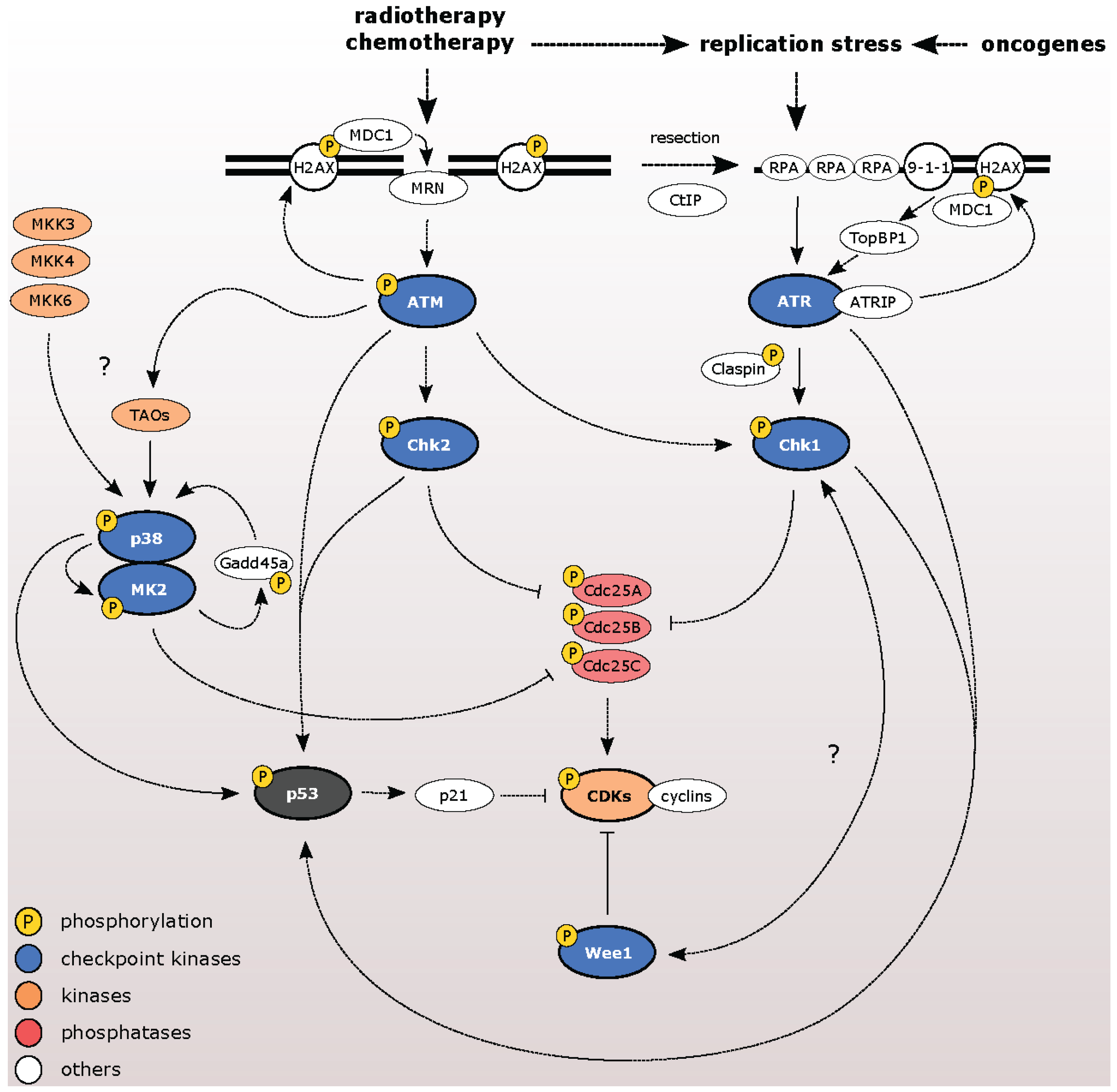
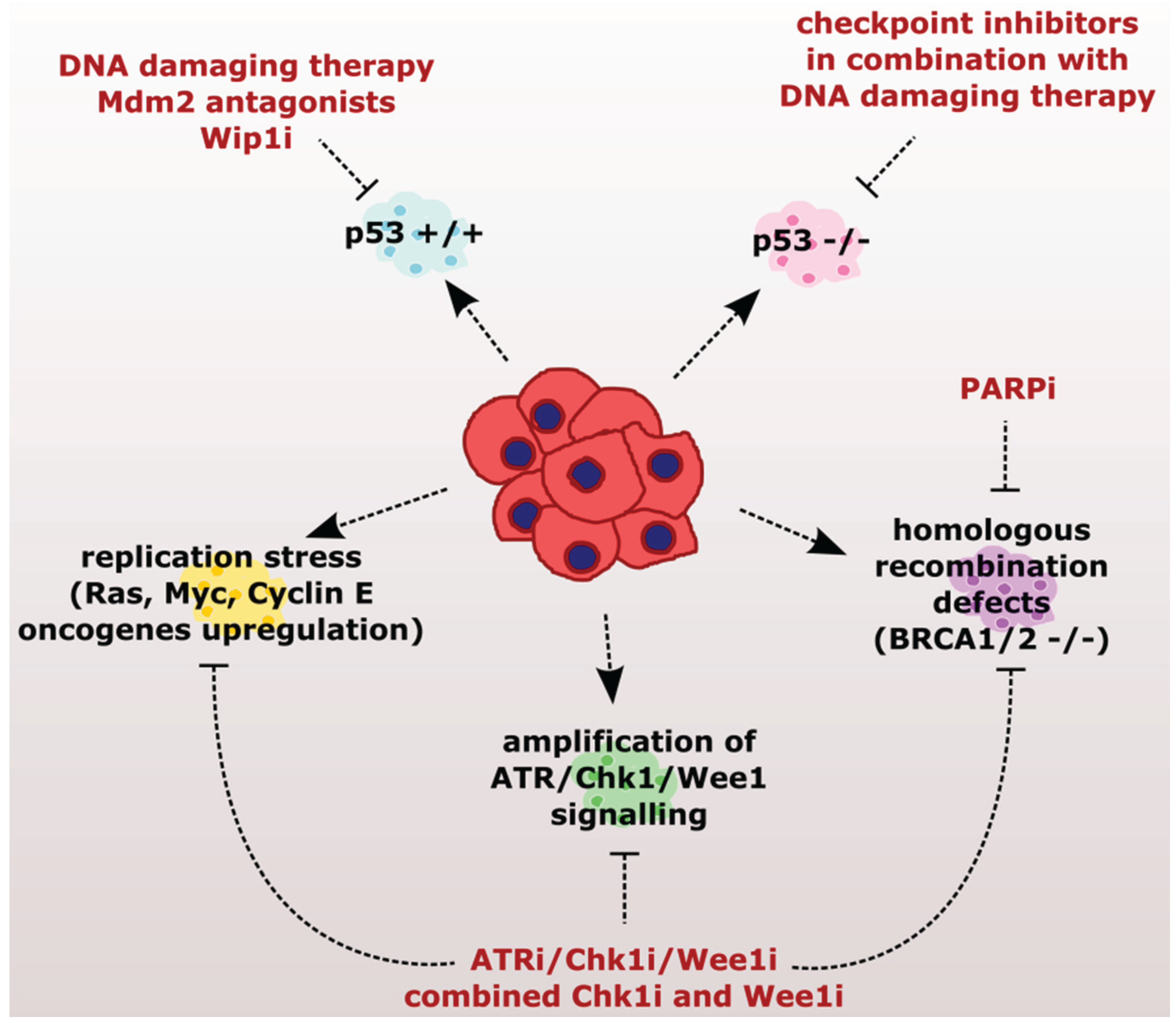
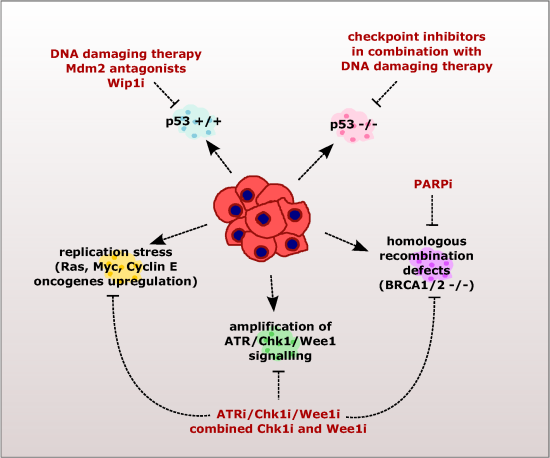
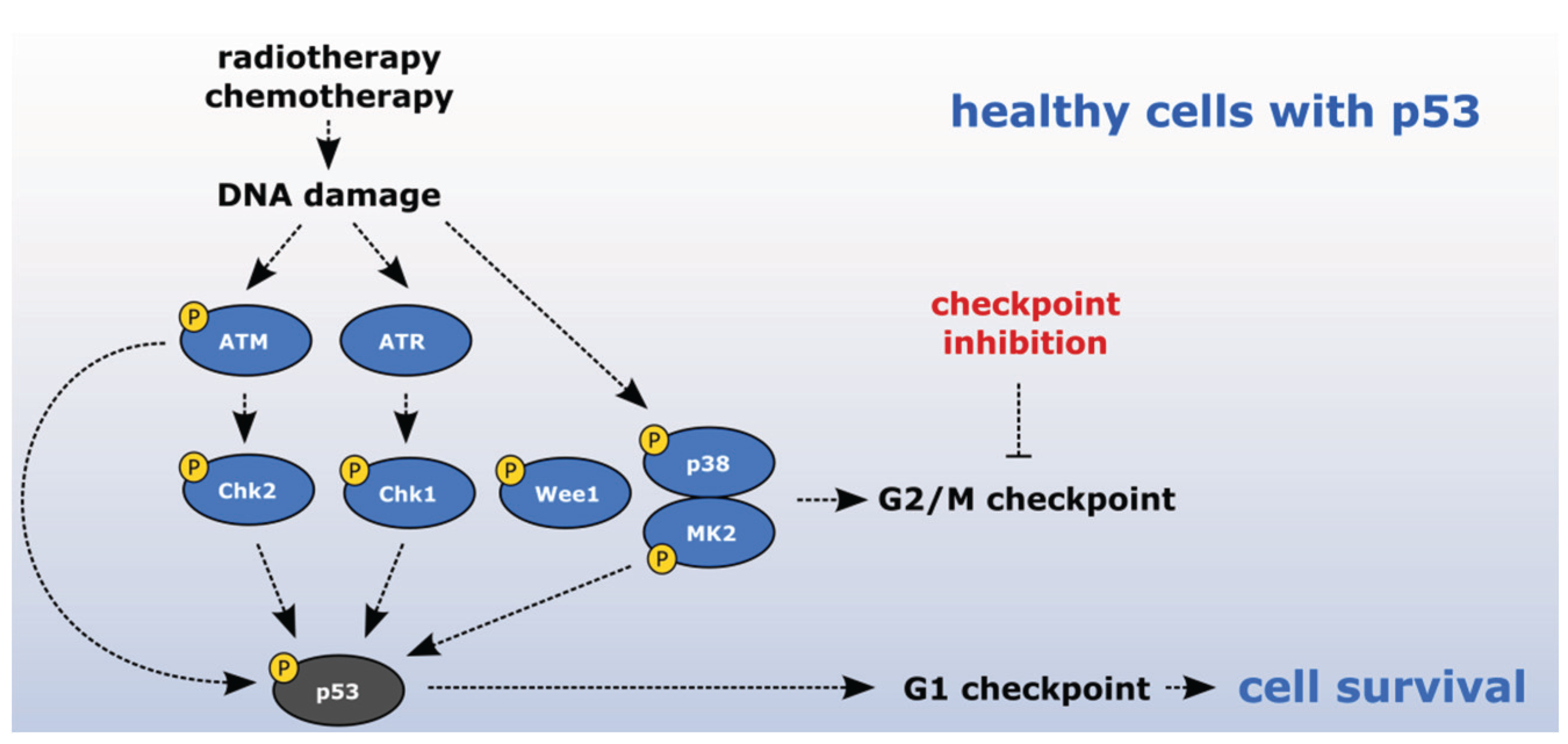
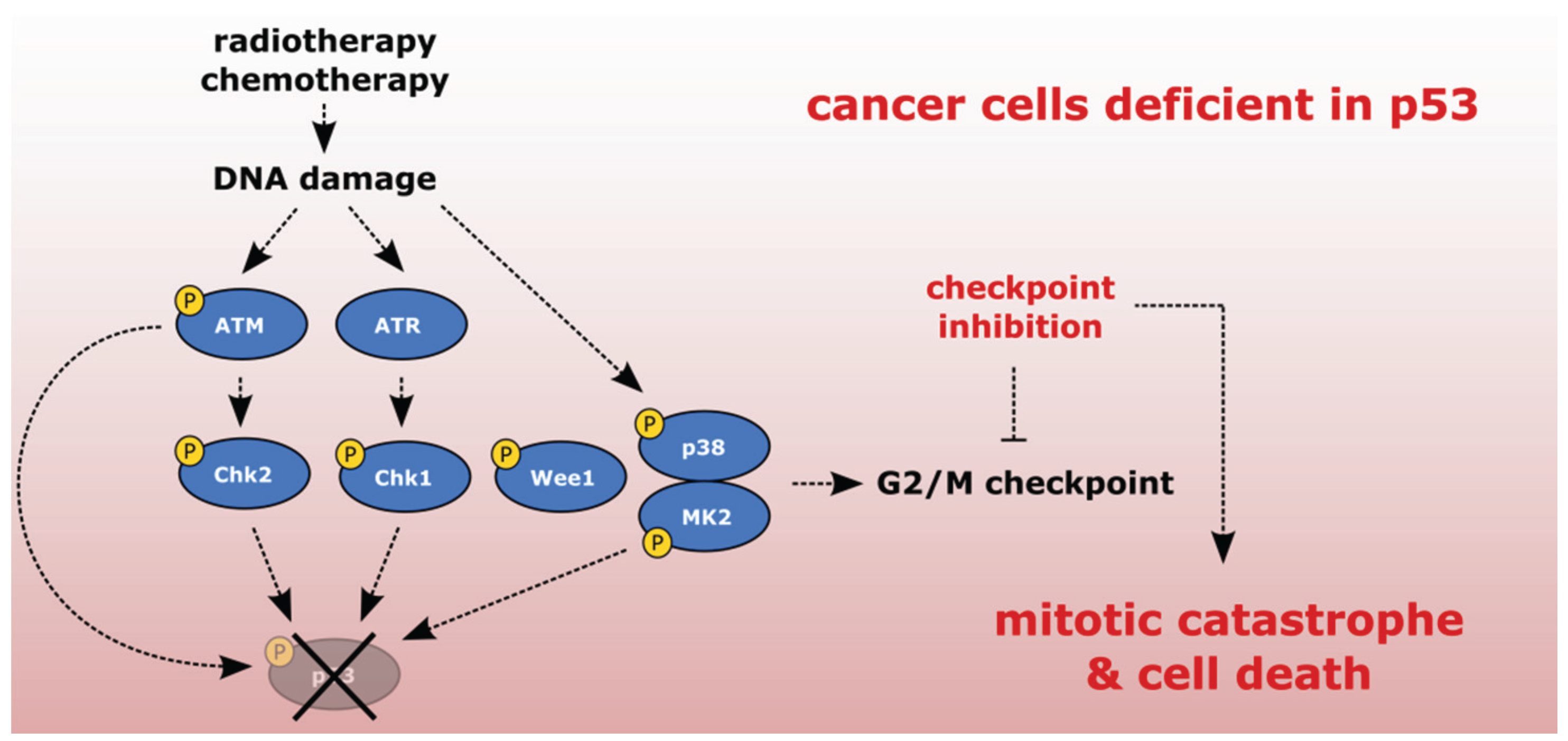
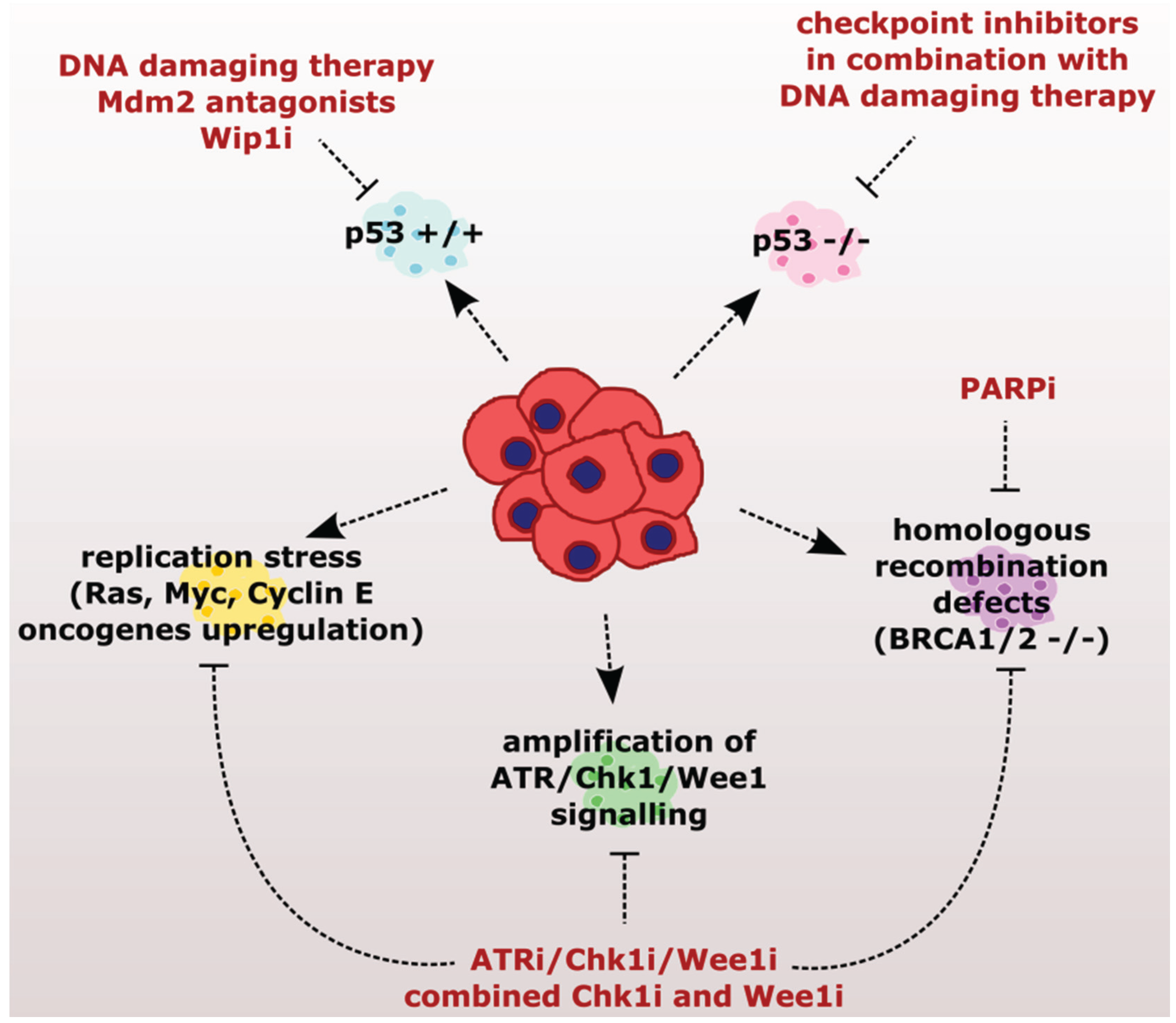
2. Checkpoint Inhibition as a Directed Anti-Cancer Therapy
2.1. Sensitizing Cancer Cells to DNA Damaging Agents with Checkpoint Inhibitors
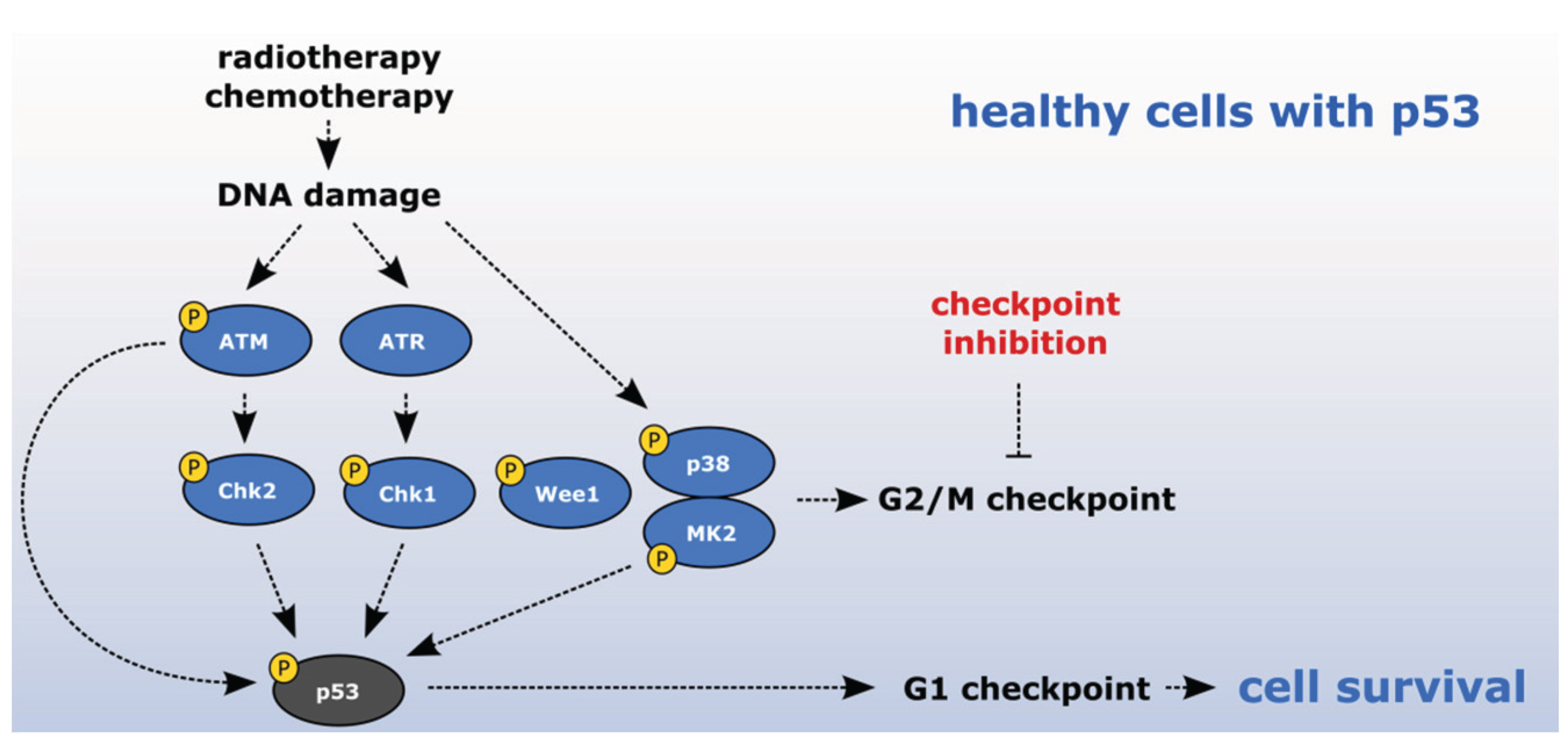

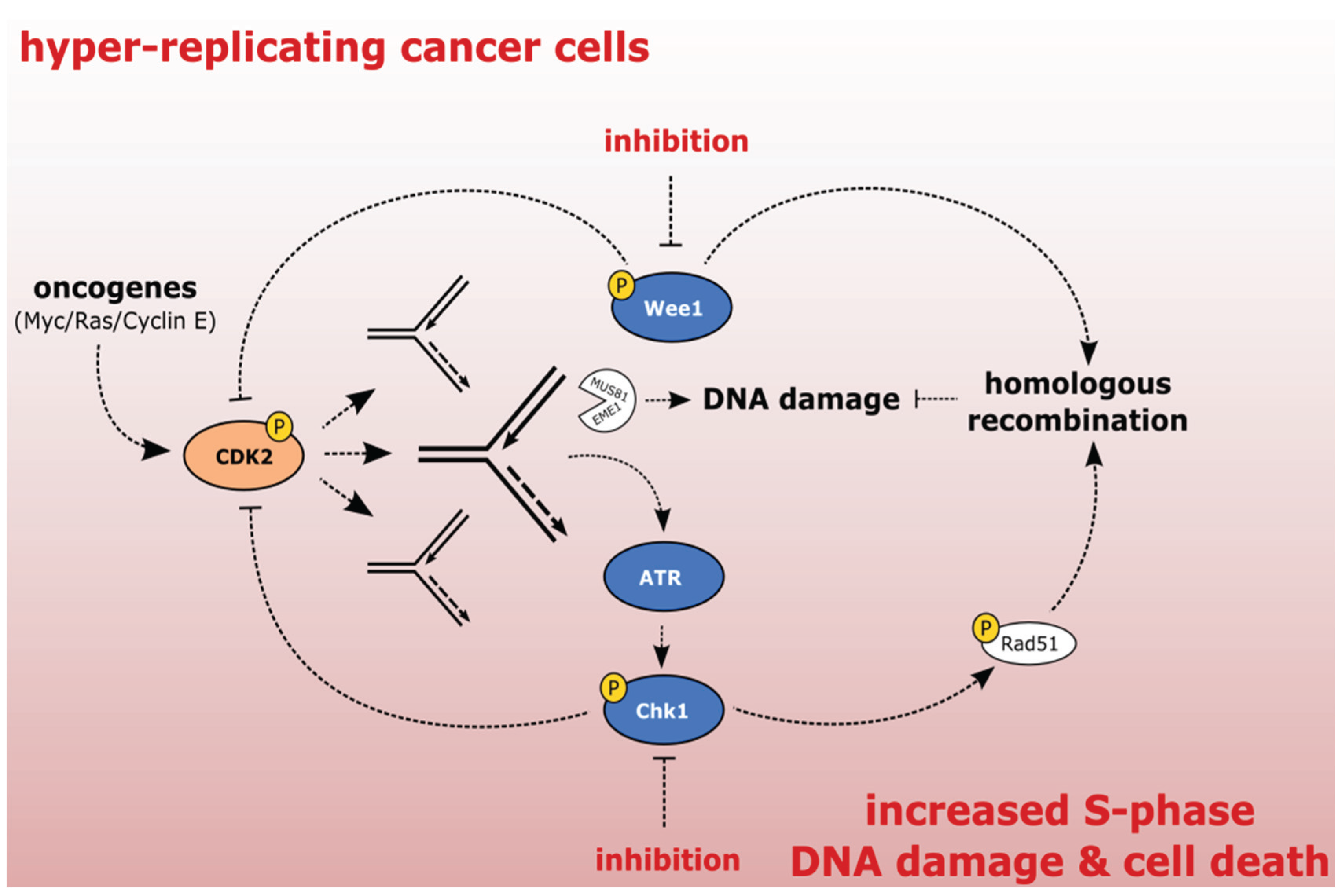
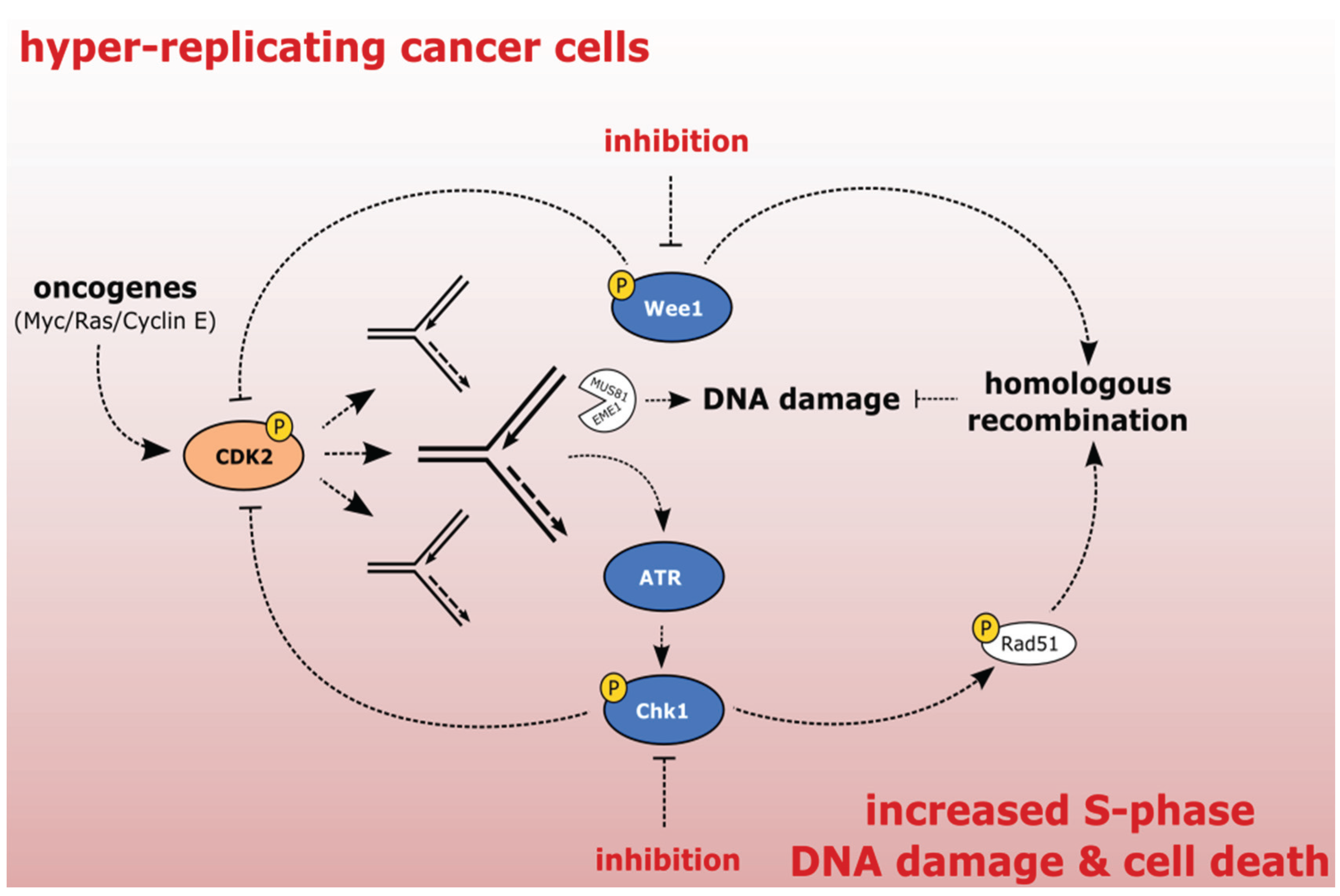
2.2. Exploiting the Addiction of Hyper-Replicating Cancer Cells to ATR-Chk1-Wee1 Signaling
2.3. Exploiting the Deficient HR Pathway for Increased Sensitivity of Cancer Cells
2.4. Exploiting the Deficient G2 Checkpoint in Targeting Cancer Cells
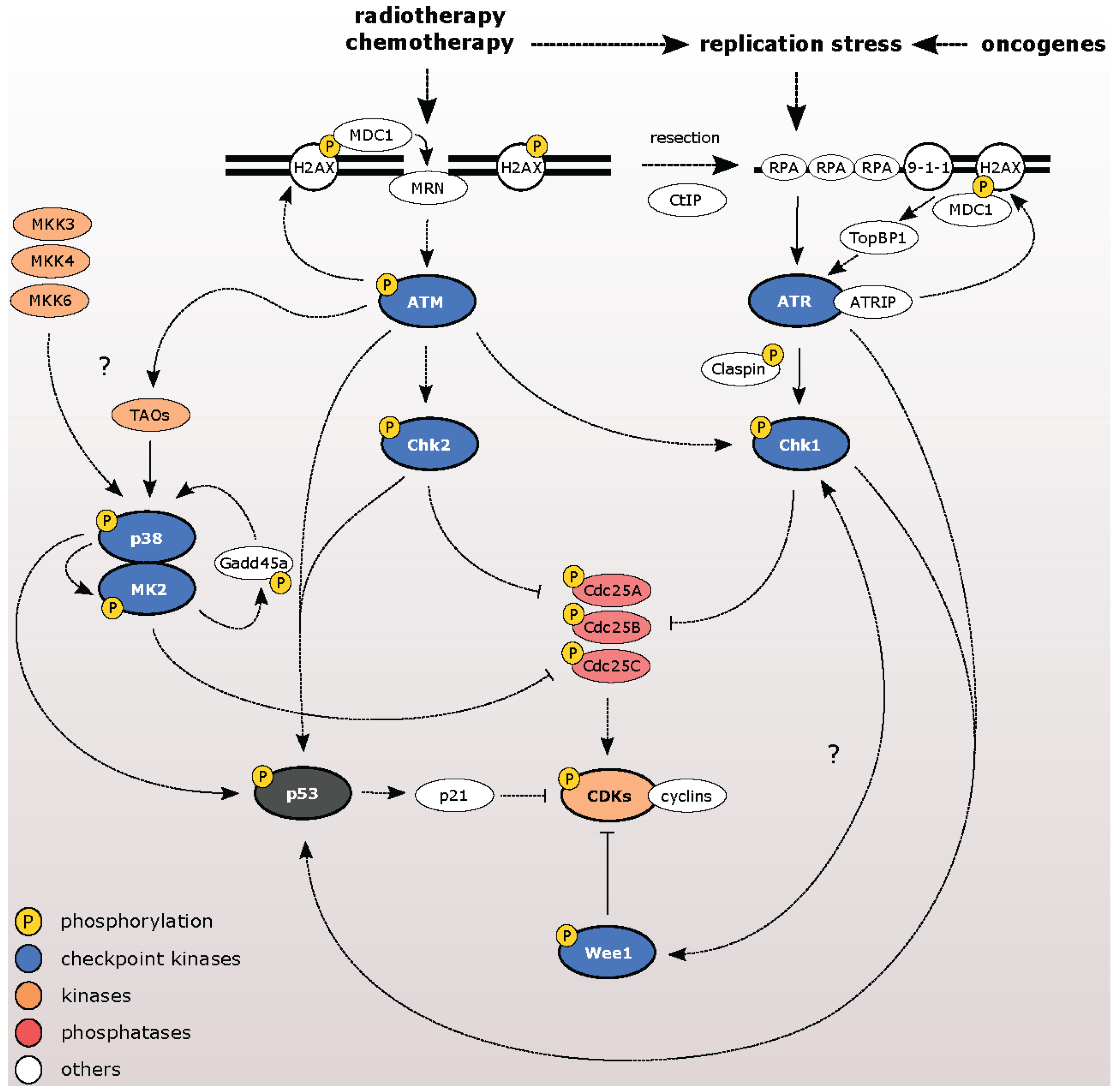
3. Pharmacological Inhibitors of Checkpoint Kinases
3.1. ATM Kinase
3.2. ATR Kinase
3.3. Chk1 and Chk2 Kinases
3.4. Wee1 Kinase
3.5. p38/MK2 Kinases
4. p53 Pathway Modulators
5. Targeting Cyclin-Dependent Kinases in Cancer Therapy
6. Conclusions

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Inhibitor | Targeted kinase(s) | Reference |
|---|---|---|
| KU-55933 | ATM (IC50 = 13 nM) | [49,53] |
| DNA-PK (IC50 = 2.5 μM) | ||
| PI3K (IC50 = 16.6 μM) | ||
| mTOR (IC50 = 9.3 μM) | ||
| KU-60019 | ATM (IC50 = 6.3 nM) | [51,53,54,55] |
| KU-59403 | ATM (IC50 = 3 nM) | [52] |
| DNA-PK (IC50 = 9.1 μM) | ||
| PI3K (IC50 = 10 μM) | ||
| mTOR (IC50 = 14 μM) | ||
| VE-821 | ATR (IC50 = 13 nM) | [77,78,80,81,82,83] |
| ATM (IC50 = 16 μM) | ||
| DNA-PK (IC50 = 2.2 μM) | ||
| PI3K (IC50 = 3.9 μM) | ||
| VE-822 (CX970) | ATR (IC50 = 19 nM) | [84,85] |
| AZ20 | ATR (IC50 = 5 nM) | [86] |
| AZD6738 | ATR (IC50 = 1 nM) | [87] |
| UCN-1 (staurosporine) | Chk1 (IC50 = 11 nM) | [97,98,99,100,101,102] |
| MK2 (IC50 = 95 nM) | ||
| Chk2 (IC50 = 1 μM) | ||
| PKC (IC50 = 7 nM) | ||
| CDK1 (IC50 = 31 nM) | ||
| CDK2 (IC50 = 30 nM) | ||
| LY2603618 | Chk1 (IC50 = 7 nM) | [106,107] |
| XL844 (EXEL-9844) | Chk1 (IC50 = 2.2 nM) | [104] |
| Chk2 (IC50 = 200 nM) | ||
| AZD7762 | Chk1 (IC50 = 5 nM) | [18] |
| Chk2 (IC50 = 5 nM) | ||
| PF00477736 | Chk1 (Ki = 0.49 nM) | [17] |
| Chk2 (Ki= 47 nM) | ||
| MK-8776 (SCH-900776) | Chk1 (IC50 = 3 nM) | [19,28,109] |
| Chk2 (IC50 = 1.5 μM) | ||
| PD0166285 | Wee1 (IC50 = 24 nM) | [115,116] |
| Myt1 (IC50 = 72 nM) | ||
| PD407824 | Wee1 (IC50 = 97 nM) | [117,118] |
| Chk1 (IC50 = 47 nM) | ||
| MK-1775 (AZD1775) | Wee1 (IC50 = 5.2 nM) | [119,120,121,122,123] |
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Lukas, J.; Lukas, C.; Bartek, J. More than just a focus: The chromatin response to DNA damage and its role in genome integrity maintenance. Nat. Cell Biol. 2011, 13, 1161–1169. [Google Scholar] [CrossRef] [PubMed]
- Medema, R.H.; Macurek, L. Checkpoint control and cancer. Oncogene 2012, 31, 2601–2613. [Google Scholar] [CrossRef] [PubMed]
- Lemaire, M.; Mondesert, O.; Bugler, B.; Ducommun, B. Ability of human Cdc25B phosphatase splice variants to replace the function of the fission yeast Cdc25 cell cycle regulator. FEMS Yeast Res. 2004, 5, 205–211. [Google Scholar] [CrossRef] [PubMed]
- Manke, I.A.; Nguyen, A.; Lim, D.; Stewart, M.Q.; Elia, A.E.; Yaffe, M.B. Mapkap kinase-2 is a cell cycle checkpoint kinase that regulates the G2/M transition and S phase progression in response to UV irradiation. Mol. Cell 2005, 17, 37–48. [Google Scholar] [CrossRef] [PubMed]
- Uchida, S.; Watanabe, N.; Kudo, Y.; Yoshioka, K.; Matsunaga, T.; Ishizaka, Y.; Nakagama, H.; Poon, R.Y.; Yamashita, K. SCFβ(TrCP) mediates stress-activated MAPK-induced Cdc25B degradation. J. Cell Sci. 2011, 124, 2816–2825. [Google Scholar] [CrossRef] [PubMed]
- Imbriano, C.; Gurtner, A.; Cocchiarella, F.; di Agostino, S.; Basile, V.; Gostissa, M.; Dobbelstein, M.; del Sal, G.; Piaggio, G.; Mantovani, R. Direct P53 transcriptional repression: In vivo analysis of ccaat-containing G2/M promoters. Mol. Cell. Biol. 2005, 25, 3737–3751. [Google Scholar] [CrossRef] [PubMed]
- McKenzie, L.; King, S.; Marcar, L.; Nicol, S.; Dias, S.S.; Schumm, K.; Robertson, P.; Bourdon, J.C.; Perkins, N.; Fuller-Pace, F.; et al. p53-dependent repression of polo-like kinase-1 (PLK1). Cell Cycle 2010, 9, 4200–4212. [Google Scholar] [CrossRef] [PubMed]
- Lord, C.J.; Ashworth, A. The DNA damage response and cancer therapy. Nature 2012, 481, 287–294. [Google Scholar] [CrossRef] [PubMed]
- Bouwman, P.; Jonkers, J. Molecular pathways: How can BRCA-mutated tumors become resistant to PARP inhibitors? Clin. Cancer Res. 2014, 20, 540–547. [Google Scholar] [CrossRef] [PubMed]
- Banerjee, S.; Kaye, S.B.; Ashworth, A. Making the best of PARP inhibitors in ovarian cancer. Nat. Rev. Clin. Oncol. 2010, 7, 508–519. [Google Scholar] [CrossRef] [PubMed]
- Lord, C.J.; Ashworth, A. Mechanisms of resistance to therapies targeting BRCA-mutant cancers. Nat. Med. 2013, 19, 1381–1388. [Google Scholar] [CrossRef] [PubMed]
- Bartkova, J.; Horejsi, Z.; Koed, K.; Kramer, A.; Tort, F.; Zieger, K.; Guldberg, P.; Sehested, M.; Nesland, J.M.; Lukas, C.; et al. DNA damage response as a candidate anti-cancer barrier in early human tumorigenesis. Nature 2005, 434, 864–870. [Google Scholar] [CrossRef] [PubMed]
- Gorgoulis, V.G.; Vassiliou, L.V.; Karakaidos, P.; Zacharatos, P.; Kotsinas, A.; Liloglou, T.; Venere, M.; Ditullio, R.A., Jr.; Kastrinakis, N.G.; Levy, B.; et al. Activation of the DNA damage checkpoint and genomic instability in human precancerous lesions. Nature 2005, 434, 907–913. [Google Scholar] [CrossRef] [PubMed]
- Castedo, M.; Perfettini, J.L.; Roumier, T.; Andreau, K.; Medema, R.; Kroemer, G. Cell death by mitotic catastrophe: A molecular definition. Oncogene 2004, 23, 2825–2837. [Google Scholar] [CrossRef] [PubMed]
- Zhou, B.B.; Bartek, J. Targeting the checkpoint kinases: Chemosensitization versus chemoprotection. Nat. Rev. Cancer 2004, 4, 216–225. [Google Scholar] [CrossRef] [PubMed]
