
Impact of Oxidative Stress on Exercising Skeletal Muscle



{kind=link}
{kind=link}
Abstract
:1. Introduction
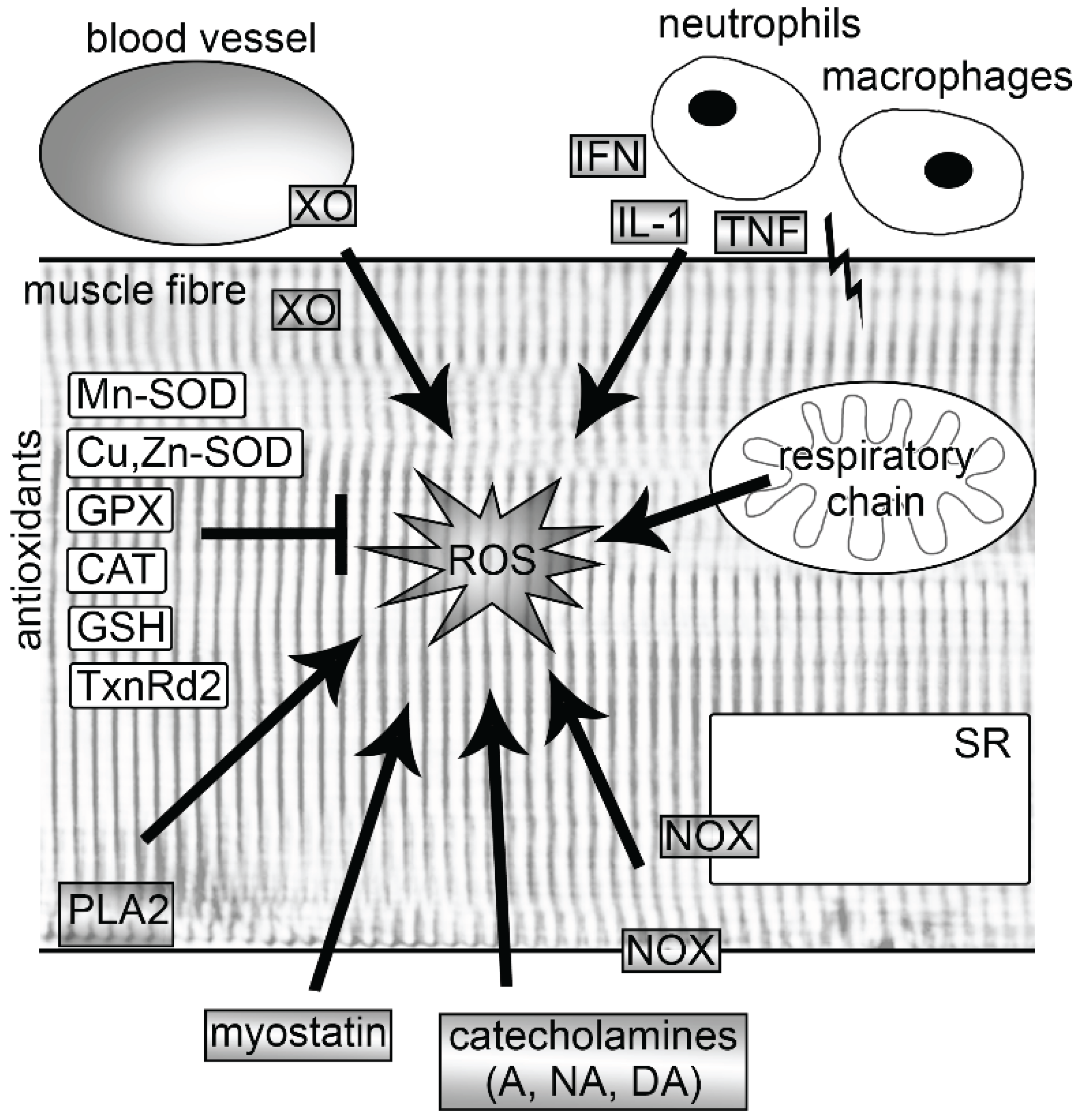
2. Sources of ROS in Muscle
2.1. Mitochondria
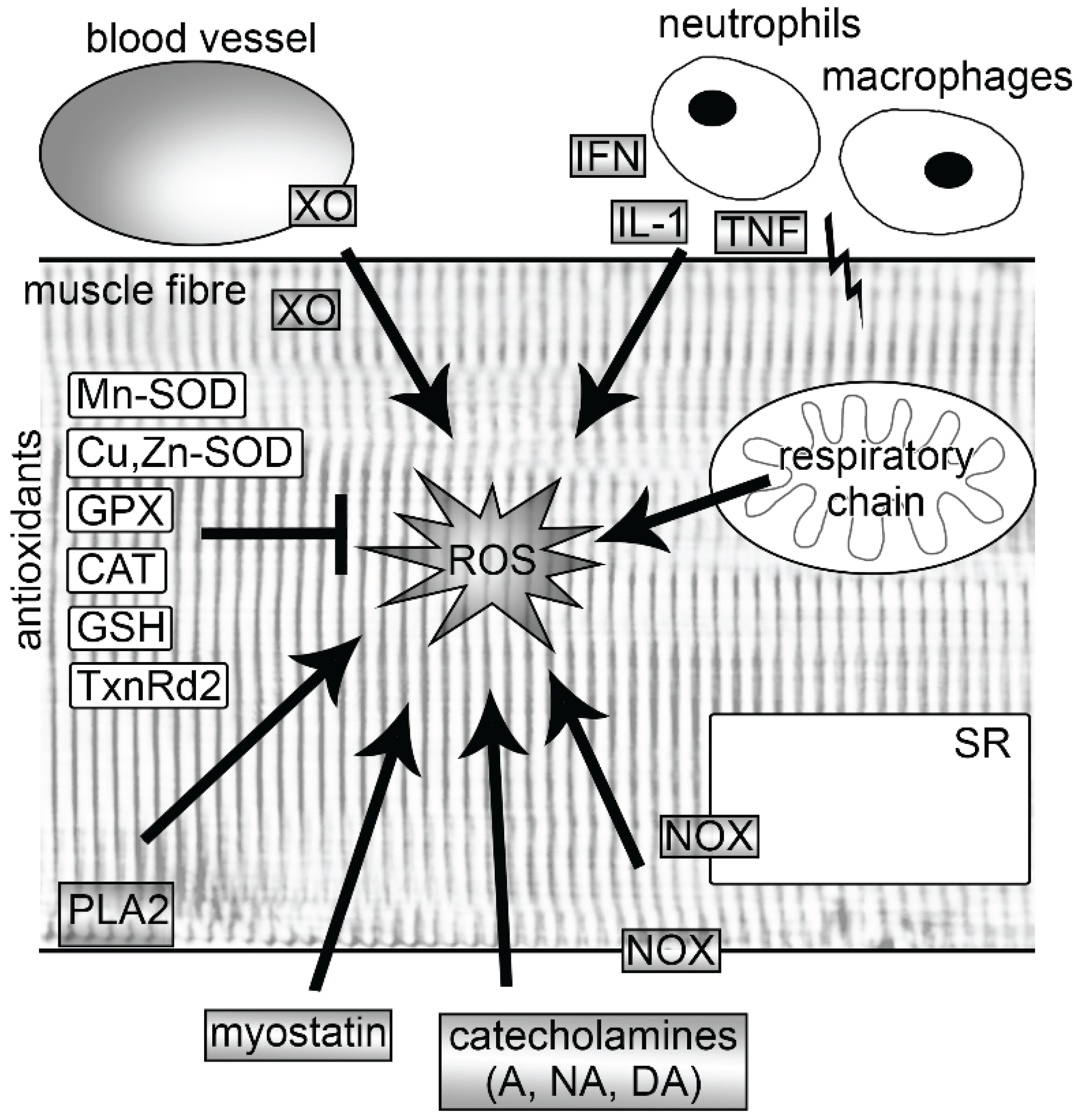
2.2. NADPH Oxidases
2.3. Xanthine Oxidase
2.4. Myostatin
2.5. Phospholipase A2
3. Effects of ROS on Force Generation and Muscle Atrophy

3.1. Contractile Dysfunction
3.2. Muscle Atrophy
4. Antioxidants in Muscle
4.1. Enzymatic and Nonenzymatic Antioxidants
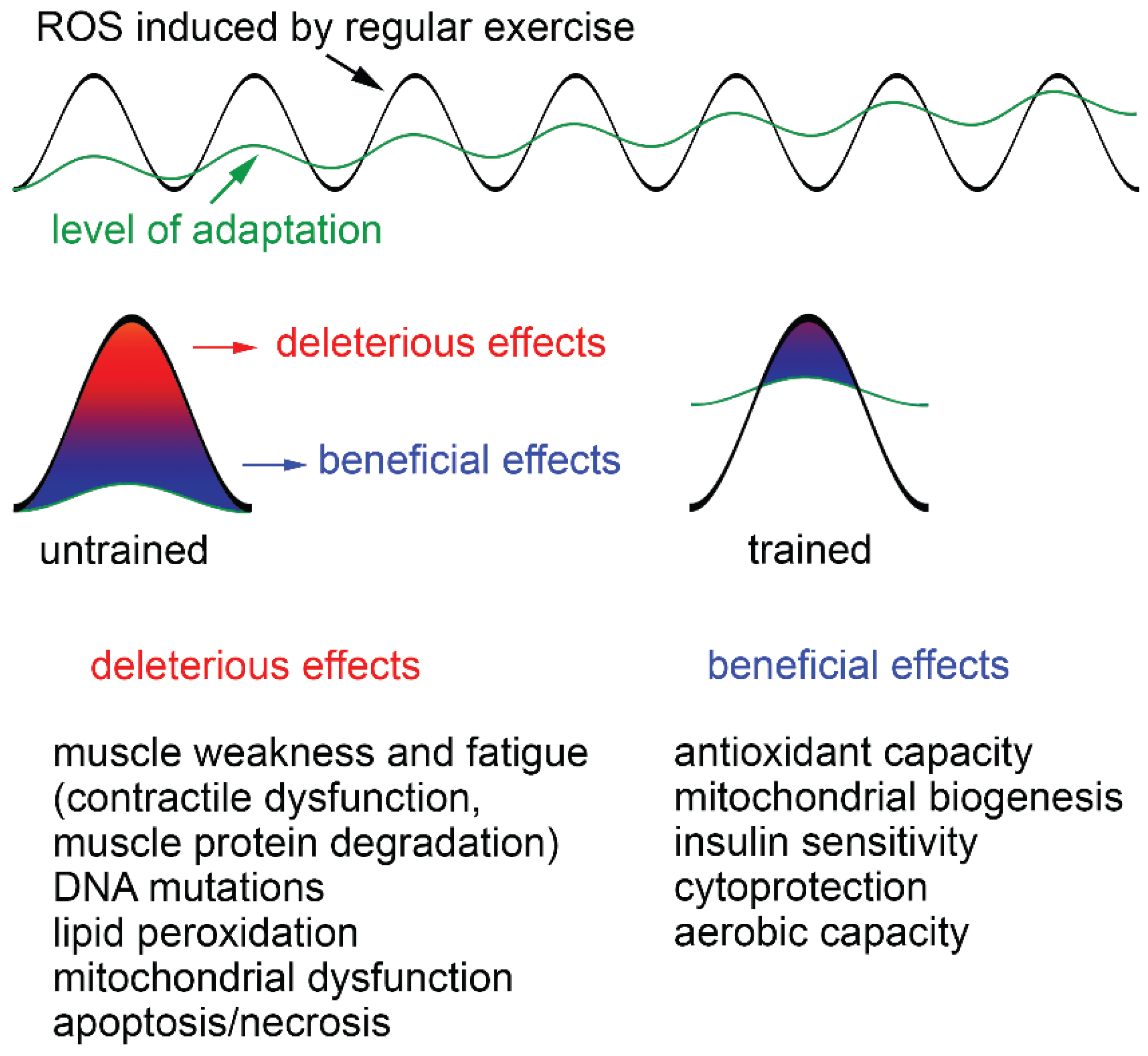
4.2. Adaptive Responses to Exercise
4.3. Exogenous Antioxidants and Exercise
5. Training-Induced Muscular Adaptation, PGC-1α and ROS
5.1. Role of PGC-1α in Exercise
5.2. PGC-1α Regulates ROS Defense
6. Conclusions
Author Contributions
Abbreviations
| AMPK | adenosine monophosphate-activated protein kinase |
| ARE | antioxidant-responsive element |
| CAT | catalase |
| CHOP | C/EBP homology protein |
| CREB | Cre-binding protein |
| Cu, Zn-SOD | copper-zinc superoxide dismutase |
| eNOS | endothelial nitric oxide synthase |
| ERR-α | estrogen-related receptor α |
| FAS | fatty acid synthase |
| GPX | glutathione peroxidase |
| GR | glutathione reductase |
| GSH | glutathione |
| GSSG | glutathione disulfide |
| HNE | 4-hydroxynonenal |
| IFN-γ | interferon-γ |
| IL-1 | interleukin-1 |
| iNOS | inducible nitric oxide synthase |
| LXRα | nuclear receptor liver x receptor α |
| MAFbx | muscle atrophy F-box |
| MAPK | mitogen-activated protein kinase |
| MEF2 | myocyte enhancer factor 2 |
| Mn-SOD | manganese superoxide dismutase |
| MuRF-1 | muscle RING-finger protein 1 |
| NADPH | nicotinamide adenine dinucleotide phosphate |
| NF-κB | nuclear factor-κB |
| NOX | nicotinamide adenine dinucleotide phosphate oxidase |
| NRF | nuclear respiratory factor |
| Nrf2 | nuclear factor erythroid-derived 2-like 2 |
| OT | overloaded training |
| PGC-1α | peroxisome proliferator-activated receptor-γ coactivator-1α |
| PLA2 | phospholipase A2 |
| PPARγ | peroxisome proliferator-activated receptor γ |
| ROS | reactive oxygen species |
| RyR | ryanodine receptor |
| SIRT3 | silent information regulator 3 |
| TBARS | thiobarbituric acid reactive substance |
| TFAM | mitochondrial transcription factor A |
| TNF | tumor necrosis factor |
| TxnRd2, | thioredoxin reductase-2 |
| UCP | uncoupling protein |
| VEGF | vascular endothelial growth factor |
| XO | xanthine oxidase |
Conflicts of Interest
References
- Dillard, C.J.; Litov, R.E.; Savin, W.M.; Dumelin, E.E.; Tappel, A.L. Effects of exercise, vitamin E, and ozone on pulmonary function and lipid peroxidation. J. Appl. Physiol. Respir. Environ. Exerc. Physiol. 1978, 45, 927–932. [Google Scholar] [PubMed]
- Davies, K.J.; Quintanilha, A.T.; Brooks, G.A.; Packer, L. Free radicals and tissue damage produced by exercise. Biochem. Biophys. Res. Commun. 1982, 107, 1198–1205. [Google Scholar] [CrossRef] [PubMed]
- Vollaard, N.B.; Shearman, J.P.; Cooper, C.E. Exercise-induced oxidative stress: Myths, realities and physiological relevance. Sports Med. 2005, 35, 1045–1062. [Google Scholar] [CrossRef] [PubMed]
- Fisher-Wellman, K.; Bloomer, R.J. Acute exercise and oxidative stress: A 30 year history. Dyn. Med. 2009. [Google Scholar] [CrossRef]
- Sahlin, K.; Shabalina, I.G.; Mattsson, C.M.; Bakkman, L.; Fernström, M.; Rozhdestvenskaya, Z.; Enqvist, J.K.; Nedergaard, J.; Ekblom, B.; Tonkonogi, M. Ultraendurance exercise increases the production of reactive oxygen species in isolated mitochondria from human skeletal muscle. J. Appl. Physiol. 2010, 108, 780–787. [Google Scholar] [CrossRef] [PubMed]
- Hey-Mogensen, M.; Højlund, K.; Vind, B.F.; Wang, L.; Dela, F.; Beck-Nielsen, H.; Fernström, M.; Sahlin, K. Effect of physical training on mitochondrial respiration and reactive oxygen species release in skeletal muscle in patients with obesity and type 2 diabetes. Diabetologia 2010, 53, 1976–1985. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, S.; Lertwattanarak, R.; Lefort, N.; Molina-Carrion, M.; Joya-Galeana, J.; Bowen, B.P.; Garduno-Garcia Jde, J.; Abdul-Ghani, M.; Richardson, A.; DeFronzo, R.A.; et al. Reduction in reactive oxygen species production by mitochondria from elderly subjects with normal and impaired glucose tolerance. Diabetes 2011, 60, 2051–2060. [Google Scholar] [CrossRef] [PubMed]
- Powers, S.K.; Jackson, M.J. Exercise-induced oxidative stress: Cellular mechanisms and impact on muscle force production. Physiol. Rev. 2008, 88, 1243–1276. [Google Scholar] [CrossRef] [PubMed]
- Peake, J.; Suzuki, K. Neutrophil activation, antioxidant supplements and exercise-induced oxidative stress. Exerc. Immunol. Rev. 2004, 10, 129–141. [Google Scholar] [PubMed]
- Moylan, J.S.; Reid, M.B. Oxidative stress, chronic disease, and muscle wasting. Muscle Nerve 2007, 35, 411–429. [Google Scholar] [CrossRef] [PubMed]
