
Mechanisms of Alpha-Synuclein Action on Neurotransmission: Cell-Autonomous and Non-Cell Autonomous Role

{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. αSyn Pathological Species
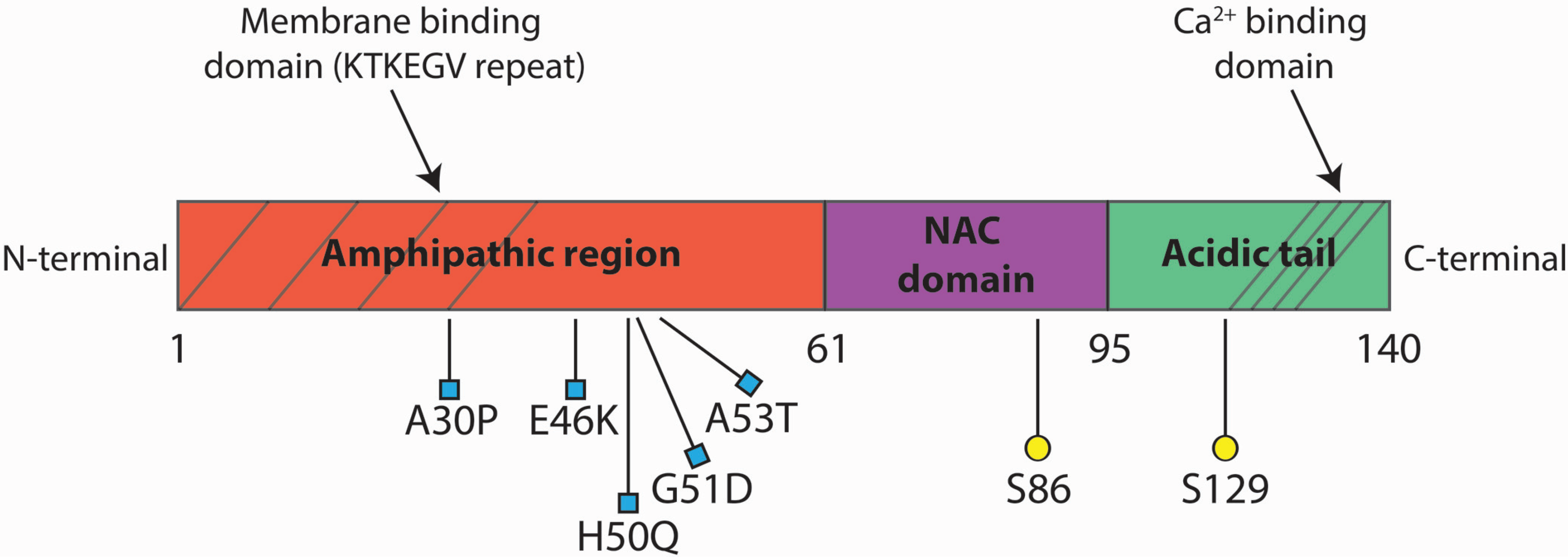
2.1. αSyn Aggregation
2.2. αSyn Mutations
2.3. αSyn Dosage
2.4. αSyn Post-Translational Modifications
3. Physiopathological Role of αSyn in Neurosecretion
3.1. Synaptic Vesicles and Transmitter Release
3.2. Calcium Entry
3.3. Membranes
4. Extracellular αSyn
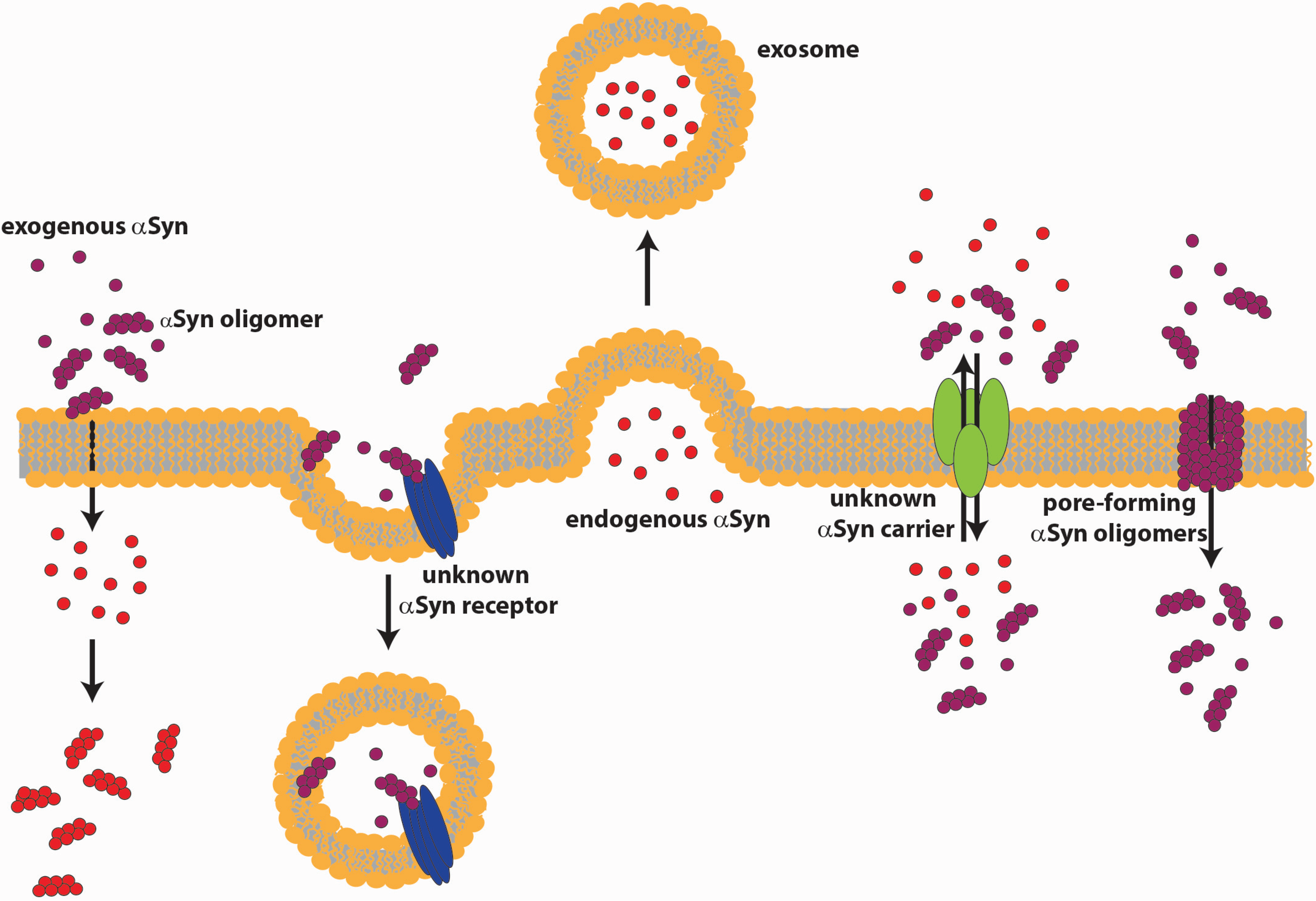
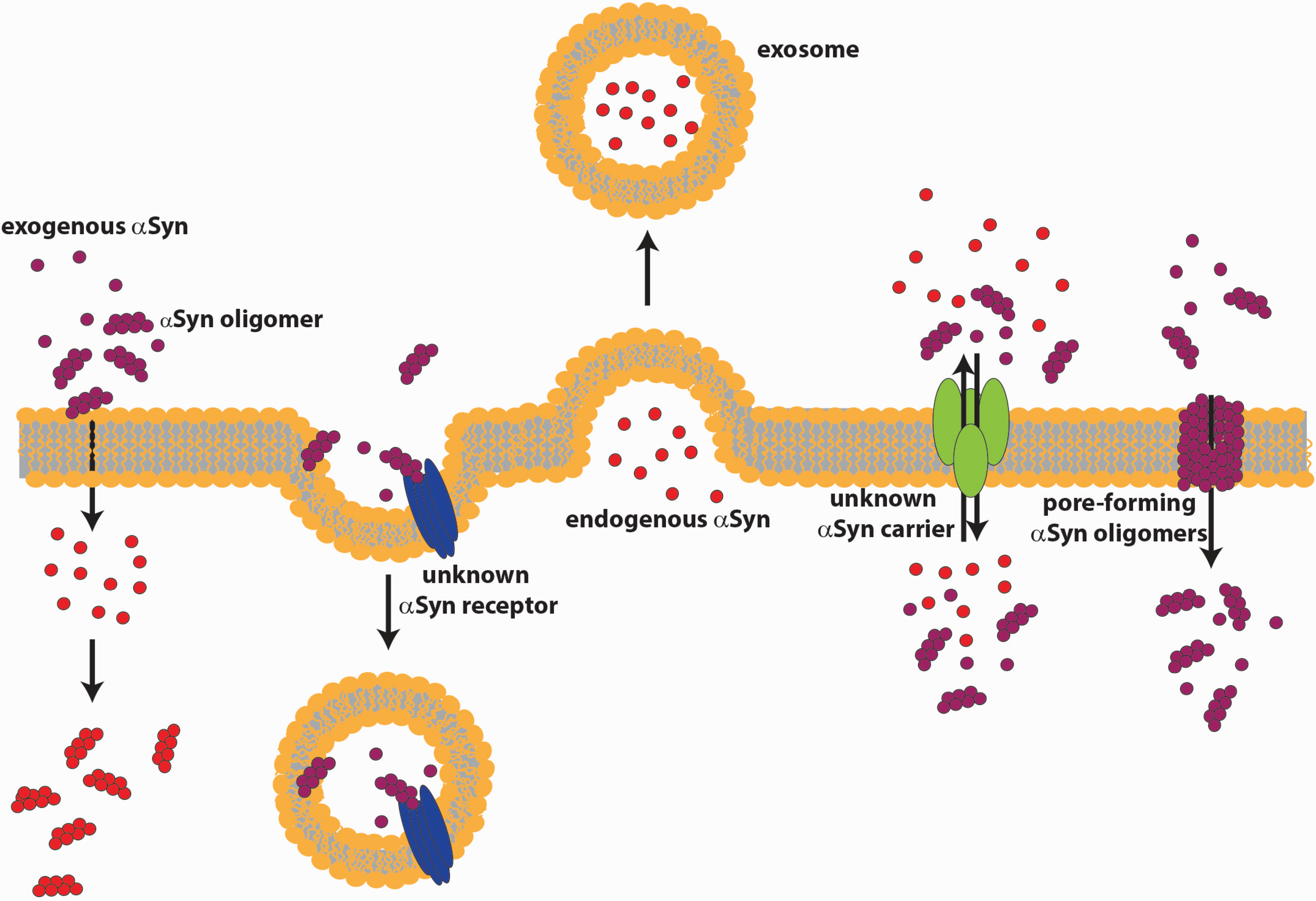
4.1. αSyn Release
4.2. αSyn Internalization
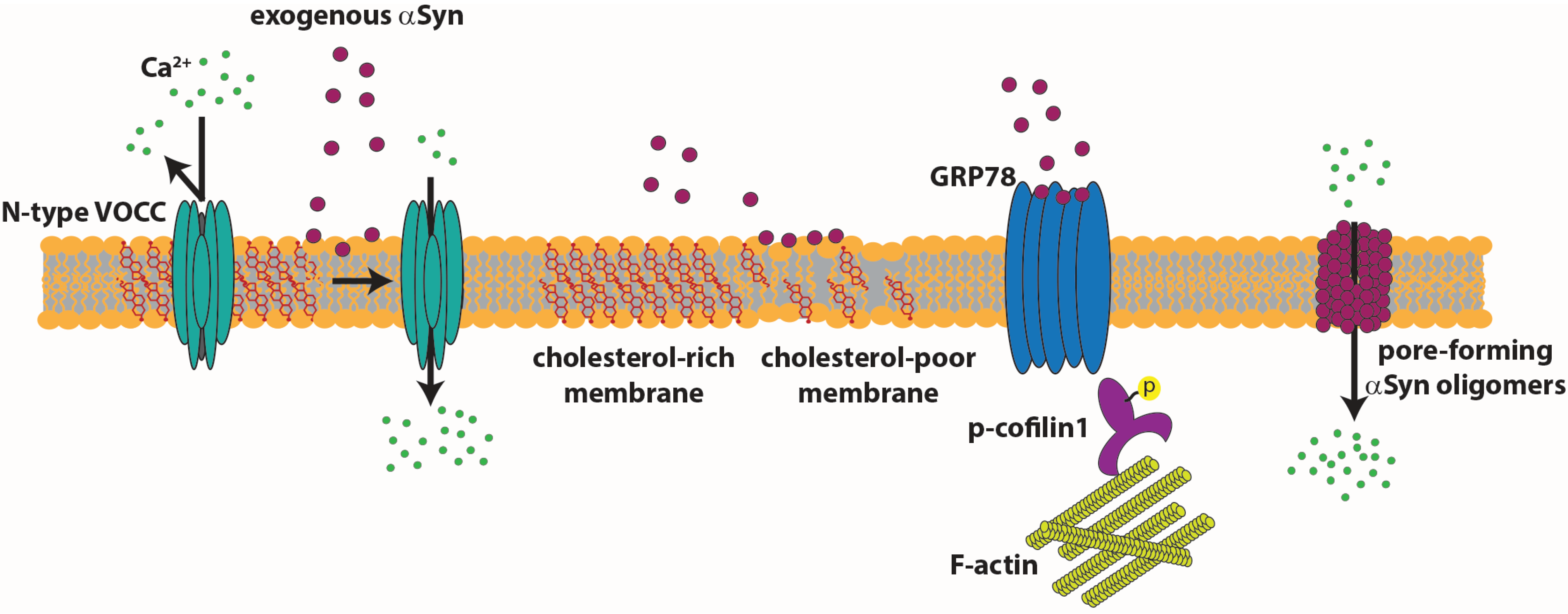
4.3. Extracellular αSyn Action
5. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Maroteaux, L.; Campanelli, J.T.; Scheller, R.H. Synuclein: A neuron-specific protein localized to the nucleus and presynaptic nerve terminal. J. Neurosci. 1988, 8, 2804–2815. [Google Scholar]
- Spillantini, M.G.; Schmidt, M.L.; Lee, V.M.-Y.; Trojanowski, J.Q.; Jakes, R.; Goedert, M. α-Synuclein in Lewy bodies. Nature 1997, 388, 839–840. [Google Scholar]
- Shults, C.W. Lewy bodies. Proc. Natl. Acad. Sci. USA 2006, 103, 1661–1668. [Google Scholar]
- Jensen, P.H.; Nielsen, M.S.; Jakes, R.; Dotti, C.G.; Goedert, M. Binding of α-synuclein to brain vesicles is abolished by familial Parkinson’s disease mutation. J. Biol. Chem. 1998, 273, 26292–26294. [Google Scholar]
- Volles, M.J.; Lansbury, P.T. Relationships between the sequence of alpha-synuclein and its membrane affinity, fibrillization propensity, and yeast toxicity. J. Mol. Biol. 2007, 366, 1510–1522. [Google Scholar]
- Uéda, K.; Fukushima, H.; Masliah, E.; Xia, Y.; Iwai, A.; Yoshimoto, M.; Otero, D.A.; Kondo, J.; Ihara, Y.; Saitoh, T. Molecular cloning of cDNA encoding an unrecognized component of amyloid in Alzheimer disease. Proc. Natl. Acad. Sci. USA 1993, 90, 11282–11286. [Google Scholar]
- Han, H.; Weinreb, P.H.; Lansbury, P.T. The core Alzheimer’s peptide NAC forms amyloid fibrils which seed and are seeded by beta-amyloid: Is NAC a common trigger or target in neurodegenerative disease? Chem. Biol. 1995, 2, 163–169. [Google Scholar]
- Alderson, T.R.; Markley, J.L. Biophysical characterization of α-synuclein and its controversial structure. Intrinsically Disord. Proteins 2013, 1, e26255-1–e26255-22. [Google Scholar]
- Nielsen, M.S.; Vorum, H.; Lindersson, E.; Jensen, P.H. Ca2+ binding to α-synuclein regulates ligand binding and oligomerization. J. Biol. Chem. 2001, 276, 22680–22684. [Google Scholar] [PubMed]
- Crowther, R.A.; Jakes, R.; Spillantini, M.G.; Goedert, M. Synthetic filaments assembled from C-terminally truncated alpha-synuclein. FEBS Lett. 1998, 436, 309–312. [Google Scholar] [PubMed]
- Murray, I.V.J.; Giasson, B.I.; Quinn, S.M.; Koppaka, V.; Axelsen, P.H.; Ischiropoulos, H.; Trojanowski, J.Q.; Lee, V.M.-Y. Role of α-synuclein carboxy-terminus on fibril formation in vitro. Biochemistry 2003, 42, 8530–8540. [Google Scholar] [PubMed]
- Giasson, B.I.; Duda, J.E.; Murray, I.V.; Chen, Q.; Souza, J.M.; Hurtig, H.I.; Ischiropoulos, H.; Trojanowski, J.Q.; Lee, V.M. Oxidative damage linked to neurodegeneration by selective α-synuclein nitration in synucleinopathy lesions. Science 2000, 290, 985–989. [Google Scholar] [PubMed]
- Fujiwara, H.; Hasegawa, M.; Dohmae, N.; Kawashima, A.; Masliah, E.; Goldberg, M.S.; Shen, J.; Takio, K.; Iwatsubo, T. α-Synuclein is phosphorylated in synucleinopathy lesions. Nat. Cell Biol. 2002, 4, 160–164. [Google Scholar] [PubMed]
- Cookson, M.R. The biochemistry of Parkinson’s disease. Annu. Rev. Biochem. 2005, 74, 29–52. [Google Scholar] [PubMed]
- Uversky, V.N. A protein-chameleon: Conformational plasticity of alpha-synuclein, a disordered protein involved in neurodegenerative disorders. J. Biomol. Struct. Dyn. 2003, 21, 211–234. [Google Scholar] [PubMed]
- Uversky, V.N. Intrinsically disordered proteins from A to Z. Int. J. Biochem. Cell Biol. 2011, 43, 1090–1103. [Google Scholar] [PubMed]
- Breydo, L.; Wu, J.W.; Uversky, V.N. α-Synuclein misfolding and Parkinson’s disease. Biochim. Biophys. Acta 2012, 1822, 261–285. [Google Scholar] [PubMed]
- Uversky, V.N.; Li, J.; Fink, A.L. Evidence for a partially folded intermediate in α-synuclein fibril formation. J. Biol. Chem. 2001, 276, 10737–10744. [Google Scholar] [PubMed]
- Uversky, V.N.; Cooper, E.M.; Bower, K.S.; Li, J.; Fink, A.L. Accelerated α-synuclein fibrillation in crowded milieu. FEBS Lett. 2002, 515, 99–103. [Google Scholar] [PubMed]
- Chandra, S.; Gallardo, G.; Fernández-Chacón, R.; Schlüter, O.M.; Südhof, T.C. α-Synuclein cooperates with CSPα in preventing neurodegeneration. Cell 2005, 123, 383–396. [Google Scholar] [PubMed]
- Lykkebo, S.; Jensen, P.H. α-Synuclein and presynaptic function. NeuroMol. Med. 2002, 2, 115–129. [Google Scholar]
- Abeliovich, A.; Schmitz, Y.; Fariñas, I.; Choi-Lundberg, D.; Ho, W.-H.; Castillo, P.E.; Shinsky, N.; Verdugo, J.M.G.; Armanini, M.; Ryan, A.; et al. Mice lacking α-synuclein display functional deficits in the nigrostriatal dopamine system. Neuron 2000, 25, 239–252. [Google Scholar] [PubMed]
- Goedert, M. α-Synuclein and neurodegenerative diseases. Nat. Rev. Neurosci. 2001, 2, 492–501. [Google Scholar] [PubMed]
- Winner, B.; Jappelli, R.; Maji, S.K.; Desplats, P.A.; Boyer, L.; Aigner, S.; Hetzer, C.; Loher, T.; Vilar, M.; Campioni, S.; et al. In vivo demonstration that α-synuclein oligomers are toxic. Proc. Natl. Acad. Sci. USA 2011, 108, 4194–4199. [Google Scholar] [PubMed]
- Lee, H.-J.; Patel, S.; Lee, S.-J. Intravesicular localization and exocytosis of α-synuclein and its aggregates. J. Neurosci. 2005, 25, 6016–6024. [Google Scholar] [PubMed]
- Li, J.; Uversky, V.N.; Fink, A.L. Effect of familial Parkinson’s disease point mutations A30P and A53T on the structural properties, aggregation, and fibrillation of human α-synuclein. Biochemistry 2001, 40, 11604–11613. [Google Scholar] [PubMed]
- Uversky, V.N.; Lee, H.-J.; Li, J.; Fink, A.L.; Lee, S.-J. Stabilization of partially folded conformation during α-synuclein oligomerization in both purified and cytosolic preparations. J. Biol. Chem. 2001, 276, 43495–43498. [Google Scholar] [PubMed]
- Uversky, V.N.; Li, J.; Fink, A.L. Metal-triggered structural transformations, aggregation, and fibrillation of human α-synuclein a possible molecular link between Parkinson’s disease and heavy metal exposure. J. Biol. Chem. 2001, 276, 44284–44296. [Google Scholar] [PubMed]