- Tse, A.N.; Rendahl, K.G.; Sheikh, T.; Cheema, H.; Aardalen, K.; Embry, M.; Ma, S.; Moler, E.J.; Ni, Z.J.; Lopes de Menezes, D.E.; et al. CHIR-124, a novel potent inhibitor of Chk1, potentiates the cytotoxicity of topoisomerase I poisons in vitro and in vivo. Clin. Cancer Res. 2007, 13, 591–602. [Google Scholar] [CrossRef] [PubMed]
- Blasina, A.; Hallin, J.; Chen, E.; Arango, M.E.; Kraynov, E.; Register, J.; Grant, S.; Ninkovic, S.; Chen, P.; Nichols, T.; et al. Breaching the DNA damage checkpoint via PF-00477736, a novel small-molecule inhibitor of checkpoint kinase 1. Mol. Cancer Ther. 2008, 7, 2394–2404. [Google Scholar] [CrossRef] [PubMed]
- Zabludoff, S.D.; Deng, C.; Grondine, M.R.; Sheehy, A.M.; Ashwell, S.; Caleb, B.L.; Green, S.; Haye, H.R.; Horn, C.L.; Janetka, J.W.; et al. AZD7762, a novel checkpoint kinase inhibitor, drives checkpoint abrogation and potentiates DNA-targeted therapies. Mol. Cancer Ther. 2008, 7, 2955–2966. [Google Scholar] [CrossRef] [PubMed]
- Guzi, T.J.; Paruch, K.; Dwyer, M.P.; Labroli, M.; Shanahan, F.; Davis, N.; Taricani, L.; Wiswell, D.; Seghezzi, W.; Penaflor, E.; et al. Targeting the replication checkpoint using SCH 900776, a potent and functionally selective Chk1 inhibitor identified via high content screening. Mol. Cancer Ther. 2011, 10, 591–602. [Google Scholar] [CrossRef] [PubMed]
- Macheret, M.; Halazonetis, T.D. DNA replication stress as a hallmark of cancer. Annu. Rev. Pathol. 2015, 10, 425–448. [Google Scholar] [CrossRef] [PubMed]
- Toledo, L.I.; Altmeyer, M.; Rask, M.B.; Lukas, C.; Larsen, D.H.; Povlsen, L.K.; Bekker-Jensen, S.; Mailand, N.; Bartek, J.; Lukas, J. ATR prohibits replication catastrophe by preventing global exhaustion of RPA. Cell 2013, 155, 1088–1103. [Google Scholar] [CrossRef] [PubMed]
- Sorensen, C.S.; Syljuasen, R.G. Safeguarding genome integrity: The checkpoint kinases ATR, Chk1 and Wee1 restrain Cdk activity during normal DNA replication. Nucleic Acids Res. 2012, 40, 477–486. [Google Scholar] [CrossRef] [PubMed]
- Petermann, E.; Helleday, T. Pathways of mammalian replication fork restart. Nat. Rev. Mol. Cell Biol. 2010, 11, 683–687. [Google Scholar] [CrossRef] [PubMed]
- Fugger, K.; Chu, W.K.; Haahr, P.; Kousholt, A.N.; Beck, H.; Payne, M.J.; Hanada, K.; Hickson, I.D.; Sorensen, C.S. FBH1 co-operates with MUS81 in inducing DNA double-strand breaks and cell death following replication stress. Nat. Commun. 2013. [Google Scholar] [CrossRef] [PubMed]
- Brown, E.J.; Baltimore, D. ATR disruption leads to chromosomal fragmentation and early embryonic lethality. Genes Dev. 2000, 14, 397–402. [Google Scholar] [PubMed]
- Takai, H.; Tominaga, K.; Motoyama, N.; Minamishima, Y.A.; Nagahama, H.; Tsukiyama, T.; Ikeda, K.; Nakayama, K.; Nakanishi, M.; Nakayama, K. Aberrant cell cycle checkpoint function and early embryonic death in Chk1(−/−) mice. Genes Dev. 2000, 14, 1439–1447. [Google Scholar] [PubMed]
- Tominaga, Y.; Li, C.; Wang, R.H.; Deng, C.X. Murine WEE1 plays a critical role in cell cycle regulation and pre-implantation stages of embryonic development. Int. J. Biol. Sci. 2006, 2, 161–170. [Google Scholar] [CrossRef] [PubMed]
- Montano, R.; Chung, I.; Garner, K.M.; Parry, D.; Eastman, A. Preclinical development of the novel Chk1 inhibitor SCH900776 in combination with DNA-damaging agents and antimetabolites. Mol. Cancer Ther. 2012, 11, 427–438. [Google Scholar] [CrossRef] [PubMed]
- Brooks, K.; Oakes, V.; Edwards, B.; Ranall, M.; Leo, P.; Pavey, S.; Pinder, A.; Beamish, H.; Mukhopadhyay, P.; Lambie, D.; et al. A potent Chk1 inhibitor is selectively cytotoxic in melanomas with high levels of replicative stress. Oncogene 2013, 32, 788–796. [Google Scholar] [CrossRef] [PubMed]
- Ferrao, P.T.; Bukczynska, E.P.; Johnstone, R.W.; McArthur, G.A. Efficacy of Chk inhibitors as single agents in MYC-driven lymphoma cells. Oncogene 2012, 31, 1661–1672. [Google Scholar] [CrossRef] [PubMed]
- Krajewska, M.; Fehrmann, R.S.; Schoonen, P.M.; Labib, S.; de Vries, E.G.; Franke, L.; van Vugt, M.A. ATR inhibition preferentially targets homologous recombination-deficient tumor cells. Oncogene 2015, 34, 3474–3481. [Google Scholar] [CrossRef] [PubMed]
- Mak, J.P.; Man, W.Y.; Ma, H.T.; Poon, R.Y. Pharmacological targeting the ATR-Chk1-Wee1 axis involves balancing cell growth stimulation and apoptosis. Oncotarget 2014, 5, 10546–10557. [Google Scholar] [PubMed]
- Huertas, P.; Cortes-Ledesma, F.; Sartori, A.A.; Aguilera, A.; Jackson, S.P. Cdk targets SAE2 to control DNA-end resection and homologous recombination. Nature 2008, 455, 689–692. [Google Scholar] [CrossRef] [PubMed]
- Huertas, P.; Jackson, S.P. Human ctip mediates cell cycle control of DNA end resection and double strand break repair. J. Biol. Chem. 2009, 284, 9558–9565. [Google Scholar] [CrossRef] [PubMed]
- Sorensen, C.S.; Hansen, L.T.; Dziegielewski, J.; Syljuasen, R.G.; Lundin, C.; Bartek, J.; Helleday, T. The cell-cycle checkpoint kinase Chk1 is required for mammalian homologous recombination repair. Nat. Cell Biol. 2005, 7, 195–201. [Google Scholar] [CrossRef] [PubMed]
- Krajewska, M.; Heijink, A.M.; Bisselink, Y.J.; Seinstra, R.I.; Sillje, H.H.; de Vries, E.G.; van Vugt, M.A. Forced activation of Cdk1 via Wee1 inhibition impairs homologous recombination. Oncogene 2013, 32, 3001–3008. [Google Scholar] [CrossRef] [PubMed]
- Thacker, J. The RAD51 gene family, genetic instability and cancer. Cancer Lett. 2005, 219, 125–135. [Google Scholar] [CrossRef] [PubMed]
- Paul, A.; Paul, S. The breast cancer susceptibility genes (BRCA) in breast and ovarian cancers. Frontiers in bioscience 2014, 19, 605–618. [Google Scholar] [CrossRef]
- Qiu, L.; Burgess, A.; Fairlie, D.P.; Leonard, H.; Parsons, P.G.; Gabrielli, B.G. Histone deacetylase inhibitors trigger a G2 checkpoint in normal cells that is defective in tumor cells. Mol. Biol. Cell 2000, 11, 2069–2083. [Google Scholar] [CrossRef] [PubMed]
- Brooks, K.; Ranall, M.; Spoerri, L.; Stevenson, A.; Gunasingh, G.; Pavey, S.; Meunier, F.; Gonda, T.J.; Gabrielli, B. Decatenation checkpoint-defective melanomas are dependent on PI3K for survival. Pigment Cell Melanoma Res. 2014, 27, 813–821. [Google Scholar] [CrossRef] [PubMed]