- Gomes, E.C.; Silva, A.N.; de Oliveira, M.R. Oxidants, antioxidants, and the beneficial roles of exercise-induced production of reactive species. Oxid. Med. Cell Longev. 2012, 2012, 756132. [Google Scholar] [PubMed]
- Duarte, J.A.; Appell, H.J.; Carvalho, F.; Bastos, M.L.; Soares, J.M. Endothelium-derived oxidative stress may contribute to exercise-induced muscle damage. Int. J. Sports Med. 1993, 14, 440–443. [Google Scholar] [CrossRef] [PubMed]
- Murphy, M.P. How mitochondria produce reactive oxygen species. Biochem. J. 2009, 417, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Brand, M.D. The sites and topology of mitochondrial superoxide production. Exp. Gerontol. 2010, 45, 466–472. [Google Scholar] [CrossRef] [PubMed]
- St-Pierre, J.; Buckingham, J.A.; Roebuck, S.J.; Brand, M.D. Topology of superoxide production from different sites in the mitochondrial electron transport chain. J. Biol. Chem. 2002, 277, 44784–44790. [Google Scholar] [CrossRef] [PubMed]
- Perevoshchikova, I.V.; Quinlan, C.L.; Orr, A.L.; Gerencser, A.A.; Brand, M.D. Sites of superoxide and hydrogen peroxide production during fatty acid oxidation in rat skeletal muscle mitochondria. Free Radic. Biol. Med. 2013, 61, 298–309. [Google Scholar] [CrossRef] [PubMed]
- Hey-Mogensen, M.; Goncalves, R.L.; Orr, A.L.; Brand, M.D. Production of superoxide/H2O2 by dihydroorotate dehydrogenase in rat skeletal muscle mitochondria. Free Radic. Biol. Med. 2014, 72, 149–155. [Google Scholar] [CrossRef] [PubMed]
- Barja, G. Mitochondrial oxygen radical generation and leak: Sites of production in states 4 and 3, organ specificity, and relation to aging and longevity. J. Bioenerg. Biomembr. 1999, 31, 347–366. [Google Scholar] [CrossRef] [PubMed]
- Muller, F.L.; Liu, Y.; van Remmen, H. Complex III releases superoxide to both sides of the inner mitochondrial membrane. J. Biol. Chem. 2004, 279, 49064–49073. [Google Scholar] [CrossRef] [PubMed]
- Goncalves, R.L.; Quinlan, C.L.; Perevoshchikova, I.V.; Hey-Mogensen, M.; Brand, M.D. Sites of superoxide and hydrogen peroxide production by muscle mitochondria assessed ex vivo under conditions mimicking rest and exercise. J. Biol. Chem. 2015, 290, 209–227. [Google Scholar] [CrossRef] [PubMed]
- Bedard, K.; Krause, K.H. The NOX family of ROS-generating NADPH oxidases: Physiology and pathophysiology. Physiol. Rev. 2007, 87, 245–313. [Google Scholar] [CrossRef] [PubMed]
- Brandes, R.P.; Weissmann, N.; Schröder, K. Nox family NADPH oxidases: Molecular mechanisms of activation. Free Radic. Biol. Med. 2014, 76, 208–226. [Google Scholar] [CrossRef] [PubMed]
- Xia, R.; Webb, J.A.; Gnall, L.L.; Cutler, K.; Abramson, J.J. Skeletal muscle sarcoplasmic reticulum contains a NADH-dependent oxidase that generates superoxide. Am. J. Physiol. Cell Physiol. 2003, 285, C215–C221. [Google Scholar] [CrossRef] [PubMed]
- Shkryl, V.M.; Martins, A.S.; Ullrich, N.D.; Nowycky, M.C.; Niggli, E.; Shirokova, N. Reciprocal amplification of ROS and Ca2+ signals in stressed mdx dystrophic skeletal muscle fibers. Pflugers Arch. 2009, 458, 915–928. [Google Scholar] [CrossRef] [PubMed]
- Sakellariou, G.K.; Vasilaki, A.; Palomero, J.; Kayani, A.; Zibrik, L.; McArdle, A.; Jackson, M.J. Studies of mitochondrial and nonmitochondrial sources implicate nicotinamide adenine dinucleotide phosphate oxidase(s) in the increased skeletal muscle superoxide generation that occurs during contractile activity. Antioxid. Redox Signal. 2013, 18, 603–621. [Google Scholar] [CrossRef] [PubMed]
- Cherednichenko, G.; Zima, A.V.; Feng, W.; Schaefer, S.; Blatter, L.A.; Pessah, I.N. NADH oxidase activity of rat cardiac sarcoplasmic reticulum regulates calcium-induced calcium release. Circ. Res. 2004, 94, 478–486. [Google Scholar] [CrossRef] [PubMed]
- Espinosa, A.; Leiva, A.; Peña, M.; Müller, M.; Debandi, A.; Hidalgo, C.; Carrasco, M.A.; Jaimovich, E. Myotube depolarization generates reactive oxygen species through NAD(P)H oxidase; ROS-elicited Ca2+ stimulates ERK, CREB, early genes. J. Cell Physiol. 2006, 209, 379–388. [Google Scholar] [CrossRef] [PubMed]
- Hidalgo, C.; Sánchez, G.; Barrientos, G.; Aracena-Parks, P. A transverse tubule NADPH oxidase activity stimulates calcium release from isolated triads via ryanodine receptor type 1 S-glutathionylation. J. Biol. Chem. 2006, 281, 26473–26482. [Google Scholar] [CrossRef] [PubMed]
- Espinosa, A.; García, A.; Härtel, S.; Hidalgo, C.; Jaimovich, E. NADPH oxidase and hydrogen peroxide mediate insulin-induced calcium increase in skeletal muscle cells. J. Biol. Chem. 2009, 284, 2568–2575. [Google Scholar] [CrossRef] [PubMed]
- Harrison, R. Structure and function of xanthine oxidoreductase: Where are we now? Free Radic. Biol. Med. 2002, 33, 774–797. [Google Scholar] [CrossRef] [PubMed]
