
Seeking a Mechanism for the Toxicity of Oligomeric α-Synuclein

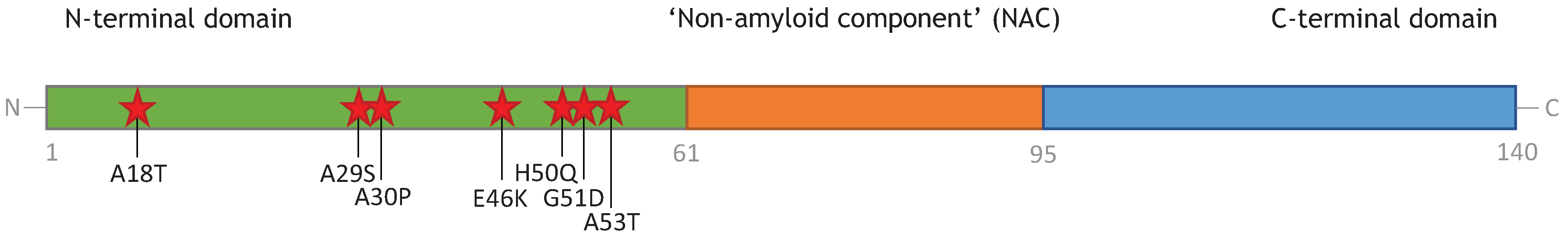{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Pathology of Synucleinopathies
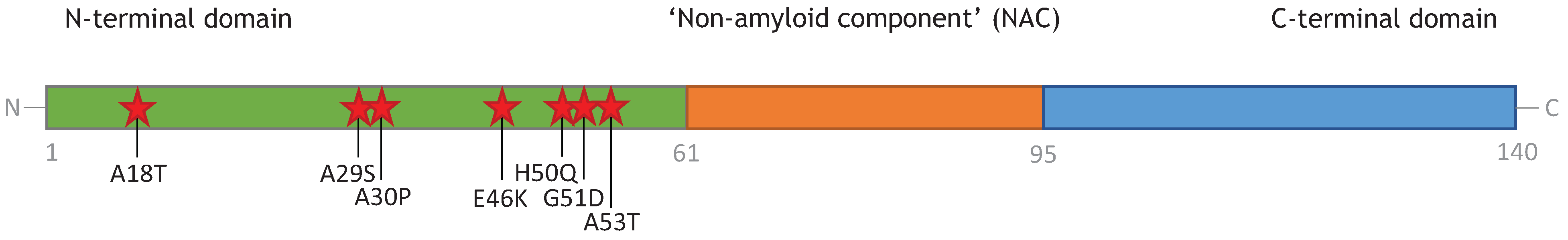
3. Monomeric α-Synuclein
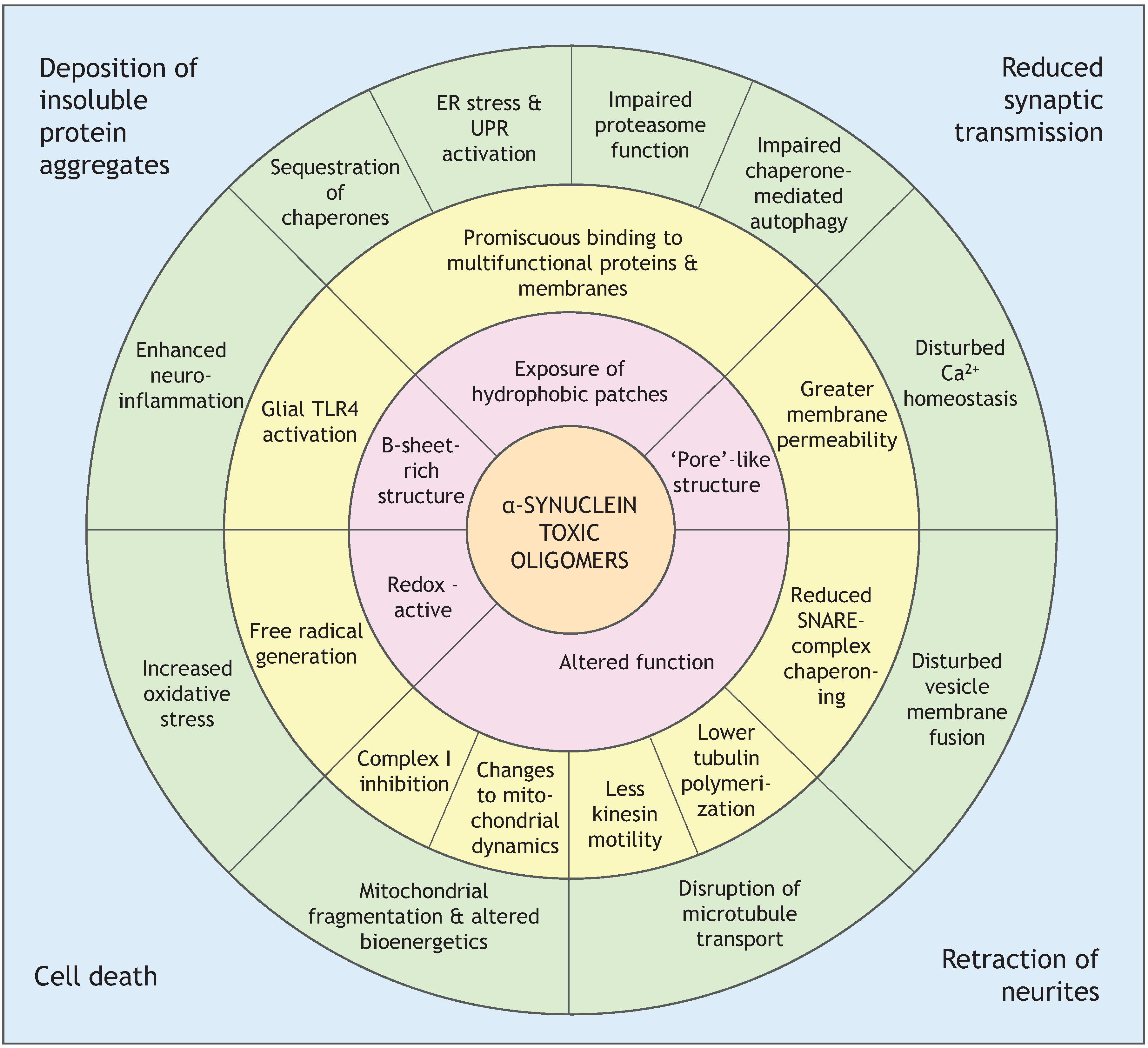
4. Oligomers of α-Synuclein and Their Toxicity
4.1. Dimers, Trimers, and Tetramers
4.2. Soluble β-Rich Oligomers
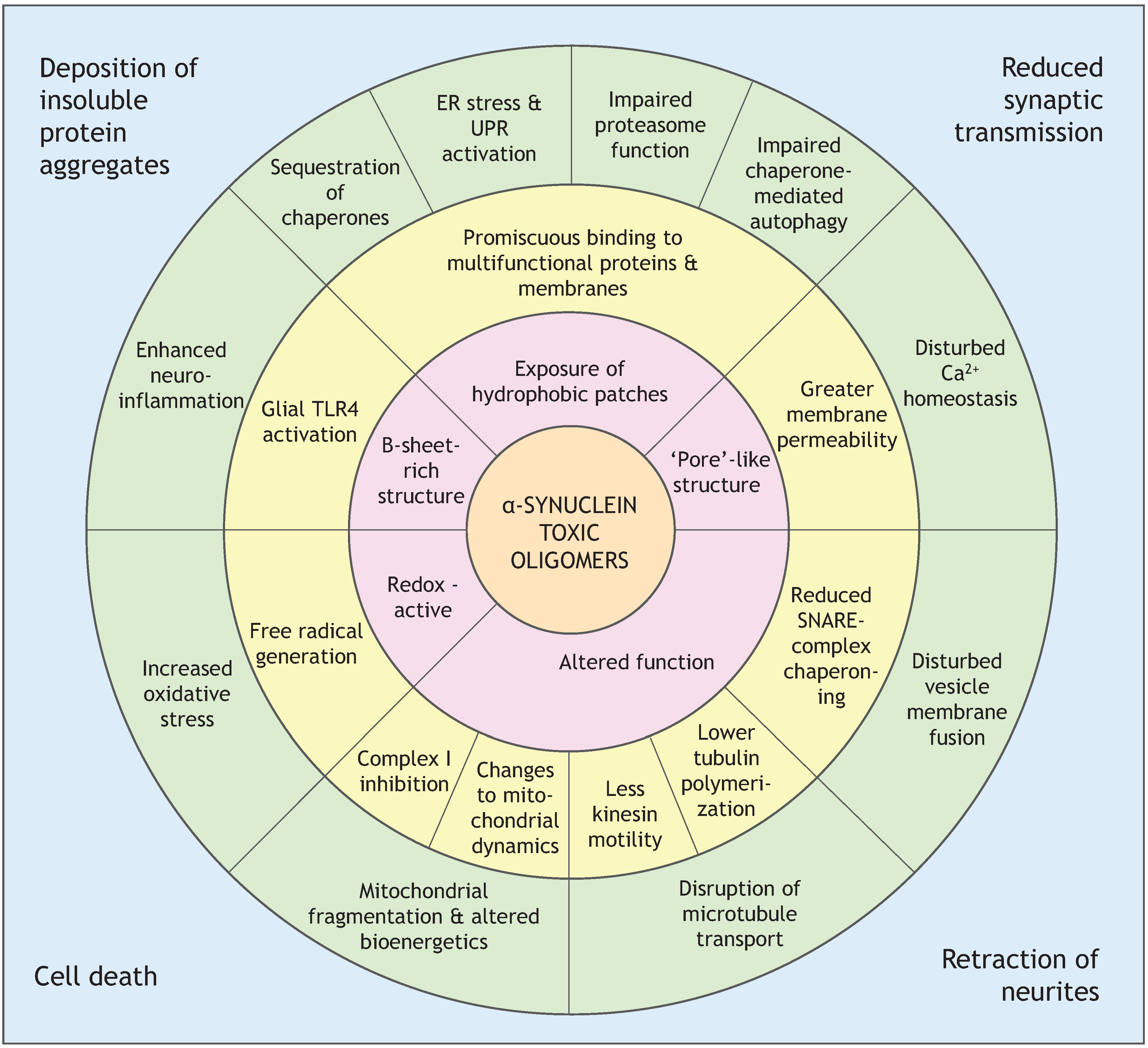
4.2.1. Membrane Permeability
4.2.2. Mitochondrial Dysfunction
4.2.3. Altered Cytoskeleton Formation
4.2.4. Enhanced Formation of Reactive Oxygen Species (ROS) and Neuroinflammation
4.2.5. Endoplasmic Reticulum Stress
4.2.6. Impaired Protein Degradation Systems
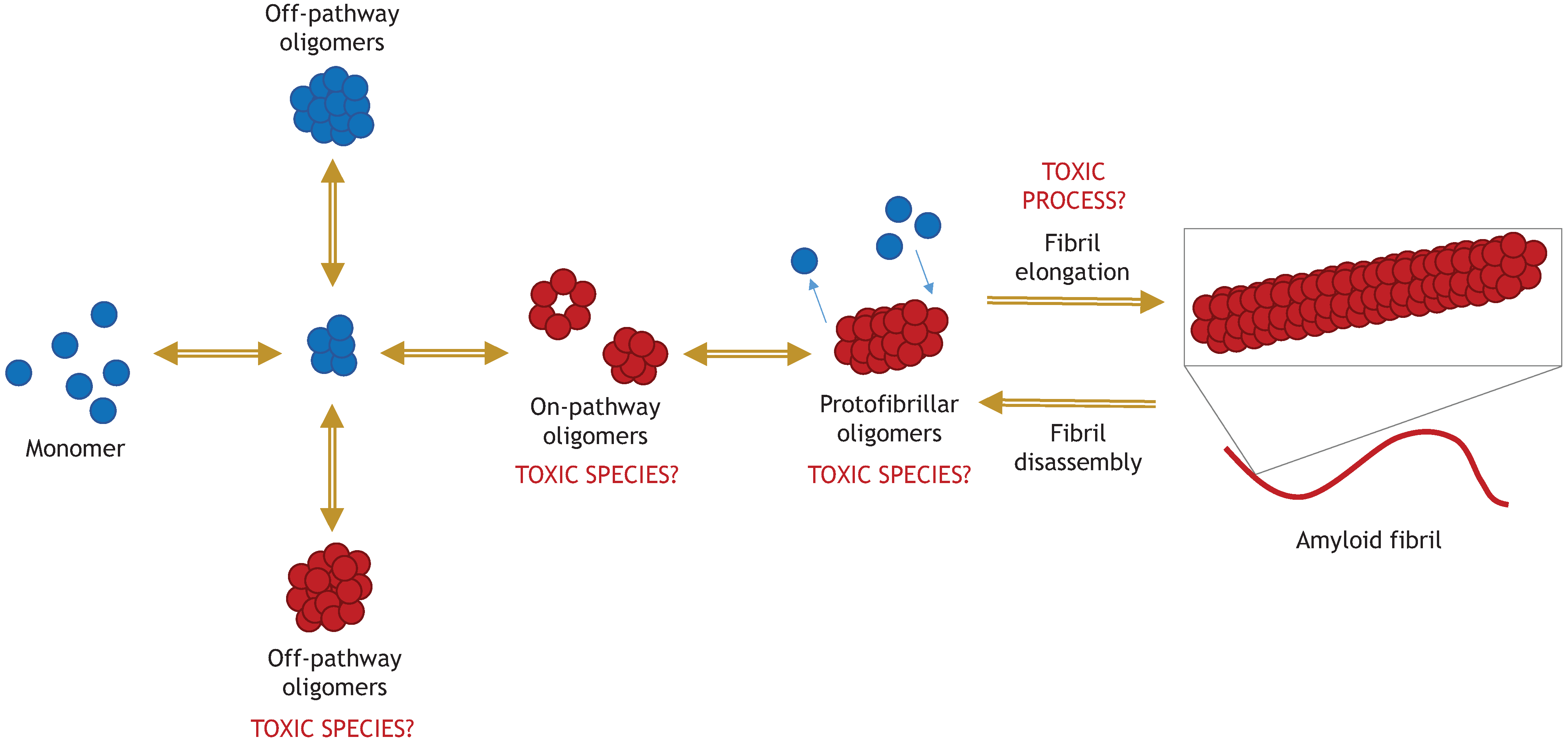
5. Amyloid Fibrils of α-Synuclein
5.1. “Toxic Oligomers Exist in a Dynamic Equilibrium with Fibril Assembly/Disassembly”
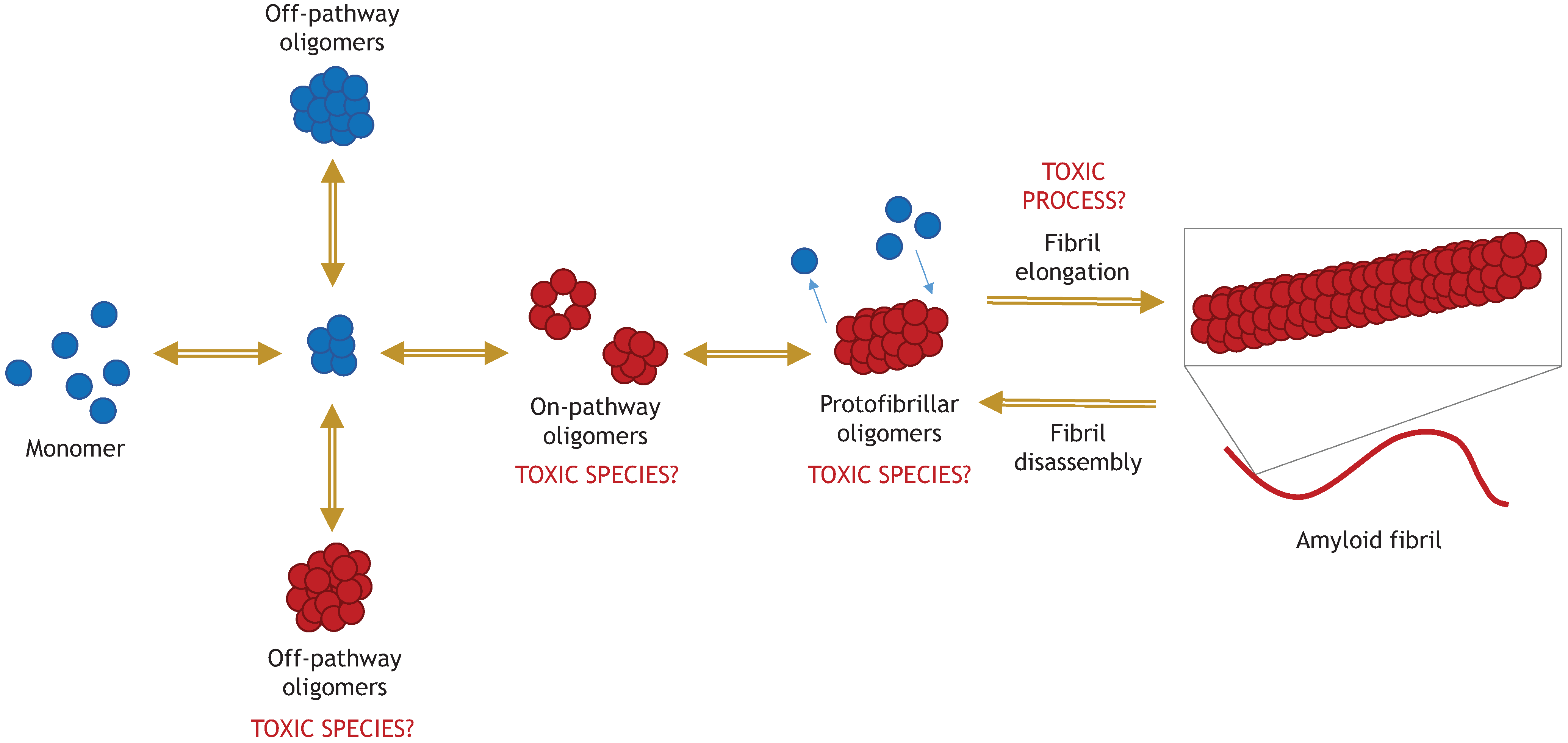
5.2. “The Process of Active Fibril Elongation is Toxic”
6. Models for Studying α-Synuclein Toxic Oligomers
7. Conclusions
Author Contributions
Conflicts of Interest
References
- Conway, K.A.; Lee, S.; Rochet, J.; Ding, T.T.; Williamson, R.E.; Lansbury, P.T. Acceleration of oligomerization, not fibrillization, is a shared property of both α-synuclein mutations linked to early-onset Parkinson’s disease: Implications for pathogenesis and therapy. PNAS 2000, 97, 571–576. [Google Scholar] [CrossRef] [PubMed]