- Uversky, V.N.; Li, J.; Fink, A.L. Pesticides directly accelerate the rate of α-synuclein fibril formation: A possible factor in Parkinson’s disease. FEBS Lett. 2001, 500, 105–108. [Google Scholar] [PubMed]
- Wood, S.J.; Wypych, J.; Steavenson, S.; Louis, J.-C.; Citron, M.; Biere, A.L. α-Synuclein fibrillogenesis is nucleation-dependent implications for the pathogenesis of parkinson’s disease. J. Biol. Chem. 1999, 274, 19509–19512. [Google Scholar]
- Cheng, F.; Vivacqua, G.; Yu, S. The role of alpha-synuclein in neurotransmission and synaptic plasticity. J. Chem. Neuroanat. 2011, 42, 242–248. [Google Scholar]
- Elkon, H.; Don, J.; Melamed, E.; Ziv, I.; Shirvan, A.; Offen, D. Mutant and wild-type α-synuclein interact with mitochondrial cytochrome C oxidase. J. Mol. Neurosci. 2002, 18, 229–238. [Google Scholar]
- Junn, E.; Mouradian, M.M. Human α-synuclein over-expression increases intracellular reactive oxygen species levels and susceptibility to dopamine. Neurosci. Lett. 2002, 320, 146–150. [Google Scholar]
- Volles, M.J.; Lee, S.-J.; Rochet, J.-C.; Shtilerman, M.D.; Ding, T.T.; Kessler, J.C.; Lansbury, P.T. Vesicle permeabilization by protofibrillar α-synuclein: Implications for the pathogenesis and treatment of Parkinson’s disease. Biochemistry 2001, 40, 7812–7819. [Google Scholar]
- Volles, M.J.; Lansbury, P.T. Vesicle permeabilization by protofibrillar α-synuclein is sensitive to Parkinson’s disease-linked mutations and occurs by a pore-like mechanism. Biochemistry 2002, 41, 4595–4602. [Google Scholar] [CrossRef]
- Emmanouilidou, E.; Stefanis, L.; Vekrellis, K. Cell-produced α-synuclein oligomers are targeted to, and impair, the 26S proteasome. Neurobiol. Aging 2010, 31, 953–968. [Google Scholar] [CrossRef] [PubMed]
- Polymeropoulos, M.H.; Lavedan, C.; Leroy, E.; Ide, S.E.; Dehejia, A.; Dutra, A.; Pike, B.; Root, H.; Rubenstein, J.; Boyer, R.; et al. Mutation in the α-synuclein gene identified in families with Parkinson’s disease. Science 1997, 276, 2045–2047. [Google Scholar] [CrossRef] [PubMed]
- Krüger, R.; Kuhn, W.; Müller, T.; Woitalla, D.; Graeber, M.; Kösel, S.; Przuntek, H.; Epplen, J.T.; Schols, L.; Riess, O. AlaSOPro mutation in the gene encoding α-synuclein in Parkinson’s disease. Nat. Genet. 1998, 18, 106–108. [Google Scholar] [CrossRef] [PubMed]
- Zarranz, J.J.; Alegre, J.; Gómez-Esteban, J.C.; Lezcano, E.; Ros, R.; Ampuero, I.; Vidal, L.; Hoenicka, J.; Rodriguez, O.; Atarés, B.; et al. The new mutation, E46K, of α-synuclein causes parkinson and Lewy body dementia. Ann. Neurol. 2004, 55, 164–173. [Google Scholar] [CrossRef] [PubMed]
- Appel-Cresswell, S.; Vilarino-Guell, C.; Encarnacion, M.; Sherman, H.; Yu, I.; Shah, B.; Weir, D.; Thompson, C.; Szu-Tu, C.; Trinh, J.; et al. Alpha-synuclein p.H50Q, a novel pathogenic mutation for Parkinson’s disease. Mov. Disord. 2013, 28, 811–813. [Google Scholar] [CrossRef] [PubMed]
- Kiely, A.P.; Asi, Y.T.; Kara, E.; Limousin, P.; Ling, H.; Lewis, P.; Proukakis, C.; Quinn, N.; Lees, A.J.; Hardy, J.; et al. α-Synucleinopathy associated with G51D SNCA mutation: A link between Parkinson’s disease and multiple system atrophy? Acta Neuropathol. 2013, 125, 753–769. [Google Scholar] [CrossRef] [PubMed]
- Lesage, S.; Anheim, M.; Letournel, F.; Bousset, L.; Honoré, A.; Rozas, N.; Pieri, L.; Madiona, K.; Dürr, A.; Melki, R.; et al. G51D α-synuclein mutation causes a novel parkinsonian-pyramidal syndrome. Ann. Neurol. 2013, 73, 459–471. [Google Scholar] [CrossRef] [PubMed]
- Proukakis, C.; Dudzik, C.G.; Brier, T.; MacKay, D.S.; Cooper, J.M.; Millhauser, G.L.; Houlden, H.; Schapira, A.H. A novel α-synuclein missense mutation in Parkinson disease. Neurology 2013, 80, 1062–1064. [Google Scholar] [CrossRef] [PubMed]
- Spira, P.J.; Sharpe, D.M.; Halliday, G.; Cavanagh, J.; Nicholson, G.A. Clinical and pathological features of a Parkinsonian syndrome in a family with an Ala53Thr α-synuclein mutation. Ann. Neurol. 2001, 49, 313–319. [Google Scholar] [CrossRef] [PubMed]
- Conway, K.A.; Harper, J.D.; Lansbury, P.T. Fibrils formed in vitro from α-synuclein and two mutant forms linked to Parkinson’s disease are typical amyloid. Biochem. 2000, 39, 2552–2563. [Google Scholar] [CrossRef]
- Rutherford, N.J.; Moore, B.D.; Golde, T.E.; Giasson, B.I. Divergent effects of the H50Q and G51D SNCA mutations on the aggregation of α-synuclein. J. Neurochem. 2014, 131, 859–867. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, Y.; Engelender, S.; Igarashi, S.; Rao, R.K.; Wanner, T.; Tanzi, R.E.; Sawa, A.; Dawson, V.L.; Dawson, T.M.; Ross, C.A. Inducible expression of mutant α-synuclein decreases proteasome activity and increases sensitivity to mitochondria-dependent apoptosis. Hum. Mol. Genet. 2001, 10, 919–926. [Google Scholar] [CrossRef] [PubMed]
- Nonaka, T.; Hasegawa, M. A cellular model to monitor proteasome dysfunction by α-synuclein. Biochem. 2009, 48, 8014–8022. [Google Scholar] [CrossRef]
- Ko, L.; Mehta, N.D.; Farrer, M.; Easson, C.; Hussey, J.; Yen, S.; Hardy, J.; Yen, S.-H.C. Sensitization of neuronal cells to oxidative stress with mutated human α-synuclein. J. Neurochem. 2000, 75, 2546–2554. [Google Scholar] [CrossRef] [PubMed]
- Kanda, S.; Bishop, J.F.; Eglitis, M.A.; Yang, Y.; Mouradian, M.M. Enhanced vulnerability to oxidative stress by α-synuclein mutations and C-terminal truncation. Neuroscience 2000, 97, 279–284. [Google Scholar] [CrossRef] [PubMed]
- Tabrizi, S.J.; Orth, M.; Wilkinson, J.M.; Taanman, J.-W.; Warner, T.T.; Cooper, J.M.; Schapira, A.H.V. Expression of mutant α-synuclein causes increased susceptibility to dopamine toxicity. Hum. Mol. Genet. 2000, 9, 2683–2689. [Google Scholar] [CrossRef] [PubMed]
- Singleton, A.B.; Farrer, M.; Johnson, J.; Singleton, A.; Hague, S.; Kachergus, J.; Hulihan, M.; Peuralinna, T.; Dutra, A.; Nussbaum, R.; et al. α-Synuclein locus triplication causes Parkinson’s disease. Science 2003, 302, 841. [Google Scholar] [CrossRef] [PubMed]
- Ross, O.A.; Braithwaite, A.T.; Skipper, L.M.; Kachergus, J.; Hulihan, M.M.; Middleton, F.A.; Nishioka, K.; Fuchs, J.; Gasser, T.; Maraganore, D.M.; et al. Genomic investigation of α-synuclein multiplication and parkinsonism. Ann. Neurol. 2008. [Google Scholar] [CrossRef]
- Devine, M.J.; Ryten, M.; Vodicka, P.; Thomson, A.J.; Burdon, T.; Houlden, H.; Cavaleri, F.; Nagano, M.; Drummond, N.J.; Taanman, J.-W.; et al. Parkinson’s disease induced pluripotent stem cells with triplication of the α-synuclein locus. Nat. Commun. 2011. [Google Scholar] [CrossRef]
