
Challenges in Antibody Development against Tn and Sialyl-Tn Antigens


 and
and
Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Glycosylation in Cancer
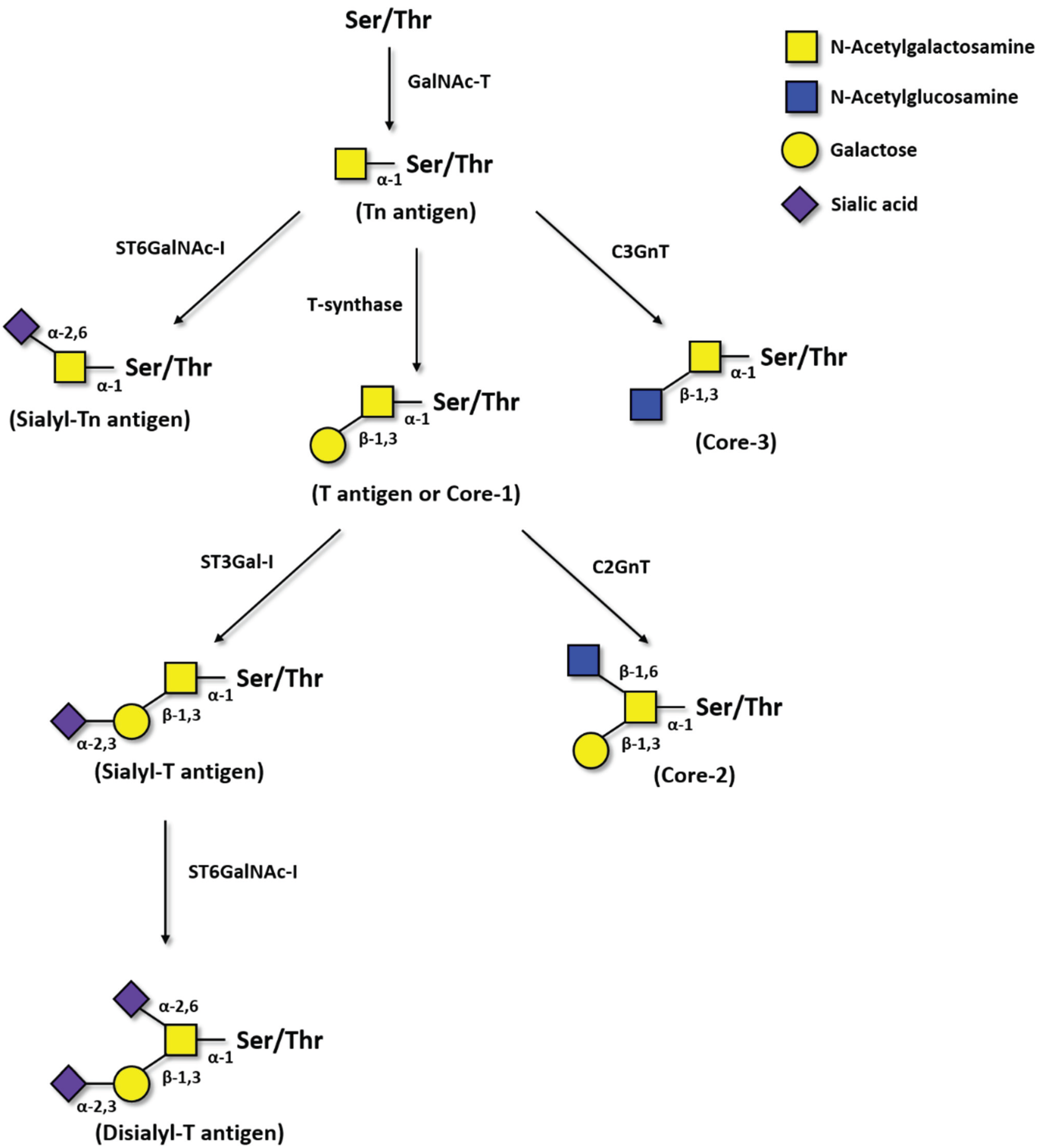
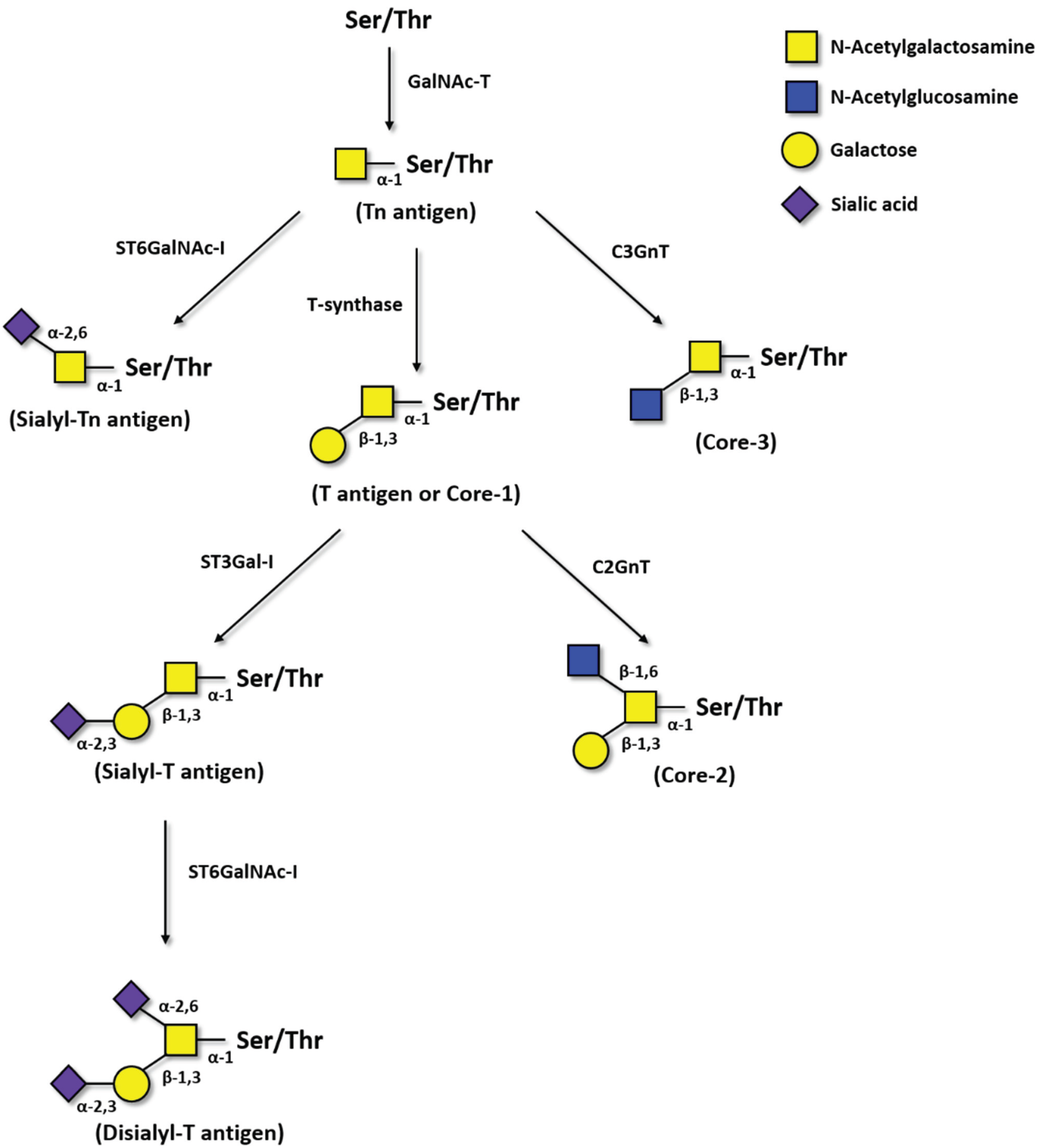
2.1. Tn Antigen
2.2. Sialyl-Tn Antigen
2.3. Pathological Role of Tn and STn in Cancer
3. Immune Response and Immunotherapies against Carbohydrates
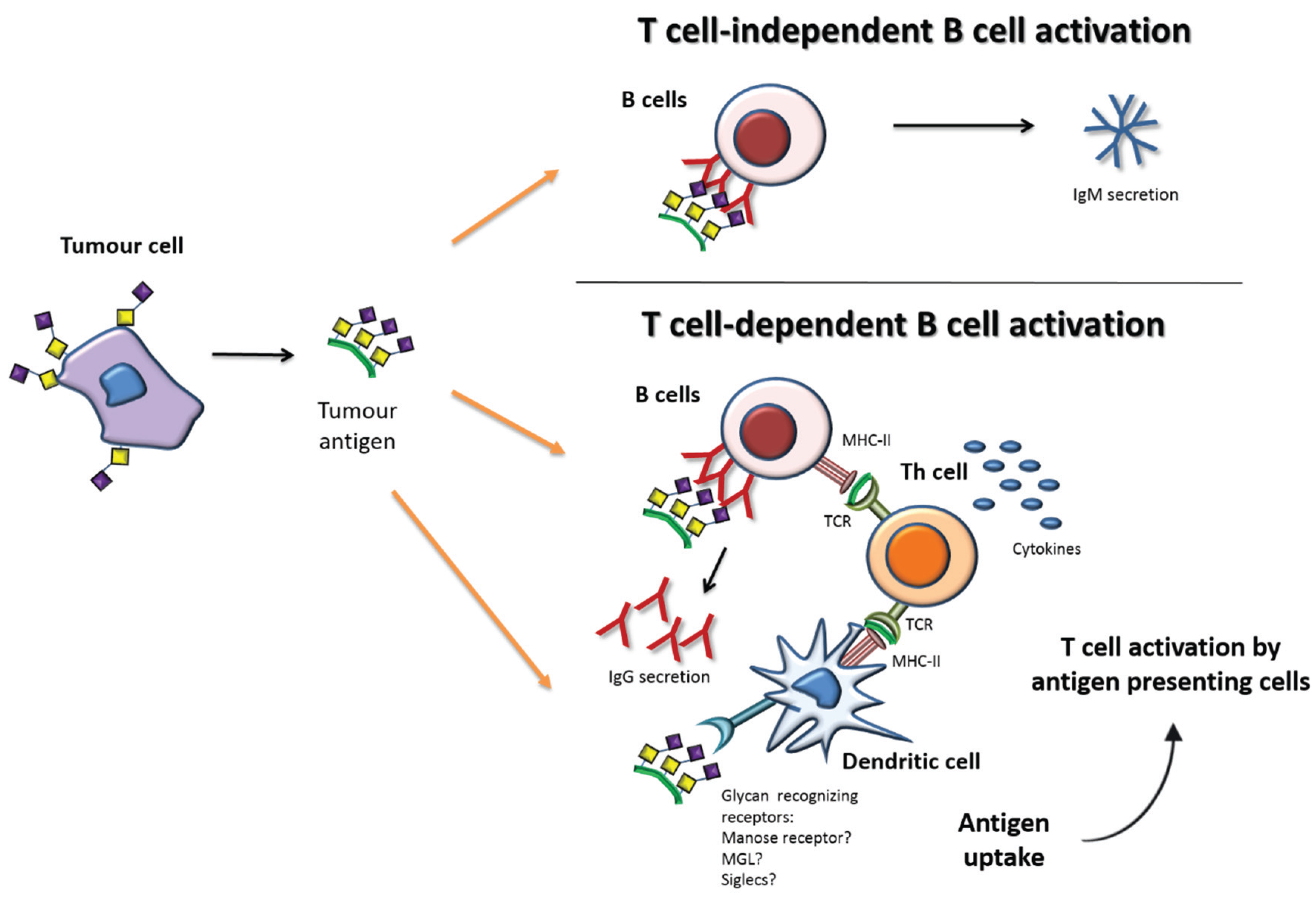
Immune Response and Tn/STn Antigens
4. Antibodies
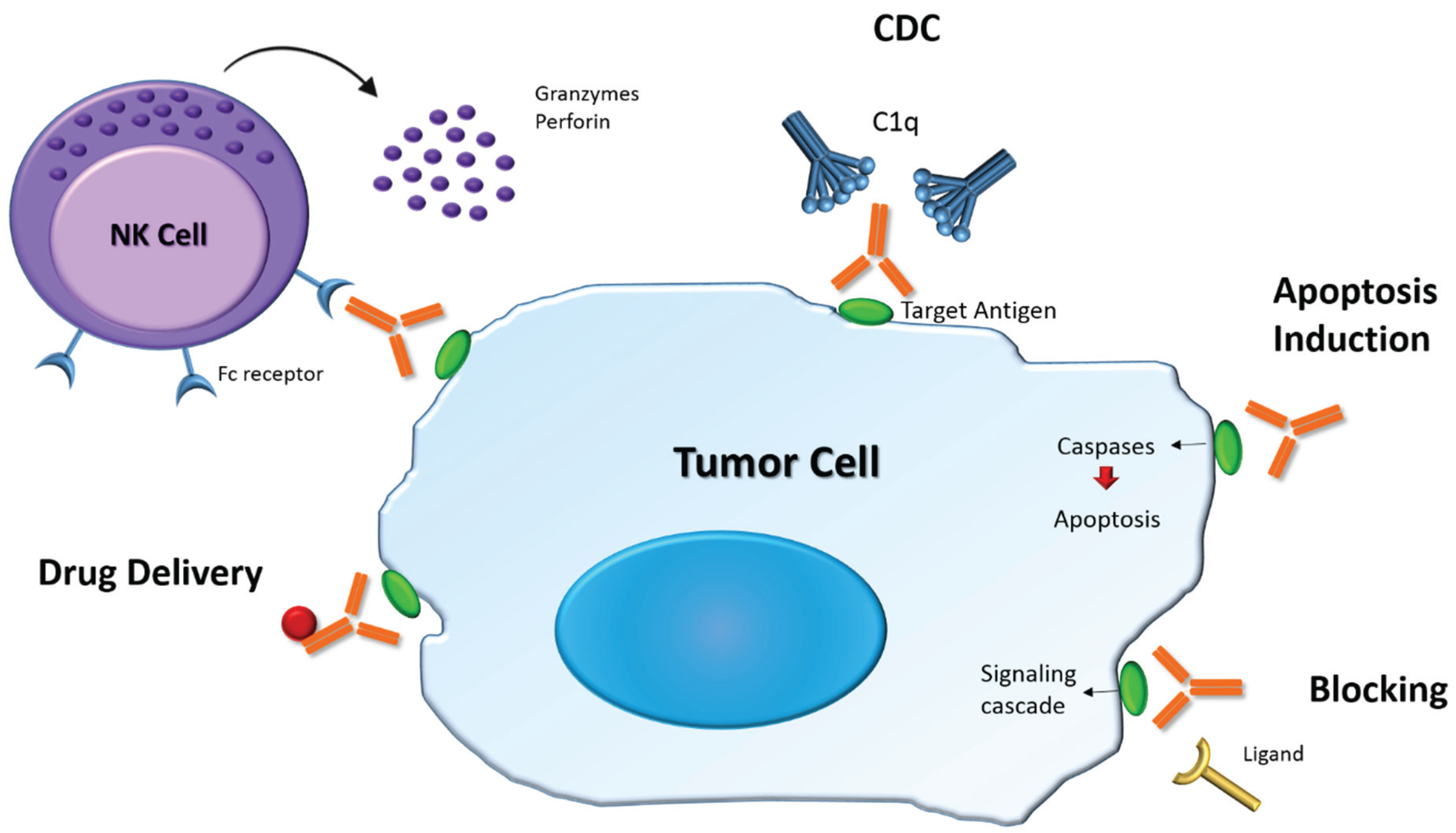
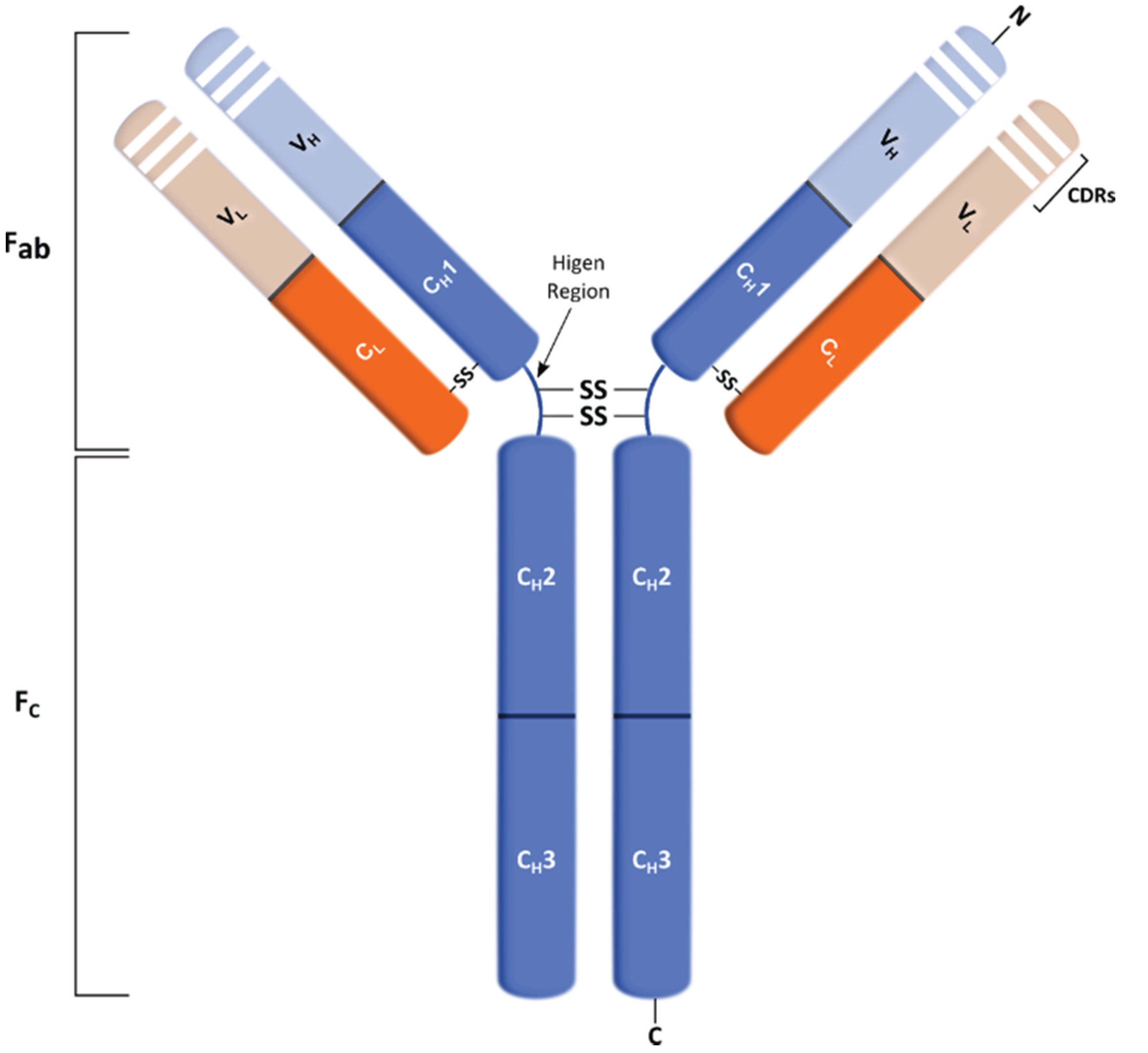
4.1. Structure and Role of Antibodies
4.2. Therapeutic Antibodies in Cancer

5. Therapeutic Antibodies against Tn and STn Antigens
5.1. Antibody Production
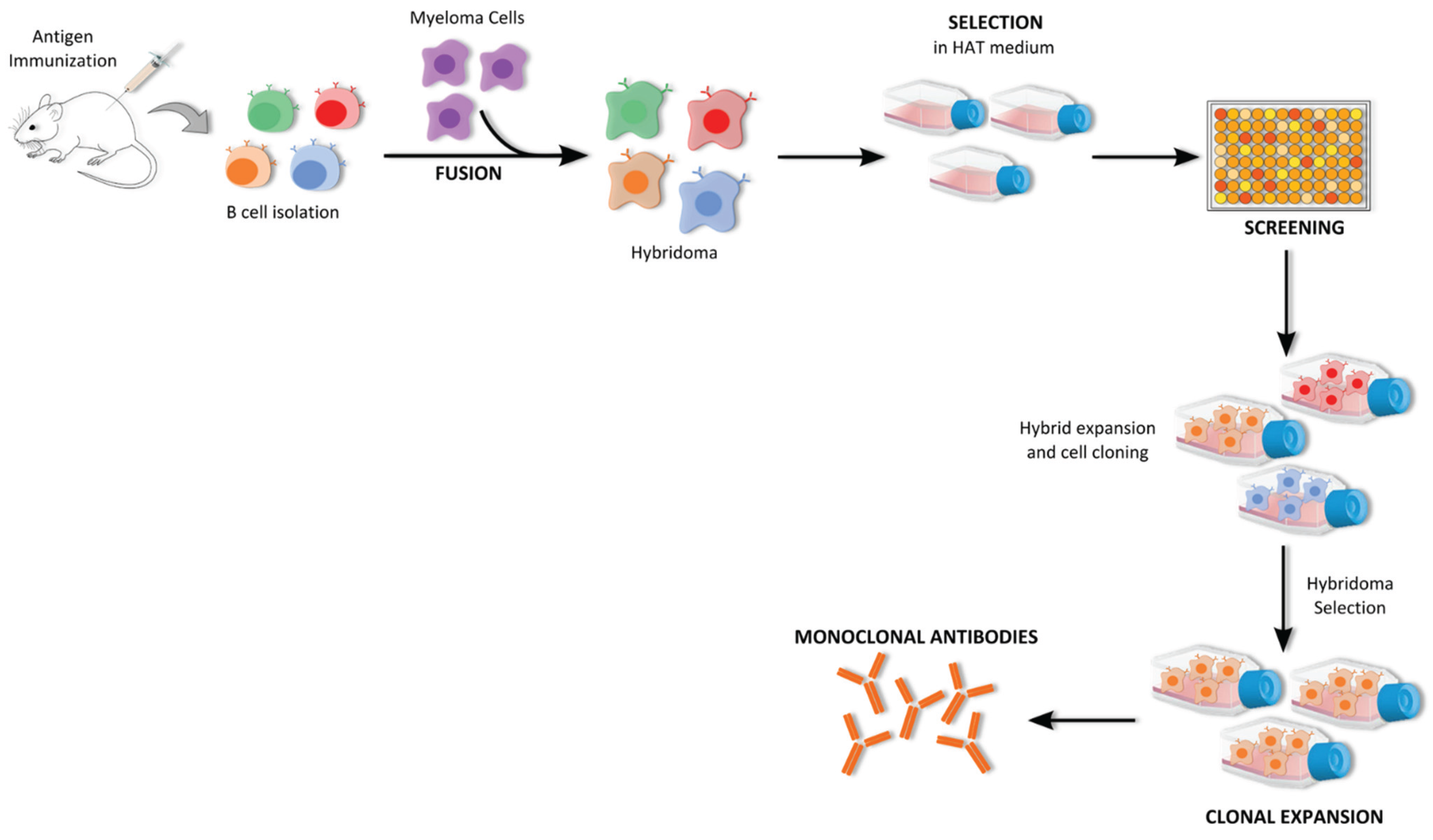
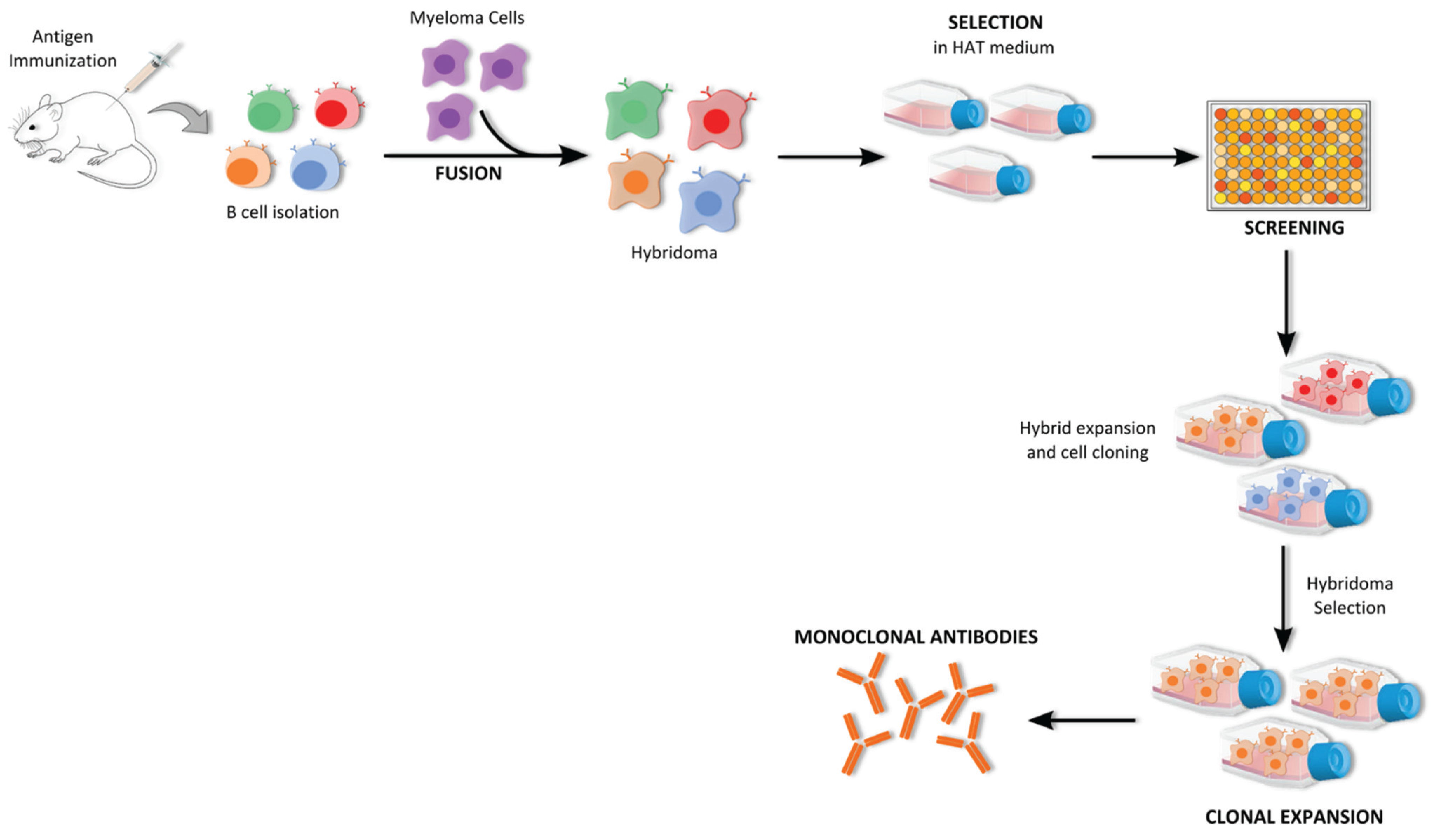
5.1.1. Hybridoma Technology
5.1.2. B Cell Immortalization
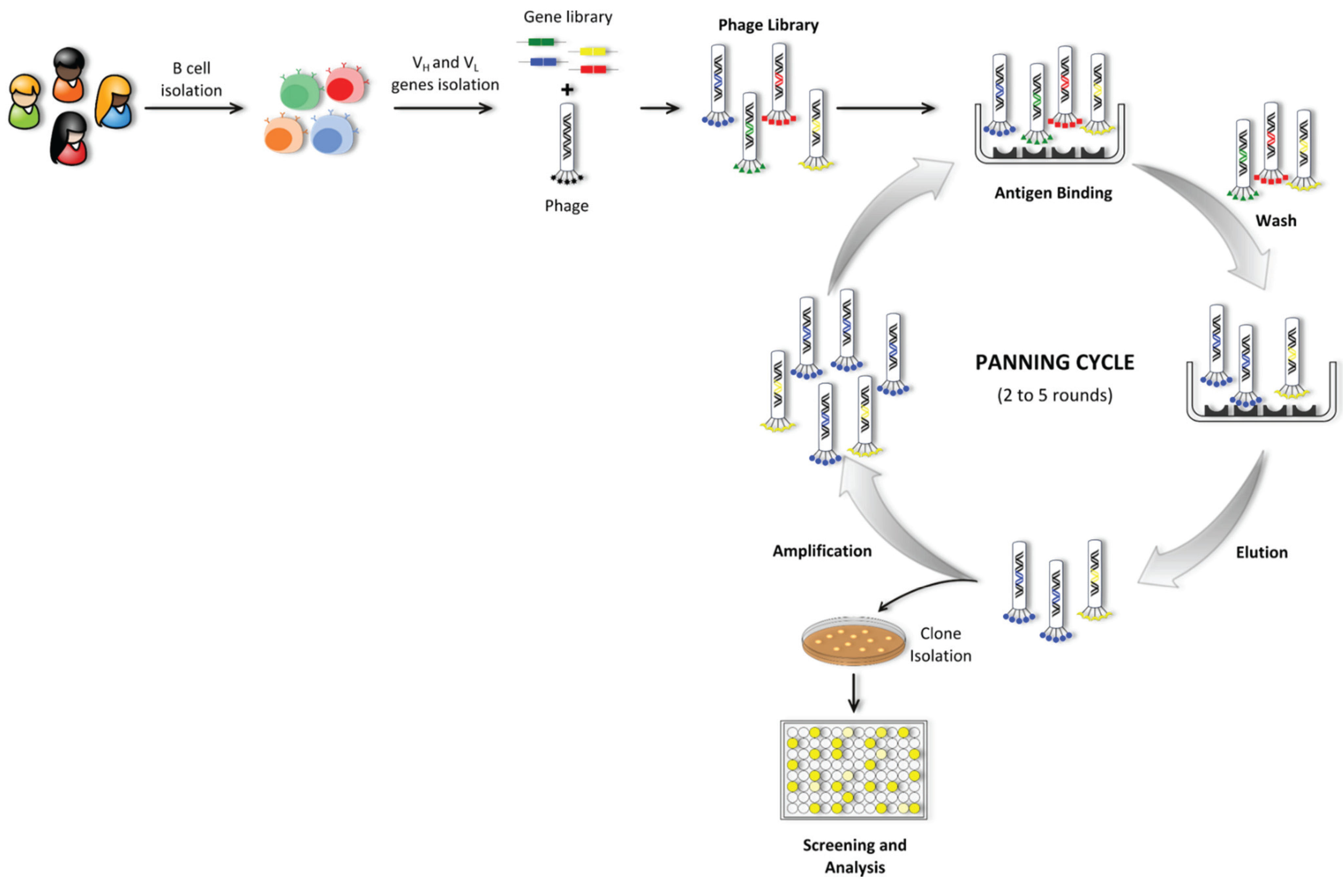
5.1.3. Phage Display Technology
5.1.4. Transgenic Mice
5.1.5. Molecularly Engineered Antibodies
6. Conclusions
Acknowledgments
Conflicts of Interest
References
- Crocker, P.R.; Feizi, T. Carbohydrate recognition systems: Functional triads in cell-cell interactions. Curr. Opin. Struct. Biol. 1996, 6, 679–691. [Google Scholar] [CrossRef]
- Feizi, T. Carbohydrate-mediated recognition systems in innate immunity. Immunol. Rev. 2000, 173, 79–88. [Google Scholar] [CrossRef] [PubMed]
- Lowe, J.B.; Marth, J.D. A genetic approach to mammalian glycan function. Annu. Rev. Biochem. 2003, 72, 643–691. [Google Scholar] [CrossRef] [PubMed]
- Kudelka, M.R.; Ju, T.; Heimburg-Molinaro, J.; Cummings, R.D. Simple sugars to complex disease—Mucin-type O-glycans in cancer. Adv. Cancer Res. 2015, 126, 53–135. [Google Scholar] [PubMed]
- Tuccillo, F.M.; de Laurentiis, A.; Palmieri, C.; Fiume, G.; Bonelli, P.; Borrelli, A.; Tassone, P.; Scala, I.; Buonaguro, F.M.; Quinto, I.; et al. Aberrant glycosylation as biomarker for cancer: Focus on CD43. Biomed. Res. Int. 2014. [Google Scholar] [CrossRef] [PubMed]
- Varki, A.; Kannagi, R.; Toole, B.P. Glycosylation changes in cancer. In Essentials of Glycobiology, 2nd ed.; Varki, A., Cummings, R.D., Esko, J.D., Freeze, H.H., Stanley, P., Bertozzi, C.R., Hart, G.W., Etzler, M.E., Eds.; Cold Spring Harbor Laboratory Press: La Jolla, CA, USA, 2009; Volume 2. [Google Scholar]
- Friedenreich, V. Production of a specific receptor quality in red cell corpuscles by bacterial activity. In The Thomsen Haemagglutination Phenomenon; Levin and Munksgaard: Copenhagen, Denmark, 1930. [Google Scholar]
- Moreau, R.; Dausset, J.; Bernard, J.; Moullec, J. Acquired hemolytic anemia with polyagglutinability of erythrocytes by a new factor present in normal blood. Bull. Mem. Soc. Med. Hop. Paris 1957, 73, 569–587. [Google Scholar] [PubMed]
- Springer, G.F. T and Tn, general carcinoma autoantigens. Science 1984, 224, 1198–1206. [Google Scholar] [CrossRef] [PubMed]
- Wang, P.H.; Lee, W.L.; Juang, C.M.; Yang, Y.H.; Lo, W.H.; Lai, C.R.; Hsieh, S.L.; Yuan, C.C. Altered mRNA expressions of sialyltransferases in ovarian cancers. Gynecol. Oncol. 2005, 99, 631–639. [Google Scholar] [CrossRef] [PubMed]
- Videira, P.A.; Correia, M.; Malagolini, N.; Crespo, H.J.; Ligeiro, D.; Calais, F.M.; Trindade, H.; Dall’Olio, F. ST3Gal.I sialyltransferase relevance in bladder cancer tissues and cell lines. BMC Cancer 2009. [Google Scholar] [CrossRef] [PubMed]
- Schneider, F.; Kemmner, W.; Haensch, W.; Franke, G.; Gretschel, S.; Karsten, U.; Schlag, P.M. Overexpression of sialyltransferase CMP-sialic acid: Galbeta1,3GalNAc-R alpha6-Sialyltransferase is related to poor patient survival in human colorectal carcinomas. Cancer Res. 2001, 61, 4605–4611. [Google Scholar] [PubMed]
- Videira, P.A.; Amado, I.F.; Crespo, H.J.; Alguero, M.C.; Dall’Olio, F.; Cabral, M.G.; Trindade, H. Surface α2-3- and α2-6-sialylation of human monocytes and derived dendritic cells and its influence on endocytosis. Glycoconjugate. J. 2008, 25, 259–268. [Google Scholar] [CrossRef] [PubMed]
- Priatel, J.J.; Chui, D.; Hiraoka, N.; Simmons, C.J.; Richardson, K.B.; Page, D.M.; Fukuda, M.; Varki, N.M.; Marth, J.D. The ST3Gal-I sialyltransferase controls CD8+ T lymphocyte homeostasis by modulating O-glycan biosynthesis. Immunity 2000, 12, 273–283. [Google Scholar] [CrossRef]
- Osako, M.; Yonezawa, S.; Siddiki, B.; Huang, J.; Ho, J.J.; Kim, Y.S.; Sato, E. Immunohistochemical study of mucin carbohydrates and core proteins in human pancreatic tumors. Cancer 1993, 71, 2191–2199. [Google Scholar] [CrossRef]
