
Genome Instability in Development and Aging: Insights from Nucleotide Excision Repair in Humans, Mice, and Worms

{kind=link}
{kind=link}
Abstract
:1. Introduction
2. DNA Damage Responses in Living Organisms
2.1. DNA Repair Systems
2.2. NER Deficiencies in Humans: Cancer versus Development and Aging
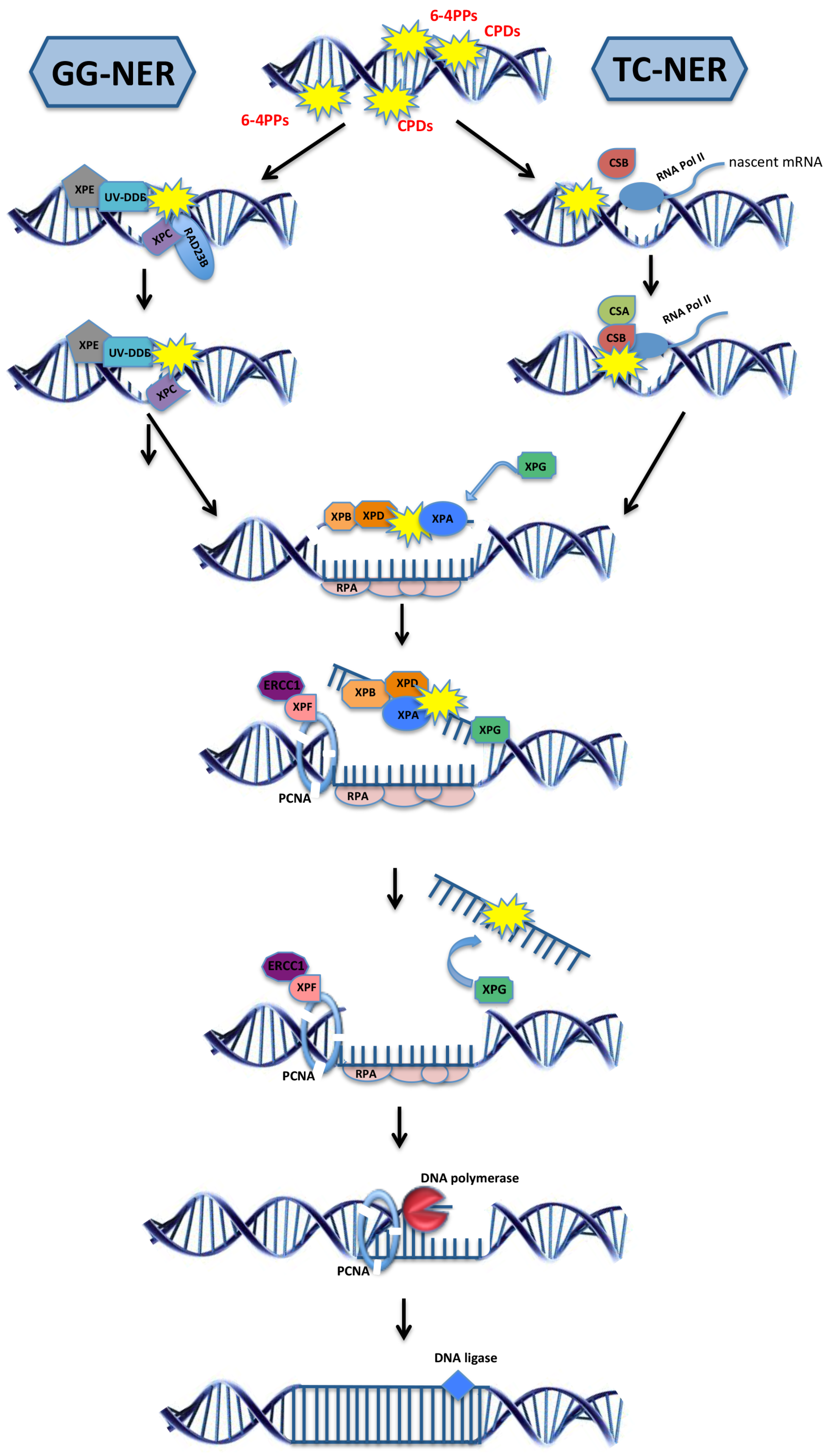
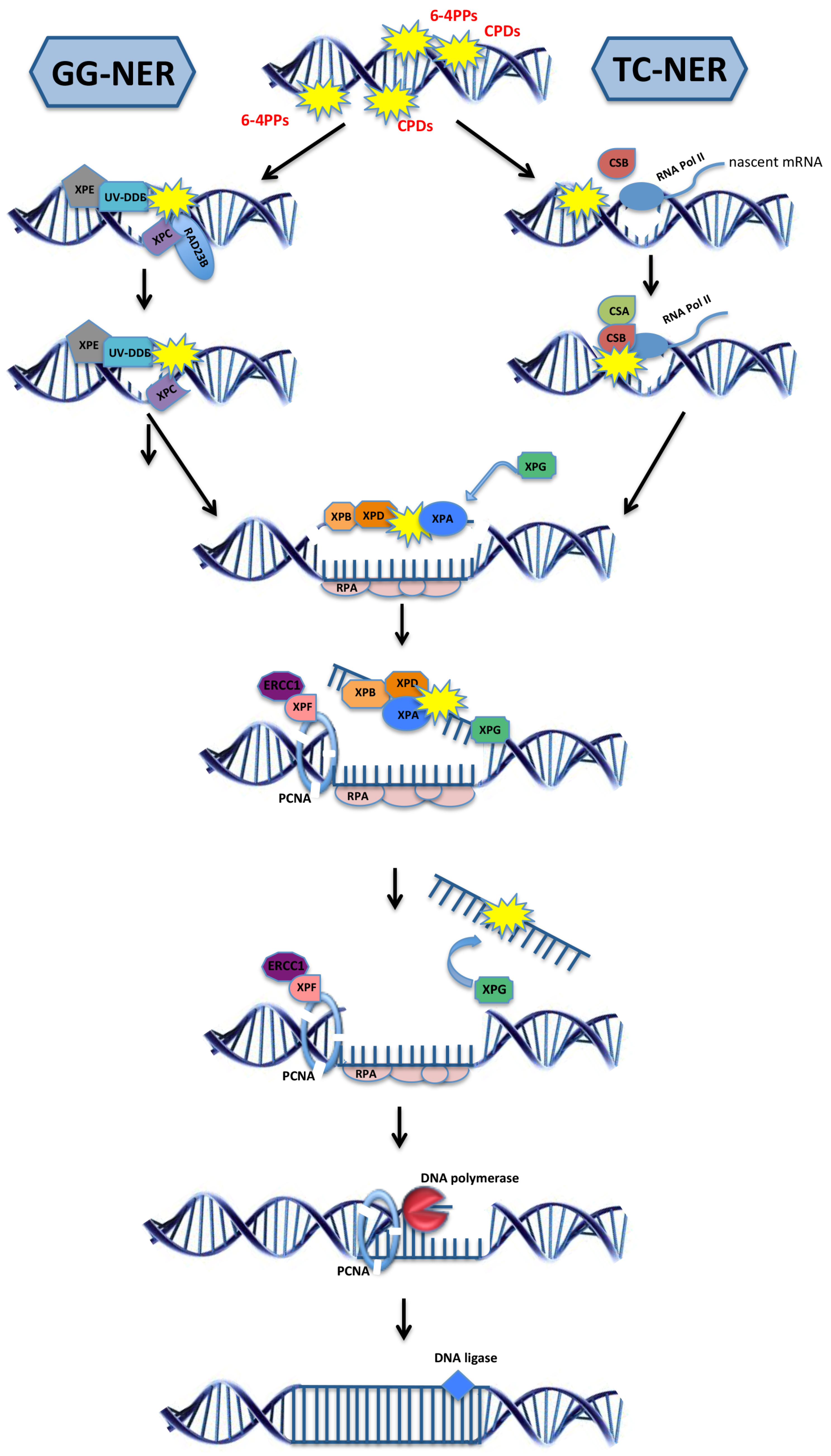
2.3. Model Organisms as a Tool to Study NER-Deficiency Syndromes
2.3.1. NER Mutant Mice: Linking DNA Damage to Aging and Longevity
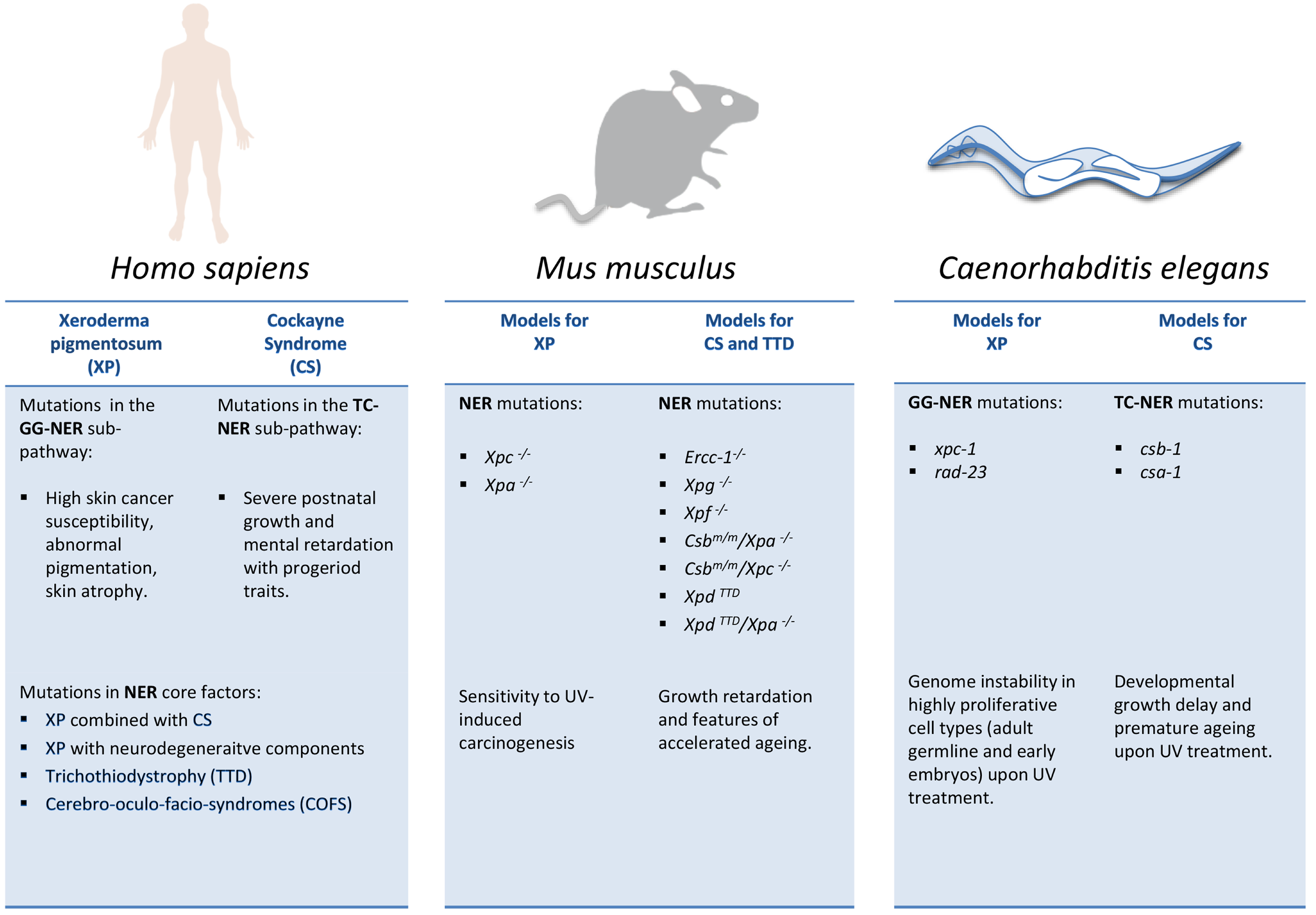
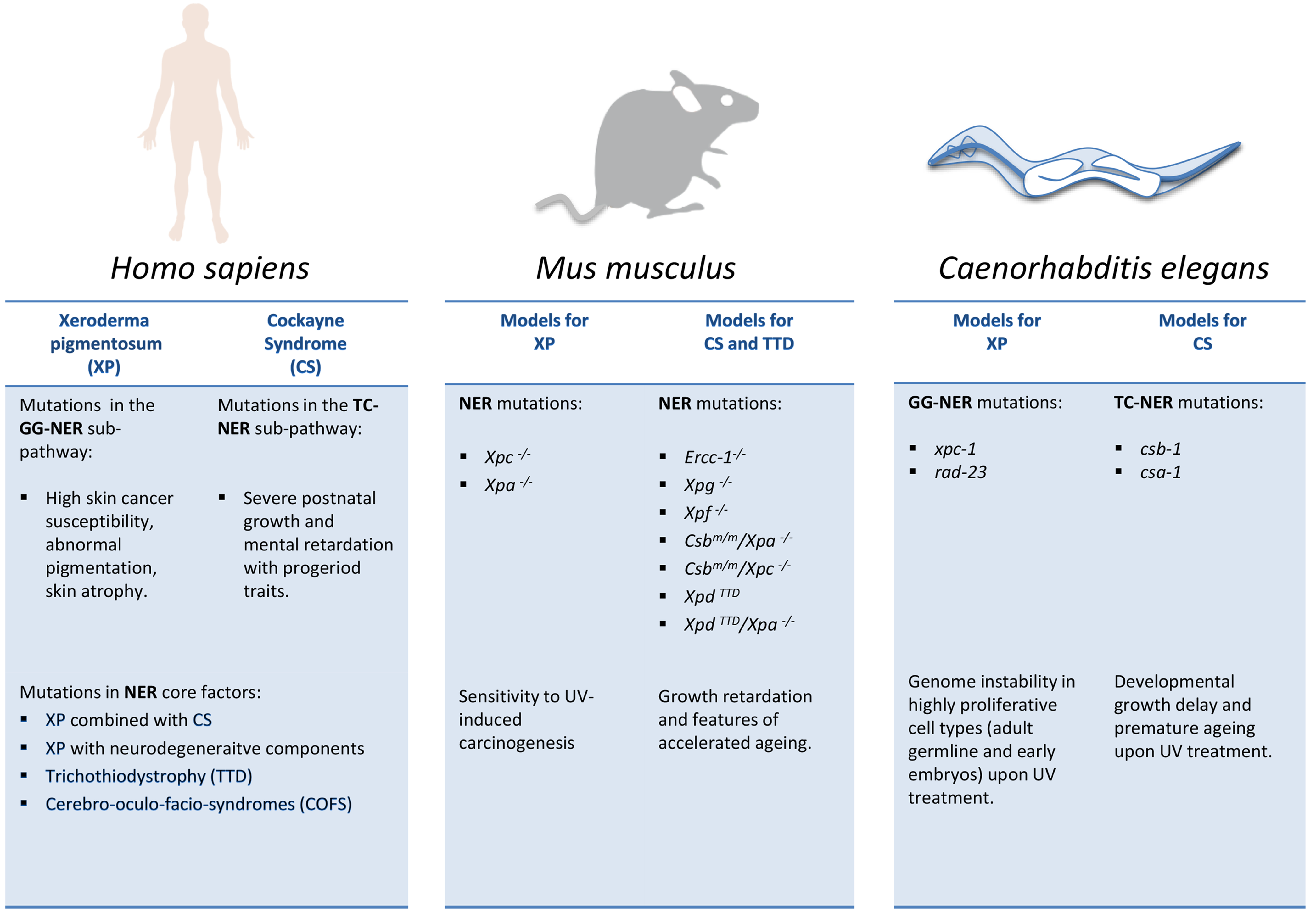
2.3.2. C. elegans as a Metazoan Model for NER Deficiencies
3. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Kirkwood, T.B. Understanding the odd science of aging. Cell 2005, 120, 437–447. [Google Scholar] [CrossRef] [PubMed]
- Schumacher, B.; Garinis, G.A.; Hoeijmakers, J.H.J. Age to survive: DNA damage and aging. Trends Genet. 2008, 24, 77–85. [Google Scholar] [CrossRef] [PubMed]
- Vijg, J.; Suh, Y. Genome instability and aging. Annu. Rev. Physiol. 2013, 75, 645–668. [Google Scholar] [CrossRef] [PubMed]
- Baker, D.J.; Jeganathan, K.B.; Cameron, J.D.; Thompson, M.; Juneja, S.; Kopecka, A.; Kumar, R.; Jenkins, R.B.; de Groen, P.C.; Roche, P.; et al. BubR1 insufficiency causes early onset of aging-associated phenotypes and infertility in mice. Nat. Genet. 2004, 36, 744–749. [Google Scholar] [CrossRef] [PubMed]
- Hoeijmakers, J.H.J. DNA damage, aging, and cancer. N. Engl. J. Med. 2009, 361, 1475–1485. [Google Scholar] [CrossRef] [PubMed]
- DiGiovanna, J.J.; Kraemer, K.H. Shining a light on xeroderma pigmentosum. J. Invest. Dermatol. 2012, 132, 785–796. [Google Scholar] [CrossRef] [PubMed]
- Tamura, D.; DiGiovanna, J.J.; Khan, S.G.; Kraemer, K.H. Living with xeroderma pigmentosum: Comprehensive photoprotection for highly photosensitive patients. Photodermatol. Photoimmunol. Photomed. 2014, 30, 146–152. [Google Scholar] [CrossRef] [PubMed]
- Fousteri, M.; Vermeulen, W.; van Zeeland, A.A.; Mullenders, L.H. Cockayne syndrome A and B proteins differentially regulate recruitment of chromatin remodeling and repair factors to stalled RNA polymerase II in vivo. Mol. Cell 2006, 23, 471–482. [Google Scholar] [CrossRef] [PubMed]