- Flierl, A.; Oliveira, L.M.A.; Falomir-Lockhart, L.J.; Mak, S.K.; Hesley, J.; Soldner, F.; Arndt-Jovin, D.J.; Jaenisch, R.; Langston, J.W.; Jovin, T.M.; et al. Higher vulnerability and stress sensitivity of neuronal precursor cells carrying an alpha-synuclein gene triplication. PLOS ONE 2014. [Google Scholar] [CrossRef]
- Edwards, T.L.; Scott, W.K.; Almonte, C.; Burt, A.; Powell, E.H.; Beecham, G.W.; Wang, L.; Züchner, S.; Konidari, I.; Wang, G.; et al. Genome-wide association study confirms SNPs in SNCA and the MAPT region as common risk factors for Parkinson disease. Ann. Hum. Genet. 2010, 74, 97–109. [Google Scholar] [CrossRef] [PubMed]
- Simón-Sánchez, J.; Schulte, C.; Bras, J.M.; Sharma, M.; Gibbs, J.R.; Berg, D.; Paisan-Ruiz, C.; Lichtner, P.; Scholz, S.W.; Hernandez, D.G.; et al. Genome-wide association study reveals genetic risk underlying Parkinson’s disease. Nat. Genet. 2009, 41, 1308–1312. [Google Scholar] [CrossRef] [PubMed]
- Chu, Y.; Kordower, J.H. Age-associated increases of α-synuclein in monkeys and humans are associated with nigrostriatal dopamine depletion: Is this the target for Parkinson’s disease? Neurobiol. Dis. 2007, 25, 134–149. [Google Scholar] [CrossRef] [PubMed]
- Uversky, V.N.; Yamin, G.; Souillac, P.O.; Goers, J.; Glaser, C.B.; Fink, A.L. Methionine oxidation inhibits fibrillation of human alpha-synuclein in vitro. FEBS Lett. 2002, 517, 239–244. [Google Scholar] [CrossRef] [PubMed]
- Hokenson, M.J.; Uversky, V.N.; Goers, J.; Yamin, G.; Munishkina, L.A.; Fink, A.L. Role of individual methionines in the fibrillation of methionine-oxidized alpha-synuclein. Biochemistry 2004, 43, 4621–4633. [Google Scholar] [CrossRef] [PubMed]
- Mishizen-Eberz, A.J.; Guttmann, R.P.; Giasson, B.I.; Day, G.A.; Hodara, R.; Ischiropoulos, H.; Lee, V.M.-Y.; Trojanowski, J.Q.; Lynch, D.R. Distinct cleavage patterns of normal and pathologic forms of alpha-synuclein by calpain I in vitro. J. Neurochem. 2003, 86, 836–847. [Google Scholar] [CrossRef] [PubMed]
- Okochi, M.; Walter, J.; Koyama, A.; Nakajo, S.; Baba, M.; Iwatsubo, T.; Meijer, L.; Kahle, P.J.; Haass, C. Constitutive phosphorylation of the Parkinson’s disease associated α-synuclein. J. Biol. Chem. 2000, 275, 390–397. [Google Scholar] [CrossRef] [PubMed]
- Pronin, A.N.; Morris, A.J.; Surguchov, A.; Benovic, J.L. Synucleins are a novel class of substrates for G protein-coupled receptor kinases. J. Biol. Chem. 2000, 275, 26515–26522. [Google Scholar] [CrossRef] [PubMed]
- Waxman, E.A.; Giasson, B.I. Characterization of kinases involved in the phosphorylation of aggregated α-synuclein. J. Neurosci. Res. 2011, 89, 231–247. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Feany, M.B. α-Synuclein phosphorylation controls neurotoxicity and inclusion formation in a Drosophila model of Parkinson disease. Nat. Neurosci. 2005, 8, 657–663. [Google Scholar] [CrossRef] [PubMed]
- Visanji, N.P.; Wislet-Gendebien, S.; Oschipok, L.W.; Zhang, G.; Aubert, I.; Fraser, P.E.; Tandon, A. Effect of Ser-129 phosphorylation on interaction of α-synuclein with synaptic and cellular membranes. J. Biol. Chem. 2011, 286, 35863–35873. [Google Scholar] [CrossRef] [PubMed]
- Paleologou, K.E.; Oueslati, A.; Shakked, G.; Rospigliosi, C.C.; Kim, H.-Y.; Lamberto, G.R.; Fernandez, C.O.; Schmid, A.; Chegini, F.; Gai, W.P.; et al. Phosphorylation at S87 is enhanced in synucleinopathies, inhibits α-synuclein oligomerization, and influences synuclein-membrane interactions. J. Neurosci. 2010, 30, 3184–3198. [Google Scholar] [CrossRef] [PubMed]
- Hasegawa, M.; Fujiwara, H.; Nonaka, T.; Wakabayashi, K.; Takahashi, H.; Lee, V.M.-Y.; Trojanowski, J.Q.; Mann, D.; Iwatsubo, T. Phosphorylated alpha-synuclein is ubiquitinated in alpha-synucleinopathy lesions. J. Biol. Chem. 2002, 277, 49071–49076. [Google Scholar] [CrossRef] [PubMed]
- Sadowski, M.; Sarcevic, B. Mechanisms of mono- and poly-ubiquitination: Ubiquitination specificity depends on compatibility between the E2 catalytic core and amino acid residues proximal to the lysine. Cell Div. 2010. [Google Scholar] [CrossRef]
- Nonaka, T.; Iwatsubo, T.; Hasegawa, M. Ubiquitination of α-synuclein. Biochem. 2005, 44, 361–368. [Google Scholar] [CrossRef]
- Steffan, J.S.; Agrawal, N.; Pallos, J.; Rockabrand, E.; Trotman, L.C.; Slepko, N.; Illes, K.; Lukacsovich, T.; Zhu, Y.-Z.; Cattaneo, E.; et al. SUMO modification of huntingtin and Huntington’s disease pathology. Science 2004, 304, 100–104. [Google Scholar] [CrossRef] [PubMed]
- Krumova, P.; Meulmeester, E.; Garrido, M.; Tirard, M.; Hsiao, H.-H.; Bossis, G.; Urlaub, H.; Zweckstetter, M.; Kügler, S.; Melchior, F.; et al. Sumoylation inhibits α-synuclein aggregation and toxicity. J. Cell Biol. 2011, 194, 49–60. [Google Scholar] [CrossRef] [PubMed]
- Kunadt, M.; Eckermann, K.; Stuendl, A.; Gong, J.; Russo, B.; Strauss, K.; Rai, S.; Kügler, S.; Lockhart, L.F.; Schwalbe, M.; et al. Extracellular vesicle sorting of α-synuclein is regulated by sumoylation. Acta Neuropathol. 2015, 129, 695–713. [Google Scholar] [CrossRef] [PubMed]
- Jakes, R.; Spillantini, M.G.; Goedert, M. Identification of two distinct synucleins from human brain. FEBS Lett. 1994, 345, 27–32. [Google Scholar] [CrossRef] [PubMed]
- McLean, P.J.; Ribich, S.; Hyman, B.T. Subcellular localization of alpha-synuclein in primary neuronal cultures: Effect of missense mutations. J. Neural Transm. Suppl. 2000, 7, 53–63. [Google Scholar]
- Zhong, S.; Luo, X.; Chen, X.; Cai, Q.; Liu, J.; Chen, X.; Yao, Z. Expression and subcellular location of α-synuclein during mouse-embryonic development. Cell. Mol. Neurobiol. 2009, 30, 469–482. [Google Scholar] [CrossRef] [PubMed]
- Mori, F.; Tanji, K.; Yoshimoto, M.; Takahashi, H.; Wakabayashi, K. Immunohistochemical comparison of alpha- and beta-synuclein in adult rat central nervous system. Brain Res. 2002, 941, 118–126. [Google Scholar] [CrossRef] [PubMed]
- Vivacqua, G.; Casini, A.; Vaccaro, R.; Fornai, F.; Yu, S.; D’Este, L. Different sub-cellular localization of alpha-synuclein in the C57BL6J mouse’s central nervous system by two novel monoclonal antibodies. J. Chem. Neuroanat. 2011, 41, 97–110. [Google Scholar] [CrossRef] [PubMed]
- Richter-Landsberg, C.; Gorath, M.; Trojanowski, J.Q.; Lee, V.M. Alpha-synuclein is developmentally expressed in cultured rat brain oligodendrocytes. J. Neurosci. Res. 2000, 62, 9–14. [Google Scholar] [CrossRef] [PubMed]