- Kayed, R.; Head, E.; Thompson, J.L.; Mcintire, T.M.; Milton, S.C.; Cotman, C.W.; Glabe, C.G. Common structure of soluble amyloid oligomers implies common mechanism of pathogenesis. Science 2003, 300, 486–489. [Google Scholar] [CrossRef] [PubMed]
- Spillantini, M.G.; Schmidt, M.L.; Lee, V.M.-Y.; Trojanowski, J.Q. α-Synuclein in Lewy bodies. Nature 1997, 388, 839–840. [Google Scholar] [CrossRef] [PubMed]
- Irwin, D.J.; Lee, V.M.-Y.; Trojanowski, J.Q. Parkinson’s disease dementia: Convergence of α-synuclein, tau and amyloid-β pathologies. Nat. Rev. Neurosci. 2013, 14, 626–636. [Google Scholar] [CrossRef] [PubMed]
- Lashuel, H.A.; Overk, C.R.; Oueslati, A.; Masliah, E. The many faces of α-synuclein: From structure and toxicity to therapeutic target. Nat. Rev. Neurosci. 2013, 14, 38–48. [Google Scholar] [CrossRef] [PubMed]
- Hamilton, R.L. Lewy bodies in Alzheimer’s disease: A neuropathological review of 145 cases using alpha-synuclein immunohistochemistry. Brain Pathol. 2000, 10, 378–384. [Google Scholar] [CrossRef] [PubMed]
- Parkkinen, L.; Soininen, H.; Alafuzoff, I. Regional distribution of alpha-synuclein pathology in unimpaired aging and Alzheimer disease. J. Neuropathol. Exp. Neurol. 2003, 62, 363–367. [Google Scholar] [PubMed]
- Jellinger, K.A. Lewy body-related alpha-synucleinopathy in the aged human brain. J. Neural Transm. 2004, 111, 1219–1235. [Google Scholar] [CrossRef] [PubMed]