- Laugel, V.; Dalloz, C.; Durand, M.; Sauvanaud, F.; Kristensen, U.; Vincent, M.C.; Pasquier, L.; Odent, S.; Cormier-Daire, V.; Gener, B.; et al. Mutation update for the CSB/ERCC6 and CSA/ERCC8 genes involved in Cockayne syndrome. Hum. Mutat. 2010, 31, 113–126. [Google Scholar] [CrossRef] [PubMed]
- Cleaver, J.E.; Lam, E.T.; Revet, I. Disorders of nucleotide excision repair: The genetic and molecular basis of heterogeneity. Nat. Rev. Genet. 2009, 10, 756–768. [Google Scholar] [CrossRef] [PubMed]
- Egly, J.M.; Coin, F.A. History of TFIIH: Two decades of molecular biology on a pivotal transcription/repair factor. DNA Repair 2011, 10, 714–721. [Google Scholar] [CrossRef] [PubMed]
- Wolters, S.; Schumacher, B. Genome maintenance and transcription integrity in aging and disease. Front. Genet. 2013. [Google Scholar] [CrossRef] [PubMed]
- De Vries, A.; van Oostrom, C.T.; Hofhuis, F.M.; Dortant, P.M.; Berg, R.J.; de Gruijl, F.R.; Wester, P.W.; van Kreijl, C.F.; Capel, P.J.; van Steeg, H.; et al. Increased susceptibility to ultraviolet-B and carcinogens of mice lacking the DNA excision repair gene XPA. Nature 1995, 377, 169–173. [Google Scholar] [CrossRef] [PubMed]
- Van der Horst, G.T.; van Steeg, H.; Berg, R.J.; van Gool, A.J.; de Wit, J.; Weeda, G.; Morreau, H.; Beems, R.B.; van Kreijl, C.F.; de Gruijl, F.R.; et al. Defective transcription-coupled repair in Cockayne syndrome B mice is associated with skin cancer predisposition. Cell 1997, 89, 425–435. [Google Scholar] [CrossRef]
- Sands, A.T.; Abuin, A.; Sanchez, A.; Conti, C.J.; Bradley, A. High susceptibility to ultraviolet-induced carcinogenesis in mice lacking XPC. Nature 1995, 377, 162–165. [Google Scholar] [CrossRef] [PubMed]
- Weeda, G.; Donker, I.; de Wit, J.; Morreau, H.; Janssens, R.; Vissers, C.J.; Nigg, A.; van Steeg, H.; Bootsma, D.; Hoeijmakers, J.H. Disruption of mouse ERCC1 results in a novel repair syndrome with growth failure, nuclear abnormalities and senescence. Curr. Biol. 1997, 7, 427–439. [Google Scholar] [CrossRef]