- Pavey, S.; Spoerri, L.; Haass, N.K.; Gabrielli, B. DNA repair and cell cycle checkpoint defects as drivers and therapeutic targets in melanoma. Pigment Cell Melanoma Res. 2013, 26, 805–816. [Google Scholar] [CrossRef] [PubMed]
- Grenon, M.; Gilbert, C.; Lowndes, N.F. Checkpoint activation in response to double-strand breaks requires the MRE11/RAD50/XRS2 complex. Nat. Cell Biol. 2001, 3, 844–847. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.H.; Paull, T.T. Activation and regulation of ATM kinase activity in response to DNA double-strand breaks. Oncogene 2007, 26, 7741–7748. [Google Scholar] [CrossRef] [PubMed]
- Burma, S.; Chen, B.P.; Murphy, M.; Kurimasa, A.; Chen, D.J. ATM phosphorylates histone H2AX in response to DNA double-strand breaks. J. Biol. Chem. 2001, 276, 42462–42467. [Google Scholar] [CrossRef] [PubMed]
- Melander, F.; Bekker-Jensen, S.; Falck, J.; Bartek, J.; Mailand, N.; Lukas, J. Phosphorylation of SDT repeats in the MDC1 N terminus triggers retention of NBS1 at the DNA damage-modified chromatin. J. Cell Biol. 2008, 181, 213–226. [Google Scholar] [CrossRef] [PubMed]
- Ahn, J.Y.; Schwarz, J.K.; Piwnica-Worms, H.; Canman, C.E. Threonine 68 phosphorylation by ataxia telangiectasia mutated is required for efficient activation of Chk2 in response to ionizing radiation. Cancer Res. 2000, 60, 5934–5936. [Google Scholar] [PubMed]
- Powell, S.N.; DeFrank, J.S.; Connell, P.; Eogan, M.; Preffer, F.; Dombkowski, D.; Tang, W.; Friend, S. Differential sensitivity of p53(−) and p53(+) cells to caffeine-induced radiosensitization and override of G2 delay. Cancer Res. 1995, 55, 1643–1648. [Google Scholar] [CrossRef]
- Price, B.D.; Youmell, M.B. The phosphatidylinositol 3-kinase inhibitor wortmannin sensitizes murine fibroblasts and human tumor cells to radiation and blocks induction of p53 following DNA damage. Cancer Res. 1996, 56, 246–250. [Google Scholar] [PubMed]
- Hickson, I.; Zhao, Y.; Richardson, C.J.; Green, S.J.; Martin, N.M.; Orr, A.I.; Reaper, P.M.; Jackson, S.P.; Curtin, N.J.; Smith, G.C. Identification and characterization of a novel and specific inhibitor of the ataxia-telangiectasia mutated kinase ATM. Cancer Res. 2004, 64, 9152–9159. [Google Scholar] [CrossRef] [PubMed]
- Rainey, M.D.; Charlton, M.E.; Stanton, R.V.; Kastan, M.B. Transient inhibition of ATM kinase is sufficient to enhance cellular sensitivity to ionizing radiation. Cancer Res. 2008, 68, 7466–7474. [Google Scholar] [CrossRef] [PubMed]
- Golding, S.E.; Rosenberg, E.; Valerie, N.; Hussaini, I.; Frigerio, M.; Cockcroft, X.F.; Chong, W.Y.; Hummersone, M.; Rigoreau, L.; Menear, K.A.; et al. Improved ATM kinase inhibitor KU-60019 radiosensitizes glioma cells, compromises insulin, AKT and ERK prosurvival signaling, and inhibits migration and invasion. Mol. Cancer Ther. 2009, 8, 2894–2902. [Google Scholar] [CrossRef] [PubMed]
- Batey, M.A.; Zhao, Y.; Kyle, S.; Richardson, C.; Slade, A.; Martin, N.M.; Lau, A.; Newell, D.R.; Curtin, N.J. Preclinical evaluation of a novel ATM inhibitor, KU59403, in vitro and in vivo in p53 functional and dysfunctional models of human cancer. Mol. Cancer Ther. 2013, 12, 959–967. [Google Scholar] [CrossRef] [PubMed]
- Raso, A.; Vecchio, D.; Cappelli, E.; Ropolo, M.; Poggi, A.; Nozza, P.; Biassoni, R.; Mascelli, S.; Capra, V.; Kalfas, F.; et al. Characterization of glioma stem cells through multiple stem cell markers and their specific sensitization to double-strand break-inducing agents by pharmacological inhibition of ataxia telangiectasia mutated protein. Brain Pathol. 2012, 22, 677–688. [Google Scholar] [CrossRef] [PubMed]
- Golding, S.E.; Rosenberg, E.; Adams, B.R.; Wignarajah, S.; Beckta, J.M.; O’Connor, M.J.; Valerie, K. Dynamic inhibition of ATM kinase provides a strategy for glioblastoma multiforme radiosensitization and growth control. Cell Cycle 2012, 11, 1167–1173. [Google Scholar] [CrossRef] [PubMed]
- Vecchio, D.; Daga, A.; Carra, E.; Marubbi, D.; Baio, G.; Neumaier, C.E.; Vagge, S.; Corvo, R.; Brisigotti, M.P.; Ravetti, J.L.; et al. Predictability, efficacy and safety of radiosensitization of glioblastoma-initiating cells by the ATM inhibitor KU-60019. Int. J. Cancer 2014, 135, 479–491. [Google Scholar] [CrossRef] [PubMed]
- Biddlestone-Thorpe, L.; Sajjad, M.; Rosenberg, E.; Beckta, J.M.; Valerie, N.C.; Tokarz, M.; Adams, B.R.; Wagner, A.F.; Khalil, A.; Gilfor, D.; et al. Atm kinase inhibition preferentially sensitizes p53-mutant glioma to ionizing radiation. Clin. Cancer Res. 2013, 19, 3189–3200. [Google Scholar] [CrossRef] [PubMed]
- Hoeijmakers, J.H. Genome maintenance mechanisms for preventing cancer. Nature 2001, 411, 366–374. [Google Scholar] [CrossRef] [PubMed]
- Jazayeri, A.; Falck, J.; Lukas, C.; Bartek, J.; Smith, G.C.; Lukas, J.; Jackson, S.P. ATM- and cell cycle-dependent regulation of ATR in response to DNA double-strand breaks. Nat. Cell Biol. 2006, 8, 37–45. [Google Scholar] [CrossRef] [PubMed]
- Sartori, A.A.; Lukas, C.; Coates, J.; Mistrik, M.; Fu, S.; Bartek, J.; Baer, R.; Lukas, J.; Jackson, S.P. Human ctip promotes DNA end resection. Nature 2007, 450, 509–514. [Google Scholar] [CrossRef] [PubMed]
- Kousholt, A.N.; Fugger, K.; Hoffmann, S.; Larsen, B.D.; Menzel, T.; Sartori, A.A.; Sorensen, C.S. CtIP-dependent DNA resection is required for DNA damage checkpoint maintenance but not initiation. J. Cell Biol. 2012, 197, 869–876. [Google Scholar] [CrossRef] [PubMed]
- Wold, M.S. Replication protein a: A heterotrimeric, single-stranded DNA-binding protein required for eukaryotic DNA metabolism. Annu. Rev. Biochem. 1997, 66, 61–92. [Google Scholar] [CrossRef] [PubMed]
- Cortez, D.; Guntuku, S.; Qin, J.; Elledge, S.J. ATR and ATRIP: Partners in checkpoint signaling. Science 2001, 294, 1713–1716. [Google Scholar] [CrossRef] [PubMed]
- Cotta-Ramusino, C.; McDonald, E.R., 3rd; Hurov, K.; Sowa, M.E.; Harper, J.W.; Elledge, S.J. A DNA damage response screen identifies RHINO, a 9-1-1 and TopBP1 interacting protein required for ATR signaling. Science 2011, 332, 1313–1317. [Google Scholar] [CrossRef] [PubMed]