- Cao, Y.; Karsten, U.R.; Liebrich, W.; Haensch, W.; Springer, G.F.; Schlag, P.M. Expression of thomsen-friedenreich-related antigens in primary and metastatic colorectal carcinomas. A reevaluation. Cancer 1995, 76, 1700–1708. [Google Scholar] [CrossRef]
- David, L.; Nesland, J.M.; Clausen, H.; Carneiro, F.; Sobrinho-Simoes, M. Simple mucin-type carbohydrate antigens (Tn, sialosyl-Tn and T) in gastric mucosa, carcinomas and metastases. APMIS Suppl. 1992, 27, 162–172. [Google Scholar] [PubMed]
- Akita, K.; Fushiki, S.; Fujimoto, T.; Inoue, M.; Oguri, K.; Okayama, M.; Yamashina, I.; Nakada, H. Developmental expression of a unique carbohydrate antigen, Tn antigen, in mouse central nervous tissues. J. Neurosci. Res. 2001, 65, 595–603. [Google Scholar] [CrossRef] [PubMed]
- Vavasseur, F.; Yang, J.M.; Dole, K.; Paulsen, H.; Brockhausen, I. Synthesis of O-glycan core 3: Characterization of UDP-GlcNAc: GalNAc-R β3-N-acetyl-glucosaminyltransferase activity from colonic mucosal tissues and lack of the activity in human cancer cell lines. Glycobiology 1995, 5, 351–357. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.M.; Byrd, J.C.; Siddiki, B.B.; Chung, Y.S.; Okuno, M.; Sowa, M.; Kim, Y.S.; Matta, K.L.; Brockhausen, I. Alterations of O-glycan biosynthesis in human colon cancer tissues. Glycobiology 1994, 4, 873–884. [Google Scholar] [CrossRef] [PubMed]
- Ju, T.; Lanneau, G.S.; Gautam, T.; Wang, Y.; Xia, B.; Stowell, S.R.; Willard, M.T.; Wang, W.; Xia, J.Y.; Zuna, R.E.; et al. Human tumor antigens Tn and sialyl-Tn arise from mutations in Cosmc. Cancer Res. 2008, 68, 1636–1646. [Google Scholar] [CrossRef] [PubMed]
- Mi, R.; Song, L.; Wang, Y.; Ding, X.; Zeng, J.; Lehoux, S.; Aryal, R.P.; Wang, J.; Crew, V.K.; van Die, I.I.; et al. Epigenetic silencing of the chaperone Cosmc in human leukocytes expressing Tn antigen. J. Biol. Chem. 2012, 287, 41523–41533. [Google Scholar] [CrossRef] [PubMed]
- Gill, D.J.; Tham, K.M.; Chia, J.; Wang, S.C.; Steentoft, C.; Clausen, H.; Bard-Chapeau, E.A.; Bard, F.A. Initiation of GalNAc-type O-glycosylation in the endoplasmic reticulum promotes cancer cell invasiveness. Proc. Natl. Acad. Sci. USA 2013, 110, 3152–3161. [Google Scholar] [CrossRef] [PubMed]
- Itzkowitz, S.H.; Bloom, E.J.; Lau, T.S.; Kim, Y.S. Mucin associated Tn and sialosyl-Tn antigen expression in colorectal polyps. Gut 1992, 33, 518–523. [Google Scholar] [CrossRef] [PubMed]
- Itzkowitz, S.H.; Yuan, M.; Montgomery, C.K.; Kjeldsen, T.; Takahashi, H.K.; Bigbee, W.L.; Kim, Y.S. Expression of Tn, sialosyl-Tn, and T antigens in human colon cancer. Cancer Res. 1989, 49, 197–204. [Google Scholar] [PubMed]
- Julien, S.; Krzewinski-Recchi, M.A.; Harduin-Lepers, A.; Gouyer, V.; Huet, G.; Le Bourhis, X.; Delannoy, P. Expression of Sialyl-Tn antigen in breast cancer cells transfected with the human CMP-Neu5Ac: GalNAc α2,6-sialyltransferase (ST6GalNAc I) cDNA. Glycoconjugate J. 2001, 18, 883–893. [Google Scholar] [CrossRef]
- Marcos, N.T.; Bennett, E.P.; Gomes, J.; Magalhaes, A.; Gomes, C.; David, L.; Dar, I.; Jeanneau, C.; DeFrees, S.; Krustrup, D.; et al. ST6GalNAc-I controls expression of sialyl-Tn antigen in gastrointestinal tissues. Front. Biosci. 2011, 3, 1443–1455. [Google Scholar] [CrossRef]
- Vazquez-Martin, C.; Cuevas, E.; Gil-Martin, E.; Fernandez-Briera, A. Correlation analysis between tumorous associated antigen sialyl-Tn expression and ST6GalNAc I activity in human colon adenocarcinoma. Oncology 2004, 67, 159–165. [Google Scholar] [PubMed]
- Ferreira, J.A.; Videira, P.A.; Lima, L.; Pereira, S.; Silva, M.; Carrascal, M.; Severino, P.F.; Fernandes, E.; Almeida, A.; Costa, C.; et al. Overexpression of tumour-associated carbohydrate antigen sialyl-Tn in advanced bladder tumours. Mol. Oncol. 2013, 7, 719–731. [Google Scholar] [CrossRef] [PubMed]
- Pinho, S.; Marcos, N.T.; Ferreira, B.; Carvalho, A.S.; Oliveira, M.J.; Santos-Silva, F.; Harduin-Lepers, A.; Reis, C.A. Biological significance of cancer-associated sialyl-Tn antigen: Modulation of malignant phenotype in gastric carcinoma cells. Cancer Lett. 2007, 249, 157–170. [Google Scholar] [CrossRef] [PubMed]
- Julien, S.; Lagadec, C.; Krzewinski-Recchi, M.A.; Courtand, G.; Le Bourhis, X.; Delannoy, P. Stable expression of sialyl-Tn antigen in T47-D cells induces a decrease of cell adhesion and an increase of cell migration. Breast Cancer Res. Treat. 2005, 90, 77–84. [Google Scholar] [CrossRef] [PubMed]