- Larson, M.E.; Sherman, M.A.; Greimel, S.; Kuskowski, M.; Schneider, J.A.; Bennett, D.A.; Lesné, S.E. Soluble α-synuclein is a novel modulator of Alzheimer’s disease pathophysiology. J. Neurosci. 2012, 32, 10253–10266. [Google Scholar] [CrossRef] [PubMed]
- Chartier-Harlin, M.; Kachergus, J.; Roumier, C.; Mouroux, V.; Douay, X.; Lincoln, S.; Levecque, C.; Larvor, L.; Andrieux, J.; Hulihan, M.; et al. α-Synuclein locus duplication as a cause of familial Parkinson’s disease. Lancet 2004, 364, 1167–1169. [Google Scholar] [CrossRef] [PubMed]
- Ahn, T.B.; Kim, S.Y.; Kim, J.Y.; Park, S.S.; Lee, D.S.; Min, H.J.; Kim, Y.K.; Kim, S.E.; Kim, J.M.; Kim, H.J.; et al. α-Synuclein gene duplication is present in sporadic Parkinson disease. Neurology 2008, 70, 43–49. [Google Scholar] [CrossRef] [PubMed]
- Kasten, M.; Klein, C. The many faces of alpha-synuclein mutations. Mov. Disord. 2013, 28, 697–701. [Google Scholar] [CrossRef] [PubMed]
- Krüger, R.; Kuhn, W.; Müller, T.; Woitalla, D.; Graeber, M.; Kösel, S.; Przuntek, H.; Epplen, J.; Schöls, L.; Riess, O. Ala30Pro mutation in the gene encoding alpha-synuclein in Parkinson’s disease. Nat. Genet. 1998, 18, 106–108. [Google Scholar] [CrossRef] [PubMed]
- Zarranz, J.; Alegre, J.; Gómez-Esteban, J.; Lezcano, E.; Ros, R.; Ampuero, I.; Vidal, L.; Hoenicka, J.; Rodriguez, O.; Atarés, B.; et al. The new mutation, E46K, of alpha-synuclein causes Parkinson and Lewy body dementia. Ann. Neurol. 2004, 55, 164–173. [Google Scholar] [CrossRef] [PubMed]