- Kumagai, A.; Lee, J.; Yoo, H.Y.; Dunphy, W.G. TopBP1 activates the ATR-ATRIP complex. Cell 2006, 124, 943–955. [Google Scholar] [CrossRef] [PubMed]
- Delacroix, S.; Wagner, J.M.; Kobayashi, M.; Yamamoto, K.; Karnitz, L.M. The Rad9-Hus1-Rad1 (9-1-1) clamp activates checkpoint signaling via TopBP1. Genes Dev. 2007, 21, 1472–1477. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; Kumagai, A.; Dunphy, W.G. The Rad9-Hus1-Rad1 checkpoint clamp regulates interaction of TopBP1 with ATR. J. Biol. Chem. 2007, 282, 28036–28044. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Gong, Z.; Chen, J. MDC1 collaborates with TopBP1 in DNA replication checkpoint control. J. Cell Biol. 2011, 193, 267–273. [Google Scholar] [CrossRef] [PubMed]
- Jeong, S.Y.; Kumagai, A.; Lee, J.; Dunphy, W.G. Phosphorylated claspin interacts with a phosphate-binding site in the kinase domain of Chk1 during ATR-mediated activation. J. Biol. Chem. 2003, 278, 46782–46788. [Google Scholar] [CrossRef] [PubMed]
- Kumagai, A.; Dunphy, W.G. Repeated phosphopeptide motifs in claspin mediate the regulated binding of Chk1. Nat. Cell Biol. 2003, 5, 161–165. [Google Scholar] [CrossRef] [PubMed]
- Liu, Q.; Guntuku, S.; Cui, X.S.; Matsuoka, S.; Cortez, D.; Tamai, K.; Luo, G.; Carattini-Rivera, S.; DeMayo, F.; Bradley, A.; et al. Chk1 is an essential kinase that is regulated by ATR and required for the G2/M DNA damage checkpoint. Genes Dev. 2000, 14, 1448–1459. [Google Scholar] [PubMed]
- Gilad, O.; Nabet, B.Y.; Ragland, R.L.; Schoppy, D.W.; Smith, K.D.; Durham, A.C.; Brown, E.J. Combining ATR suppression with oncogenic Ras synergistically increases genomic instability, causing synthetic lethality or tumorigenesis in a dosage-dependent manner. Cancer Res. 2010, 70, 9693–9702. [Google Scholar] [CrossRef] [PubMed]
- Murga, M.; Campaner, S.; Lopez-Contreras, A.J.; Toledo, L.I.; Soria, R.; Montaña, M.F.; D’Artista, L.; Schleker, T.; Guerra, C.; Garcia, E.; et al. Exploiting oncogene-induced replicative stress for the selective killing of MYC-driven tumors. Nat. Struct. Mol. Biol. 2011, 18, 1331–1335. [Google Scholar] [CrossRef] [PubMed]
- Schoppy, D.W.; Ragland, R.L.; Gilad, O.; Shastri, N.; Peters, A.A.; Murga, M.; Fernandez-Capetillo, O.; Diehl, J.A.; Brown, E.J. Oncogenic stress sensitizes murine cancers to hypomorphic suppression of ATR. J. Clin. Investig. 2012, 122, 241–252. [Google Scholar] [CrossRef] [PubMed]
- Flynn, R.L.; Cox, K.E.; Jeitany, M.; Wakimoto, H.; Bryll, A.R.; Ganem, N.J.; Bersani, F.; Pineda, J.R.; Suvà, M.L.; Benes, C.H.; et al. Alternative lengthening of telomeres renders cancer cells hypersensitive to ATR inhibitors. Science 2015, 347, 273–277. [Google Scholar] [CrossRef] [PubMed]
- Mohni, K.N.; Thompson, P.S.; Luzwick, J.W.; Glick, G.G.; Pendleton, C.S.; Lehmann, B.D.; Pietenpol, J.A.; Cortez, D. A synthetic lethal screen identifies DNA repair pathways that sensitize cancer cells to combined ATR inhibition and cisplatin treatments. PLoS ONE 2015, 10, e0125482. [Google Scholar] [CrossRef] [PubMed]
- Sultana, R.; Abdel-Fatah, T.; Perry, C.; Moseley, P.; Albarakti, N.; Mohan, V.; Seedhouse, C.; Chan, S.; Madhusudan, S. Ataxia telangiectasia mutated and RAD3 related (ATR) protein kinase inhibition is synthetically lethal in XRCC1 deficient ovarian cancer cells. PLoS ONE 2013, 8, e57098. [Google Scholar] [CrossRef] [PubMed]
- Charrier, J.D.; Durrant, S.J.; Golec, J.M.; Kay, D.P.; Knegtel, R.M.; MacCormick, S.; Mortimore, M.; O’Donnell, M.E.; Pinder, J.L.; Reaper, P.M.; et al. Discovery of potent and selective inhibitors of ataxia telangiectasia mutated and Rad3 related (ATR) protein kinase as potential anticancer agents. J. Med. Chem. 2011, 54, 2320–2330. [Google Scholar] [CrossRef] [PubMed]
- Reaper, P.M.; Griffiths, M.R.; Long, J.M.; Charrier, J.D.; Maccormick, S.; Charlton, P.A.; Golec, J.M.; Pollard, J.R. Selective killing of ATM- or p53-deficient cancer cells through inhibition of ATR. Nat. Chem. Biol. 2011, 7, 428–430. [Google Scholar] [CrossRef] [PubMed]
- Prevo, R.; Fokas, E.; Reaper, P.M.; Charlton, P.A.; Pollard, J.R.; McKenna, W.G.; Muschel, R.J.; Brunner, T.B. The novel ATR inhibitor VE-821 increases sensitivity of pancreatic cancer cells to radiation and chemotherapy. Cancer Biol. Ther. 2012, 13, 1072–1081. [Google Scholar] [CrossRef] [PubMed]
- Huntoon, C.J.; Flatten, K.S.; Wahner Hendrickson, A.E.; Huehls, A.M.; Sutor, S.L.; Kaufmann, S.H.; Karnitz, L.M. Atr inhibition broadly sensitizes ovarian cancer cells to chemotherapy independent of BRCA status. Cancer Res. 2013, 73, 3683–3691. [Google Scholar] [CrossRef] [PubMed]
- Vavrova, J.; Zarybnicka, L.; Lukasova, E.; Rezacova, M.; Novotna, E.; Sinkorova, Z.; Tichy, A.; Pejchal, J.; Durisova, K. Inhibition of ATR kinase with the selective inhibitor VE-821 results in radiosensitization of cells of promyelocytic leukaemia (HL-60). Radiat. Environ. Biophys. 2013, 52, 471–479. [Google Scholar] [CrossRef] [PubMed]
- Salovska, B.; Fabrik, I.; Durisova, K.; Link, M.; Vavrova, J.; Rezacova, M.; Tichy, A. Radiosensitization of human leukemic HL-60 cells by ATR kinase inhibitor (VE-821): Phosphoproteomic analysis. Int. J. Mol. Sci. 2014, 15, 12007–12026. [Google Scholar] [CrossRef] [PubMed]
- Abdel-Fatah, T.M.; Middleton, F.K.; Arora, A.; Agarwal, D.; Chen, T.; Moseley, P.M.; Perry, C.; Doherty, R.; Chan, S.; Green, A.R.; et al. Untangling the ATR-CHEK1 network for prognostication, prediction and therapeutic target validation in breast cancer. Mol. Oncol. 2015, 9, 569–585. [Google Scholar] [CrossRef] [PubMed]
- Fokas, E.; Prevo, R.; Pollard, J.R.; Reaper, P.M.; Charlton, P.A.; Cornelissen, B.; Vallis, K.A.; Hammond, E.M.; Olcina, M.M.; Gillies McKenna, W.; et al. Targeting atr in vivo using the novel inhibitor VE-822 results in selective sensitization of pancreatic tumors to radiation. Cell Death Dis. 2012, 3, e441. [Google Scholar] [CrossRef] [PubMed]