- Judge, A.R.; Dodd, S.L. Xanthine oxidase and activated neutrophils cause oxidative damage to skeletal muscle after contractile claudication. Am. J. Physiol. Heart Circ. Physiol. 2004, 286, H252–H256. [Google Scholar] [CrossRef] [PubMed]
- Radák, Z.; Zhao, Z.; Koltai, E.; Ohno, H.; Atalay, M. Oxygen consumption and usage during physical exercise: The balance between oxidative stress and ROS-dependent adaptive signaling. Antioxid. Redox Signal. 2013, 18, 1208–1246. [Google Scholar] [CrossRef] [PubMed]
- Kang, C.; Chung, E.; Diffee, G.; Ji, L.L. Exercise training attenuates aging-associated mitochondrial dysfunction in rat skeletal muscle: Role of PGC-1α. Exp. Gerontol. 2013, 48, 1343–1350. [Google Scholar] [CrossRef] [PubMed]
- Sriram, S.; Subramanian, S.; Sathiakumar, D.; Venkatesh, R.; Salerno, M.S.; McFarlane, C.D.; Kambadur, R.; Sharma, M. Modulation of reactive oxygen species in skeletal muscle by myostatin is mediated through NF-κB. Aging Cell 2011, 10, 931–948. [Google Scholar] [CrossRef] [PubMed]
- Sriram, S.; Subramanian, S.; Juvvuna, P.K.; Ge, X.; Lokireddy, S.; McFarlane, C.D.; Wahli, W.; Kambadur, R.; Sharma, M. Myostatin augments muscle-specific ring finger protein-1 expression through an NF-κB independent mechanism in SMAD3 null muscle. Mol. Endocrinol. 2014, 28, 317–330. [Google Scholar] [CrossRef] [PubMed]
- Zuo, L.; Christofi, F.L.; Wright, V.P.; Bao, S.; Clanton, T.L. Lipoxygenase-dependent superoxide release in skeletal muscle. J. Appl. Physiol. 2004, 97, 661–668. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.; Bey, E.A.; Wientjes, F.B.; Cathcart, M.K. Cytosolic phospholipase A2 (cPLA2) regulation of human monocyte NADPH oxidase activity. cPLA2 affects translocation but not phosphorylation of p67(phox) and p47(phox). J. Biol. Chem. 2002, 277, 25385–25392. [Google Scholar] [PubMed]
- Ježek, J.; Jaburek, M.; Zelenka, J.; Ježek, P. Mitochondrial phospholipase A2 activated by reactive oxygen species in heart mitochondria induces mild uncoupling. Physiol. Res. 2010, 59, 737–747. [Google Scholar] [PubMed]
- Gong, M.C.; Arbogast, S.; Guo, Z.; Mathenia, J.; Su, W.; Reid, M.B. Calcium-independent phospholipase A2 modulates cytosolic oxidant activity and contractile function in murine skeletal muscle cells. J. Appl. Physiol. 2006, 100, 399–405. [Google Scholar] [CrossRef] [PubMed]
- Smith, M.A.; Reid, M.B. Redox modulation of contractile function in respiratory and limb skeletal muscle. Respir. Physiol. Neurobiol. 2006, 151, 229–241. [Google Scholar] [CrossRef] [PubMed]
- Radák, Z.; Taylor, A.W.; Ohno, H.; Goto, S. Adaptation to exercise-induced oxidative stress: From muscle to brain. Exerc. Immunol. Rev. 2001, 7, 90–107. [Google Scholar] [PubMed]
- Reid, M.B. Free radicals and muscle fatigue: Of ROS, canaries, and the IOC. Free Radic. Biol. Med. 2008, 44, 169–179. [Google Scholar] [CrossRef] [PubMed]
- Allen, D.G.; Lamb, G.D.; Westerblad, H. Skeletal muscle fatigue: Cellular mechanisms. Physiol. Rev. 2008, 88, 287–332. [Google Scholar] [CrossRef] [PubMed]
- Lamb, G.D.; Westerblad, H. Acute effects of reactive oxygen and nitrogen species on the contractile function of skeletal muscle. J. Physiol. 2011, 589, 2119–2127. [Google Scholar] [CrossRef] [PubMed]
- Lamb, G.D.; Posterino, G.S. Effects of oxidation and reduction on contractile function in skeletal muscle fibres of the rat. J. Physiol. 2003, 546, 149–163. [Google Scholar] [CrossRef] [PubMed]
- Posterino, G.S.; Cellini, M.A.; Lamb, G.D. Effects of oxidation and cytosolic redox conditions on excitation-contraction coupling in rat skeletal muscle. J. Physiol. 2003, 547, 807–823. [Google Scholar] [CrossRef] [PubMed]
- Cheng, A.J.; Bruton, J.D.; Lanner, J.T.; Westerblad, H. Antioxidant treatments do not improve force recovery after fatiguing stimulation of mouse skeletal muscle fibres. J. Physiol. 2015, 593, 457–472. [Google Scholar] [CrossRef] [PubMed]
- Andrade, F.H.; Reid, M.B.; Allen, D.G.; Westerblad, H. Effect of hydrogen peroxide and dithiothreitol on contractile function of single skeletal muscle fibres from the mouse. J. Physiol. 1998, 509, 565–575. [Google Scholar] [CrossRef] [PubMed]
- Prochniewicz, E.; Spakowicz, D.; Thomas, D.D. Changes in actin structural transitions associated with oxidative inhibition of muscle contraction. Biochemistry 2008, 47, 11811–11817. [Google Scholar] [CrossRef] [PubMed]
- Mollica, J.P.; Dutka, T.L.; Merry, T.L.; Lamboley, C.R.; McConell, G.K.; McKenna, M.J.; Murphy, R.M.; Lamb, G.D. S-glutathionylation of troponin I (fast) increases contractile apparatus Ca2+ sensitivity in fast-twitch muscle fibres of rats and humans. J. Physiol. 2012, 590, 1443–1463. [Google Scholar] [CrossRef] [PubMed]