- Julien, S.; Adriaenssens, E.; Ottenberg, K.; Furlan, A.; Courtand, G.; Vercoutter-Edouart, A.S.; Hanisch, F.G.; Delannoy, P.; Le Bourhis, X. ST6GalNAc I expression in MDA-MB-231 breast cancer cells greatly modifies their O-glycosylation pattern and enhances their tumorigenicity. Glycobiology 2006, 16, 54–64. [Google Scholar] [CrossRef] [PubMed]
- Tamura, F.; Sato, Y.; Hirakawa, M.; Yoshida, M.; Ono, M.; Osuga, T.; Okagawa, Y.; Uemura, N.; Arihara, Y.; Murase, K.; et al. RNAi-mediated gene silencing of ST6GalNAc I suppresses the metastatic potential in gastric cancer cells. Gastric Cancer 2014. [Google Scholar] [CrossRef] [PubMed]
- Ghazizadeh, M.; Ogawa, H.; Sasaki, Y.; Araki, T.; Aihara, K. Mucin carbohydrate antigens (T, Tn, and sialyl-Tn) in human ovarian carcinomas: Relationship with histopathology and prognosis. Hum. Pathol. 1997, 28, 960–966. [Google Scholar] [CrossRef]
- Carrilho, C.; Cantel, M.; Gouveia, P.; David, L. Simple mucin-type carbohydrate antigens (Tn, sialosyl-Tn, T and sialosyl-T) and gp 230 mucin-like glycoprotein are candidate markers for neoplastic transformation of the human cervix. Virchows Arch. 2000, 437, 173–179. [Google Scholar] [CrossRef] [PubMed]
- Sasaki, M.; Yamato, T.; Nakanuma, Y. Expression of sialyl-Tn, Tn and T antigens in primary liver cancer. Pathol. Int. 1999, 49, 325–331. [Google Scholar] [CrossRef] [PubMed]
- Terashima, S.; Takano, Y.; Ohori, T.; Kanno, T.; Kimura, T.; Motoki, R.; Kawaguchi, T. Sialyl-Tn antigen as a useful predictor of poor prognosis in patients with advanced stomach cancer. Surg. Today 1998, 28, 682–686. [Google Scholar] [CrossRef] [PubMed]
- Soares, R.; Marinho, A.; Schmitt, F. Expression of sialyl-Tn in breast cancer. Correlation with prognostic parameters. Pathol. Res. Pract. 1996, 192, 1181–1186. [Google Scholar] [CrossRef]
- Miles, D.W.; Happerfield, L.C.; Smith, P.; Gillibrand, R.; Bobrow, L.G.; Gregory, W.M.; Rubens, R.D. Expression of sialyl-Tn predicts the effect of adjuvant chemotherapy in node-positive breast cancer. Br. J. Cancer 1994, 70, 1272–1275. [Google Scholar] [CrossRef] [PubMed]
- Mapara, M.Y.; Sykes, M. Tolerance and cancer: Mechanisms of tumor evasion and strategies for breaking tolerance. J. Clin. Oncol. 2004, 22, 1136–1151. [Google Scholar] [CrossRef] [PubMed]
- Galli-Stampino, L.; Meinjohanns, E.; Frische, K.; Meldal, M.; Jensen, T.; Werdelin, O.; Mouritsen, S. T-cell recognition of tumor-associated carbohydrates: The nature of the glycan moiety plays a decisive role in determining glycopeptide immunogenicity. Cancer Res. 1997, 57, 3214–3222. [Google Scholar] [PubMed]
- Glithero, A.; Tormo, J.; Haurum, J.S.; Arsequell, G.; Valencia, G.; Edwards, J.; Springer, S.; Townsend, A.; Pao, Y.L.; Wormald, M.; et al. Crystal structures of two H-2Db/glycopeptide complexes suggest a molecular basis for CTL cross-reactivity. Immunity 1999, 10, 63–74. [Google Scholar] [CrossRef]
- Haurum, J.S.; Arsequell, G.; Lellouch, A.C.; Wong, S.Y.; Dwek, R.A.; McMichael, A.J.; Elliott, T. Recognition of carbohydrate by major histocompatibility complex class I-restricted, glycopeptide-specific cytotoxic T lymphocytes. J. Exp. Med. 1994, 180, 739–744. [Google Scholar] [CrossRef] [PubMed]
- Haurum, J.S.; Tan, L.; Arsequell, G.; Frodsham, P.; Lellouch, A.C.; Moss, P.A.; Dwek, R.A.; McMichael, A.J.; Elliott, T. Peptide anchor residue glycosylation: Effect on class I major histocompatibility complex binding and cytotoxic T lymphocyte recognition. Eur. J. Immunol. 1995, 25, 3270–3276. [Google Scholar] [CrossRef] [PubMed]
- Jensen, T.; Galli-Stampino, L.; Mouritsen, S.; Frische, K.; Peters, S.; Meldal, M.; Werdelin, O. T cell recognition of Tn-glycosylated peptide antigens. Eur. J. Immunol. 1996, 26, 1342–1349. [Google Scholar] [CrossRef] [PubMed]
- Julien, S.; Videira, P.A.; Delannoy, P. Sialyl-Tn in cancer: (How) did we miss the target? Biomolecules 2012, 2, 435–466. [Google Scholar] [CrossRef] [PubMed]
- Adderson, E.E. Antibody repertoires in infants and adults: Effects of T-independent and T-dependent immunizations. Springer Semin. Immun. 2001, 23, 387–403. [Google Scholar] [CrossRef]
- Amon, R.; Reuven, E.M.; Leviatan Ben-Arye, S.; Padler-Karavani, V. Glycans in immune recognition and response. Carbohyd. Res. 2014, 389, 115–122. [Google Scholar] [CrossRef] [PubMed]