- Tian, M.; Shinkura, R.; Shinkura, N.; Alt, F.W. Growth retardation, early death, and DNA repair defects in mice deficient for the nucleotide excision repair enzyme XPF. Mol. Cell. Biol. 2004, 24, 1200–1205. [Google Scholar] [CrossRef] [PubMed]
- De Boer, J.; Andressoo, J.O.; de Wit, J.; Huijmans, J.; Beems, R.B.; van Steeg, H.; Weeda, G.; van der Horst, G.T.; van Leeuwen, W.; Themmen, A.P.; et al. Premature aging in mice deficient in DNA repair and transcription. Science 2002, 296, 1276–1279. [Google Scholar] [CrossRef] [PubMed]
- Van der Pluijm, I.; Garinis, G.A.; Brandt, R.M.; Gorgels, T.G.; Wijnhoven, S.W.; Diderich, K.E.; de Wit, J.; Mitchell, J.R.; van Oostrom, C.; Beems, R.; et al. Impaired genome maintenance suppresses the growth hormone—Insulin-like growth factor 1 axis in mice with Cockayne syndrome. PLoS Biol. 2006, 5, e2. [Google Scholar] [CrossRef] [PubMed]
- Cleaver, J.E.; Bezrookove, V.; Revet, I.; Huang, E.J. Conceptual developments in the causes of Cockayne syndrome. Mech. Aging Dev. 2013, 134, 284–290. [Google Scholar] [CrossRef] [PubMed]
- Kamileri, I.; Karakasilioti, I.; Sideri, A.; Kosteas, T.; Tatarakis, A.; Talianidis, I.; Garinis, G.A. Defective transcription initiation causes postnatal growth failure in a mouse model of nucleotide excision repair (NER) progeria. Proc. Natl. Acad. Sci. USA 2012, 109, 2995–3000. [Google Scholar] [CrossRef] [PubMed]
- Le May, N.; Mota-Fernandes, D.; Velez-Cruz, R.; Iltis, I.; Biard, D.; Egly, J.M. NER factors are recruited to active promoters and facilitate chromatin modification for transcription in the absence of exogenous genotoxic attack. Mol. Cell 2010, 38, 54–66. [Google Scholar] [CrossRef] [PubMed]
- Fang, E.F.; Scheibye-Knudsen, M.; Brace, L.E.; Kassahun, H.; SenGupta, T.; Nilsen, H.; Mitchell, J.R.; Croteau, D.L.; Bohr, V.A. Defective Mitophagy in XPA via PARP-1 Hyperactivation and NAD. Cell 2014, 157, 882–896. [Google Scholar] [CrossRef] [PubMed]
- Niedernhofer, L.J.; Garinis, G.A.; Raams, A.; Lalai, A.S.; Robinson, A.R.; Appeldoorn, E.; Odijk, H.; Oostendorp, R.; Ahmad, A.; van Leeuwen, W.; et al. A new progeroid syndrome reveals that genotoxic stress suppresses the somatotroph axis. Nature 2006, 444, 1038–1043. [Google Scholar] [CrossRef] [PubMed]