- Wakabayashi, K.; Hayashi, S.; Yoshimoto, M.; Kudo, H.; Takahashi, H. NACP/α-synuclein-positive filamentous inclusions in astrocytes and oligodendrocytes of Parkinson’s disease brains. Acta Neuropathol. 2000, 99, 14–20. [Google Scholar] [CrossRef] [PubMed]
- Tanji, K.; Mori, F.; Imaizumi, T.; Yoshida, H.; Matsumiya, T.; Tamo, W.; Yoshimoto, M.; Odagiri, H.; Sasaki, M.; Takahashi, H.; Satoh, K.; Wakabayashi, K. Upregulation of alpha-synuclein by lipopolysaccharide and interleukin-1 in human macrophages. Pathol. Int. 2002, 52, 572–577. [Google Scholar] [CrossRef] [PubMed]
- Tamo, W.; Imaizumi, T.; Tanji, K.; Yoshida, H.; Mori, F.; Yoshimoto, M.; Takahashi, H.; Fukuda, I.; Wakabayashi, K.; Satoh, K. Expression of α-synuclein, the precursor of non-amyloid β component of Alzheimer’s disease amyloid, in human cerebral blood vessels. Neurosci. Lett. 2002, 326, 5–8. [Google Scholar] [CrossRef] [PubMed]
- Hashimoto, M.; Yoshimoto, M.; Sisk, A.; Hsu, L.J.; Sundsmo, M.; Kittel, A.; Saitoh, T.; Miller, A.; Masliah, E. NACP, a synaptic protein involved in Alzheimer’s disease, is differentially regulated during megakaryocyte differentiation. Biochem. Biophys. Res. Commun. 1997, 237, 611–616. [Google Scholar] [CrossRef] [PubMed]
- Goers, J.; Manning-Bog, A.B.; McCormack, A.L.; Millett, I.S.; Doniach, S.; di Monte, D.A.; Uversky, V.N.; Fink, A.L. Nuclear localization of α-synuclein and its interaction with histones. Biochemistry 2003, 42, 8465–8471. [Google Scholar] [CrossRef] [PubMed]
- Yu, S.; Li, X.; Liu, G.; Han, J.; Zhang, C.; Li, Y.; Xu, S.; Liu, C.; Gao, Y.; Yang, H.; et al. Extensive nuclear localization of alpha-synuclein in normal rat brain neurons revealed by a novel monoclonal antibody. Neuroscience 2007, 145, 539–555. [Google Scholar] [CrossRef] [PubMed]
- Kahle, P.J.; Neumann, M.; Ozmen, L.; Müller, V.; Jacobsen, H.; Schindzielorz, A.; Okochi, M.; Leimer, U.; van der Putten, H.; Probst, A.; et al. Subcellular localization of wild-type and Parkinson’s disease-associated mutant α-synuclein in human and transgenic mouse brain. J. Neurosci. 2000, 20, 6365–6373. [Google Scholar] [PubMed]
- Hsu, L.J.; Mallory, M.; Xia, Y.; Veinbergs, I.; Hashimoto, M.; Yoshimoto, M.; Thal, L.J.; Saitoh, T.; Masliah, E. Expression pattern of synucleins (non-Abeta component of Alzheimer’s disease amyloid precursor protein/α-synuclein) during murine brain development. J. Neurochem. 1998, 71, 338–344. [Google Scholar] [CrossRef] [PubMed]
- Iwai, A.; Masliah, E.; Yoshimoto, M.; Ge, N.; Flanagan, L.; de Silva, H.A.; Kittel, A.; Saitoh, T. The precursor protein of non-A beta component of Alzheimer’s disease amyloid is a presynaptic protein of the central nervous system. Neuron 1995, 14, 467–475. [Google Scholar] [CrossRef] [PubMed]
- Davidson, W.S.; Jonas, A.; Clayton, D.F.; George, J.M. Stabilization of α-synuclein secondary structure upon binding to synthetic membranes. J. Biol. Chem. 1998, 273, 9443–9449. [Google Scholar] [CrossRef] [PubMed]
- Clayton, D.F.; George, J.M. Synucleins in synaptic plasticity and neurodegenerative disorders. J. Neurosci. Res. 1999, 58, 120–129. [Google Scholar] [CrossRef] [PubMed]
- George, J.M.; Jin, H.; Woods, W.S.; Clayton, D.F. Characterization of a novel protein regulated during the critical period for song learning in the zebra finch. Neuron 1995, 15, 361–372. [Google Scholar] [CrossRef] [PubMed]
- Hartman, V.N.; Miller, M.A.; Clayton, D.F.; Liu, W.C.; Kroodsma, D.E.; Brenowitz, E.A. Testosterone regulates α-synuclein mRNA in the avian song system. Neuroreport 2001, 12, 943–946. [Google Scholar] [CrossRef] [PubMed]
- Snead, D.; Eliezer, D. α-Synuclein function and dysfunction on cellular membranes. Exp. Neurobiol. 2014, 23, 292–313. [Google Scholar] [CrossRef] [PubMed]
- Murphy, D.D.; Rueter, S.M.; Trojanowski, J.Q.; Lee, V.M.-Y. Synucleins are developmentally expressed, and α-synuclein regulates the size of the presynaptic vesicular pool in primary hippocampal neurons. J. Neurosci. 2000, 20, 3214–3220. [Google Scholar] [PubMed]
- Yavich, L.; Tanila, H.; Vepsäläinen, S.; Jäkälä, P. Role of alpha-synuclein in presynaptic dopamine recruitment. J. Neurosci. 2004, 24, 11165–11170. [Google Scholar] [CrossRef] [PubMed]
- Yavich, L.; Oksman, M.; Tanila, H.; Kerokoski, P.; Hiltunen, M.; van Groen, T.; Puoliväli, J.; Männistö, P.T.; García-Horsman, A.; MacDonald, E.; et al. Locomotor activity and evoked dopamine release are reduced in mice overexpressing A30P-mutated human α-synuclein. Neurobiol. Dis. 2005, 20, 303–313. [Google Scholar] [CrossRef] [PubMed]
- Yavich, L.; Jäkälä, P.; Tanila, H. Abnormal compartmentalization of norepinephrine in mouse dentate gyrus in α-synuclein knockout and A30P transgenic mice. J. Neurochem. 2006, 99, 724–732. [Google Scholar] [CrossRef] [PubMed]
- Gureviciene, I.; Gurevicius, K.; Tanila, H. Role of α-synuclein in synaptic glutamate release. Neurobiol. Dis. 2007, 28, 83–89. [Google Scholar] [CrossRef] [PubMed]
- Larsen, K.E.; Schmitz, Y.; Troyer, M.D.; Mosharov, E.; Dietrich, P.; Quazi, A.Z.; Savalle, M.; Nemani, V.; Chaudhry, F.A.; Edwards, R.H.; et al. α-Synuclein overexpression in PC12 and chromaffin cells impairs catecholamine release by interfering with a late step in exocytosis. J. Neurosci. 2006, 26, 11915–11922. [Google Scholar] [CrossRef] [PubMed]
- Nemani, V.M.; Lu, W.; Berge, V.; Nakamura, K.; Onoa, B.; Lee, M.K.; Chaudhry, F.A.; Nicoll, R.A.; Edwards, R.H. Increased expression of α-synuclein reduces neurotransmitter release by inhibiting synaptic vesicle reclustering after endocytosis. Neuron 2010, 65, 66–79. [Google Scholar] [CrossRef] [PubMed]
- Cabin, D.E.; Shimazu, K.; Murphy, D.; Cole, N.B.; Gottschalk, W.; McIlwain, K.L.; Orrison, B.; Chen, A.; Ellis, C.E.; Paylor, R.; et al. Synaptic vesicle depletion correlates with attenuated synaptic responses to prolonged repetitive stimulation in mice lacking alpha-synuclein. J. Neurosci. 2002, 22, 8797–8807. [Google Scholar] [PubMed]
- Chandra, S.; Fornai, F.; Kwon, H.-B.; Yazdani, U.; Atasoy, D.; Liu, X.; Hammer, R.E.; Battaglia, G.; German, D.C.; Castillo, P.E.; et al. Double-knockout mice for alpha- and beta-synucleins: Effect on synaptic functions. Proc. Natl. Acad. Sci. USA 2004, 101, 14966–14971. [Google Scholar] [CrossRef] [PubMed]
- Burre, J.; Sharma, M.; Tsetsenis, T.; Buchman, V.; Etherton, M.; Sudhof, T.C. α-Synuclein promotes SNARE-complex assembly in vivo and in vitro. Science 2010, 329, 1663–1667. [Google Scholar] [CrossRef] [PubMed]