- Lakshminarayanan, V.; Thompson, P.; Wolfert, M.A.; Buskas, T.; Bradley, J.M.; Pathangey, L.B.; Madsen, C.S.; Cohen, P.A.; Gendler, S.J.; Boons, G.J. Immune recognition of tumor-associated mucin MUC1 is achieved by a fully synthetic aberrantly glycosylated MUC1 tripartite vaccine. Proc. Natl. Acad. Sci. USA 2012, 109, 261–266. [Google Scholar] [CrossRef] [PubMed]
- Abdel-Aal, A.B.; Lakshminarayanan, V.; Thompson, P.; Supekar, N.; Bradley, J.M.; Wolfert, M.A.; Cohen, P.A.; Gendler, S.J.; Boons, G.J. Immune and anticancer responses elicited by fully synthetic aberrantly glycosylated MUC1 tripartite vaccines modified by a TLR2 or TLR9 agonist. Chembiochem 2014, 15, 1508–1513. [Google Scholar] [CrossRef] [PubMed]
- Gaidzik, N.; Kaiser, A.; Kowalczyk, D.; Westerlind, U.; Gerlitzki, B.; Sinn, H.P.; Schmitt, E.; Kunz, H. Synthetic antitumor vaccines containing MUC1 glycopeptides with two immunodominant domains-induction of a strong immune response against breast tumor tissues. Angew. Chem. Int. Ed. 2011, 50, 9977–9981. [Google Scholar] [CrossRef] [PubMed]
- Gaidzik, N.; Westerlind, U.; Kunz, H. The development of synthetic antitumour vaccines from mucin glycopeptide antigens. Chem. Soc. Rev. 2013, 42, 4421–4442. [Google Scholar] [CrossRef] [PubMed]
- Palitzsch, B.; Glaffig, M.; Kunz, H. Mucin glycopeptide-protein conjugates—Promising antitumor vaccine candidates. Isr. J. Chem. 2015, 55, 256–267. [Google Scholar] [CrossRef]
- Blixt, O.; Bueti, D.; Burford, B.; Allen, D.; Julien, S.; Hollingsworth, M.; Gammerman, A.; Fentiman, I.; Taylor-Papadimitriou, J.; Burchell, J.M. Autoantibodies to aberrantly glycosylated MUC1 in early stage breast cancer are associated with a better prognosis. Breast Cancer Res. 2011. [Google Scholar] [CrossRef] [PubMed]
- Pedersen, J.W.; Blixt, O.; Bennett, E.P.; Tarp, M.A.; Dar, I.; Mandel, U.; Poulsen, S.S.; Pedersen, A.E.; Rasmussen, S.; Jess, P.; et al. Seromic profiling of colorectal cancer patients with novel glycopeptide microarray. Int. J. Cancer 2011, 128, 1860–1871. [Google Scholar] [CrossRef] [PubMed]
- Burford, B.; Gentry-Maharaj, A.; Graham, R.; Allen, D.; Pedersen, J.W.; Nudelman, A.S.; Blixt, O.; Fourkala, E.O.; Bueti, D.; Dawnay, A.; et al. Autoantibodies to MUC1 glycopeptides cannot be used as a screening assay for early detection of breast, ovarian, lung or pancreatic cancer. Br. J. Cancer 2013, 108, 2045–2055. [Google Scholar] [CrossRef] [PubMed]
- Ibrahim, N.K.; Murray, J.L.; Zhou, D.; Mittendorf, E.A.; Sample, D.; Tautchin, M.; Miles, D. Survival advantage in patients with metastatic breast cancer receiving endocrine therapy plus sialyl Tn-KHL vaccine: Post hoc analysis of a large randomized trial. J. Cancer 2013, 4, 577–584. [Google Scholar] [CrossRef] [PubMed]
- Miles, D.; Roche, H.; Martin, M.; Perren, T.J.; Cameron, D.A.; Glaspy, J.; Dodwell, D.; Parker, J.; Mayordomo, J.; Tres, A.; et al. Phase III multicenter clinical trial of the sialyl-Tn (STn)-keyhole limpet hemocyanin (KLH) vaccine for metastatic breast cancer. Oncologist 2011, 16, 1092–1100. [Google Scholar] [CrossRef] [PubMed]
- Julien, S.; Picco, G.; Sewell, R.; Vercoutter-Edouart, A.S.; Tarp, M.; Miles, D.; Clausen, H.; Taylor-Papadimitriou, J.; Burchell, J.M. Sialyl-Tn vaccine induces antibody-mediated tumour protection in a relevant murine model. Br. J. Cancer 2009, 100, 1746–1754. [Google Scholar] [CrossRef] [PubMed]
- Holmberg, L.A.; Sandmaier, B.M. Vaccination with theratope (STn-KLH) as treatment for breast cancer. Expert Rev. Vaccines 2004, 3, 655–663. [Google Scholar] [CrossRef] [PubMed]
- Freire, T.; Lo-Man, R.; Bay, S.; Leclerc, C. Tn glycosylation of the MUC6 protein modulates its immunogenicity and promotes the induction of Th17-biased T cell responses. J. Biol. Chem. 2011, 286, 7797–7811. [Google Scholar] [CrossRef] [PubMed]
- Saeland, E.; van Vliet, S.J.; Backstrom, M.; van den Berg, V.C.; Geijtenbeek, T.B.; Meijer, G.A.; van Kooyk, Y. The C-type lectin MGL expressed by dendritic cells detects glycan changes on MUC1 in colon carcinoma. Cancer Immunol. Immunother. 2007, 56, 1225–1236. [Google Scholar] [CrossRef] [PubMed]
- Ohno, S.; Ohno, Y.; Nakada, H.; Suzuki, N.; Soma, G.; Inoue, M. Expression of Tn and sialyl-Tn antigens in endometrial cancer: Its relationship with tumor-produced cyclooxygenase-2, tumor- infiltrated lymphocytes and patient prognosis. Anticancer Res. 2006, 26, 4047–4053. [Google Scholar] [PubMed]