- Polymeropoulos, M.H.; Lavedan, C.; Leroy, E.; Ide, S.E.; Dehejia, A.; Dutra, A.; Pike, B.; Root, H.; Rubenstein, J.; Boyer, R.; et al. Mutation in the α-synuclein gene identified in families with Parkinson’s Disease. Science 1997, 276, 2045–2048. [Google Scholar] [CrossRef] [PubMed]
- Appel-Cresswell, S.; Vilarino-Guell, C.; Encarnacion, M.; Sherman, H.; Yu, I.; Shah, B.; Weir, D.; Thompson, C.; Szu-Tu, C.; Trinh, J.; et al. Alpha-synuclein p.H50Q, a novel pathogenic mutation for Parkinson’s disease. Mov. Disord. 2013, 28, 811–813. [Google Scholar] [CrossRef] [PubMed]
- Lesage, S.; Anheim, M.; Letournel, F.; Bousset, L.; Honoré, A.; Rozas, N.; Pieri, L.; Madiona, K.; Dürr, A.; Melki, R.; et al. G51D α-synuclein mutation causes a novel parkinsonian-pyramidal syndrome. Ann. Neurol. 2013, 73, 459–471. [Google Scholar] [CrossRef] [PubMed]
- Hoffman-Zacharska, D.; Koziorowski, D.; Ross, O.A.; Milewski, M.; Poznański, J.; Jurek, M.; Wszolek, Z.K.; Soto-Ortolaza, A.; Sławek, J.; Janik, P.; et al. Novel A18T and pA29S substitutions in α-synuclein may be associated with sporadic Parkinson’s disease. Parkinsonism Relat. Disord. 2013, 19, 1057–1060. [Google Scholar] [CrossRef] [PubMed]
- Fredenburg, R.A.; Rospigliosi, C.; Meray, R.K.; Kessler, J.C.; Lashuel, H.A.; Eliezer, D.; Lansbury, P.T.J. The impact of the E46K mutation on the properties of alpha-synuclein in its monomeric and oligomeric states. Biochemistry 2007, 46, 7107–7118. [Google Scholar] [CrossRef] [PubMed]