- Van de Ven, M.; Andressoo, J.O.; Holcomb, V.B.; von Lindern, M.; Jong, W.M.; Zeeuw, C.I.; Suh, Y.; Hasty, P.; Hoeijmakers, J.H.; van der Horst, G.T.; et al. Adaptive Stress Response in Segmental Progeria Resembles Long-Lived Dwarfism and Calorie Restriction in Mice. PLoS Genet. 2006, 2, e192. [Google Scholar] [CrossRef] [PubMed]
- Mostoslavsky, R.; Chua, K.F.; Lombard, D.B.; Pang, W.W.; Fischer, M.R.; Gellon, L.; Liu, P.; Mostoslavsky, G.; Franco, S.; Murphy, M.M.; et al. Genomic instability and aging-like phenotype in the absence of mammalian SIRT6. Cell 2006, 124, 315–329. [Google Scholar] [CrossRef] [PubMed]
- Mariño, G.; Ugalde, A.P.; Fernández, A.F.; Osorio, F.G.; Fueyo, A.; Freije, J.M.P.; López-Otín, C. Insulin-like growth factor 1 treatment extends longevity in a mouse model of human premature aging by restoring somatotroph axis function. Proc. Natl. Acad. Sci. USA 2010, 107, 16268–16273. [Google Scholar] [CrossRef] [PubMed]
- Song, Z.; Wang, J.; Guachalla, L.M.; Terszowski, G.; Rodewald, H.R.; Ju, Z.; Rudolph, K.L. Alterations of the systemic environment are the primary cause of impaired B and T lymphopoiesis in telomere-dysfunctional mice. Blood 2009, 115, 1481–1489. [Google Scholar] [CrossRef] [PubMed]
- Carter, C.S.; Ramsey, M.M.; Sonntag, W.E. A critical analysis of the role of growth hormone and IGF-1 in aging and lifespan. Trends Genet. 2002, 18, 295–301. [Google Scholar] [CrossRef]
- Bartke, A. New findings in transgenic, gene knockout and mutant mice. Exp. Gerontol. 2006, 41, 1217–1219. [Google Scholar] [CrossRef] [PubMed]
- Holzenberger, M.; Dupont, J.; Ducos, B.; Leneuve, P.; Géloën, A.; Even, P.C.; Cervera, P.; le Bouc, Y. IGF-1 receptor regulates lifespan and resistance to oxidative stress in mice. Nature 2003, 421, 182–187. [Google Scholar] [CrossRef] [PubMed]
- Schumacher, B.; van der Pluijm, I.; Moorhouse, M.J.; Kosteas, T.; Robinson, A.R.; Suh, Y.; Breit, T.M.; van Steeg, H.; Niedernhofer, L.J.; van Ijcken, W.; et al. Delayed and accelerated aging share common longevity assurance mechanisms. PLoS Genet. 2008, 4, e1000161. [Google Scholar] [CrossRef] [PubMed]