- Toda, M.; Akita, K.; Inoue, M.; Taketani, S.; Nakada, H. Down-modulation of B cell signal transduction by ligation of mucins to CD22. Biochem. Biophys. Res. Commun. 2008, 372, 45–50. [Google Scholar] [CrossRef] [PubMed]
- Takamiya, R.; Ohtsubo, K.; Takamatsu, S.; Taniguchi, N.; Angata, T. The interaction between Siglec-15 and tumor-associated sialyl-Tn antigen enhances TGF-β secretion from monocytes/macrophages through the DAP12-Syk pathway. Glycobiology 2013, 23, 178–187. [Google Scholar] [CrossRef] [PubMed]
- Carrascal, M.A.; Severino, P.F.; Guadalupe Cabral, M.; Silva, M.; Ferreira, J.A.; Calais, F.; Quinto, H.; Pen, C.; Ligeiro, D.; Santos, L.L.; et al. Sialyl Tn-expressing bladder cancer cells induce a tolerogenic phenotype in innate and adaptive immune cells. Mol. Oncol. 2014, 8, 753–765. [Google Scholar] [CrossRef] [PubMed]
- Scott, A.M.; Allison, J.P.; Wolchok, J.D. Monoclonal antibodies in cancer therapy. Cancer Immun. Res. 2012, 12, 14. [Google Scholar]
- Pucca, M.B.; Bertolini, T.B.; Barbosa, J.E.; Galina, S.V.R.; Porto, G.S. Therapeutic monoclonal antibodies: ScFv patents as a marker of a new class of potential biopharmaceuticals. Braz. J. Pharm. Sci. 2011, 47, 31–38. [Google Scholar]
- Ali, M.; Hitomi, K.; Nakano, H. Generation of monoclonal antibodies using simplified single-cell reverse transcription-polymerase chain reaction and cell-free protein synthesis. J. Biosci. Bioeng. 2006, 101, 284–286. [Google Scholar] [CrossRef] [PubMed]
- Babcook, J.S.; Leslie, K.B.; Olsen, O.A.; Salmon, R.A.; Schrader, J.W. A novel strategy for generating monoclonal antibodies from single, isolated lymphocytes producing antibodies of defined specificities. Proc. Natl. Acad. Sci. USA 1996, 93, 7843–7848. [Google Scholar] [CrossRef] [PubMed]
- Smith, K.; Garman, L.; Wrammert, J.; Zheng, N.Y.; Capra, J.D.; Ahmed, R.; Wilson, P.C. Rapid generation of fully human monoclonal antibodies specific to a vaccinating antigen. Nat. Protoc. 2009, 4, 372–384. [Google Scholar] [CrossRef] [PubMed]
- Köhler, G.; Milstein, C. Continuous cultures of fused cells secreting antibody of predefined specificity. Nature 1975, 256, 495–497. [Google Scholar] [CrossRef] [PubMed]
- Ecker, D.M.; Jones, S.D.; Levine, H.L. The therapeutic monoclonal antibody market. MAbs 2015, 7, 9–14. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.K. The history of monoclonal antibody development—Progress, remaining challenges and future innovations. Ann. Med. Surg. 2014, 3, 113–116. [Google Scholar] [CrossRef] [PubMed]
- Brekke, O.H.; Sandlie, I. Therapeutic antibodies for human diseases at the dawn of the twenty-first century. Nat. Rev. Drug Discov. 2003, 2, 52–62. [Google Scholar] [CrossRef] [PubMed]
- Weiner, L.M.; Surana, R.; Wang, S. Monoclonal antibodies: Versatile platforms for cancer immunotherapy. Nat. Rev. Immunol. 2010, 10, 317–327. [Google Scholar] [CrossRef] [PubMed]
- Kubota, T.; Niwa, R.; Satoh, M.; Akinaga, S.; Shitara, K.; Hanai, N. Engineered therapeutic antibodies with improved effector functions. Cancer Sci. 2009, 100, 1566–1572. [Google Scholar] [CrossRef] [PubMed]
- Carter, P. Improving the efficacy of antibody-based cancer therapies. Nat. Rev. Cancer 2001, 1, 118–129. [Google Scholar] [CrossRef] [PubMed]
- Ludwig, D.L.; Pereira, D.S.; Zhu, Z.; Hicklin, D.J.; Bohlen, P. Monoclonal antibody therapeutics and apoptosis. Oncogene 2003, 22, 9097–9106. [Google Scholar] [CrossRef] [PubMed]
- Chames, P.; van Regenmortel, M.; Weiss, E.; Baty, D. Therapeutic antibodies: Successes, limitations and hopes for the future. Br. J. Pharmacol. 2009, 157, 220–233. [Google Scholar] [CrossRef] [PubMed]
- Scott, A.M.; Wolchok, J.D.; Old, L.J. Antibody therapy of cancer. Nat. Rev. Cancer 2012, 12, 278–287. [Google Scholar] [CrossRef] [PubMed]
- Van Cutsem, E.; Kohne, C.H.; Hitre, E.; Zaluski, J.; Chang Chien, C.R.; Makhson, A.; D’Haens, G.; Pinter, T.; Lim, R.; Bodoky, G.; et al. Cetuximab and chemotherapy as initial treatment for metastatic colorectal cancer. N. Engl. J. Med. 2009, 360, 1408–1417. [Google Scholar] [CrossRef] [PubMed]
- Hudis, C.A. Trastuzumab—Mechanism of action and use in clinical practice. N. Engl. J. Med. 2007, 357, 39–51. [Google Scholar] [CrossRef] [PubMed]
- Weiner, G.J. Rituximab: Mechanism of action. Semin. Hematol. 2010, 47, 115–123. [Google Scholar] [CrossRef] [PubMed]
