
Transcription of Interleukin-8: How Altered Regulation Can Affect Cystic Fibrosis Lung Disease



Abstract
:1. Introduction
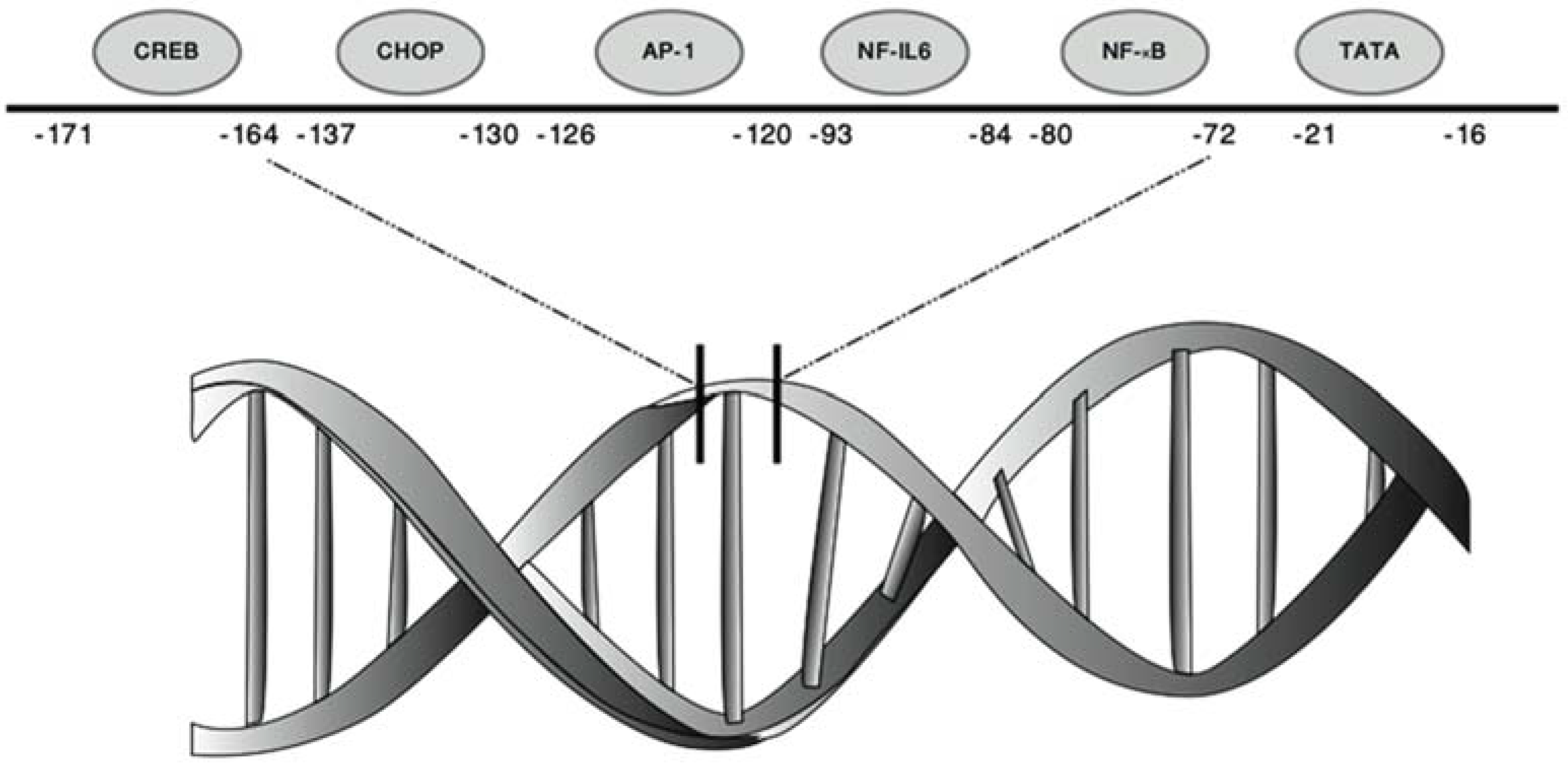
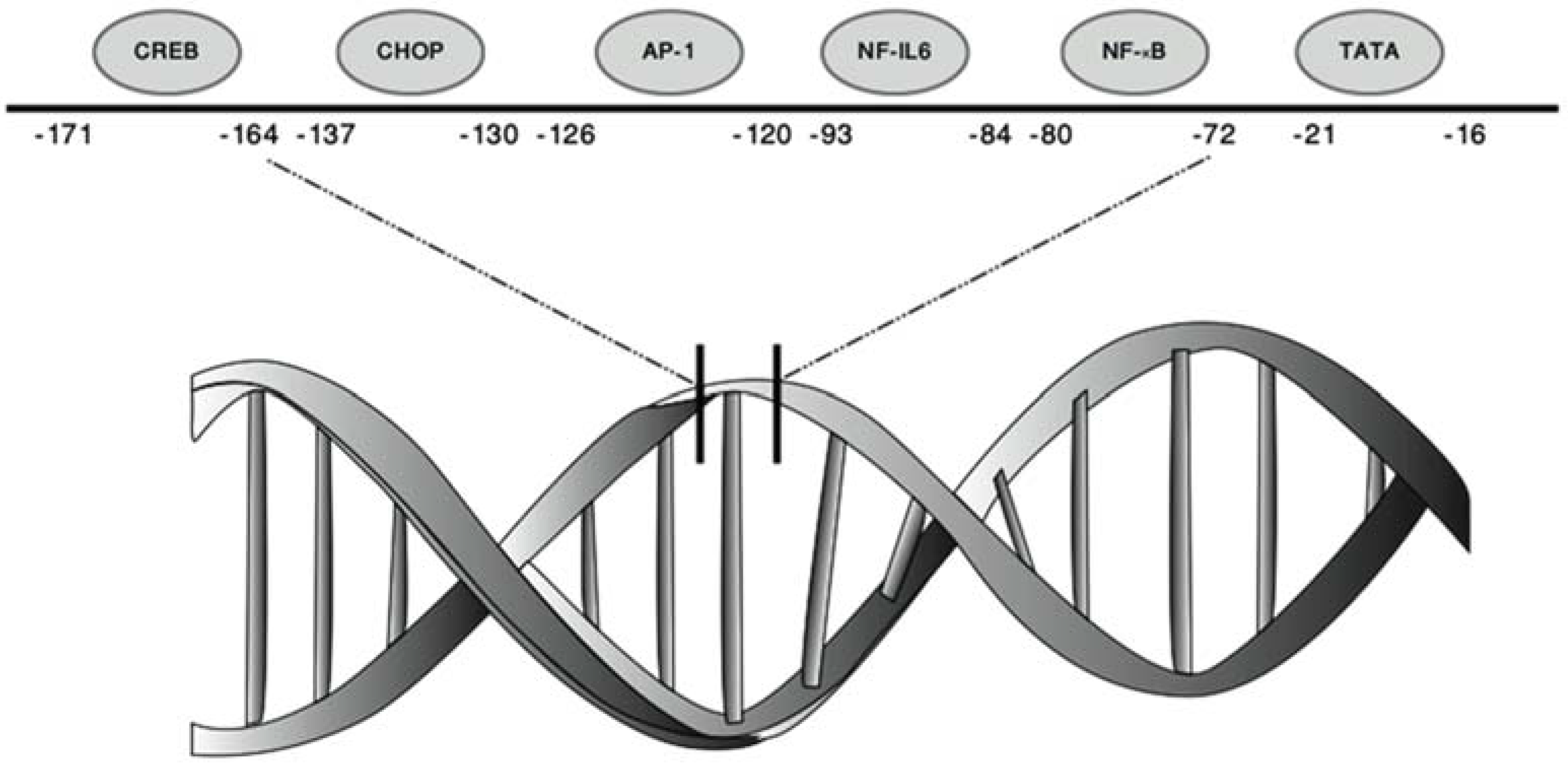
2. Transcriptional Regulation of CXCL8
2.1. Repression of the CXCL8 Promoter
2.2. Transcriptional Activation of CXCL8 by Inducible Transcription Factors
2.3. CXCL8 mRNA Stabilization

{kind=link}
{kind=link}
| Transcription Factors | Transcription Factor Consensus Sequence | Subunits/Isoforms | Structure | Position bp: Base Pair |
|---|---|---|---|---|
| NF-κB | GGAATTTCC | NF-κB1 (p50 & p105) | Dimeric | −80/−72 bp |
| NF-κB2 (p52 & p100) | ||||
| C-Rel | ||||
| RelA (p65) | ||||
| RelB | ||||
| NF-IL6/C/EBPβ | AGTTGCAAAT | 3 Isoforms: | Binds DNA as a dimer | −93/−84 bp |
| P17676-1 | Forms stable heterodimers with CEBPA, CEBPD and CEBPG | |||
| P17676-2 | ||||
| P17676-3 | ||||
| AP-1 | TGACTCA | c-Jun | Homodimer/heterodimer | −126/−120 bp |
| JunD | ||||
| JunB | ||||
| Atf-2 | ||||
| c-Fos | ||||
| Fra-1 | ||||
| Fra-2 | ||||
| CHOP | GTGTGATG | Isoforms: | Heterodimer | −137/−130 bp |
| P35638-1 | ||||
| P35638-2 | ||||
| CREB | TTTCGTCA | Isoforms: | Binds DNA as a dimer | −171/−164 bp |
| P16220-1 | ||||
| P16220-2 | ||||
| P16220-3 |
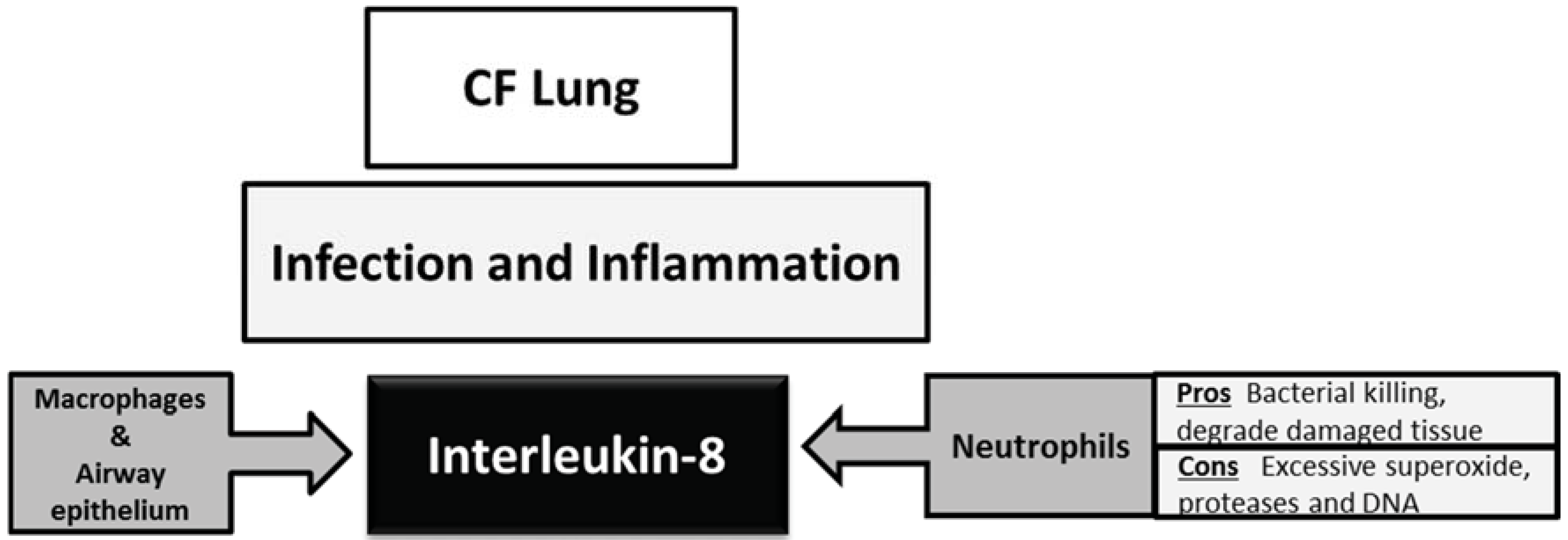
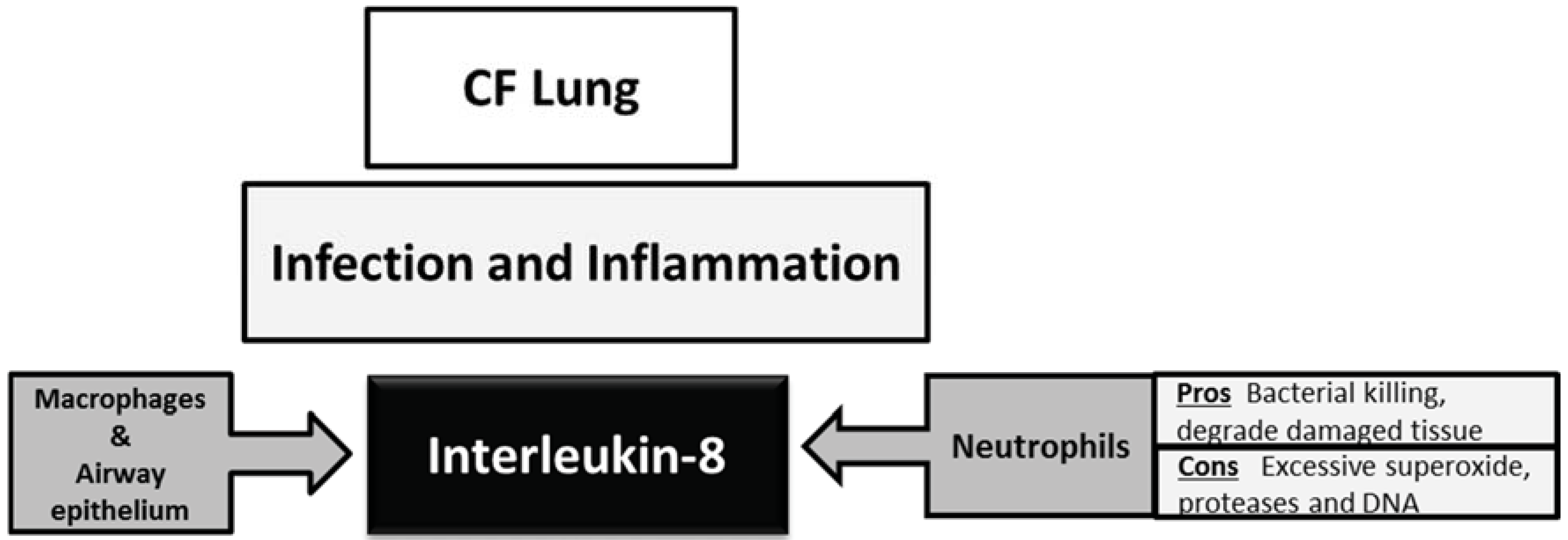
3. Interleukin-8 in the Cystic Fibrosis Lung
4. Strategies Targeting CXCL8 in Cystic Fibrosis
5. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Bacon, K.; Baggiolini, M.; Broxmeyer, H.; Horuk, R.; Lindley, I.; Mantovani, A.; Maysushima, K.; Murphy, P.; Nomiyama, H.; Oppenheim, J.; et al. Chemokine/chemokine receptor nomenclature. J. Interferon Cytokine Res. 2002, 22, 1067–1068. [Google Scholar] [PubMed]
- Colobran, R.; Pujol-Borrell, R.; Armengol, M.P.; Juan, M. The chemokine network. I. How the genomic organization of chemokines contains clues for deciphering their functional complexity. Clin. Exp. Immunol. 2007, 148, 208–217. [Google Scholar] [CrossRef] [PubMed]
- Lindley, I.; Aschauer, H.; Seifert, J.M.; Lam, C.; Brunowsky, W.; Kownatzki, E.; Thelen, M.; Peveri, P.; Dewald, B.; von Tscharner, V.; et al. Synthesis and expression in Escherichia coli of the gene encoding monocyte-derived neutrophil-activating factor: Biological equivalence between natural and recombinant neutrophil-activating factor. Proc. Natl. Acad. Sci. USA 1988, 85, 9199–9203. [Google Scholar] [CrossRef] [PubMed]
- Gimbrone, M.A., Jr.; Obin, M.S.; Brock, A.F.; Luis, E.A.; Hass, P.E.; Hébert, C.A.; Yip, Y.K.; Leung, D.W.; Lowe, D.G.; Kohr, W.J.; et al. Endothelial interleukin-8: A novel inhibitor of leukocyte-endothelial interactions. Science 1989, 246, 1601–1603. [Google Scholar] [CrossRef] [PubMed]