- Greten-Harrison, B.; Polydoro, M.; Morimoto-Tomita, M.; Diao, L.; Williams, A.M.; Nie, E.H.; Makani, S.; Tian, N.; Castillo, P.E.; Buchman, V.L.; et al. αβγ-Synuclein triple knockout mice reveal age-dependent neuronal dysfunction. Proc. Natl. Acad. Sci. USA 2010, 107, 19573–19578. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Ninan, I.; Antonova, I.; Battaglia, F.; Trinchese, F.; Narasanna, A.; Kolodilov, N.; Dauer, W.; Hawkins, R.D.; Arancio, O. α-Synuclein produces a long-lasting increase in neurotransmitter release. EMBO J. 2004, 23, 4506–4516. [Google Scholar] [CrossRef] [PubMed]
- Martín, E.D.; González-García, C.; Milán, M.; Fariñas, I.; Ceña, V. Stressor-related impairment of synaptic transmission in hippocampal slices from alpha-synuclein knockout mice. Eur. J. Neurosci. 2004, 20, 3085–3091. [Google Scholar] [CrossRef] [PubMed]
- Lundblad, M.; Decressac, M.; Mattsson, B.; Björklund, A. Impaired neurotransmission caused by overexpression of α-synuclein in nigral dopamine neurons. Proc. Natl. Acad. Sci. USA 2012, 109, 3213–3219. [Google Scholar] [CrossRef] [PubMed]
- Ahn, B.-H.; Rhim, H.; Kim, S.Y.; Sung, Y.-M.; Lee, M.-Y.; Choi, J.-Y.; Wolozin, B.; Chang, J.-S.; Lee, Y.H.; Kwon, T.K.; et al. α-Synuclein interacts with phospholipase D isozymes and inhibits pervanadate-induced phospholipase D activation in human embryonic kidney-293 Cells. J. Biol. Chem. 2002, 277, 12334–12342. [Google Scholar] [CrossRef] [PubMed]
- Cockcroft, S. Signalling roles of mammalian phospholipase D1 and D2. Cell. Mol. Life Sci. 2001, 58, 1674–1687. [Google Scholar] [CrossRef] [PubMed]
- Jenco, J.M.; Rawlingson, A.; Daniels, B.; Morris, A.J. Regulation of phospholipase D2: Selective inhibition of mammalian phospholipase D isoenzymes by α- and β-synucleins. Biochem. 1998, 37, 4901–4909. [Google Scholar] [CrossRef]
- Bellani, S.; Sousa, V.L.; Ronzitti, G.; Valtorta, F.; Meldolesi, J.; Chieregatti, E. The regulation of synaptic function by α-synuclein. Commun. Integr. Biol. 2010, 3, 106–109. [Google Scholar] [CrossRef] [PubMed]
- Sousa, V.L.; Bellani, S.; Giannandrea, M.; Yousuf, M.; Valtorta, F.; Meldolesi, J.; Chieregatti, E. α-Synuclein and its A30P mutant affect actin cytoskeletal structure and dynamics. Mol. Biol. Cell 2009, 20, 3725–3739. [Google Scholar] [CrossRef] [PubMed]
- Berridge, M.J.; Bootman, M.D.; Roderick, H.L. Calcium signalling: Dynamics, homeostasis and remodelling. Nat. Rev. Mol. Cell Biol. 2003, 4, 517–529. [Google Scholar] [CrossRef] [PubMed]
- Patel, S.; Docampo, R. Acidic calcium stores open for business: Expanding the potential for intracellular Ca2+ signaling. Trends Cell Biol. 2010, 20, 277–286. [Google Scholar] [CrossRef] [PubMed]
- Burgoyne, R.D. Neuronal calcium sensor proteins: Generating diversity in neuronal Ca2+ signalling. Nat. Rev. Neurosci. 2007, 8, 182–193. [Google Scholar] [CrossRef] [PubMed]
- Thayer, S.A.; Usachev, Y.M.; Pottorf, W.J. Modulating Ca2+ clearance from neurons. Front. Biosci. J. Virtual Libr. 2002, 7, d1255–1279. [Google Scholar] [CrossRef]
- Zündorf, G.; Reiser, G. Calcium dysregulation and homeostasis of neural calcium in the molecular mechanisms of neurodegenerative diseases provide multiple targets for neuroprotection. Antioxid. Redox Signal. 2011, 14, 1275–1288. [Google Scholar] [CrossRef]
- Damier, P.; Hirsch, E.C.; Agid, Y.; Graybiel, A.M. The substantia nigra of the human brain. I. Nigrosomes and the nigral matrix, a compartmental organization based on calbindin D(28K) immunohistochemistry. Brain J. Neurol. 1999, 122, 1421–1436. [Google Scholar] [CrossRef]
- Kim, B.G.; Shin, D.H.; Jeon, G.S.; Seo, J.H.; Kim, Y.W.; Jeon, B.S.; Cho, S.S. Relative sparing of calretinin containing neurons in the substantia nigra of 6-OHDA treated rat parkinsonian model. Brain Res. 2000, 855, 162–165. [Google Scholar] [CrossRef] [PubMed]
- Soós, J.; Engelhardt, J.I.; Siklós, L.; Havas, L.; Majtényi, K. The expression of PARP, NF-κB and parvalbumin is increased in Parkinson disease. Neuroreport 2004, 15, 1715–1718. [Google Scholar] [CrossRef] [PubMed]
- Liang, C.L.; Sinton, C.M.; Sonsalla, P.K.; German, D.C. Midbrain dopaminergic neurons in the mouse that contain calbindin-D28k exhibit reduced vulnerability to MPTP-induced neurodegeneration. Neuroregeneration 1996, 5, 313–318. [Google Scholar] [CrossRef]
- Surmeier, D.J.; Guzman, J.N.; Sanchez-Padilla, J. Calcium, cellular aging, and selective neuronal vulnerability in Parkinson’s disease. Cell Calcium 2010, 47, 175–182. [Google Scholar] [CrossRef] [PubMed]
- Adamczyk, A.; Strosznajder, J.B. α-Synuclein potentiates Ca2+ influx through voltage-dependent Ca2+ channels. NeuroReport 2006, 17, 1883–1886. [Google Scholar] [CrossRef] [PubMed]
- Tamamizu-Kato, S.; Kosaraju, M.G.; Kato, H.; Raussens, V.; Ruysschaert, J.-M.; Narayanaswami, V. Calcium-triggered membrane interaction of the alpha-synuclein acidic tail. Biochemistry 2006, 45, 10947–10956. [Google Scholar] [CrossRef] [PubMed]
- Danzer, K.M.; Haasen, D.; Karow, A.R.; Moussaud, S.; Habeck, M.; Giese, A.; Kretzschmar, H.; Hengerer, B.; Kostka, M. Different species of α-synuclein oligomers induce calcium influx and seeding. J. Neurosci. 2007, 27, 9220–9232. [Google Scholar] [CrossRef] [PubMed]
- Feng, L.R.; Federoff, H.J.; Vicini, S.; Maguire-Zeiss, K.A. α-Synuclein mediates alterations in membrane conductance: A potential role for α-synuclein oligomers in cell vulnerability. Eur. J. Neurosci. 2010, 32, 10–17. [Google Scholar] [CrossRef] [PubMed]
- Lashuel, H.A.; Petre, B.M.; Wall, J.; Simon, M.; Nowak, R.J.; Walz, T.; Lansbury, P.T. α-Synuclein, especially the Parkinson’s disease-associated mutants, forms pore-like annular and tubular protofibrils. J. Mol. Biol. 2002, 322, 1089–1102. [Google Scholar] [CrossRef] [PubMed]
- Furukawa, K.; Matsuzaki-Kobayashi, M.; Hasegawa, T.; Kikuchi, A.; Sugeno, N.; Itoyama, Y.; Wang, Y.; Yao, P.J.; Bushlin, I.; Takeda, A. Plasma membrane ion permeability induced by mutant α-synuclein contributes to the degeneration of neural cells. J. Neurochem. 2006, 97, 1071–1077. [Google Scholar] [CrossRef] [PubMed]
- Hettiarachchi, N.T.; Parker, A.; Dallas, M.L.; Pennington, K.; Hung, C.-C.; Pearson, H.A.; Boyle, J.P.; Robinson, P.; Peers, C. α-Synuclein modulation of Ca2+ signaling in human neuroblastoma (SH-SY5Y) cells. J. Neurochem. 2009, 111, 1192–1201. [Google Scholar] [CrossRef] [PubMed]