- Brucale, M.; Sandal, M.; di Maio, S.; Rampioni, A.; Tessari, I.; Tosatto, L.; Bisaglia, M.; Bubacco, L.; Samorì, B. Pathogenic mutations shift the equilibria of alpha-synuclein single molecules towards structured conformers. Chembiochem 2009, 10, 176–183. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Uversky, V.N.; Fink, A.L. Effect of familial Parkinson’s Disease point mutations A30P and A53T on the structural properties, aggregation, and fibrillation of human α-synuclein. Biochemistry 2001, 40, 11604–11613. [Google Scholar] [CrossRef] [PubMed]
- Maraganore, D.M.; de Andrade, M.; Elbaz, A.; Farrer, M.J.; Ioannidis, J.P.; Krüger, R.; Rocca, W.A.; Schneider, N.K.; Lesnick, T.G.; Lincoln, S.J.; et al. Collaborative analysis of alpha-synuclein gene promoter variability and Parkinson Disease. JAMA 2006, 296, 661–670. [Google Scholar] [CrossRef] [PubMed]
- Jowaed, A.; Schmitt, I.; Kaut, O.; Wüllner, U. Methylation regulates alpha-synuclein expression and is decreased in Parkinson’s disease patients’ brains. J. Neurosci. 2010, 30, 6355–6359. [Google Scholar] [CrossRef] [PubMed]
- Olivares, D.; Huang, X.; Branden, L.; Greig, N.H.; Rogers, J.T. Physiological and pathological role of alpha-synuclein in Parkinson’s disease through iron mediated oxidative stress; the role of a putative iron-responsive element. Int. J. Mol. Sci. 2009, 10, 1226–1260. [Google Scholar] [CrossRef] [PubMed]