- Cohen, E.; Paulsson, J.F.; Blinder, P.; Burstyn-Cohen, T.; Du, D.; Estepa, G.; Adame, A.; Pham, H.M.; Holzenberger, M.; Kelly, J.W.; et al. Reduced IGF-1 signaling delays age-associated proteotoxicity in mice. Cell 2009, 139, 1157–1169. [Google Scholar] [CrossRef] [PubMed]
- Freude, S.; Hettich, M.M.; Schumann, C.; Stöhr, O.; Koch, L.; Köhler, C.; Udelhoven, M.; Leeser, U.; Müller, M.; Kubota, N.; et al. Neuronal IGF-1 resistance reduces Aβ accumulation and protects against premature death in a model of Alzheimer’s disease. FASEB J. 2009, 23, 3315–3324. [Google Scholar] [CrossRef] [PubMed]
- Ikeno, Y.; Hubbard, G.B.; Lee, S.; Cortez, L.A.; Lew, C.M.; Webb, C.R.; Berryman, D.E.; List, E.O.; Kopchick, J.J.; Bartke, A. Reduced incidence and delayed occurrence of fatal neoplastic diseases in growth hormone receptor/binding protein knockout mice. J. Gerontol. A Biol. Sci. Med. Sci. 2009, 64, 522–529. [Google Scholar] [CrossRef] [PubMed]
- Ikeno, Y.; Bronson, R.T.; Hubbard, G.B.; Lee, S.; Bartke, A. Delayed occurrence of fatal neoplastic diseases in ames dwarf mice: Correlation to extended longevity. J. Gerontol. A Biol. Sci. Med. Sci. 2003, 58, 291–296. [Google Scholar] [CrossRef] [PubMed]
- Wijnhoven, S.W.; Beems, R.B.; Roodbergen, M.; van den Berg, J.; Lohman, P.H.; Diderich, K.; van der Horst, G.T.; Vijg, J.; Hoeijmakers, J.H.; van Steeg, H. Accelerated aging pathology in AD libitum fed Xpd(TTD) mice is accompanied by features suggestive of caloric restriction. DNA Repair 2005, 4, 1314–1324. [Google Scholar] [CrossRef] [PubMed]
- Garinis, G.A.; Uittenboogaard, L.M.; Stachelscheid, H.; Fousteri, M.; van Ijcken, W.; Breit, T.M.; van Steeg, H.; Mullenders, L.H.F.; van der Horst, G.T.J.; Brüning, J.C.; et al. Persistent transcription-blocking DNA lesions trigger somatic growth attenuation associated with longevity. Nat. Cell Biol. 2009, 11, 604–615. [Google Scholar] [CrossRef] [PubMed]
- Sulston, J.E.; Horvitz, H.R. Post-embryonic cell lineages of the nematode, Caenorhabditis elegans. Dev. biol. 1977, 56, 110–156. [Google Scholar] [CrossRef]