- Martin, Z.S.; Neugebauer, V.; Dineley, K.T.; Kayed, R.; Zhang, W.; Reese, L.C.; Taglialatela, G. Alpha synuclein oligomers oppose long-term potentiation and impair memory through a calcineurin-dependent mechanism: Relevance to human synucleopathic diseases. J. Neurochem. 2012, 120, 440–452. [Google Scholar] [CrossRef] [PubMed]
- Eliezer, D.; Kutluay, E.; Bussell, R., Jr.; Browne, G. Conformational properties of α-synuclein in its free and lipid-associated states. J. Mol. Biol. 2001, 307, 1061–1073. [Google Scholar] [CrossRef] [PubMed]
- Chandra, S.; Chen, X.; Rizo, J.; Jahn, R.; Südhof, T.C. A broken alpha-helix in folded α-synuclein. J. Biol. Chem. 2003, 278, 15313–15318. [Google Scholar] [CrossRef] [PubMed]
- Kubo, S.; Nemani, V.M.; Chalkley, R.J.; Anthony, M.D.; Hattori, N.; Mizuno, Y.; Edwards, R.H.; Fortin, D.L. A combinatorial code for the interaction of α-synuclein with membranes. J. Biol. Chem. 2005, 280, 31664–31672. [Google Scholar] [PubMed]
- Ulmer, T.S.; Bax, A.; Cole, N.B.; Nussbaum, R.L. Structure and dynamics of micelle-bound human α-synuclein. J. Biol. Chem. 2005, 280, 9595–9603. [Google Scholar]
- Bussell, R.; Eliezer, D. Effects of Parkinson’s disease-linked mutations on the structure of lipid-associated alpha-synuclein. Biochem. 2004, 43, 4810–4818. [Google Scholar]
- Fortin, D.L.; Nemani, V.M.; Voglmaier, S.M.; Anthony, M.D.; Ryan, T.A.; Edwards, R.H. Neural activity controls the synaptic accumulation of alpha-synuclein. J. Neurosci. 2005, 25, 10913–10921. [Google Scholar]
- Cole, N.B.; Murphy, D.D.; Grider, T.; Rueter, S.; Brasaemle, D.; Nussbaum, R.L. Lipid droplet binding and oligomerization properties of the Parkinson’s disease protein α-synuclein. J. Biol. Chem. 2002, 277, 6344–6352. [Google Scholar] [PubMed]
- Kim, Y.S.; Laurine, E.; Woods, W.; Lee, S.-J. A novel mechanism of interaction between α-synuclein and biological membranes. J. Mol. Biol. 2006, 360, 386–397. [Google Scholar] [PubMed]
- Edidin, M. The state of lipid rafts: From model membranes to cells. Annu. Rev. Biophys. Biomol. Struct. 2003, 32, 257–283. [Google Scholar] [PubMed]
- Fortin, D.L.; Troyer, M.D.; Nakamura, K.; Kubo, S.; Anthony, M.D.; Edwards, R.H. Lipid rafts mediate the synaptic localization of α-synuclein. J. Neurosci. 2004, 24, 6715–6723. [Google Scholar] [PubMed]
- Castagnet, P.I.; Golovko, M.Y.; Barceló-Coblijn, G.C.; Nussbaum, R.L.; Murphy, E.J. Fatty acid incorporation is decreased in astrocytes cultured from alpha-synuclein gene-ablated mice. J. Neurochem. 2005, 94, 839–849. [Google Scholar] [PubMed]
- Golovko, M.Y.; Faergeman, N.J.; Cole, N.B.; Castagnet, P.I.; Nussbaum, R.L.; Murphy, E.J. Alpha-synuclein gene deletion decreases brain palmitate uptake and alters the palmitate metabolism in the absence of alpha-synuclein palmitate binding. Biochemistry 2005, 44, 8251–8259. [Google Scholar] [CrossRef] [PubMed]
- Sharon, R.; Bar-Joseph, I.; Mirick, G.E.; Serhan, C.N.; Selkoe, D.J. Altered fatty acid composition of dopaminergic neurons expressing alpha-synuclein and human brains with alpha-synucleinopathies. J. Biol. Chem. 2003, 278, 49874–49881. [Google Scholar] [CrossRef] [PubMed]
- Golovko, M.Y.; Rosenberger, T.A.; Feddersen, S.; Cole, N.B.; Pribill, I.; Berger, J.; Nussbaum, R.L.; Murphy, E.J. Acyl-CoA synthetase activity links wild-type but not mutant α-synuclein to brain arachidonate metabolism. Biochemistry 2006, 45, 6956–6966. [Google Scholar] [CrossRef] [PubMed]
- Golovko, M.Y.; Barceló-Coblijn, G.; Castagnet, P.I.; Austin, S.; Combs, C.K.; Murphy, E.J. The role of α-synuclein in brain lipid metabolism: A downstream impact on brain inflammatory response. Mol. Cell. Biochem. 2009, 326, 55–66. [Google Scholar] [CrossRef] [PubMed]
- Kamp, F.; Beyer, K. Binding of alpha-synuclein affects the lipid packing in bilayers of small vesicles. J. Biol. Chem. 2006, 281, 9251–9259. [Google Scholar] [CrossRef] [PubMed]
- Dikiy, I.; Eliezer, D. Folding and misfolding of alpha-synuclein on membranes. Biochim. Biophys. Acta 2012, 1818, 1013–1018. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.-J.; Choi, C.; Lee, S.-J. Membrane-bound α-synuclein has a high aggregation propensity and the ability to seed the aggregation of the cytosolic form. J. Biol. Chem. 2002, 277, 671–678. [Google Scholar] [CrossRef] [PubMed]
- Borghi, R.; Marchese, R.; Negro, A.; Marinelli, L.; Forloni, G.; Zaccheo, D.; Abbruzzese, G.; Tabaton, M. Full length α-synuclein is present in cerebrospinal fluid from Parkinson’s disease and normal subjects. Neurosci. Lett. 2000, 287, 65–67. [Google Scholar] [CrossRef] [PubMed]
- El-Agnaf, O.M.A.; Salem, S.A.; Paleologou, K.E.; Cooper, L.J.; Fullwood, N.J.; Gibson, M.J.; Curran, M.D.; Court, J.A.; Mann, D.M.A.; Ikeda, S.; et al. α-Synuclein implicated in Parkinson’s disease is present in extracellular biological fluids, including human plasma. FASEB J. 2003. [Google Scholar] [CrossRef]
- Lee, P.H.; Lee, G.; Park, H.J.; Bang, O.Y.; Joo, I.S.; Huh, K. The plasma alpha-synuclein levels in patients with Parkinson’s disease and multiple system atrophy. J. Neural Transm. 2006, 113, 1435–1439. [Google Scholar] [CrossRef] [PubMed]
- Tokuda, T.; Salem, S.A.; Allsop, D.; Mizuno, T.; Nakagawa, M.; Qureshi, M.M.; Locascio, J.J.; Schlossmacher, M.G.; El-Agnaf, O.M.A. Decreased alpha-synuclein in cerebrospinal fluid of aged individuals and subjects with Parkinson’s disease. Biochem. Biophys. Res. Commun. 2006, 349, 162–166. [Google Scholar] [CrossRef] [PubMed]
- Braak, H.; del Tredici, K.; Rüb, U.; de Vos, R.A.I.; Jansen Steur, E.N.H.; Braak, E. Staging of brain pathology related to sporadic Parkinson’s disease. Neurobiol. Aging 2003, 24, 197–211. [Google Scholar] [CrossRef] [PubMed]
- Braak, H.; Ghebremedhin, E.; Rüb, U.; Bratzke, H.; del Tredici, K. Stages in the development of Parkinson’s disease-related pathology. Cell Tissue Res. 2004, 318, 121–134. [Google Scholar] [CrossRef] [PubMed]
- Rubartelli, A.; Cozzolino, F.; Talio, M.; Sitia, R. A novel secretory pathway for interleukin-1 beta, a protein lacking a signal sequence. EMBO J. 1990, 9, 1503–1510. [Google Scholar] [PubMed]
- Lecellier, C.-H.; Vermeulen, W.; Bachelerie, F.; Giron, M.-L.; Saib, A. Intra- and intercellular trafficking of the foamy virus auxiliary bet protein. J. Virol. 2002, 76, 3388–3394. [Google Scholar] [CrossRef] [PubMed]
- Emmanouilidou, E.; Elenis, D.; Papasilekas, T.; Stranjalis, G.; Gerozissis, K.; Ioannou, P.C.; Vekrellis, K. Assessment of α-synuclein secretion in mouse and human brain parenchyma. PLOS ONE 2011. [Google Scholar] [CrossRef]