- Jackson, M.J.; Papa, S.; Bolaños, J.; Bruckdorfer, R.; Carlsen, H.; Elliott, R.M.; Flier, J.; Griffiths, H.R.; Heales, S.; Holst, B.; et al. Antioxidants, reactive oxygen and nitrogen species, gene induction and mitochondrial function. Mol. Aspects Med. 2002, 23, 209–285. [Google Scholar] [CrossRef] [PubMed]
- Gumucio, J.P.; Mendias, C.L. Atrogin-1, MuRF-1, and sarcopenia. Endocrine 2013, 43, 12–21. [Google Scholar] [CrossRef] [PubMed]
- Witt, S.H.; Granzier, H.; Witt, C.C.; Labeit, S. MURF-1 and MURF-2 target a specific subset of myofibrillar proteins redundantly: Towards understanding MURF-dependent muscle ubiquitination. J. Mol. Biol. 2005, 350, 713–722. [Google Scholar] [CrossRef] [PubMed]
- Cohen, S.; Brault, J.J.; Gygi, S.P.; Glass, D.J.; Valenzuela, D.M.; Gartner, C.; Latres, E.; Goldberg, A.L. During muscle atrophy, thick, but not thin, filament components are degraded by MuRF1-dependent ubiquitylation. J. Cell Biol. 2009, 185, 1083–1095. [Google Scholar] [CrossRef] [PubMed]
- Ji, L.L. Antioxidants and oxidative stress in exercise. Proc. Soc. Exp. Biol. Med. 1999, 222, 283–292. [Google Scholar] [CrossRef] [PubMed]
- Smith, C.V.; Jones, D.P.; Guenther, T.M.; Lash, L.H.; Lauterburg, B.H. Compartmentation of glutathione: Implications for the study of toxicity and disease. Toxicol. Appl. Pharmacol. 1996, 140, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Hayes, J.D.; Flanagan, J.U.; Jowsey, I.R. Glutathione transferases. Ann. Rev. Pharmacol. Toxicol. 2005, 45, 51–88. [Google Scholar]
- Meister, A.; Anderson, M.E. Glutathione. Annu. Rev. Biochem. 1983, 52, 711–760. [Google Scholar] [CrossRef] [PubMed]
- Halliwell, B.; Gutteridge, J. Free Radicals in Biology and Medicine; Oxford University Press: New York, NY, USA, 2007. [Google Scholar]
- Bellomo, G.; Mirabelli, F.; DiMonte, D.; Richelmi, P.; Thor, H.; Orrenius, C.; Orrenius, S. Formation and reduction of glutathione-protein mixed disulfides during oxidative stress. A study with isolated hepatocytes and menadione (2-methyl-1,4-naphthoquinone). Biochem. Pharmacol. 1987, 36, 1313–1320. [Google Scholar] [CrossRef] [PubMed]
- Ji, L.L.; Fu, R.; Mitchell, E.W. Glutathione and antioxidant enzymes in skeletal muscle: Effects of fiber type and exercise intensity. J. Appl. Physiol. 1992, 73, 1854–1859. [Google Scholar] [PubMed]
- Lew, H.; Pyke, S.; Quintanilha, A. Changes in the glutathione status of plasma, liver and muscle following exhaustive exercise in rats. FEBS Lett. 1985, 185, 262–266. [Google Scholar] [PubMed]
- Marin, E.; Kretzschmar, M.; Arokoski, J.; Hänninen, O.; Klinger, W. Enzymes of glutathione synthesis in dog skeletal muscles and their response to training. Acta Physiol. Scand. 1993, 147, 369–373. [Google Scholar] [CrossRef] [PubMed]
- Leeuwenburgh, C.; Hollander, J.; Leichtweis, S.; Griffiths, M.; Gore, M.; Ji, L.L. Adaptations of glutathione antioxidant system to endurance training are tissue and muscle fiber specific. Am. J. Physiol. 1997, 272, R363–R369. [Google Scholar] [PubMed]
- Ji, L.L. Exercise and oxidative stress: Role of the cellular antioxidant systems. Exerc. Sport Sci. Rev. 1995, 23, 135–166. [Google Scholar] [PubMed]
- Sen, C.K. Oxidants and antioxidants in exercise. J. Appl. Physiol. 1995, 79, 675–686. [Google Scholar] [PubMed]
- Fisher-Wellman, K.H.; Mattox, T.A.; Thayne, K.; Katunga, L.A.; la Favor, J.D.; Neufer, P.D.; Hickner, R.C.; Wingard, C.J.; Anderson, E.J. Novel role for thioredoxin reductase-2 in mitochondrial redox adaptations to obesogenic diet and exercise in heart and skeletal muscle. J. Physiol. 2013, 591, 3471–3486. [Google Scholar] [CrossRef] [PubMed]
- Ji, L.L. Modulation of skeletal muscle antioxidant defense by exercise: Role of redox signaling. Free Radic. Biol. Med. 2008, 44, 142–152. [Google Scholar] [CrossRef] [PubMed]
- Powers, S.K.; Criswell, D.; Lawler, J.; Martin, D.; Ji, L.L.; Herb, R.A.; Dudley, G. Regional training-induced alterations in diaphragmatic oxidative and antioxidant enzymes. Respir. Physiol. 1994, 95, 227–237. [Google Scholar] [CrossRef] [PubMed]
- Gore, M.; Fiebig, R.; Hollander, J.; Leeuwenburgh, C.; Ohno, H.; Ji, L.L. Endurance training alters antioxidant enzyme gene expression in rat skeletal muscle. Can. J. Physiol. Pharmacol. 1998, 76, 1139–1145. [Google Scholar] [CrossRef] [PubMed]
- Hollander, J.; Fiebig, R.; Gore, M.; Bejma, J.; Ookawara, T.; Ohno, H.; Ji, L.L. Superoxide dismutase gene expression in skeletal muscle: Fiber-specific adaptation to endurance training. Am. J. Physiol. 1999, 277, R856–R862. [Google Scholar] [PubMed]
- Lambertucci, R.H.; Levada-Pires, A.C.; Rossoni, L.V.; Curi, R.; Pithon-Curi, T.C. Effects of aerobic exercise training on antioxidant enzyme activities and mRNA levels in soleus muscle from young and aged rats. Mech. Ageing Dev. 2007, 128, 267–275. [Google Scholar] [CrossRef] [PubMed]