- Baggiolini, M. Chemokines and leukocyte traffic. Nature 1998, 392, 565–568. [Google Scholar] [CrossRef] [PubMed]
- Ahuja, S.K.; Murphy, P.M. The CXC chemokines growth-regulated oncogene (GRO) α, GROβ, GROγ, neutrophil-activating peptide-2, and epithelial cell-derived neutrophil-activating peptide-78 are potent agonists for the type B, but not the type A, human interleukin-8 receptor. J. Biol. Chem. 1996, 271, 20545–20550. [Google Scholar] [CrossRef] [PubMed]
- Winzen, R.; Thakur, B.K.; Dittrich-Breiholz, O.; Shah, M.; Redich, N.; Dhamija, S.; Kracht, M.; Holtmann, H. Functional analysis of KSRP interaction with the AU-rich element of interleukin-8 and identification of inflammatory mRNA targets. Mol. Cell. Biol. 2007, 27, 8388–8400. [Google Scholar] [CrossRef] [PubMed]
- Hu, N.; Zhang, J.; Cui, W.; Kong, G.; Zhang, S.; Yue, L.; Bai, X.; Zhang, Z.; Zhang, W.; Zhang, X.; et al. miR-520b Regulates migration of breast cancer cells by targeting hepatitis B X-interacting protein and interleukin-8. J. Biol. Chem. 2011, 286, 13714–13722. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, Y. The role of chemokines in neutrophil biology. Front. Biosci. 2008, 13, 2400–2407. [Google Scholar] [CrossRef] [PubMed]
- Hoffman, E.; Dittrich-Brehiholz, O.; Holtmann, H.; Kracht, M. Multiple Control of Interleukin-8 gene expression. J. Leukoc. Biol. 2002, 72, 847–855. [Google Scholar]
- Kasahara, T.; Mukaida, N.; Yamashita, K.; Yagisawa, H.; Akahoshi, T.; Matsushima, K.N. IL-1 and TNF-α induction of IL-8 and monocyte chemotactic and activating factor (MCAF) mRNA expression in a human astrocytoma cell line. Immunology 1991, 74, 60–67. [Google Scholar] [PubMed]
- Bezzerri, V.; Borgatti, M.; Finotti, A.; Tamanini, A.; Gambari, R.; Cabrini, G. Mapping the transcriptional machinery of the IL-8 gene in human bronchial epithelial cells. J. Immunol. 2011, 187, 6069–6081. [Google Scholar] [CrossRef] [PubMed]
- Ashburner, B.P.; Westerheide, S.D.; Baldwin, A.S., Jr. The p65 (RelA) subunit of NF-κB interacts with the histone deacetylase (HDAC) corepressors HDAC1 and HDAC2 to negatively regulate gene expression. Mol. Cell. Biol. 2001, 21, 7065–7077. [Google Scholar] [CrossRef] [PubMed]
- Vanden Berghe, W.; de Bosscher, K.; Boone, E.; Plaisance, S.; Haegeman, G. The nuclear factor-κB engages CBP/p300 and histone acetyltransferase activity for transcriptional activation of the interleukin-6 gene promoter. J. Biol. Chem. 1999, 274, 32091–32098. [Google Scholar] [CrossRef] [PubMed]
- Wu, G.D.; Lai, E.J.; Huang, N.; Wen, X. Oct-1 and CCAAT/enhancer-binding protein (C/EBP) bind to overlapping elements within the interleukin-8 promoter. The role of Oct-1 as a transcriptional repressor. J. Biol. Chem. 1997, 272, 2396–2403. [Google Scholar] [PubMed]