- Paillusson, S.; Clairembault, T.; Biraud, M.; Neunlist, M.; Derkinderen, P. Activity-dependent secretion of alpha-synuclein by enteric neurons. J. Neurochem. 2013, 125, 512–517. [Google Scholar] [CrossRef] [PubMed]
- Emmanouilidou, E.; Melachroinou, K.; Roumeliotis, T.; Garbis, S.D.; Ntzouni, M.; Margaritis, L.H.; Stefanis, L.; Vekrellis, K. Cell-produced α-synuclein is secreted in a calcium-dependent manner by exosomes and impacts neuronal survival. J. Neurosci. 2010, 30, 6838–6851. [Google Scholar] [CrossRef] [PubMed]
- Hasegawa, T.; Konno, M.; Baba, T.; Sugeno, N.; Kikuchi, A.; Kobayashi, M.; Miura, E.; Tanaka, N.; Tamai, K.; Furukawa, K.; et al. The AAA-ATPase VPS4 regulates extracellular secretion and lysosomal targeting of α-synuclein. PLOS ONE 2011, 6, e29460. [Google Scholar] [CrossRef] [PubMed]
- Denzer, K.; Kleijmeer, M.J.; Heijnen, H.F.; Stoorvogel, W.; Geuze, H.J. Exosome: From internal vesicle of the multivesicular body to intercellular signaling device. J. Cell Sci. 2000, 113, 3365–3374. [Google Scholar] [PubMed]
- Davies, B.A.; Babst, M.; Katzmann, D.J. Regulation of Vps4 during MVB Sorting and cytokinesis. Traffic Cph. Den. 2011, 12, 1298–1305. [Google Scholar] [CrossRef]
- Sung, J.Y.; Park, S.M.; Lee, C.-H.; Um, J.W.; Lee, H.J.; Kim, J.; Oh, Y.J.; Lee, S.-T.; Paik, S.R.; Chung, K.C. Proteolytic cleavage of extracellular secreted α-synuclein via matrix metalloproteinases. J. Biol. Chem. 2005, 280, 25216–25224. [Google Scholar] [CrossRef] [PubMed]
- Sung, J.Y.; Kim, J.; Paik, S.R.; Park, J.H.; Ahn, Y.S.; Chung, K.C. Induction of neuronal cell death by Rab5A-dependent endocytosis of α-synuclein. J. Biol. Chem. 2001, 276, 27441–27448. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Wang, T.; Pei, Z.; Miller, D.S.; Wu, X.; Block, M.L.; Wilson, B.; Zhang, W.; Zhou, Y.; Hong, J.-S.; et al. Aggregated α-synuclein activates microglia: A process leading to disease progression in Parkinson’s disease. FASEB J. 2005, 19, 533–542. [Google Scholar] [CrossRef] [PubMed]
- Park, J.-Y.; Kim, K.S.; Lee, S.-B.; Ryu, J.-S.; Chung, K.C.; Choo, Y.-K.; Jou, I.; Kim, J.; Park, S.M. On the mechanism of internalization of α-synuclein into microglia: Roles of ganglioside GM1 and lipid raft. J. Neurochem. 2009, 110, 400–411. [Google Scholar] [CrossRef] [PubMed]
- Ahn, K.J.; Paik, S.R.; Chung, K.C.; Kim, J. Amino acid sequence motifs and mechanistic features of the membrane translocation of alpha-synuclein. J. Neurochem. 2006, 97, 265–279. [Google Scholar] [CrossRef] [PubMed]
- Crews, L.; Tsigelny, I.; Hashimoto, M.; Masliah, E. Role of synucleins in Alzheimer’s disease. Neurotox. Res. 2009, 16, 306–317. [Google Scholar] [CrossRef] [PubMed]
- Danzer, K.M.; Krebs, S.K.; Wolff, M.; Birk, G.; Hengerer, B. Seeding induced by α-synuclein oligomers provides evidence for spreading of α-synuclein pathology. J. Neurochem. 2009, 111, 192–203. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.-J.; Suk, J.-E.; Bae, E.-J.; Lee, J.-H.; Paik, S.R.; Lee, S.-J. Assembly-dependent endocytosis and clearance of extracellular a-synuclein. Int. J. Biochem. Cell Biol. 2008, 40, 1835–1849. [Google Scholar] [CrossRef] [PubMed]
- Hansen, C.; Angot, E.; Bergström, A.-L.; Steiner, J.A.; Pieri, L.; Paul, G.; Outeiro, T.F.; Melki, R.; Kallunki, P.; Fog, K.; et al. α-Synuclein propagates from mouse brain to grafted dopaminergic neurons and seeds aggregation in cultured human cells. J. Clin. Investig. 2011, 121, 715–725. [Google Scholar] [CrossRef] [PubMed]
- Cheung, A.Y.; de Vries, S.C. Membrane trafficking: Intracellular highways and country roads. Plant Physiol. 2008, 147, 1451–1453. [Google Scholar] [CrossRef] [PubMed]
- Matsui, T.; Itoh, T.; Fukuda, M. Small GTPase Rab12 regulates constitutive degradation of transferrin receptor. Traffic 2011, 12, 1432–1443. [Google Scholar] [CrossRef] [PubMed]
- Chai, Y.-J.; Kim, D.; Park, J.; Zhao, H.; Lee, S.-J.; Chang, S. The secreted oligomeric form of α-synuclein affects multiple steps of membrane trafficking. FEBS Lett. 2013, 587, 452–459. [Google Scholar] [CrossRef] [PubMed]
- Wilms, H.; Rosenstiel, P.; Sievers, J.; Deuschl, G.; Zecca, L.; Lucius, R. Activation of microglia by human neuromelanin is NF-κB-dependent and involves p38 mitogen-activated protein kinase: Implications for Parkinson’s disease. FASEB J. 2003, 17, 3500–3502. [Google Scholar]
- Klegeris, A.; Giasson, B.I.; Zhang, H.; Maguire, J.; Pelech, S.; McGeer, P.L. Alpha-synuclein and its disease-causing mutants induce ICAM-1 and IL-6 in human astrocytes and astrocytoma cells. Fed. Am. Soc. Exp. Biol. 2006, 20, 2000–2008. [Google Scholar] [PubMed]
- Lee, H.-J.; Suk, J.-E.; Patrick, C.; Bae, E.-J.; Cho, J.-H.; Rho, S.; Hwang, D.; Masliah, E.; Lee, S.-J. Direct transfer of α-synuclein from neuron to astroglia causes inflammatory responses in synucleinopathies. J. Biol. Chem. 2010, 285, 9262–9272. [Google Scholar] [CrossRef] [PubMed]
- Luk, K.C.; Song, C.; O’Brien, P.; Stieber, A.; Branch, J.R.; Brunden, K.R.; Trojanowski, J.Q.; Lee, V.M.-Y. Exogenous alpha-synuclein fibrils seed the formation of Lewy body-like intracellular inclusions in cultured cells. Proc. Natl. Acad. Sci. USA 2009, 106, 20051–20056. [Google Scholar] [CrossRef] [PubMed]
- Desplats, P.; Lee, H.-J.; Bae, E.-J.; Patrick, C.; Rockenstein, E.; Crews, L.; Spencer, B.; Masliah, E.; Lee, S.-J. Inclusion formation and neuronal cell death through neuron-to-neuron transmission of α-synuclein. Proc. Natl. Acad. Sci. USA 2009, 106, 13010–13015. [Google Scholar] [CrossRef] [PubMed]
- Luk, K.C.; Kehm, V.; Carroll, J.; Zhang, B.; O’Brien, P.; Trojanowski, J.Q.; Lee, V.M.-Y. Pathological α-synuclein transmission initiates Parkinson-like neurodegeneration in nontransgenic mice. Science 2012, 338, 949–953. [Google Scholar] [CrossRef] [PubMed]
- Sacino, A.N.; Brooks, M.; McGarvey, N.H.; McKinney, A.B.; Thomas, M.A.; Levites, Y.; Ran, Y.; Golde, T.E.; Giasson, B.I. Induction of CNS α-synuclein pathology by fibrillar and non-amyloidogenic recombinant α-synuclein. Acta Neuropathol. Commun. 2013. [Google Scholar] [CrossRef]
- Diógenes, M.J.; Dias, R.B.; Rombo, D.M.; Miranda, H.V.; Maiolino, F.; Guerreiro, P.; Näsström, T.; Franquelim, H.G.; Oliveira, L.M.; Castanho, M.A.; et al. Extracellular alpha-synuclein oligomers modulate synaptic transmission and impair LTP via NMDA-receptor activation. J. Neurosci. 2012, 32, 11750–11762. [Google Scholar] [CrossRef] [PubMed]