- Grosso, H.; Woo, J.M.; Lee, K.W.; Im, J.Y.; Masliah, E.; Junn, E.; Mouradian, M.M. Transglutaminase 2 exacerbates α-synuclein toxicity in mice and yeast. FASEB J. 2014, 28, 4280–4291. [Google Scholar] [CrossRef] [PubMed]
- Fauvet, B.; Mbefo, M.K.; Fares, M.-B.; Desobry, C.; Michael, S.; Ardah, M.T.; Tsika, E.; Coune, P.; Prudent, M.; Lion, N.; et al. α-Synuclein in central nervous system and from erythrocytes, mammalian cells, and Escherichia coli exists predominantly as disordered monomer. J. Biol. Chem. 2012, 287, 15345–15364. [Google Scholar] [CrossRef] [PubMed]
- Breydo, L.; Wu, J.W.; Uversky, V.N. α-synuclein misfolding and Parkinson’s disease. Biochim. Biophys. Acta 2012, 1822, 261–285. [Google Scholar] [CrossRef] [PubMed]
- Visanji, N.P.; Wislet-Gendebien, S.; Oschipok, L.W.; Zhang, G.; Aubert, I.; Fraser, P.E.; Tandon, A. Effect of Ser-129 phosphorylation on interaction of α-synuclein with synaptic and cellular membranes. J. Biol. Chem. 2011, 286, 35863–35873. [Google Scholar] [CrossRef] [PubMed]
- Lorenzen, N.; Lemminger, L.; Pedersen, J.N.; Nielsen, S.B.; Otzen, D.E. The N-terminus of α-synuclein is essential for both monomeric and oligomeric interactions with membranes. FEBS Lett. 2014, 588, 497–502. [Google Scholar] [CrossRef] [PubMed]
- Middleton, E.R.; Rhoades, E. Effects of curvature and composition on α-synuclein binding to lipid vesicles. Biophys. J. 2010, 99, 2279–2288. [Google Scholar] [CrossRef] [PubMed]