- Nourbakhsh, M.; Kalble, S.; Dorrie, A.; Hauser, H.; Resch, K.; Kracht, M. The NF-κB repressing factor is involved in basal repression and interleukin (IL)-1-induced activation of IL-8 transcription by binding to a conserved NF-κB-flanking sequence element. Biol. Chem. 2001, 276, 4501–4508. [Google Scholar] [CrossRef] [PubMed]
- Vij, N.; Amoako, M.O.; Mazur, S.; Zeitlin, P.L. CHOP transcription factor mediates IL-8 signaling in cystic fibrosis bronchial epithelial cells. Am. J. Respir. Cell Mol. Biol. 2008, 38, 176–184. [Google Scholar] [CrossRef] [PubMed]
- Hisatsune, J.; Nakayama, M.; Isomoto, H.; Kurazono, H.; Mukaida, N.; Mukhopadhyay, A.K.; Azuma, T.; Yamaoka, Y.; Sap, J.; Yamasaki, E.; et al. Molecular characterization of Helicobacter pylori VacA induction of IL-8 in U937 cells reveals a prominent role for p38MAPK in activating transcription factor-2, cAMP response element binding protein, and NF-κB activation. J. Immunol. 2008, 180, 5017–5027. [Google Scholar] [CrossRef] [PubMed]
- Baeuerle, P.A.; Baltimore, D. NF-κB: Ten years after. Cell 1996, 87, 13–20. [Google Scholar] [CrossRef]
- Mukaida, N.; Okamoto, S.; Ishikawa, Y.; Matsushima, K.J. Molecular mechanism of interleukin-8 gene expression. J. Leukoc. Biol. 1994, 56, 554–558. [Google Scholar] [PubMed]
- Karin, M.; Liu, Z. AP-1 function and regulation. Curr. Opin. Cell Biol. 1997, 9, 240–246. [Google Scholar] [CrossRef]
- Whitmarsh, A.J.; Davis, R.J. Transcription factor AP-1 regulation by mitogen-activated protein kinase signal transduction pathways. J. Mol. Med. 1996, 74, 589–607. [Google Scholar] [CrossRef] [PubMed]
- Holtmann, H.; Winzen, R.; Holland, P.; Eickemeier, S.; Hoffmann, E.; Wallach, D.; Malinin, N.L.; Cooper, J.A.; Resch, K.; Kracht, M. Induction of interleukin-8 synthesis integrates effects on transcription and mRNA degradation from at least three different cytokine- or stress-activated signal transduction pathways. Mol. Cell. Biol. 1999, 19, 6742–6753. [Google Scholar] [PubMed]
- Krause, A.; Holtmann, H.; Eickemeier, S.; Winzen, R.; Szamel, M.; Resch, K.; Saklatvala, J.; Kracht, M. Stress-activated protein kinase/Jun N-terminal kinase is required for interleukin (IL)-1-induced IL-6 and IL-8 gene expression in the human epidermal carcinoma cell line KB. J. Biol. Chem. 1998, 273, 23681–23689. [Google Scholar] [CrossRef] [PubMed]
- Matsusaka, T.; Fujikawa, K.; Nishio, Y.; Mukaida, N.; Matsushima, K.; Kishimoto, T.; Akira, S. Transcription factors NF-IL6 and NF-κB synergistically activate transcription of the inflammatory cytokines, interleukin 6 and interleukin 8. Proc. Natl. Acad. Sci. USA 1993, 90, 10193–10197. [Google Scholar] [CrossRef] [PubMed]