- Hodi, F.S.; O’Day, S.J.; McDermott, D.F.; Weber, R.W.; Sosman, J.A.; Haanen, J.B.; Gonzalez, R.; Robert, C.; Schadendorf, D.; Hassel, J.C.; et al. Improved survival with ipilimumab in patients with metastatic melanoma. N. Engl. J. Med. 2010, 363, 711–723. [Google Scholar] [CrossRef] [PubMed]
- Wandall, H.H.; Blixt, O.; Tarp, M.A.; Pedersen, J.W.; Bennett, E.P.; Mandel, U.; Ragupathi, G.; Livingston, P.O.; Hollingsworth, M.A.; Taylor-Papadimitriou, J.; et al. Cancer biomarkers defined by autoantibody signatures to aberrant O-glycopeptide epitopes. Cancer Res. 2010, 70, 1306–1313. [Google Scholar] [CrossRef] [PubMed]
- Colcher, D.; Hand, P.H.; Nuti, M.; Schlom, J. A spectrum of monoclonal antibodies reactive with human mammary tumor cells. Proc. Natl. Acad. Sci. USA 1981, 78, 3199–3203. [Google Scholar] [CrossRef] [PubMed]
- Kjeldsen, T.; Clausen, H.; Hirohashi, S.; Ogawa, T.; Iijima, H.; Hakomori, S. Preparation and characterization of monoclonal antibodies directed to the tumor-associated O-linked sialosyl-2-6 α-N-acetylgalactosaminyl (sialosyl-Tn) epitope. Cancer Res. 1988, 48, 2214–2220. [Google Scholar] [PubMed]
- Muraro, R.; Kuroki, M.; Wunderlich, D.; Poole, D.J.; Colcher, D.; Thor, A.; Greiner, J.W.; Simpson, J.F.; Molinolo, A.; Noguchi, P.; et al. Generation and characterization of B72.3 second generation monoclonal antibodies reactive with the tumor-associated glycoprotein 72 antigen. Cancer Res. 1988, 48, 4588–4596. [Google Scholar] [PubMed]
- Rogers, B.E.; Roberson, P.L.; Shen, S.; Khazaeli, M.B.; Carpenter, M.; Yokoyama, S.; Brechbiel, M.W.; LoBuglio, A.F.; Buchsbaum, D.J. Intraperitoneal radioimmunotherapy with a humanized anti-TAG-72 (CC49) antibody with a deleted CH2 region. Cancer Biother. Radiopharm. 2005, 20, 502–513. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Walberg, L.A.; Ogata, S.; Itzkowitz, S.H.; Koganty, R.R.; Reddish, M.; Gandhi, S.S.; Longenecker, B.M.; Lloyd, K.O.; Livingston, P.O. Immune sera and monoclonal antibodies define two configurations for the sialyl Tn tumor antigen. Cancer Res. 1995, 55, 3364–3368. [Google Scholar] [PubMed]
- Kurosaka, A.; Fukui, S.; Kitagawa, H.; Nakada, H.; Numata, Y.; Funakoshi, I.; Kawasaki, T.; Yamashina, I. Mucin-carbohydrate directed monoclonal antibody. FEBS Lett. 1987, 215, 137–139. [Google Scholar] [CrossRef]
- Pant, K.D.; Jain, A.; McCracken, J.D.; Thompson, K. Immunohistochemical examination of anti-STn monoclonal antibodies LLU9B4, B72.3 and B35.2 for their potential use as tumor markers. Dig. Dis. Sci. 2008, 53, 2189–2194. [Google Scholar] [CrossRef] [PubMed]
- An, Y.; Han, W.; Chen, X.; Zhao, X.; Lu, D.; Feng, J.; Yang, D.; Song, L.; Yan, X. A novel anti-sTn monoclonal antibody 3P9 inhibits human xenografted colorectal carcinomas. J. Immunother. 2013, 36, 20–28. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, H.K.; Metoki, R.; Hakomori, S. Immunoglobulin G3 monoclonal antibody directed to Tn antigen (tumor-associated α-N-acetylgalactosaminyl epitope) that does not cross-react with blood group A antigen. Cancer Res. 1988, 48, 4361–4367. [Google Scholar] [PubMed]
- Numata, Y.; Nakada, H.; Fukui, S.; Kitagawa, H.; Ozaki, K.; Inoue, M.; Kawasaki, T.; Funakoshi, I.; Yamashina, I. A monoclonal antibody directed to Tn antigen. Biochem. Biophys. Res. Commun. 1990, 170, 981–985. [Google Scholar] [CrossRef]
- King, M.J.; Parsons, S.F.; Wu, A.M.; Jones, N. Immunochemical studies on the differential binding properties of two monoclonal antibodies reacting with Tn red cells. Transfusion 1991, 31, 142–149. [Google Scholar] [CrossRef] [PubMed]
- Reis, C.A.; Sorensen, T.; Mandel, U.; David, L.; Mirgorodskaya, E.; Roepstorff, P.; Kihlberg, J.; Hansen, J.E.; Clausen, H. Development and characterization of an antibody directed to an α-N-acetyl-d-galactosamine glycosylated MUC2 peptide. Glycoconjugate J. 1998, 15, 51–62. [Google Scholar] [CrossRef]
- Ando, H.; Matsushita, T.; Wakitani, M.; Sato, T.; Kodama-Nishida, S.; Shibata, K.; Shitara, K.; Ohta, S. Mouse-human chimeric anti-Tn IgG1 induced anti-tumor activity against Jurkat cells in vitro and in vivo. Biol. Pharm. Bull. 2008, 31, 1739–1744. [Google Scholar] [CrossRef] [PubMed]
- Danussi, C.; Coslovi, A.; Campa, C.; Mucignat, M.T.; Spessotto, P.; Uggeri, F.; Paoletti, S.; Colombatti, A. A newly generated functional antibody identifies Tn antigen as a novel determinant in the cancer cell-lymphatic endothelium interaction. Glycobiology 2009, 19, 1056–1067. [Google Scholar] [CrossRef] [PubMed]