- Hall, A.B.; Newsome, D.; Wang, Y.; Boucher, D.M.; Eustace, B.; Gu, Y.; Hare, B.; Johnson, M.A.; Milton, S.; Murphy, C.E.; et al. Potentiation of tumor responses to DNA damaging therapy by the selective ATR inhibitor VX-970. Oncotarget 2014, 5, 5674–5685. [Google Scholar] [PubMed]
- Foote, K.M.; Blades, K.; Cronin, A.; Fillery, S.; Guichard, S.S.; Hassall, L.; Hickson, I.; Jacq, X.; Jewsbury, P.J.; McGuire, T.M.; et al. Discovery of 4-{4-[(3R)-3-Methylmorpholin-4-yl]-6-[1-(methylsulfonyl)cyclopropyl]pyrimidin-2-yl}-1H-indole (AZ20): A potent and selective inhibitor of ATR protein kinase with monotherapy in vivo antitumor activity. J. Med. Chem. 2013, 56, 2125–2138. [Google Scholar] [CrossRef] [PubMed]
- Guichard, S.M.; Brown, E.; Odedra, R.; Hughes, A.; Heathcote, D.; Barnes, J.; Lau, A.; Powell, S.; Jones, C.D.; Nissink, W.; et al. The pre-clinical in vitro and in vivo activity of AZD6738: A potent and selective inhibitor of ATR kinase. Cancer Res. 2013. [Google Scholar] [CrossRef]
- Liu, Y.; Vidanes, G.; Lin, Y.C.; Mori, S.; Siede, W. Characterization of a Saccharomyces cerevisiae homologue of Schizosaccharomyces pombe Chk1 involved in DNA-damage-induced M-phase arrest. Mol. Gen. Genet. 2000, 262, 1132–1146. [Google Scholar] [CrossRef] [PubMed]
- Okita, N.; Minato, S.; Ohmi, E.; Tanuma, S.; Higami, Y. DNA damage-induced Chk1 autophosphorylation at Ser296 is regulated by an intramolecular mechanism. FEBS Lett. 2012, 586, 3974–3979. [Google Scholar] [CrossRef] [PubMed]
- Kasahara, K.; Goto, H.; Enomoto, M.; Tomono, Y.; Kiyono, T.; Inagaki, M. 14-3-3gamma mediates Cdc25A proteolysis to block premature mitotic entry after DNA damage. EMBO J. 2010, 29, 2802–2812. [Google Scholar] [CrossRef] [PubMed]
- Busino, L.; Donzelli, M.; Chiesa, M.; Guardavaccaro, D.; Ganoth, D.; Dorrello, N.V.; Hershko, A.; Pagano, M.; Draetta, G.F. Degradation of Cdc25A by beta-TRCP during S phase and in response to DNA damage. Nature 2003, 426, 87–91. [Google Scholar] [CrossRef] [PubMed]
- Jin, J.; Shirogane, T.; Xu, L.; Nalepa, G.; Qin, J.; Elledge, S.J.; Harper, J.W. Scfbeta-TRCP links Chk1 signaling to degradation of the Cdc25A protein phosphatase. Genes Dev. 2003, 17, 3062–3074. [Google Scholar] [CrossRef] [PubMed]
- Forrest, A.; Gabrielli, B. Cdc25B activity is regulated by 14-3-3. Oncogene 2001, 20, 4393–4401. [Google Scholar] [CrossRef] [PubMed]
- Graves, P.R.; Lovly, C.M.; Uy, G.L.; Piwnica-Worms, H. Localization of human Cdc25C is regulated both by nuclear export and 14-3-3 protein binding. Oncogene 2001, 20, 1839–1851. [Google Scholar] [CrossRef] [PubMed]
- Schwarz, J.K.; Lovly, C.M.; Piwnica-Worms, H. Regulation of the Chk2 protein kinase by oligomerization-mediated cis- and trans-phosphorylation. Mol. Cancer Res. 2003, 1, 598–609. [Google Scholar] [PubMed]
- Jack, M.T.; Woo, R.A.; Hirao, A.; Cheung, A.; Mak, T.W.; Lee, P.W. Chk2 is dispensable for p53-mediated G1 arrest but is required for a latent p53-mediated apoptotic response. Proc. Natl. Acad. Sci. USA 2002, 99, 9825–9829. [Google Scholar] [CrossRef] [PubMed]
- Ma, C.X.; Janetka, J.W.; Piwnica-Worms, H. Death by releasing the breaks: Chk1 inhibitors as cancer therapeutics. Trends Mol. Med. 2011, 17, 88–96. [Google Scholar] [CrossRef] [PubMed]
- Reinhardt, H.C.; Aslanian, A.S.; Lees, J.A.; Yaffe, M.B. p53-deficient cells rely on ATM- and ATR-mediated checkpoint signaling through the p38MAPK/MK2 pathway for survival after DNA damage. Cancer Cell 2007, 11, 175–189. [Google Scholar] [CrossRef] [PubMed]
- Bunch, R.T.; Eastman, A. Enhancement of cisplatin-induced cytotoxicity by 7-hydroxystaurosporine (UCN-01), a new G2-checkpoint inhibitor. Clin. Cancer Res. 1996, 2, 791–797. [Google Scholar] [PubMed]
- Tse, A.N.; Schwartz, G.K. Potentiation of cytotoxicity of topoisomerase i poison by concurrent and sequential treatment with the checkpoint inhibitor UCN-01 involves disparate mechanisms resulting in either p53-independent clonogenic suppression or p53-dependent mitotic catastrophe. Cancer Res. 2004, 64, 6635–6644. [Google Scholar] [CrossRef] [PubMed]
- Fuse, E.; Tanii, H.; Kurata, N.; Kobayashi, H.; Shimada, Y.; Tamura, T.; Sasaki, Y.; Tanigawara, Y.; Lush, R.D.; Headlee, D.; et al. Unpredicted clinical pharmacology of UCN-01 caused by specific binding to human alpha1-acid glycoprotein. Cancer Res. 1998, 58, 3248–3253. [Google Scholar] [PubMed]
- Kortmansky, J.; Shah, M.A.; Kaubisch, A.; Weyerbacher, A.; Yi, S.; Tong, W.; Sowers, R.; Gonen, M.; O’Reilly, E.; Kemeny, N.; et al. Phase I trial of the cyclin-dependent kinase inhibitor and protein kinase C inhibitor 7-hydroxystaurosporine in combination with fluorouracil in patients with advanced solid tumors. J. Clin. Oncol. 2005, 23, 1875–1884. [Google Scholar] [CrossRef] [PubMed]
- Dent, P.; Tang, Y.; Yacoub, A.; Dai, Y.; Fisher, P.B.; Grant, S. Chk1 inhibitors in combination chemotherapy: Thinking beyond the cell cycle. Mol. Interv. 2011, 11, 133–140. [Google Scholar] [CrossRef] [PubMed]
- Matthews, D.J.; Yakes, F.M.; Chen, J.; Tadano, M.; Bornheim, L.; Clary, D.O.; Tai, A.; Wagner, J.M.; Miller, N.; Kim, Y.D.; et al. Pharmacological abrogation of S-phase checkpoint enhances the anti-tumor activity of gemcitabine in vivo. Cell Cycle 2007, 6, 104–110. [Google Scholar] [CrossRef] [PubMed]
- Sausville, E.; Lorusso, P.; Carducci, M.; Carter, J.; Quinn, M.F.; Malburg, L.; Azad, N.; Cosgrove, D.; Knight, R.; Barker, P.; et al. Phase I dose-escalation study of AZD7762, a checkpoint kinase inhibitor, in combination with gemcitabine in US patients with advanced solid tumors. Cancer Chemother. Pharmacol. 2014, 73, 539–549. [Google Scholar] [CrossRef] [PubMed]
- Weiss, G.J.; Donehower, R.C.; Iyengar, T.; Ramanathan, R.K.; Lewandowski, K.; Westin, E.; Hurt, K.; Hynes, S.M.; Anthony, S.P.; McKane, S. Phase I dose-escalation study to examine the safety and tolerability of ly2603618, a checkpoint 1 kinase inhibitor, administered 1 day after pemetrexed 500 mg/m2 every 21 days in patients with cancer. Investig. New Drug 2013, 31, 136–144. [Google Scholar] [CrossRef] [PubMed]