- Ristow, M.; Zarse, K.; Oberbach, A.; Klöting, N.; Birringer, M.; Kiehntopf, M.; Stumvoll, M.; Kahn, C.R.; Blüher, M. Antioxidants prevent health-promoting effects of physical exercise in humans. Proc. Natl. Acad. Sci. USA 2009, 106, 8665–8670. [Google Scholar] [CrossRef] [PubMed]
- Ji, L.L.; Fu, R. Responses of glutathione system and antioxidant enzymes to exhaustive exercise and hydroperoxide. J. Appl. Physiol. 1992, 72, 549–554. [Google Scholar] [PubMed]
- Lawler, J.M.; Powers, S.K.; Visser, T.; van Dijk, H.; Kordus, M.J.; Ji, L.L. Acute exercise and skeletal muscle antioxidant and metabolic enzymes: Effects of fiber type and age. Am. J. Physiol. 1993, 265, R1344–R1350. [Google Scholar] [PubMed]
- Hollander, J.; Fiebig, R.; Gore, M.; Ookawara, T.; Ohno, H.; Ji, L.L. Superoxide dismutase gene expression is activated by a single bout of exercise in rat skeletal muscle. Pflugers Arch. 2001, 442, 426–434. [Google Scholar] [CrossRef] [PubMed]
- Hitomi, Y.; Watanabe, S.; Kizaki, T.; Sakurai, T.; Takemasa, T.; Haga, S.; Ookawara, T.; Suzuki, K.; Ohno, H. Acute exercise increases expression of extracellular superoxide dismutase in skeletal muscle and the aorta. Redox. Rep. 2008, 13, 213–226. [Google Scholar] [CrossRef] [PubMed]
- Radák, Z.; Asano, K.; Inoue, M.; Kizaki, T.; Oh-Ishi, S.; Suzuki, K.; Taniguchi, N.; Ohno, H. Superoxide dismutase derivative reduces oxidative damage in skeletal muscle of rats during exhaustive exercise. J. Appl. Physiol. 1995, 79, 129–135. [Google Scholar] [PubMed]
- Alessio, H.M.; Goldfarb, A.H. Lipid peroxidation and scavenger enzymes during exercise: Adaptive response to training. J. Appl. Physiol. 1988, 64, 1333–1336. [Google Scholar] [PubMed]
- Mastaloudis, A.; Leonard, S.W.; Traber, M.G. Oxidative stress in athletes during extreme endurance exercise. Free Radic. Biol. Med. 2001, 31, 911–922. [Google Scholar] [CrossRef] [PubMed]
- Steensberg, A.; Morrow, J.; Toft, A.D.; Bruunsgaard, H.; Pedersen, B.K. Prolonged exercise, lymphocyte apoptosis and F2-isoprostanes. Eur. J. Appl. Physiol. 2002, 87, 38–42. [Google Scholar] [CrossRef] [PubMed]
- Ashton, T.; Rowlands, C.C.; Jones, E.; Young, I.S.; Jackson, S.K.; Davies, B.; Peters, J.R. Electron spin resonance spectroscopic detection of oxygen-centred radicals in human serum following exhaustive exercise. Eur. J. Appl. Physiol. Occup. Physiol. 1998, 77, 498–502. [Google Scholar] [CrossRef] [PubMed]
- Vasankari, T.; Kujala, U.; Heinonen, O.; Kapanen, J.; Ahotupa, M. Measurement of serum lipid peroxidation during exercise using three different methods: Diene conjugation, thiobarbituric acid reactive material and fluorescent chromolipids. Clin. Chim. Acta 1995, 234, 63–69. [Google Scholar] [CrossRef] [PubMed]
- Umegaki, K.; Daohua, P.; Sugisawa, A.; Kimura, M.; Higuchi, M. Influence of one bout of vigorous exercise on ascorbic acid in plasma and oxidative damage to DNA in blood cells and muscle in untrained rats. J. Nutr. Biochem. 2000, 11, 401–407. [Google Scholar] [CrossRef] [PubMed]
- Niess, A.M.; Hartmann, A.; Grünert-Fuchs, M.; Poch, B.; Speit, G. DNA damage after exhaustive treadmill running in trained and untrained men. Int. J. Sports Med. 1996, 17, 397–403. [Google Scholar] [CrossRef] [PubMed]
- Powers, S.K.; Criswell, D.; Lawler, J.; Ji, L.L.; Martin, D.; Herb, R.A.; Dudley, G. Influence of exercise and fiber type on antioxidant enzyme activity in rat skeletal muscle. Am. J. Physiol. 1994, 266, R375–R380. [Google Scholar] [PubMed]
- Radák, Z.; Naito, H.; Kaneko, T.; Tahara, S.; Nakamoto, H.; Takahashi, R.; Cardozo-Pelaez, F.; Goto, S. Exercise training decreases DNA damage and increases DNA repair and resistance against oxidative stress of proteins in aged rat skeletal muscle. Pflugers Arch. 2002, 445, 273–278. [Google Scholar] [CrossRef] [PubMed]
- Radák, Z.; Chung, H.Y.; Goto, S. Systemic adaptation to oxidative challenge induced by regular exercise. Free Radic. Biol. Med. 2008, 44, 153–159. [Google Scholar] [CrossRef] [PubMed]
- Alleman, R.J.; Katunga, L.A.; Nelson, M.A.M.; Brown, D.A.; Anderson, E.J. The “Goldilocks Zone” from a redox perspective—Adaptive vs. deleterious responses to oxidative stress in striated muscle. Front. Physiol. 2014. [Google Scholar] [CrossRef]
- Kalogeris, T.; Bao, Y.; Korthuis, R.J. Mitochondrial reactive oxygen species: A double edged sword in ischemia/reperfusion vs. preconditioning. Redox Biol. 2014, 2, 702–714. [Google Scholar] [CrossRef] [PubMed]
- Dodd, S.L.; Gagnon, B.J.; Senf, S.M.; Hain, B.A.; Judge, A.R. Ros-mediated activation of NF-kappaB and Foxo during muscle disuse. Muscle Nerve 2010, 41, 110–113. [Google Scholar] [CrossRef] [PubMed]