- Avery, L.; Horvitz, H.R. A cell that dies during wild-type C. elegans development can function as a neuron in a CED-3 mutant. Cell 1987, 51, 1071–1078. [Google Scholar]
- Fire, A.; Xu, S.; Montgomery, M.K.; Kostas, S.A.; Driver, S.E.; Mello, C.C. Potent and specific genetic interference by double-stranded RNA in Caenorhabditis elegans. Nature 1998, 391, 806–811. [Google Scholar] [CrossRef] [PubMed]
- Hartman, P.S.; Herman, R.K. Radiation-sensitive mutants of Caenorhabditis elegans. Genetics 1982, 102, 159–178. [Google Scholar] [PubMed]
- Gumienny, T.L.; Lambie, E.; Hartwieg, E.; Horvitz, H.R.; Hengartner, M.O. Genetic control of programmed cell death in the Caenorhabditis elegans hermaphrodite germline. Development 1999, 126, 1011–1022. [Google Scholar] [PubMed]
- Gartner, A.; Milstein, S.; Ahmed, S.; Hodgkin, J.; Hengartner, M.O. A conserved checkpoint pathway mediates DNA damage—Induced apoptosis and cell cycle arrest in C. elegans. Mol. Cell 2000, 5, 435–443. [Google Scholar] [CrossRef]
- Schumacher, B.; Hofmann, K.; Boulton, S.; Gartner, A. The C. elegans homolog of the p53 tumor suppressor is required for DNA damage-induced apoptosis. Curr. Biol. 2001, 11, 1722–1727. [Google Scholar] [PubMed]
- Derry, W.B.; Putzke, A.P.; Rothman, J.H. Caenorhabditis elegans p53: Role in apoptosis, meiosis, and stress resistance. Science 2001, 294, 591–595. [Google Scholar] [CrossRef] [PubMed]
- Hofmann, E.R.; Milstein, S.; Boulton, S.J.; Ye, M.; Hofmann, J.J.; Stergiou, L.; Gartner, A.; Vidal, M.; Hengartner, M.O. Caenorhabditis elegans HUS-1 is a DNA damage checkpoint protein required for genome stability and EGL-1-mediated apoptosis. Curr. Biol. 2002, 12, 1908–1918. [Google Scholar] [CrossRef]
- Schumacher, B.; Schertel, C.; Wittenburg, N.; Tuck, S.; Mitani, S.; Gartner, A.; Conradt, B.; Shaham, S. C. elegans CED-13 can promote apoptosis and is induced in response to DNA damage. Cell Death Differ. 2005, 12, 153–161. [Google Scholar] [PubMed]