- King, C.; Diaz, H.; Barnard, D.; Barda, D.; Clawson, D.; Blosser, W.; Cox, K.; Guo, S.; Marshall, M. Characterization and preclinical development of ly2603618: A selective and potent Chk1 inhibitor. Investig. New Drugs 2014, 32, 213–226. [Google Scholar] [CrossRef] [PubMed]
- Calvo, E.; Chen, V.; Marshall, M.; Ohnmacht, U.; Hynes, S.; Kumm, E.; Diaz, H.B.; Barnard, D.; Merzoug, F.; Huber, L.; et al. Preclinical analyses and phase I evaluation of ly2603618 administered in combination with pemetrexed and cisplatin in patients with advanced cancer. Investig. New Drug 2014, 32, 955–968. [Google Scholar] [CrossRef] [PubMed]
- Daud, A.I.; Ashworth, M.T.; Strosberg, J.; Goldman, J.W.; Mendelson, D.; Springett, G.; Venook, A.P.; Loechner, S.; Rosen, L.S.; Shanahan, F.; et al. Phase I dose-escalation trial of checkpoint kinase 1 inhibitor MK-8776 as monotherapy and in combination with gemcitabine in patients with advanced solid tumors. J. Clin. Oncol. 2015, 33, 1060–1066. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, N.; Broome, M.; Hunter, T. Regulation of the human WEE1HU Cdk tyrosine 15-kinase during the cell cycle. EMBO J. 1995, 14, 1878–1891. [Google Scholar] [PubMed]
- Watanabe, N.; Arai, H.; Nishihara, Y.; Taniguchi, M.; Watanabe, N.; Hunter, T.; Osada, H. M-phase kinases induce phospho-dependent ubiquitination of somatic Wee1 by SCFβ-TrCP. Proc. Natl. Acad. Sci. USA 2004, 101, 4419–4424. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; Kumagai, A.; Dunphy, W.G. Positive regulation of Wee1 by Chk1 and 14-3-3 proteins. Mol. Biol. Cell 2001, 12, 551–563. [Google Scholar] [CrossRef] [PubMed]
- Parker, L.L.; Piwnica-Worms, H. Inactivation of the p34Cdc2-cyclin B complex by the human Wee1 tyrosine kinase. Science 1992, 257, 1955–1957. [Google Scholar] [CrossRef] [PubMed]
- Saini, P.; Li, Y.; Dobbelstein, M. Wee1 is required to sustain ATR/Chk1 signaling upon replicative stress. Oncotarget 2015, 6, 13072–13087. [Google Scholar] [PubMed]
- Panek, R.L.; Lu, G.H.; Klutchko, S.R.; Batley, B.L.; Dahring, T.K.; Hamby, J.M.; Hallak, H.; Doherty, A.M.; Keiser, J.A. In vitro pharmacological characterization of PD 166285, a new nanomolar potent and broadly active protein tyrosine kinase inhibitor. J. Pharmacol. Exp. Ther. 1997, 283, 1433–1444. [Google Scholar] [PubMed]
- Wang, Y.; Li, J.; Booher, R.N.; Kraker, A.; Lawrence, T.; Leopold, W.R.; Sun, Y. Radiosensitization of p53 mutant cells by PD0166285, a novel G(2) checkpoint abrogator. Cancer Res. 2001, 61, 8211–8217. [Google Scholar] [PubMed]
- Palmer, B.D.; Thompson, A.M.; Booth, R.J.; Dobrusin, E.M.; Kraker, A.J.; Lee, H.H.; Lunney, E.A.; Mitchell, L.H.; Ortwine, D.F.; Smaill, J.B.; et al. 4-Phenylpyrrolo[3,4-c]carbazole-1,3(2H,6H)-dione inhibitors of the checkpoint kinase Wee1. Structure-activity relationships for chromophore modification and phenyl ring substitution. J. Med. Chem. 2006, 49, 4896–4911. [Google Scholar] [CrossRef] [PubMed]
- Arora, S.; Bisanz, K.M.; Peralta, L.A.; Basu, G.D.; Choudhary, A.; Tibes, R.; Azorsa, D.O. RNAi screening of the kinome identifies modulators of cisplatin response in ovarian cancer cells. Gynecol. Oncol. 2010, 118, 220–227. [Google Scholar] [CrossRef] [PubMed]
- Hirai, H.; Iwasawa, Y.; Okada, M.; Arai, T.; Nishibata, T.; Kobayashi, M.; Kimura, T.; Kaneko, N.; Ohtani, J.; Yamanaka, K.; et al. Small-molecule inhibition of Wee1 kinase by MK-1775 selectively sensitizes p53-deficient tumor cells to DNA-damaging agents. Mol. Cancer Ther. 2009, 8, 2992–3000. [Google Scholar] [CrossRef] [PubMed]
- Mizuarai, S.; Yamanaka, K.; Itadani, H.; Arai, T.; Nishibata, T.; Hirai, H.; Kotani, H. Discovery of gene expression-based pharmacodynamic biomarker for a p53 context-specific anti-tumor drug Wee1 inhibitor. Mol. Cancer 2009, 8, 34. [Google Scholar] [CrossRef] [PubMed]
- Hirai, H.; Arai, T.; Okada, M.; Nishibata, T.; Kobayashi, M.; Sakai, N.; Imagaki, K.; Ohtani, J.; Sakai, T.; Yoshizumi, T.; et al. MK-1775, a small molecule Wee1 inhibitor, enhances anti-tumor efficacy of various DNA-damaging agents, including 5-fluorouracil. Cancer Biol Ther 2010, 9, 514–522. [Google Scholar] [CrossRef] [PubMed]
- Bridges, K.A.; Hirai, H.; Buser, C.A.; Brooks, C.; Liu, H.; Buchholz, T.A.; Molkentine, J.M.; Mason, K.A.; Meyn, R.E. MK-1775, a novel Wee1 kinase inhibitor, radiosensitizes p53-defective human tumor cells. Clin. Cancer Res. 2011, 17, 5638–5648. [Google Scholar] [CrossRef] [PubMed]
- Rajeshkumar, N.V.; de Oliveira, E.; Ottenhof, N.; Watters, J.; Brooks, D.; Demuth, T.; Shumway, S.D.; Mizuarai, S.; Hirai, H.; Maitra, A.; et al. MK-1775, a potent Wee1 inhibitor, synergizes with gemcitabine to achieve tumor regressions, selectively in p53-deficient pancreatic cancer xenografts. Clin. Cancer Res. 2011, 17, 2799–2806. [Google Scholar] [CrossRef] [PubMed]
- Do, K.; Wilsker, D.; Ji, J.; Zlott, J.; Freshwater, T.; Kinders, R.J.; Collins, J.; Chen, A.P.; Doroshow, J.H.; Kummar, S. Phase I study of single-agent AZD1775 (MK-1775), a Wee1 kinase inhibitor, in patients with refractory solid tumors. J. Clin. Oncol. 2015. [Google Scholar] [CrossRef] [PubMed]
- Carrassa, L.; Chila, R.; Lupi, M.; Ricci, F.; Celenza, C.; Mazzoletti, M.; Broggini, M.; Damia, G. Combined inhibition of chk1 and wee1: In vitro synergistic effect translates to tumor growth inhibition in vivo. Cell Cycle 2012, 11, 2507–2517. [Google Scholar] [CrossRef] [PubMed]
- Russell, M.R.; Levin, K.; Rader, J.; Belcastro, L.; Li, Y.; Martinez, D.; Pawel, B.; Shumway, S.D.; Maris, J.M.; Cole, K.A. Combination therapy targeting the Chk1 and Wee1 kinases shows therapeutic efficacy in neuroblastoma. Cancer Res. 2013, 73, 776–784. [Google Scholar] [CrossRef] [PubMed]
- Chila, R.; Basana, A.; Lupi, M.; Guffanti, F.; Gaudio, E.; Rinaldi, A.; Cascione, L.; Restelli, V.; Tarantelli, C.; Bertoni, F.; et al. Combined inhibition of Chk1 and Wee1 as a new therapeutic strategy for mantle cell lymphoma. Oncotarget 2015, 6, 3394–3408. [Google Scholar] [PubMed]