- Derbre, F.; Ferrando, B.; Gomez-Cabrera, M.C.; Sanchis-Gomar, F.; Martinez-Bello, V.E.; Olaso-Gonzalez, G.; Diaz, A.; Gratas-Delamarche, A.; Cerda, M.; Viña, J. Inhibition of xanthine oxidase by allopurinol prevents skeletal muscle atrophy: Role of p38 MAPKinase and E3 ubiquitin ligases. PLoS ONE 2012, 7, e46668. [Google Scholar] [CrossRef] [PubMed]
- Kröller-Schön, S.; Jansen, T.; Hauptmann, F.; Schüler, A.; Heeren, T.; Hausding, M.; Oelze, M.; Viollet, B.; Keaney, J.F., Jr.; Wenzel, P.; et al. α1AMP-activated protein kinase mediates vascular protective effects of exercise. Arterioscler. Thromb. Vasc. Biol. 2012, 32, 1632–1641. [Google Scholar] [CrossRef] [PubMed]
- Morgan, M.J.; Liu, Z.G. Crosstalk of reactive oxygen species and NF-κB signaling. Cell Res. 2011, 21, 103–115. [Google Scholar] [CrossRef] [PubMed]
- McArdle, F.; Spiers, S.; Aldemir, H.; Vasilaki, A.; Beaver, A.; Iwanejko, L.; McArdle, A.; Jackson, M.J. Preconditioning of skeletal muscle against contraction-induced damage: The role of adaptations to oxidants in mice. J. Physiol. 2004, 561, 233–244. [Google Scholar] [CrossRef] [PubMed]
- Eckl, P.M.; Ortner, A.; Esterbauer, H. Genotoxic properties of 4-hydroxyalkenals and analogous aldehydes. Mutat. Res. 1993, 290, 183–192. [Google Scholar] [CrossRef] [PubMed]
- Poli, G.; Schaur, R.J.; Siems, W.G.; Leonarduzzi, G. 4-hydroxynonenal: A membrane lipid oxidation product of medicinal interest. Med. Res. Rev. 2008, 28, 569–631. [Google Scholar] [CrossRef] [PubMed]
- Ishii, T.; Itoh, K.; Ruiz, E.; Leake, D.S.; Unoki, H.; Yamamoto, M.; Mann, G.E. Role of Nrf2 in the regulation of CD36 and stress protein expression in murine macrophages: Activation by oxidatively modified LDL and 4-hydroxynonenal. Circ. Res. 2004, 94, 609–616. [Google Scholar] [CrossRef] [PubMed]
- Levonen, A.L.; Landar, A.; Ramachandra, A.; Cease, E.K.; Dickinson, D.A.; Zanoni, G.; Morrow, J.D.; Darley-Usmar, V.M. Cellular mechanisms of redox cell signalling: Role of cysteine modification in controlling antioxidant defences in response to electrophilic lipid oxidation products. Biochem. J. 2004, 378, 373–382. [Google Scholar] [CrossRef] [PubMed]
- Echtay, K.S.; Esteves, T.C.; Pakay, J.L.; Jekabsons, M.B.; Lambert, A.J.; Portero-Otín, M.; Pamplona, R.; Vidal-Puig, A.J.; Wang, S.; Roebuck, S.J.; et al. A signalling role for 4-hydroxy-2-nonenal in regulation of mitochondrial uncoupling. EMBO J. 2003, 22, 4103–4110. [Google Scholar] [CrossRef] [PubMed]
- Gomez-Cabrera, M.C.; Borrás, C.; Pallardó, F.V.; Sastre, J.; Ji, L.L.; Viña, J. Decreasing xanthine oxidase-mediated oxidative stress prevents useful cellular adaptations to exercise in rats. J. Physiol. 2005, 567, 113–120. [Google Scholar] [CrossRef] [PubMed]
- Gomez-Cabrera, M.C.; Domenech, E.; Viña, J. Moderate exercise is an antioxidant: Upregulation of antioxidant genes by training. Free Radic. Biol. Med. 2008, 44, 126–131. [Google Scholar] [CrossRef] [PubMed]
- Ji, L.L.; Gomez-Cabrera, M.C.; Steinhafel, N.; Viña, J. Acute exercise activates nuclear factor (NF)-kappaB signaling pathway in rat skeletal muscle. FASEB J. 2004, 18, 1499–1506. [Google Scholar] [CrossRef]
- Margonis, K.; Fatouros, I.G.; Jamurtas, A.Z.; Nikolaidis, M.G.; Douroudos, I.; Chatzinikolaou, A.; Mitrakou, A.; Mastorakos, G.; Papassotiriou, I.; Taxildaris, K.; et al. Oxidative stress biomarkers responses to physical overtraining: Implications for diagnosis. Free Radic. Biol. Med. 2007, 43, 901–910. [Google Scholar] [CrossRef] [PubMed]
- Palazzetti, S.; Richard, M.J.; Favier, A.; Margaritis, I. Overloaded training increases exercise-induced oxidative stress and damage. Can. J. Appl. Physiol. 2003, 28, 588–604. [Google Scholar] [CrossRef] [PubMed]
- Tanskanen, M.; Atalay, M.; Uusitalo, A. Altered oxidative stress in overtrained athletes. J. Sports Sci. 2010, 28, 309–317. [Google Scholar] [CrossRef] [PubMed]
- Irrcher, I.; Adhihetty, P.J.; Sheehan, T.; Joseph, A.M.; Hood, D.A. PPARγ coactivator-1α expression during thyroid hormone- and contractile activity-induced mitochondrial adaptations. Am. J. Physiol. Cell Physiol. 2003, 284, C1669–C1677. [Google Scholar] [CrossRef] [PubMed]
- Holloszy, J.O. Regulation by exercise of skeletal muscle content of mitochondria and GLUT4. J. Physiol. Pharmacol. 2008, 59, 5–18. [Google Scholar] [PubMed]
- Yan, Z.; Okutsu, M.; Akhtar, Y.N.; Lira, V.A. Regulation of exercise-induced fiber type transformation, mitochondrial biogenesis, and angiogenesis in skeletal muscle. J. Appl. Physiol. 2011, 110, 264–274. [Google Scholar] [CrossRef] [PubMed]
- Van Loon, L.J.C.; Goodpaster, B.H. Increased intramuscular lipid storage in the insulin-resistant and endurance-trained state. Pflugers Arch. 2006, 451, 606–616. [Google Scholar] [CrossRef] [PubMed]
- Hawley, J.A.; Lessard, S.J. Exercise training-induced improvements in insulin action. Acta Physiol. 2008, 192, 127–135. [Google Scholar] [CrossRef]