- Schumacher, B.; Hanazawa, M.; Lee, M.-H.; Nayak, S.; Volkmann, K.; Hofmann, R.; Hengartner, M.; Schedl, T.; Gartner, A. Translational repression of C. elegans p53 by GLD-1 regulates DNA damage-induced apoptosis. Cell 2005, 120, 357–368. [Google Scholar] [PubMed]
- Johnson, T.E.; Hartman, P.S. Radiation effects on life span in Caenorhabditis elegans. J. Gerontol. 1988, 43, B137–B141. [Google Scholar] [CrossRef] [PubMed]
- Hartman, P.S.; Hevelone, J.; Dwarakanath, V.; Mitchell, D.L. Excision repair of UV radiation-induced DNA damage in Caenorhabditis elegans. Genetics 1989, 122, 379–385. [Google Scholar] [PubMed]
- Stergiou, L.; Doukoumetzidis, K.; Sendoel, A.; Hengartner, M.O. The nucleotide excision repair pathway is required for UV-C-induced apoptosis in Caenorhabditis elegans. Cell Death Differ. 2007, 14, 1129–1138. [Google Scholar] [CrossRef] [PubMed]
- Lans, H.; Marteijn, J.A.; Schumacher, B.; Hoeijmakers, J.H.J.; Jansen, G.; Vermeulen, W. Involvement of global genome repair, transcription coupled repair, and chromatin remodeling in UV DNA damage response changes during development. PLoS Genet. 2010, 6, e1000941. [Google Scholar] [CrossRef] [PubMed]
- Mueller, M.M.; Castells-Roca, L.; Babu, V.; Ermolaeva, M.A.; Müller, R.-U.; Frommolt, P.; Williams, A.B.; Greiss, S.; Schneider, J.I.; Benzing, T.; et al. DAF-16/FOXO and EGL-27/GATA promote developmental growth in response to persistent somatic DNA damage. Nat. Cell Biol. 2014, 16, 1168–1179. [Google Scholar] [CrossRef] [PubMed]
- Babu, V.; Hofmann, K.; Schumacher, B. A C. elegans homolog of the Cockayne syndrome complementation group A gene. DNA Repair 2014, 24, 57–62. [Google Scholar]
- Ermolaeva, M.A.; Segref, A.; Dakhovnik, A.; Ou, H.-L.; Schneider, J.I.; Utermöhlen, O.; Hoppe, T.; Schumacher, B. DNA damage in germ cells induces an innate immune response that triggers systemic stress resistance. Nature 2013, 501, 416–420. [Google Scholar] [CrossRef] [PubMed]
- Ermolaeva, M.A.; Schumacher, B. Systemic DNA damage responses: Organismal adaptations to genome instability. Trends Genet. 2014, 30, 95–102. [Google Scholar] [CrossRef] [PubMed]
- Kenyon, C.; Chang, J.; Gensch, E.; Rudner, A.; Tabtiang, R. A C. elegans mutant that lives twice as long as wild type. Nature 1993, 366, 461–464. [Google Scholar]