- Magnussen, G.I.; Emilsen, E.; Giller Fleten, K.; Engesaeter, B.; Nahse-Kumpf, V.; Fjaer, R.; Slipicevic, A.; Florenes, V.A. Combined inhibition of the cell cycle related proteins Wee1 and Chk1/2 induces synergistic anti-cancer effect in melanoma. BMC Cancer 2015. [Google Scholar] [CrossRef] [PubMed]
- Raman, M.; Earnest, S.; Zhang, K.; Zhao, Y.; Cobb, M.H. TAO kinases mediate activation of p38 in response to DNA damage. EMBO J. 2007, 26, 2005–2014. [Google Scholar] [CrossRef] [PubMed]
- Brancho, D.; Tanaka, N.; Jaeschke, A.; Ventura, J.J.; Kelkar, N.; Tanaka, Y.; Kyuuma, M.; Takeshita, T.; Flavell, R.A.; Davis, R.J. Mechanism of p38 MAP kinase activation in vivo. Genes Dev. 2003, 17, 1969–1978. [Google Scholar] [CrossRef] [PubMed]
- Ben-Levy, R.; Leighton, I.A.; Doza, Y.N.; Attwood, P.; Morrice, N.; Marshall, C.J.; Cohen, P. Identification of novel phosphorylation sites required for activation of MAPKAP kinase-2. EMBO J. 1995, 14, 5920–5930. [Google Scholar] [PubMed]
- Reinhardt, H.C.; Hasskamp, P.; Schmedding, I.; Morandell, S.; van Vugt, M.A.; Wang, X.; Linding, R.; Ong, S.E.; Weaver, D.; Carr, S.A.; et al. DNA damage activates a spatially distinct late cytoplasmic cell-cycle checkpoint network controlled by MK2-mediated RNA stabilization. Mol. Cell 2010, 40, 34–49. [Google Scholar] [CrossRef] [PubMed]
- Morandell, S.; Reinhardt, H.C.; Cannell, I.G.; Kim, J.S.; Ruf, D.M.; Mitra, T.; Couvillon, A.D.; Jacks, T.; Yaffe, M.B. A reversible gene-targeting strategy identifies synthetic lethal interactions between MK2 and p53 in the DNA damage response in vivo. Cell Rep. 2013, 5, 868–877. [Google Scholar] [CrossRef] [PubMed]
- Dietlein, F.; Kalb, B.; Jokic, M.; Noll, E.M.; Strong, A.; Tharun, L.; Ozretic, L.; Kunstlinger, H.; Kambartel, K.; Randerath, W.J.; et al. A synergistic interaction between Chk1- and MK2 inhibitors in KRAS-mutant cancer. Cell 2015, 162, 146–159. [Google Scholar] [CrossRef] [PubMed]
- Petitjean, A.; Achatz, M.I.; Borresen-Dale, A.L.; Hainaut, P.; Olivier, M. Tp53 mutations in human cancers: Functional selection and impact on cancer prognosis and outcomes. Oncogene 2007, 26, 2157–2165. [Google Scholar] [CrossRef] [PubMed]
- Ventura, A.; Kirsch, D.G.; McLaughlin, M.E.; Tuveson, D.A.; Grimm, J.; Lintault, L.; Newman, J.; Reczek, E.E.; Weissleder, R.; Jacks, T. Restoration of p53 function leads to tumour regression in vivo. Nature 2007, 445, 661–665. [Google Scholar] [CrossRef] [PubMed]
- Khoo, K.H.; Verma, C.S.; Lane, D.P. Drugging the p53 pathway: Understanding the route to clinical efficacy. Nat. Rev. Drug Discov. 2014, 13, 217–236. [Google Scholar] [CrossRef] [PubMed]
- Vassilev, L.T.; Vu, B.T.; Graves, B.; Carvajal, D.; Podlaski, F.; Filipovic, Z.; Kong, N.; Kammlott, U.; Lukacs, C.; Klein, C.; et al. In vivo activation of the p53 pathway by small-molecule antagonists of MDM2. Science 2004, 303, 844–848. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Rousseau, R.F.; Middleton, S.A.; Nichols, G.L.; Newell, D.R.; Lunec, J.; Tweddle, D.A. Pre-clinical evaluation of the MDM2-p53 antagonist RG7388 alone and in combination with chemotherapy in neuroblastoma. Oncotarget 2015, 6, 10207–10221. [Google Scholar] [PubMed]
- Ding, Q.; Zhang, Z.; Liu, J.J.; Jiang, N.; Zhang, J.; Ross, T.M.; Chu, X.J.; Bartkovitz, D.; Podlaski, F.; Janson, C.; et al. Discovery of RG7388, a potent and selective p53-MDM2 inhibitor in clinical development. J. Med. Chem. 2013, 56, 5979–5983. [Google Scholar] [CrossRef] [PubMed]
- Higgins, B.; Glenn, K.; Walz, A.; Tovar, C.; Filipovic, Z.; Hussain, S.; Lee, E.; Kolinsky, K.; Tannu, S.; Adames, V.; et al. Preclinical optimization of MDM2 antagonist scheduling for cancer treatment by using a model-based approach. Clin. Cancer Res. 2014, 20, 3742–3752. [Google Scholar] [CrossRef] [PubMed]
- Bulavin, D.V.; Phillips, C.; Nannenga, B.; Timofeev, O.; Donehower, L.A.; Anderson, C.W.; Appella, E.; Fornace, A.J., Jr. Inactivation of the Wip1 phosphatase inhibits mammary tumorigenesis through p38 MAPK-mediated activation of the p16(INK4A)-p19(ARF) pathway. Nat. Genet. 2004, 36, 343–350. [Google Scholar] [CrossRef] [PubMed]
- Demidov, O.N.; Timofeev, O.; Lwin, H.N.; Kek, C.; Appella, E.; Bulavin, D.V. Wip1 phosphatase regulates p53-dependent apoptosis of stem cells and tumorigenesis in the mouse intestine. Cell Stem Cell 2007, 1, 180–190. [Google Scholar] [CrossRef] [PubMed]
- Saito-Ohara, F.; Imoto, I.; Inoue, J.; Hosoi, H.; Nakagawara, A.; Sugimoto, T.; Inazawa, J. PPM1D is a potential target for 17q gain in neuroblastoma. Cancer Res. 2003, 63, 1876–1883. [Google Scholar] [PubMed]
- Wang, P.; Rao, J.; Yang, H.; Zhao, H.; Yang, L. Ppm1d silencing by lentiviral-mediated RNA interference inhibits proliferation and invasion of human glioma cells. J. Huazhong Univ. Sci. Technol. 2011, 31, 94–99. [Google Scholar] [CrossRef] [PubMed]
- Gilmartin, A.G.; Faitg, T.H.; Richter, M.; Groy, A.; Seefeld, M.A.; Darcy, M.G.; Peng, X.; Federowicz, K.; Yang, J.; Zhang, S.Y.; et al. Allosteric Wip1 phosphatase inhibition through flap-subdomain interaction. Nat. Chem. Biol. 2014, 10, 181–187. [Google Scholar] [CrossRef] [PubMed]
- Richter, M.; Dayaram, T.; Gilmartin, A.G.; Ganji, G.; Pemmasani, S.K.; van der Key, H.; Shohet, J.M.; Donehower, L.A.; Kumar, R. Wip1 phosphatase as a potential therapeutic target in neuroblastoma. PLoS One 2015, 10, e0115635. [Google Scholar] [CrossRef] [PubMed]
- Emelyanov, A.; Bulavin, D.V. Wip1 phosphatase in breast cancer. Oncogene 2014. [Google Scholar] [CrossRef] [PubMed]
- Kleiblova, P.; Shaltiel, I.A.; Benada, J.; Sevcik, J.; Pechackova, S.; Pohlreich, P.; Voest, E.E.; Dundr, P.; Bartek, J.; Kleibl, Z.; et al. Gain-of-function mutations of Ppm1d/Wip1 impair the p53-dependent g1 checkpoint. J. Cell Biol. 2013, 201, 511–521. [Google Scholar] [CrossRef] [PubMed]
- Lapenna, S.; Giordano, A. Cell cycle kinases as therapeutic targets for cancer. Nat. Rev. Drug Discov. 2009, 8, 547–566. [Google Scholar] [CrossRef] [PubMed]