- Caristi, S.; Piraino, G.; Cucinotta, M.; Valenti, A.; Loddo, S.; Teti, D. Prostaglandin E2 induces interleukin-8 gene transcription by activating C/EBP homologous protein in human T lymphocytes. J. Biol. Chem. 2005, 280, 14433–14442. [Google Scholar] [CrossRef] [PubMed]
- Spooren, A.; Kooijman, R.; Lintermans, B.; van Craenenbroeck, K.; Vermeulen, L.; Haegeman, G.; Gerlo, S. Cooperation of NF-κB and CREB to induce synergistic IL-6 expression in astrocytes. Cell Signal. 2010, 22, 871–881. [Google Scholar] [CrossRef] [PubMed]
- Winzen, R.; Kracht, M.; Ritter, B.; Wilhelm, A.; Chen, C.Y.; Shyu, A.B.; Müller, M.; Gaestel, M.; Resch, K.; Holtmann, H. The p38 MAP kinase pathway signals for cytokine-induced mRNA stabilization via MAP kinase-activated protein kinase 2 and an AU-rich region-targeted mechanism. EMBO J. 1999, 18, 4969–4980. [Google Scholar] [CrossRef] [PubMed]
- Jobin, C.; Holt, L.; Bradham, C.A.; Streetz, K.; Brenner, D.A.; Sartor, R.B. TNF receptor-associated factor-2 is involved in both IL-1 beta and TNF-alpha signaling cascades leading to NF-κB activation and IL-8 expression in human intestinal epithelial cells. J. Immunol. 1999, 162, 4447–4454. [Google Scholar] [PubMed]
- Mall, M.A.; Hartl, D. CFTR: Cystic fibrosis and beyond. Eur. Respir. J. 2014, 44, 1042–1054. [Google Scholar] [CrossRef] [PubMed]
- Richman-Eisenstat, J.B.; Jorens, P.G.; Hébert, C.A.; Ueki, I.; Nadel, J.A. Interleukin-8: An important chemoattractant in sputum of patients with chronic inflammatory airway diseases. Am. J. Physiol. 1993, 264, L413–L418. [Google Scholar] [PubMed]
- Bonfield, T.L.; Panuska, J.R.; Konstan, M.W.; Hilliard, K.A.; Hilliard, J.B.; Ghnaim, H.; Berger, M. Inflammatory cytokines in cystic fibrosis lungs. Am. J. Respir. Crit. Care Med. 1995, 152, 2111–2118. [Google Scholar] [CrossRef] [PubMed]
- Sagel, S.D.; Kapsner, R.; Osberg, I.; Sontag, M.K.; Accurso, F.J. Airway inflammation in children with cystic fibrosis and healthy children assessed by sputum induction. Am. J. Respir. Crit. Care Med. 2001, 164, 1425–1431. [Google Scholar] [CrossRef] [PubMed]
- Mayer-Hamblett, N.; Aitken, M.L.; Accurso, F.J.; Kronmal, R.A.; Konstan, M.W.; Burns, J.L.; Sagel, S.D.; Ramsey, B.W. Association between pulmonary function and sputum biomarkers in cystic fibrosis. Am. J. Respir. Crit. Care Med. 2007, 175, 822–828. [Google Scholar] [CrossRef] [PubMed]
- Hartl, D.; Latzin, P.; Hordijk, P.; Marcos, V.; Rudolph, C.; Woischnik, M.; Krauss-Etschmann, S.; Koller, B.; Reinhardt, D.; Roscher, A.A.; et al. Cleavage of CXCR1 on neutrophils disables bacterial killing in cystic fibrosis lung disease. Nat. Med. 2007, 13, 1423–1430. [Google Scholar] [CrossRef] [PubMed]
- Hartl, D.; Gaggar, A.; Bruscia, E.; Hector, A.; Marcos, V.; Jung, A.; Greene, C.; McElvaney, G.; Mall, M.; Döring, G. Innate immunity in cystic fibrosis lung disease. J. Cyst. Fibros. 2012, 11, 363–382. [Google Scholar] [CrossRef] [PubMed]