- Bendor, J.T.; Logan, T.P.; Edwards, R.H. The function of α-synuclein. Neuron 2013, 79, 1044–1066. [Google Scholar] [CrossRef] [PubMed]
- Burré, J.; Sharma, M.; Tsetsenis, T.; Buchman, V.; Etherton, M.R.; Südhof, T.C. α-synuclein promotes SNARE-complex assembly in vivo and in vitro. Science 2010, 329, 1663–1667. [Google Scholar] [CrossRef] [PubMed]
- Burré, J.; Sharma, M.; Südhof, T.C. α-Synuclein assembles into higher-order multimers upon membrane binding to promote SNARE complex formation. Proc. Natl. Acad. Sci. USA 2014, 111, E4274–E4283. [Google Scholar] [CrossRef] [PubMed]
- Van Rooijen, B.D.; Claessens, M.M.A.E.; Subramaniam, V. Membrane permeabilization by oligomeric α-synuclein: In search of the mechanism. PLOS ONE 2010, 5, e14292. [Google Scholar] [CrossRef] [PubMed]
- Ouberai, M.M.; Wang, J.; Swann, M.J.; Galvagnion, C.; Guilliams, T.; Dobson, C.M.; Welland, M.E. α-Synuclein senses lipid packing defects and induces lateral expansion of lipids leading to membrane remodeling. J. Biol. Chem. 2013, 288, 20883–20895. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Liu, L.; Zhang, L.; Peng, Y.; Zhou, F. Redox reactions of the α-synuclein-Cu2+ complex and their effects on neuronal cell viability. Biochemistry 2011, 49, 323–343. [Google Scholar]