- Zamri, N.; Masuda, N.; Oura, F.; Yajima, Y.; Nakada, H.; Fujita-Yamaguchi, Y. Effects of two monoclonal antibodies MLS128 against Tn-antigen and 1H7 against insulin-like growth factor-I receptor on the growth of colon cancer cells. Biosci. Trends 2012, 6, 303–312. [Google Scholar] [CrossRef] [PubMed]
- Welinder, C.; Baldetorp, B.; Borrebaeck, C.; Fredlund, B.M.; Jansson, B. A new murine IgG1 anti-Tn monoclonal antibody with in vivo anti-tumor activity. Glycobiology 2011, 21, 1097–1107. [Google Scholar] [CrossRef] [PubMed]
- De Paz, J.L.; Seeberger, P.H. Recent advances and future challenges in glycan microarray technology. Methods Mol. Biol. 2012, 808, 1–12. [Google Scholar] [PubMed]
- Tarp, M.A.; Sorensen, A.L.; Mandel, U.; Paulsen, H.; Burchell, J.; Taylor-Papadimitriou, J.; Clausen, H. Identification of a novel cancer-specific immunodominant glycopeptide epitope in the MUC1 tandem repeat. Glycobiology 2007, 17, 197–209. [Google Scholar] [CrossRef] [PubMed]
- Steinitz, M.; Klein, G.; Koskimies, S.; Makel, O. EB virus-induced B lymphocyte cell lines producing specific antibody. Nature 1977, 269, 420–422. [Google Scholar] [CrossRef] [PubMed]
- Jones, P.T.; Dear, P.H.; Foote, J.; Neuberger, M.S.; Winter, G. Replacing the complementarity-determining regions in a human antibody with those from a mouse. Nature 1986, 321, 522–525. [Google Scholar] [CrossRef] [PubMed]
- Smith, G.P. Filamentous fusion phage: Novel expression vectors that display cloned antigens on the virion surface. Science 1985, 228, 1315–1317. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C. Hybridoma technology for the generation of monoclonal antibodies. Methods Mol. Biol. 2012, 901, 117–135. [Google Scholar] [PubMed]
- Loyau, J.; Rousseau, F. Cloning, reformatting, and small-scale expression of monoclonal antibody isolated from mouse, rat, or hamster hybridoma. Methods Mol. Biol. 2014, 1131, 207–228. [Google Scholar] [PubMed]
- Gorny, M.K. Human hybridoma technology. Antib. Technol. J. 2012, 2012, 1–5. [Google Scholar] [CrossRef]
- Bakhtiar, R. Therapeutic recombinant monoclonal antibodies. J. Chem. Educ. 2012, 89, 1537–1542. [Google Scholar] [CrossRef]
- Kim, H.Y.; Stojadinovic, A.; Izadjoo, M.J. Immunization, hybridoma generation, and selection for monoclonal antibody production. Methods Mol. Biol. 2014, 1131, 33–45. [Google Scholar] [PubMed]
- Yokoyama, W.M.; Christensen, M.; Santos, G.D.; Miller, D.; Ho, J.; Wu, T.; Dziegelewski, M.; Neethling, F.A. Production of monoclonal antibodies. Curr. Protoc. Immunol. 2006, 25, 1–25. [Google Scholar]
- Little, M.; Kipriyanov, S.M.; Le Gall, F.; Moldenhauer, G. Of mice and men: Hybridoma and recombinant antibodies. Immunol. Today 2000, 21, 364–370. [Google Scholar] [CrossRef]
- Tomita, M.; Tsumoto, K. Hybridoma technologies for antibody production. Immunotherapy 2011, 3, 371–380. [Google Scholar] [CrossRef] [PubMed]
- Emmons, C.; Hunsicker, L.G. Muromonab-CD3 (Orthoclone OKT3): The first monoclonal antibody approved for therapeutic use. Iowa Med. 1987, 77, 78–82. [Google Scholar] [PubMed]
- McCafferty, J.; Griffiths, A.D.; Winter, G.; Chiswell, D.J. Phage antibodies: Filamentous phage displaying antibody variable domains. Nature 1990, 348, 552–554. [Google Scholar] [CrossRef] [PubMed]
- Burton, D.R.; Barbas, C.F., III. Human antibodies from combinatorial libraries. Adv. Immunol. 1994, 57, 191–280. [Google Scholar] [PubMed]
- Wang, S. Advances in the production of human monoclonal antibodies. Antib. Technol. J. 2011, 1, 1–4. [Google Scholar] [CrossRef]
- Steinitz, M. Production of human monoclonal antibodies by the Epstein-Barr virus method. Methods Mol. Biol. 2014, 1060, 111–122. [Google Scholar] [PubMed]
- Kozbor, D.; Roder, J.C.; Chang, T.H.; Steplewski, Z.; Koprowski, H. Human anti-tetanus toxoid monoclonal antibody secreted by EBV-transformed human B cells fused with murine myeloma. Hybridoma 1982, 1, 323–328. [Google Scholar] [CrossRef] [PubMed]
- Steinitz, M.; Tamir, S. Human monoclonal antibodies produced by Epstein-Barr virus transformed cell lines bind protein A. Immunol. Lett. 1985, 9, 19–22. [Google Scholar] [CrossRef]
- Steinitz, M.; Seppala, I.; Eichmann, K.; Klein, G. Establishment of a human lymphoblastoid cell line with specific antibody production against group a streptococcal carbohydrate. Immunobiology 1979, 156, 41–47. [Google Scholar] [PubMed]