- Roum, J.H.; Borok, Z.; McElvaney, N.G.; Grimes, G.J.; Bokser, A.D.; Buhl, R.; Crystal, R.G. Glutathione aerosol suppresses lung epithelial surface inflammatory cell-derived oxidants in cystic fibrosis. J. Appl. Physiol. 1999, 87, 438–443. [Google Scholar] [PubMed]
- Nakamura, H.; Yoshimura, K.; McElvaney, N.G.; Crystal, R.G. Neutrophil elastase in respiratory epithelial lining fluid of individuals with cystic fibrosis induces interleukin-8 gene expression in a human bronchial epithelial cell line. J. Clin. Invest. 1992, 89, 1478–1484. [Google Scholar] [CrossRef] [PubMed]
- Manzenreiter, R.; Kienberger, F.; Marcos, V.; Schilcher, K.; Krautgartner, W.D.; Obermayer, A.; Huml, M.; Stoiber, W.; Hector, A.; Griese, M.; et al. Ultrastructural characterization of cystic fibrosis sputum using atomic force and scanning electron microscopy. J. Cyst. Fibros. 2012, 11, 84–92. [Google Scholar] [CrossRef] [PubMed]
- Greene, C.M.; Carroll, T.P.; Smith, S.G.; Taggart, C.C.; Devaney, J.; Griffin, S.; O’Neill, S.J.; McElvaney, N.G. TLR-induced inflammation in cystic fibrosis and non-cystic fibrosis airway epithelial cells. J. Immunol. 2005, 174, 1638–1646. [Google Scholar] [CrossRef] [PubMed]
- Muir, A.; Soong, G.; Sokol, S.; Reddy, B.; Gomez, M.I.; van Heeckeren, A.; Prince, A. Toll-like receptors in normal and cystic fibrosis airway epithelial cells. Am. J. Respir. Cell Mol. Biol. 2004, 30, 777–783. [Google Scholar] [CrossRef] [PubMed]
- Hauber, H.P.; Tulic, M.K.; Tsicopoulos, A.; Wallaert, B.; Olivenstein, R.; Daigneault, P.; Hamid, Q. Toll-like receptors 4 and 2 expression in the bronchial mucosa of patients with cystic fibrosis. Can. Respir. J. 2005, 12, 13–18. [Google Scholar] [PubMed]
- Zhang, Z.; Louboutin, J.P.; Weiner, D.J.; Goldberg, J.B.; Wilson, J.M. Human airway epithelial cells sense Pseudomonas aeruginosa infection via recognition of flagellin by Toll-like receptor 5. Infect. Immun. 2005, 73, 7151–7160. [Google Scholar] [CrossRef] [PubMed]
- De C. Ventura, G.M.; le Goffic, R.; Balloy, V.; Plotkowski, M.C.; Chignard, M.; Si-Tahar, M. TLR 5, but neither TLR2 nor TLR4, is involved in lung epithelial cell response to Burkholderia cenocepacia. FEMS Immunol. Med. Microbiol. 2008, 54, 37–44. [Google Scholar] [CrossRef] [PubMed]
- Joseph, T.; Look, D.; Ferkol, T. NF-κB activation and sustained IL-8 gene expression in primary cultures of cystic fibrosis airway epithelial cells stimulated with Pseudomonas aeruginosa. Am. J. Physiol. Lung Cell Mol. Physiol. 2005, 288, L471–L479. [Google Scholar] [CrossRef] [PubMed]
- Muselet-Charlier, C.; Roque, T.; Boncoeur, E.; Chadelat, K.; Clement, A.; Jacquot, J.; Tabary, O. Enhanced IL-1β-induced IL-8 production in cystic fibrosis lung epithelial cells is dependent of both mitogen-activated protein kinases and NF-κB signaling. Biochem. Biophys. Res. Commun. 2007, 357, 402–407. [Google Scholar] [CrossRef] [PubMed]