- Johnson, T.E. Increased life-span of age-1 mutants in Caenorhabditis elegans and lower Gompertz rate of aging. Science 1990, 249, 908–912. [Google Scholar] [CrossRef] [PubMed]
- Ogg, S.; Paradis, S.; Gottlieb, S.; Patterson, G.I.; Lee, L.; Tissenbaum, H.A.; Ruvkun, G. The Fork head transcription factor DAF-16 transduces insulin-like metabolic and longevity signals in C. elegans. Nature 1997, 389, 994–999. [Google Scholar] [PubMed]
- Murphy, C.T.; McCarroll, S.A.; Bargmann, C.I.; Fraser, A.; Kamath, R.S.; Ahringer, J.; Li, H.; Kenyon, C. Genes that act downstream of DAF-16 to influence the lifespan of Caenorhabditis elegans. Nature 2003, 424, 277–283. [Google Scholar] [CrossRef] [PubMed]
- Oh, S.W.; Mukhopadhyay, A.; Dixit, B.L.; Raha, T.; Green, M.R.; Tissenbaum, H.A. Identification of direct DAF-16 targets controlling longevity, metabolism and diapause by chromatin immunoprecipitation. Nat. Genet. 2005, 38, 251–257. [Google Scholar] [PubMed]
- Barrière, A.; Félix, M.-A. Temporal dynamics and linkage disequilibrium in natural Caenorhabditis elegans populations. Genetics 2007, 176, 999–1011. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Edifizi, D.; Schumacher, B. Genome Instability in Development and Aging: Insights from Nucleotide Excision Repair in Humans, Mice, and Worms. Biomolecules 2015, 5, 1855-1869. https://doi.org/10.3390/biom5031855
Edifizi D, Schumacher B. Genome Instability in Development and Aging: Insights from Nucleotide Excision Repair in Humans, Mice, and Worms. Biomolecules. 2015; 5(3):1855-1869. https://doi.org/10.3390/biom5031855
Chicago/Turabian StyleEdifizi, Diletta, and Björn Schumacher. 2015. "Genome Instability in Development and Aging: Insights from Nucleotide Excision Repair in Humans, Mice, and Worms" Biomolecules 5, no. 3: 1855-1869. https://doi.org/10.3390/biom5031855
APA StyleEdifizi, D., & Schumacher, B. (2015). Genome Instability in Development and Aging: Insights from Nucleotide Excision Repair in Humans, Mice, and Worms. Biomolecules, 5(3), 1855-1869. https://doi.org/10.3390/biom5031855