- Malumbres, M.; Barbacid, M. Cell cycle, Cdks and cancer: A changing paradigm. Nat. Rev. Cancer 2009, 9, 153–166. [Google Scholar] [CrossRef] [PubMed]
- Yu, Q.; Geng, Y.; Sicinski, P. Specific protection against breast cancers by cyclin D1 ablation. Nature 2001, 411, 1017–1021. [Google Scholar] [CrossRef] [PubMed]
- Landis, M.W.; Pawlyk, B.S.; Li, T.; Sicinski, P.; Hinds, P.W. Cyclin D1-dependent kinase activity in murine development and mammary tumorigenesis. Cancer Cell 2006, 9, 13–22. [Google Scholar] [CrossRef] [PubMed]
- Puyol, M.; Martin, A.; Dubus, P.; Mulero, F.; Pizcueta, P.; Khan, G.; Guerra, C.; Santamaria, D.; Barbacid, M. A synthetic lethal interaction between K-Ras oncogenes and Cdk4 unveils a therapeutic strategy for non-small cell lung carcinoma. Cancer Cell 2010, 18, 63–73. [Google Scholar] [CrossRef] [PubMed]
- Sicinska, E.; Aifantis, I.; Le Cam, L.; Swat, W.; Borowski, C.; Yu, Q.; Ferrando, A.A.; Levin, S.D.; Geng, Y.; von Boehmer, H.; et al. Requirement for cyclin D3 in lymphocyte development and T cell leukemias. Cancer Cell 2003, 4, 451–461. [Google Scholar] [CrossRef]
- Schmitz, R.; Young, R.M.; Ceribelli, M.; Jhavar, S.; Xiao, W.; Zhang, M.; Wright, G.; Shaffer, A.L.; Hodson, D.J.; Buras, E.; et al. Burkitt lymphoma pathogenesis and therapeutic targets from structural and functional genomics. Nature 2012, 490, 116–120. [Google Scholar] [CrossRef] [PubMed]
- Monahan, K.B.; Rozenberg, G.I.; Krishnamurthy, J.; Johnson, S.M.; Liu, W.; Bradford, M.K.; Horner, J.; Depinho, R.A.; Sharpless, N.E. Somatic p16(Ink4A) loss accelerates melanomagenesis. Oncogene 2010, 29, 5809–5817. [Google Scholar] [CrossRef] [PubMed]
- Curtin, J.A.; Fridlyand, J.; Kageshita, T.; Patel, H.N.; Busam, K.J.; Kutzner, H.; Cho, K.H.; Aiba, S.; Brocker, E.B.; LeBoit, P.E.; et al. Distinct sets of genetic alterations in melanoma. N. Engl. J. Med. 2005, 353, 2135–2147. [Google Scholar] [CrossRef] [PubMed]
- Smalley, K.S.; Lioni, M.; Dalla Palma, M.; Xiao, M.; Desai, B.; Egyhazi, S.; Hansson, J.; Wu, H.; King, A.J.; van Belle, P.; et al. Increased cyclin D1 expression can mediate BRAF inhibitor resistance in BRAF V600E-mutated melanomas. Mol. Cancer Ther. 2008, 7, 2876–2883. [Google Scholar] [CrossRef] [PubMed]
- Arguello, F.; Alexander, M.; Sterry, J.A.; Tudor, G.; Smith, E.M.; Kalavar, N.T.; Greene, J.F., Jr.; Koss, W.; Morgan, C.D.; Stinson, S.F.; et al. Flavopiridol induces apoptosis of normal lymphoid cells, causes immunosuppression, and has potent antitumor activity in vivo against human leukemia and lymphoma xenografts. Blood 1998, 91, 2482–2490. [Google Scholar] [PubMed]
- Parker, B.W.; Kaur, G.; Nieves-Neira, W.; Taimi, M.; Kohlhagen, G.; Shimizu, T.; Losiewicz, M.D.; Pommier, Y.; Sausville, E.A.; Senderowicz, A.M. Early induction of apoptosis in hematopoietic cell lines after exposure to flavopiridol. Blood 1998, 91, 458–465. [Google Scholar] [PubMed]
- Joshi, K.S.; Rathos, M.J.; Joshi, R.D.; Sivakumar, M.; Mascarenhas, M.; Kamble, S.; Lal, B.; Sharma, S. In vitro antitumor properties of a novel cyclin-dependent kinase inhibitor, p276–00. Mol. Cancer Ther. 2007, 6, 918–925. [Google Scholar] [CrossRef] [PubMed]
- Finn, R.S.; Dering, J.; Conklin, D.; Kalous, O.; Cohen, D.J.; Desai, A.J.; Ginther, C.; Atefi, M.; Chen, I.; Fowst, C.; et al. PD 0332991, a selective cyclin D kinase 4/6 inhibitor, preferentially inhibits proliferation of luminal estrogen receptor-positive human breast cancer cell lines in vitro. Breast Cancer Res. 2009. [Google Scholar] [CrossRef] [PubMed]
- Choi, Y.J.; Li, X.; Hydbring, P.; Sanda, T.; Stefano, J.; Christie, A.L.; Signoretti, S.; Look, A.T.; Kung, A.L.; von Boehmer, H.; et al. The requirement for cyclin D function in tumor maintenance. Cancer Cell 2012, 22, 438–451. [Google Scholar] [CrossRef] [PubMed]
- Sawai, C.M.; Freund, J.; Oh, P.; Ndiaye-Lobry, D.; Bretz, J.C.; Strikoudis, A.; Genesca, L.; Trimarchi, T.; Kelliher, M.A.; Clark, M.; et al. Therapeutic targeting of the cyclin D3:Cdk4/6 complex in T cell leukemia. Cancer Cell 2012, 22, 452–465. [Google Scholar] [CrossRef] [PubMed]
- Leonard, J.P.; LaCasce, A.S.; Smith, M.R.; Noy, A.; Chirieac, L.R.; Rodig, S.J.; Yu, J.Q.; Vallabhajosula, S.; Schoder, H.; English, P.; et al. Selective Cdk4/6 inhibition with tumor responses by PD0332991 in patients with mantle cell lymphoma. Blood 2012, 119, 4597–4607. [Google Scholar] [CrossRef] [PubMed]
- Murphy, C.G.; Dickler, M.N. The role of Cdk4/6 inhibition in breast cancer. Oncologist 2015, 20, 483–490. [Google Scholar] [CrossRef] [PubMed]
- Anders, L.; Ke, N.; Hydbring, P.; Choi, Y.J.; Widlund, H.R.; Chick, J.M.; Zhai, H.; Vidal, M.; Gygi, S.P.; Braun, P.; et al. A systematic screen for Cdk4/6 substrates links foxm1 phosphorylation to senescence suppression in cancer cells. Cancer Cell 2011, 20, 620–634. [Google Scholar] [CrossRef] [PubMed]
- Yadav, V.; Chen, S.H.; Yue, Y.G.; Buchanan, S.; Beckmann, R.P.; Peng, S.B. Co-targeting BRAF and cyclin dependent kinases 4/6 for BRAF mutant cancers. Pharmacol. Ther. 2015, 149, 139–149. [Google Scholar] [CrossRef] [PubMed]
- Derenzini, E.; Agostinelli, C.; Imbrogno, E.; Iacobucci, I.; Casadei, B.; Brighenti, E.; Righi, S.; Fuligni, F.; di Rora, A.G.L.; Ferrari, A.; et al. Constitutive activation of the DNA damage response pathway as a novel therapeutic target in diffuse large B-cell lymphoma. Oncotarget 2015, 6, 6553–6569. [Google Scholar] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Benada, J.; Macurek, L. Targeting the Checkpoint to Kill Cancer Cells. Biomolecules 2015, 5, 1912-1937. https://doi.org/10.3390/biom5031912
Benada J, Macurek L. Targeting the Checkpoint to Kill Cancer Cells. Biomolecules. 2015; 5(3):1912-1937. https://doi.org/10.3390/biom5031912
Chicago/Turabian StyleBenada, Jan, and Libor Macurek. 2015. "Targeting the Checkpoint to Kill Cancer Cells" Biomolecules 5, no. 3: 1912-1937. https://doi.org/10.3390/biom5031912
APA StyleBenada, J., & Macurek, L. (2015). Targeting the Checkpoint to Kill Cancer Cells. Biomolecules, 5(3), 1912-1937. https://doi.org/10.3390/biom5031912