- Bradbury, A.R.; Marks, J.D. Antibodies from phage antibody libraries. J. Immunol. Methods 2004, 290, 29–49. [Google Scholar] [CrossRef] [PubMed]
- Deantonio, C.; Cotella, D.; Macor, P.; Santoro, C.; Sblattero, D. Phage display technology for human monoclonal antibodies. Methods Mol. Biol. 2014, 1060, 277–295. [Google Scholar] [PubMed]
- Bahara, N.H.H.; Tye, G.J.; Choong, Y.S.; Ong, E.B.; Ismail, A.; Lim, T.S. Phage display antibodies for diagnostic applications. Biologicals 2013, 41, 209–216. [Google Scholar] [CrossRef] [PubMed]
- Shui, X.; Huang, J.; Li, Y.H.; Xie, P.L.; Li, G.C. Construction and selection of human FAb antibody phage display library of liver cancer. Hybridoma 2009, 28, 341–347. [Google Scholar] [CrossRef] [PubMed]
- Kong, M.; Wang, J.; Li, B. Application of phage display technology in targeted therapy of breast cancer. Chin. Ger. J. Clin. Oncol. 2013, 12, 246–248. [Google Scholar] [CrossRef]
- Willats, W.G. Phage display: Practicalities and prospects. Plant Mol. Biol. 2002, 50, 837–854. [Google Scholar] [CrossRef] [PubMed]
- Pansri, P.; Jaruseranee, N.; Rangnoi, K.; Kristensen, P.; Yamabhai, M. A compact phage display human SCFV library for selection of antibodies to a wide variety of antigens. BMC Biotechnol. 2009. [Google Scholar] [CrossRef] [PubMed]
- Bain, B.; Brazil, M. Adalimumab. Nat. Rev. Drug Discov. 2003, 2, 693–694. [Google Scholar] [CrossRef] [PubMed]
- Stohl, W.; Hilbert, D.M. The discovery and development of belimumab: The anti-BLyS-lupus connection. Nat. Biotechnol. 2012, 30, 69–77. [Google Scholar] [CrossRef] [PubMed]
- Bolmer, S.D.; Migone, T.S. Raxibacumab, human monoclonal antibody against anthrax toxin. In Handbook of Therapeutic Antibodies; Dübel, S., Reichert, J.M., Eds.; Wiley-VCH Verlag GmbH & Co. KGaA: Weinheim, Germany, 2014; pp. 1899–1908. [Google Scholar]
- Fukuda, M.N. Peptide-displaying phage technology in glycobiology. Glycobiology 2012, 22, 318–325. [Google Scholar] [CrossRef] [PubMed]
- Kubota, T.; Matsushita, T.; Niwa, R.; Kumagai, I.; Nakamura, K. Novel anti-Tn single-chain Fv-Fc fusion proteins derived from immunized phage library and antibody Fc domain. Anticancer Res. 2010, 30, 3397–3405. [Google Scholar] [PubMed]
- Jakobovits, A. Production of fully human antibodies by transgenic mice. Curr. Opin. Biotechnol. 1995, 6, 561–566. [Google Scholar] [CrossRef]
- Brezski, R.; Almagro, J.C. Application of antibody engineering in the development of next generation antibody-based therapeutics. In Development of Antibody-Based Therapeutics; Tabrizi, M.A., Bornstein, G.G., Klakamp, S.L., Eds.; Springer: New York, NY, USA, 2012; pp. 65–93. [Google Scholar]
- Carter, P.J. Potent antibody therapeutics by design. Nat. Rev. Immunol. 2006, 6, 343–357. [Google Scholar] [CrossRef] [PubMed]
- Sanz, L.; Cuesta, A.M.; Compte, M.; Alvarez-Vallina, L. Antibody engineering: Facing new challenges in cancer therapy. Acta Pharmacol. Sin. 2005, 26, 641–648. [Google Scholar] [CrossRef] [PubMed]
- Presta, L.G. Molecular engineering and design of therapeutic antibodies. Curr. Opin. Immunol. 2008, 20, 460–470. [Google Scholar] [CrossRef] [PubMed]
- Carter, P.J.; Senter, P.D. Antibody-drug conjugates for cancer therapy. Cancer J. 2008, 14, 154–169. [Google Scholar] [CrossRef] [PubMed]
- Chames, P.; Baty, D. Bispecific antibodies for cancer therapy. Curr. Opin. Drug Discov. Dev. 2009, 12, 276–283. [Google Scholar] [CrossRef]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Loureiro, L.R.; Carrascal, M.A.; Barbas, A.; Ramalho, J.S.; Novo, C.; Delannoy, P.; Videira, P.A. Challenges in Antibody Development against Tn and Sialyl-Tn Antigens. Biomolecules 2015, 5, 1783-1809. https://doi.org/10.3390/biom5031783
Loureiro LR, Carrascal MA, Barbas A, Ramalho JS, Novo C, Delannoy P, Videira PA. Challenges in Antibody Development against Tn and Sialyl-Tn Antigens. Biomolecules. 2015; 5(3):1783-1809. https://doi.org/10.3390/biom5031783
Chicago/Turabian StyleLoureiro, Liliana R., Mylène A. Carrascal, Ana Barbas, José S. Ramalho, Carlos Novo, Philippe Delannoy, and Paula A. Videira. 2015. "Challenges in Antibody Development against Tn and Sialyl-Tn Antigens" Biomolecules 5, no. 3: 1783-1809. https://doi.org/10.3390/biom5031783
APA StyleLoureiro, L. R., Carrascal, M. A., Barbas, A., Ramalho, J. S., Novo, C., Delannoy, P., & Videira, P. A. (2015). Challenges in Antibody Development against Tn and Sialyl-Tn Antigens. Biomolecules, 5(3), 1783-1809. https://doi.org/10.3390/biom5031783