- Melachroinou, K.; Xilouri, M.; Emmanouilidou, E.; Masgrau, R.; Papazafiri, P.; Stefanis, L.; Vekrellis, K. Deregulation of calcium homeostasis mediates secreted α-synuclein-induced neurotoxicity. Neurobiol. Aging 2013, 34, 2853–2865. [Google Scholar] [CrossRef] [PubMed]
- Ronzitti, G.; Bucci, G.; Emanuele, M.; Leo, D.; Sotnikova, T.D.; Mus, L.V.; Soubrane, C.H.; Dallas, M.L.; Thalhammer, A.; Cingolani, L.A.; et al. Exogenous α-synuclein decreases raft partitioning of Cav2.2 channels inducing dopamine release. J. Neurosci. 2014, 34, 10603–10615. [Google Scholar] [CrossRef] [PubMed]
- Lücking, C.B.; Brice, A. Alpha-synuclein and Parkinson’s disease. Cell. Mol. Life Sci. 2000, 57, 1894–1908. [Google Scholar] [CrossRef] [PubMed]
- Jensen, P.H.; Hager, H.; Nielsen, M.S.; Hojrup, P.; Gliemann, J.; Jakes, R. α-Synuclein binds to tau and stimulates the protein kinase A-catalyzed tau phosphorylation of serine residues 262 and 356. J. Biol. Chem. 1999, 274, 25481–25489. [Google Scholar] [CrossRef] [PubMed]
- Jensen, P.H.; Islam, K.; Kenney, J.; Nielsen, M.S.; Power, J.; Gai, W.P. Microtubule-associated protein 1B is a component of cortical Lewy bodies and binds alpha-synuclein filaments. J. Biol. Chem. 2000, 275, 21500–21507. [Google Scholar] [CrossRef] [PubMed]
- D’Andrea, M.R.; Ilyin, S.; Plata-Salaman, C.R. Abnormal patterns of microtubule-associated protein-2 (MAP-2) immunolabeling in neuronal nuclei and Lewy bodies in Parkinson’s disease substantia nigra brain tissues. Neurosci. Lett. 2001, 306, 137–140. [Google Scholar] [CrossRef] [PubMed]
- Esposito, A.; Dohm, C.P.; Kermer, P.; Bähr, M.; Wouters, F.S. α-Synuclein and its disease-related mutants interact differentially with the microtubule protein tau and associate with the actin cytoskeleton. Neurobiol. Dis. 2007, 26, 521–531. [Google Scholar] [CrossRef] [PubMed]
- Alim, M.A.; Ma, Q.-L.; Takeda, K.; Aizawa, T.; Matsubara, M.; Nakamura, V.; Asada, A.; Saito, T.; Kaji, H.; Yoshii, M.; et al. Demonstration of a role for a-synuclein as a functional microtubule-associated protein. J. Alzheimers Dis. 2004, 6, 435–442. [Google Scholar] [PubMed]
- Liu, G.; Wang, P.; Li, X.; Li, Y.; Xu, S.; Uéda, K.; Chan, P.; Yu, S. Alpha-synuclein promotes early neurite outgrowth in cultured primary neurons. J. Neural Transm. 2013, 120, 1331–1343. [Google Scholar] [CrossRef] [PubMed]
- Geddes, J.W. α-Synuclein: A potent inducer of tau pathology. Exp. Neurol. 2005, 192, 244–250. [Google Scholar] [CrossRef] [PubMed]
- Duka, T.; Duka, V.; Joyce, J.N.; Sidhu, A. Alpha-synuclein contributes to GSK-3beta-catalyzed tau phosphorylation in Parkinson’s disease models. Fed. Am. Soc. Exp. Biol. 2009, 23, 2820–2830. [Google Scholar] [PubMed]
- Garcia, M.L.; Cleveland, D.W. Going new places using an old MAP: Tau, microtubules and human neurodegenerative disease. Curr. Opin. Cell Biol. 2001, 13, 41–48. [Google Scholar] [CrossRef] [PubMed]
- Gąssowska, M.; Czapski, G.A.; Pająk, B.; Cieślik, M.; Lenkiewicz, A.M.; Adamczyk, A. Extracellular α-synuclein leads to microtubule destabilization via GSK-3β-dependent tau phosphorylation in PC12 cells. PLOS ONE 2014. [Google Scholar] [CrossRef]
- Bellani, S.; Mescola, A.; Ronzitti, G.; Tsushima, H.; Tilve, S.; Canale, C.; Valtorta, F.; Chieregatti, E. GRP78 clustering at the cell surface of neurons transduces the action of exogenous alpha-synuclein. Cell Death Differ. 2014, 21, 1971–1983. [Google Scholar] [CrossRef] [PubMed]
- Lakso, M.; Vartiainen, S.; Moilanen, A.-M.; Sirviö, J.; Thomas, J.H.; Nass, R.; Blakely, R.D.; Wong, G. Dopaminergic neuronal loss and motor deficits in Caenorhabditis elegans overexpressing human alpha-synuclein. J. Neurochem. 2003, 86, 165–172. [Google Scholar] [CrossRef] [PubMed]
- Feany, M.B.; Bender, W.W. A Drosophila model of Parkinson’s disease. Nature 2000, 404, 394–398. [Google Scholar] [CrossRef] [PubMed]
- Yamada, M.; Iwatsubo, T.; Mizuno, Y.; Mochizuki, H. Overexpression of α-synuclein in rat substantia nigra results in loss of dopaminergic neurons, phosphorylation of α-synuclein and activation of caspase-9: Resemblance to pathogenetic changes in Parkinson’s disease. J. Neurochem. 2004, 91, 451–461. [Google Scholar] [CrossRef] [PubMed]
- Nuber, S.; Harmuth, F.; Kohl, Z.; Adame, A.; Trejo, M.; Schönig, K.; Zimmermann, F.; Bauer, C.; Casadei, N.; Giel, C.; et al. A progressive dopaminergic phenotype associated with neurotoxic conversion of α-synuclein in BAC-transgenic rats. Brain 2013, 136, 412–432. [Google Scholar] [CrossRef] [PubMed]
- Lo Bianco, C.; Ridet, J.-L.; Schneider, B.L.; Déglon, N.; Aebischer, P. α-Synucleinopathy and selective dopaminergic neuron loss in a rat lentiviral-based model of Parkinson’s disease. Proc. Natl. Acad. Sci. USA 2002, 99, 10813–10818. [Google Scholar] [CrossRef] [PubMed]
- Kirik, D.; Annett, L.E.; Burger, C.; Muzyczka, N.; Mandel, R.J.; Björklund, A. Nigrostriatal α-synucleinopathy induced by viral vector-mediated overexpression of human α-synuclein: A new primate model of Parkinson’s disease. Proc. Natl. Acad. Sci. USA 2003, 100, 2884–2889. [Google Scholar] [CrossRef] [PubMed]
- Van derPutten, H.; Wiederhold, K.-H.; Probst, A.; Barbieri, S.; Mistl, C.; Danner, S.; Kauffmann, S.; Hofele, K.; Spooren, W.P.; Ruegg, M.A.; et al. Neuropathology in Mice Expressing Human α-synuclein. J. Neurosci. 2000, 20, 6021–6029. [Google Scholar] [PubMed]
- Gomez-Isla, T.; Irizarry, M.C.; Mariash, A.; Cheung, B.; Soto, O.; Schrump, S.; Sondel, J.; Kotilinek, L.; Day, J.; Schwarzschild, M.A.; et al. Motor dysfunction and gliosis with preserved dopaminergic markers in human α-synuclein A30P transgenic mice. Neurobiol. Aging 2003, 24, 245–258. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Emanuele, M.; Chieregatti, E. Mechanisms of Alpha-Synuclein Action on Neurotransmission: Cell-Autonomous and Non-Cell Autonomous Role. Biomolecules 2015, 5, 865-892. https://doi.org/10.3390/biom5020865
Emanuele M, Chieregatti E. Mechanisms of Alpha-Synuclein Action on Neurotransmission: Cell-Autonomous and Non-Cell Autonomous Role. Biomolecules. 2015; 5(2):865-892. https://doi.org/10.3390/biom5020865
Chicago/Turabian StyleEmanuele, Marco, and Evelina Chieregatti. 2015. "Mechanisms of Alpha-Synuclein Action on Neurotransmission: Cell-Autonomous and Non-Cell Autonomous Role" Biomolecules 5, no. 2: 865-892. https://doi.org/10.3390/biom5020865
APA StyleEmanuele, M., & Chieregatti, E. (2015). Mechanisms of Alpha-Synuclein Action on Neurotransmission: Cell-Autonomous and Non-Cell Autonomous Role. Biomolecules, 5(2), 865-892. https://doi.org/10.3390/biom5020865