- Venkatakrishnan, A.; Stecenko, A.A.; King, G.; Blackwell, T.R.; Brigham, K.L.; Christman, J.W.; Blackwell, T.S. Exaggerated activation of nuclear factor-κB and altered IκB-β processing in cystic fibrosis bronchial epithelial cells. Am. J. Respir. Cell Mol. Biol. 2000, 23, 396–403. [Google Scholar] [CrossRef] [PubMed]
- Roussel, L.; Martel, G.; Bérubé, J.; Rousseau, S. P. aeruginosa drives CXCL8 synthesis via redundant toll-like receptors and NADPH oxidase in CFTR∆F508 airway epithelial cells. J. Cyst. Fibros. 2011, 10, 107–113. [Google Scholar] [CrossRef] [PubMed]
- DiMango, E.; Ratner, A.J.; Bryan, R.; Tabibi, S.; Prince, A. Activation of NF-κB by adherent Pseudomonas aeruginosa in normal and cystic fibrosis respiratory epithelial cells. J. Clin. Invest. 1998, 101, 2598–2605. [Google Scholar] [CrossRef] [PubMed]
- Walsh, D.E.; Greene, C.M.; Carroll, T.P.; Taggart, C.C.; Gallagher, P.M.; O’Neill, S.J.; McElvaney, N.G. Interleukin-8 up-regulation by neutrophil elastase is mediated by MyD88/IRAK/TRAF-6 in human bronchial epithelium. J. Biol. Chem. 2001, 276, 35494–35499. [Google Scholar] [CrossRef] [PubMed]
- Devaney, J.M.; Greene, C.M.; Taggart, C.C.; Carroll, T.P.; O’Neill, S.J.; McElvaney, N.G. Neutrophil elastase up-regulates interleukin-8 via toll-like receptor 4. FEBS Lett. 2003, 544, 129–132. [Google Scholar] [CrossRef]
- Bergin, D.A.; Greene, C.M.; Sterchi, E.E.; Kenna, C.; Geraghty, P.; Belaaouaj, A.; Taggart, C.C.; O’Neill, S.J.; McElvaney, N.G. Activation of the epidermal growth factor receptor (EGFR) by a novel metalloprotease pathway. J. Biol. Chem. 2008, 283, 31736–31744. [Google Scholar] [CrossRef] [PubMed]
- Cosgrove, S.; Chotirmall, S.H.; Greene, C.M.; McElvaney, N.G. Pulmonary proteases in the cystic fibrosis lung induce interleukin 8 expression from bronchial epithelial cells via a heme/meprin/epidermal growth factor receptor/Toll-like receptor pathway. J. Biol. Chem. 2011, 286, 7692–7704. [Google Scholar] [CrossRef] [PubMed]
- Shao, M.X.; Nadel, J.A. Neutrophil elastase induces MUC5AC mucin production in human airway epithelial cells via a cascade involving protein kinase C, reactive oxygen species, and TNF-α-converting enzyme. J. Immunol. 2005, 175, 4009–4016. [Google Scholar] [CrossRef] [PubMed]
- Kelly, E.; Greene, C.M.; McElvaney, N.G. Targeting neutrophil elastase in cystic fibrosis. Expert Opin. Ther. Targets 2008, 12, 145–157. [Google Scholar] [CrossRef] [PubMed]
- Greene, C.M.; McElvaney, N.G. Proteases and antiproteases in chronic neutrophilic lung disease—Relevance to drug discovery. Br. J. Pharmacol. 2009, 158, 1048–1058. [Google Scholar] [CrossRef] [PubMed]