- Olesen, J.; Kiilerich, K.; Pilegaard, H. PGC-1α-mediated adaptations in skeletal muscle. Pflugers Arch. 2010, 460, 153–162. [Google Scholar] [CrossRef] [PubMed]
- Kang, C.; Ji, L.L. Role of PGC-1α in muscle function and aging. J. Sport Health Sci. 2013, 2, 81–86. [Google Scholar] [CrossRef]
- Pilegaard, H.; Osada, T.; Andersen, L.T.; Helge, J.W.; Saltin, B.; Neufer, P.D. Substrate availability and transcriptional regulation of metabolic genes in human skeletal muscle during recovery from exercise. Metabolism 2005, 54, 1048–1055. [Google Scholar] [CrossRef] [PubMed]
- Krämer, D.K.; Ahlsen, M.; Norrbom, J.; Jansson, E.; Hjeltnes, N.; Gustafsson, T.; Krook, A. Human skeletal muscle fibre type variations correlate with PPARα, PPARδ and PGC-1α mRNA. Acta Physiol. 2006, 188, 207–216. [Google Scholar]
- Mathai, A.S.; Bonen, A.; Benton, C.R.; Robinson, D.L.; Graham, T.E. Rapid exercise-induced changes in PGC-1α mRNA and protein in human skeletal muscle. J. Appl. Physiol. 2008, 105, 1098–1105. [Google Scholar] [CrossRef] [PubMed]
- Little, J.P.; Safdar, A.; Bishop, D.; Tarnopolsky, M.A.; Gibala, M.J. An acute bout of high-intensity interval training increases the nuclear abundance of PGC-1α and activates mitochondrial biogenesis in human skeletal muscle. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2011, 300, R1303–R1310. [Google Scholar] [CrossRef] [PubMed]
- Cobley, J.N.; Bartlett, J.D.; Kayani, A.; Murray, S.W.; Louhelainen, J.; Donovan, T.; Waldron, S.; Gregson, W.; Burniston, J.G.; Morton, J.P.; et al. PGC-1α transcriptional response and mitochondrial adaptation to acute exercise is maintained in skeletal muscle of sedentary elderly males. Biogerontology 2012, 13, 621–631. [Google Scholar] [CrossRef] [PubMed]
- Wright, D.C.; Han, D.H.; Garcia-Roves, P.M.; Geiger, P.C.; Jones, T.E.; Holloszy, J. Exercise-induced mitochondrial biogenesis begins before the increase in muscle PGC-1α expression. J. Biol. Chem. 2007, 282, 194–199. [Google Scholar] [CrossRef] [PubMed]
- Wu, Z.; Puigserver, P.; Andersson, U.; Zhang, C.; Adelmant, G.; Mootha, V.; Troy, A.; Cinti, S.; Lowell, B.; Scarpulla, R.C.; et al. Mechanisms controlling mitochondrial biogenesis and respiration through the thermogenic coactivator PGC-1. Cell 1999, 98, 115–124. [Google Scholar] [CrossRef] [PubMed]
- Koves, T.R.; Li, P.; An, J.; Akimoto, T.; Slentz, D.; Ilkayeva, O.; Dohm, G.L.; Yan, Z.; Newgard, C.B.; Muoio, D.M. Peroxisome proliferator-activated receptor-γ co-activator 1α-mediated metabolic remodeling of skeletal myocytes mimics exercise training and reverses lipid-induced mitochondrial inefficiency. J. Biol. Chem. 2005, 280, 33588–33598. [Google Scholar] [CrossRef] [PubMed]
- Lin, J.; Wu, H.; Tarr, P.T.; Zhang, C.Y.; Wu, Z.; Boss, O.; Michael, L.F.; Puigserver, P.; Isotani, E.; Olson, E.N.; et al. Transcriptional co-activator PGC-1α drives the formation of slow-twitch muscle fibres. Nature 2002, 418, 797–801. [Google Scholar] [CrossRef] [PubMed]
- Handschin, C.; Chin, S.; Li, P.; Liu, F.; Maratos-Flier, E.; Lebrasseur, N.K.; Yan, Z.; Spiegelman, B.M. Skeletal muscle fiber-type switching, exercise intolerance, and myopathy in PGC-1α muscle-specific knock-out animals. J. Biol. Chem. 2007, 282, 30014–30021. [Google Scholar] [CrossRef] [PubMed]
- Puigserver, P.; Spiegelman, B.M. Peroxisome proliferator-activated receptor-γ coactivator 1α (PGC-1α): Transcriptional coactivator and metabolic regulator. Endocr. Rev. 2003, 24, 78–90. [Google Scholar] [CrossRef] [PubMed]
- Scarpulla, R.C.; Vega, R.B.; Kelly, D.P. Transcriptional integration of mitochondrial biogenesis. Trends Endocrinol. Metab. 2012, 23, 459–466. [Google Scholar] [CrossRef] [PubMed]
- Oberkofler, H.; Schraml, E.; Krempler, F.; Patsch, W. Potentiation of liver X receptor transcriptional activity by peroxisome-proliferator-activated receptor γ co-activator 1α. Biochem. J. 2003, 371, 89–96. [Google Scholar] [CrossRef] [PubMed]
- Summermatter, S.; Baum, O.; Santos, G.; Hoppeler, H.; Handschin, C. Peroxisome proliferator-activated receptor γ coactivator 1α (PGC-1α) promotes skeletal muscle lipid refueling in vivo by activating de novo lipogenesis and the pentose phosphate pathway. J. Biol. Chem. 2010, 285, 32793–32800. [Google Scholar] [CrossRef] [PubMed]
- Arany, Z.; Foo, S.Y.; Ma, Y.; Ruas, J.L.; Bommi-Reddy, A.; Girnun, G.; Cooper, M.; Laznik, D.; Chinsomboon, J.; Rangwala, S.M.; et al. HIF-independent regulation of VEGF and angiogenesis by the transcriptional coactivator PGC-1α. Nature 2008, 451, 1008–1012. [Google Scholar] [CrossRef] [PubMed]
- Chinsomboon, J.; Ruas, J.; Gupta, R.K.; Thom, R.; Shoag, J.; Rowe, G.C.; Sawada, N.; Raghuram, S.; Arany, Z. The transcriptional coactivator PGC-1α mediates exercise-induced angiogenesis in skeletal muscle. Proc. Natl. Acad. Sci. USA 2009, 106, 21401–21406. [Google Scholar] [CrossRef] [PubMed]
- Chin, E.R.; Olson, E.N.; Richardson, J.A.; Yang, Q.; Humphries, C.; Shelton, J.M.; Wu, H.; Zhu, W.; Bassel-Duby, R.; Williams, R.S. A calcineurin-dependent transcriptional pathway controls skeletal muscle fiber type. Genes Dev. 1998, 12, 2499–2509. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.; Naya, F.J.; McKinsey, T.A.; Mercer, B.; Shelton, J.M.; Chin, E.R.; Simard, A.R.; Michel, R.N.; Bassel-Duby, R.; Olson, E.N.; et al. MEF2 responds to multiple calcium-regulated signals in the control of skeletal muscle fiber type. EMBO J. 2000, 19, 1963–1973. [Google Scholar] [CrossRef] [PubMed]
- Potthoff, M.J.; Wu, H.; Arnold, M.A.; Shelton, J.M.; Backs, J.; McAnally, J.; Richardson, J.A.; Bassel-Duby, R.; Olson, E.N. Histone deacetylase degradation and MEF2 activation promote the formation of slow-twitch myofibers. J. Clin. Investig. 2007, 117, 2459–2467. [Google Scholar] [CrossRef] [PubMed]
- Handschin, C.; Rhee, J.; Lin, J.; Tarr, P.T.; Spiegelman, B.M. An autoregulatory loop controls peroxisome proliferator-activated receptor γ coactivator 1α expression in muscle. Proc. Natl. Acad. Sci. USA 2003, 100, 7111–7116. [Google Scholar] [CrossRef] [PubMed]
- Kang, C.; O’Moore, K.M.; Dickman, J.R.; Ji, L.L. Exercise activation of muscle peroxisome proliferator-activated receptor-gamma coactivator-1α signaling is redox sensitive. Free Radic. Biol. Med. 2009, 47, 1394–1400. [Google Scholar] [CrossRef] [PubMed]
- Strobel, N.A.; Matsumoto, A.; Peake, J.M.; Marsh, S.A.; Peternelj, T.T.; Briskey, D.; Fassett, R.G.; Coombes, J.S.; Wadley, G.D. Altering the redox state of skeletal muscle by glutathione depletion increases the exercise-activation of PGC-1α. Physiol. Rep. 2014, 2, e12224. [Google Scholar] [CrossRef] [PubMed]
- St-Pierre, J.; Drori, S.; Uldry, M.; Silvaggi, J.M.; Rhee, J.; Jäger, S.; Handschin, C.; Zheng, K.; Lin, J.; Yang, W.; et al. Suppression of reactive oxygen species and neurodegeneration by the PGC-1 transcriptional coactivators. Cell 2006, 127, 397–408. [Google Scholar] [CrossRef] [PubMed]
- Kong, X.; Wang, R.; Xue, Y.; Liu, X.; Zhang, H.; Chen, Y.; Fang, F.; Chang, Y. Sirtuin 3, a new target of PGC-1α, plays an important role in the suppression of ROS and mitochondrial biogenesis. PLoS ONE 2010, 5, e11707. [Google Scholar] [CrossRef] [PubMed]
- Ahn, B.H.; Kim, H.S.; Song, S.; Lee, I.H.; Liu, J.; Vassilopoulos, A.; Deng, C.X.; Finkel, T. A role for the mitochondrial deacetylase Sirt3 in regulating energy homeostasis. Proc. Natl. Acad. Sci. USA 2008, 105, 14447–14452. [Google Scholar] [CrossRef] [PubMed]
- Finley, L.W.; Haas, W.; Desquiret-Dumas, V.; Wallace, D.C.; Procaccio, V.; Gygi, S.P.; Haigis, M.C. Succinate dehydrogenase is a direct target of sirtuin 3 deacetylase activity. PLoS ONE 2011, 6, e23295. [Google Scholar] [CrossRef] [PubMed]
- Qiu, X.; Brown, K.; Hirschey, M.D.; Verdin, E.; Chen, D. Calorie restriction reduces oxidative stress by SIRT3-mediated SOD2 activation. Cell Metab. 2010, 12, 662–667. [Google Scholar] [CrossRef] [PubMed]
- Tao, R.; Coleman, M.C.; Pennington, J.D.; Ozden, O.; Park, S.H.; Jiang, H.; Kim, H.S.; Flynn, C.R.; Hill, S.; Hayes McDonald, W.; et al. Sirt3-mediated deacetylation of evolutionarily conserved lysine 122 regulates MnSOD activity in response to stress. Mol. Cell 2010, 40, 893–904. [Google Scholar] [PubMed]
- Chen, Y.; Zhang, J.; Lin, Y.; Lei, Q.; Guan, K.L.; Zhao, S.; Xiong, Y. Tumour suppressor SIRT3 deacetylates and activates manganese superoxide dismutase to scavenge ROS. EMBO Rep. 2011, 12, 534–541. [Google Scholar] [CrossRef] [PubMed]
- Wenz, T.; Rossi, S.G.; Rotundo, R.L.; Spiegelman, B.M.; Moraes, C.T. Increased muscle PGC-1α expression protects from sarcopenia and metabolic disease during aging. Proc. Natl. Acad. Sci. USA 2009, 106, 20405–20410. [Google Scholar] [CrossRef] [PubMed]
- St-Pierre, J.; Lin, J.; Krauss, S.; Tarr, P.T.; Yang, R.; Newgard, C.B.; Spiegelman, B.M. Bioenergetic analysis of peroxisome proliferator-activated receptor γ coactivators 1α and 1β (PGC-1α and PGC-1β) in muscle cells. J. Biol. Chem. 2003, 278, 26597–26603. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Steinbacher, P.; Eckl, P. Impact of Oxidative Stress on Exercising Skeletal Muscle. Biomolecules 2015, 5, 356-377. https://doi.org/10.3390/biom5020356
Steinbacher P, Eckl P. Impact of Oxidative Stress on Exercising Skeletal Muscle. Biomolecules. 2015; 5(2):356-377. https://doi.org/10.3390/biom5020356
Chicago/Turabian StyleSteinbacher, Peter, and Peter Eckl. 2015. "Impact of Oxidative Stress on Exercising Skeletal Muscle" Biomolecules 5, no. 2: 356-377. https://doi.org/10.3390/biom5020356
APA StyleSteinbacher, P., & Eckl, P. (2015). Impact of Oxidative Stress on Exercising Skeletal Muscle. Biomolecules, 5(2), 356-377. https://doi.org/10.3390/biom5020356