- Carrabino, S.; Carpani, D.; Livraghi, A.; di Cicco, M.; Costantini, D.; Copreni, E.; Colombo, C.; Conese, M. Dysregulated interleukin-8 secretion and NF-κB activity in human cystic fibrosis nasal epithelial cells. J. Cyst. Fibros. 2006, 5, 113–119. [Google Scholar] [CrossRef] [PubMed]
- Bartling, T.R.; Drumm, M.L. Oxidative stress causes IL8 promoter hyperacetylation in cystic fibrosis airway cell models. Am. J. Respir. Cell Mol. Biol. 2009, 40, 58–65. [Google Scholar] [CrossRef] [PubMed]
- Bhattacharyya, S.; Gutti, U.; Mercado, J.; Moore, C.; Pollard, H.B.; Biswas, R. MAPK signaling pathways regulate IL-8 mRNA stability and IL-8 protein expression in cystic fibrosis lung epithelial cell lines. Am. J. Physiol. Lung Cell Mol. Physiol. 2011, 300, L81–L87. [Google Scholar] [CrossRef] [PubMed]
- McElvaney, O.J.; O’Reilly, N.; White, M.; Lacey, N.; Pohl, K.; Gerlza, T.; Bergin, D.A.; Kerr, H.; McCarthy, C.; O’Brien, M.E.; et al. The effect of the decoy molecule PA401 on CXCL8 levels in bronchoalveolar lavage fluid of patients with cystic fibrosis. Mol. Immunol. 2015, 63, 550–558. [Google Scholar] [CrossRef] [PubMed]
- Reeves, E.P.; Williamson, M.; O’Neill, S.J.; Greally, P.; McElvaney, N.G. Nebulized hypertonic saline decreases IL-8 in sputum of patients with cystic fibrosis. Am. J. Respir. Crit. Care Med. 2011, 183, 1517–1523. [Google Scholar] [CrossRef] [PubMed]
- Reeves, E.P.; Williamson, M.; Byrne, B.; Bergin, D.A.; Smith, S.G.; Greally, P.; O’Kennedy, R.; O’Neill, S.J.; McElvaney, N.G. IL-8 dictates glycosaminoglycan binding and stability of IL-18 in cystic fibrosis. J. Immunol. 2010, 184, 1642–1652. [Google Scholar] [CrossRef] [PubMed]
- Tabary, O.; Escotte, S.; Couetil, J.P.; Hubert, D.; Dusser, D.; Puchelle, E.; Jacquot, J. Genistein inhibits constitutive and inducible NF-κB activation and decreases IL-8 production by human cystic fibrosis bronchial gland cells. Am. J. Pathol. 1999, 155, 473–481. [Google Scholar] [CrossRef]
- Jouneau, S.; Bonizec, M.; Belleguic, C.; Desrues, B.; Brinchault, G.; Galaine, J.; Gangneux, J.P.; Martin-Chouly, C. Anti-inflammatory effect of fluvastatin on IL-8 production induced by Pseudomonas aeruginosa and Aspergillus fumigatus antigens in cystic fibrosis. PLoS ONE 2011, 6, e22655. [Google Scholar] [CrossRef] [PubMed]
- Gambari, R.; Borgatti, M.; Lampronti, I.; Fabbri, E.; Brognara, E.; Bianchi, N.; Piccagli, L.; Yuen, M.C.; Kan, C.W.; Hau, D.K.; et al. Corilagin is a potent inhibitor of NF-κB activity and downregulates TNF-α induced expression of IL-8 gene in cystic fibrosis IB3-1 cells. Int. Immunopharmacol. 2012, 13, 308–315. [Google Scholar] [CrossRef] [PubMed]
- Bezzerri, V.; Borgatti, M.; Nicolis, E.; Lampronti, I.; Dechecchi, M.C.; Mancini, I.; Rizzotti, P.; Gambari, R.; Cabrini, G. Transcription factor oligodeoxynucleotides to NF-κB inhibit transcription of IL-8 in bronchial cells. Am. J. Respir. Cell Mol. Biol. 2008, 39, 86–96. [Google Scholar] [CrossRef] [PubMed]
- De Stefano, D.; Ungaro, F.; Giovino, C.; Polimeno, A.; Quaglia, F.; Carnuccio, R. Sustained inhibition of IL-6 and IL-8 expression by decoy ODN to NF-κB delivered through respirable large porous particles in LPS-stimulated cystic fibrosis bronchial cells. J. Gene Med. 2011, 13, 200–208. [Google Scholar] [CrossRef] [PubMed]
- Finotti, A.; Borgatti, M.; Bezzerri, V.; Nicolis, E.; Lampronti, I.; Dechecchi, M.; Mancini, I.; Cabrini, G.; Saviano, M.; Avitabile, C.; et al. Effects of decoy molecules targeting NF-κB transcription factors in Cystic fibrosis IB3-1 cells: Recruitment of NF-κB to the IL-8 gene promoter and transcription of the IL-8 gene. Artif. DNA PNA XNA 2012, 3, 97–296. [Google Scholar] [CrossRef] [PubMed]
- Bhattacharyya, S.; Balakathiresan, N.S.; Dalgard, C.; Gutti, U.; Armistead, D.; Jozwik, C.; Srivastava, M.; Pollard, H.B.; Biswas, R. Elevated miR-155 promotes inflammation in cystic fibrosis by driving hyperexpression of interleukin-8. J. Biol. Chem. 2011, 286, 11604–11615. [Google Scholar] [CrossRef] [PubMed]
- Fabbri, E.; Borgatti, M.; Montagner, G.; Bianchi, N.; Finotti, A.; Lampronti, I.; Bezzerri, V.; Dechecchi, M.C.; Cabrini, G.; Gambari, R. Expression of microRNA-93 and Interleukin-8 during Pseudomonas aeruginosa-mediated induction of proinflammatory responses. Am. J. Respir. Cell Mol. Biol. 2014, 50, 1144–1155. [Google Scholar] [CrossRef] [PubMed]
- Oglesby, I.K.; Vencken, S.; Agrawal, R.; Gaughan, K.; Molloy, K.; Higgins, G.; McNally, P.; McElvaney, N.G.; Mall, M.A.; Greene, C.M. MiR-17 overexpression in cystic fibrosis airway epithelial cells decreases IL-8 production. Eur. Respir. J. 2015, in press. [Google Scholar]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jundi, K.; Greene, C.M. Transcription of Interleukin-8: How Altered Regulation Can Affect Cystic Fibrosis Lung Disease. Biomolecules 2015, 5, 1386-1398. https://doi.org/10.3390/biom5031386
Jundi K, Greene CM. Transcription of Interleukin-8: How Altered Regulation Can Affect Cystic Fibrosis Lung Disease. Biomolecules. 2015; 5(3):1386-1398. https://doi.org/10.3390/biom5031386
Chicago/Turabian StyleJundi, Karim, and Catherine M. Greene. 2015. "Transcription of Interleukin-8: How Altered Regulation Can Affect Cystic Fibrosis Lung Disease" Biomolecules 5, no. 3: 1386-1398. https://doi.org/10.3390/biom5031386
APA StyleJundi, K., & Greene, C. M. (2015). Transcription of Interleukin-8: How Altered Regulation Can Affect Cystic Fibrosis Lung Disease. Biomolecules, 5(3), 1386-1398. https://doi.org/10.3390/biom5031386