- Fellner, L.; Irschick, R.; Schanda, K.; Reindl, M.; Klimaschewski, L.; Poewe, W.; Wenning, G.K.; Stefanova, N. Toll-like receptor 4 is required for α-synuclein dependent activation of microglia and astroglia. Glia 2013, 61, 349–360. [Google Scholar] [CrossRef] [PubMed]
- Roodveldt, C.; Labrador-Garrido, A.; Gonzalez-Rey, E.; Fernandez-Montesinos, R.; Caro, M.; Lachaud, C.C.; Waudby, C.A.; Delgado, M.; Dobson, C.M.; Pozo, D. Glial innate immunity generated by non-aggregated alpha-synuclein in mouse: Differences between wild-type and Parkinson’s disease-linked mutants. PLOS ONE 2010, 5, e13481. [Google Scholar] [CrossRef] [PubMed]
- Souza, J.M.; Giasson, B.I.; Chen, Q.; Lee, V.M.; Ischiropoulos, H. Dityrosine cross-linking promotes formation of stable alpha-synuclein polymers. Implication of nitrative and oxidative stress in the pathogenesis of neurodegenerative synucleinopathies. J. Biol. Chem. 2000, 275, 18344–18349. [Google Scholar] [CrossRef] [PubMed]
- Krasnoslobodtsev, A.V.; Volkov, I.L.; Asiago, J.M.; Hindupur, J.; Rochet, J.-C.; Lyubchenko, Y.L. Alpha-synuclein misfolding assessed with single molecule AFM force spectroscopy: Effect of pathogenic mutations. Biochemistry 2013, 52, 1–22. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, K.; Nemani, V.M.; Azarbal, F.; Skibinski, G.; Levy, J.M.; Egami, K.; Munishkina, L.; Zhang, J.; Gardner, B.; Wakabayashi, J.; et al. Direct membrane association drives mitochondrial fission by the Parkinson disease-associated protein alpha-synuclein. J. Biol. Chem. 2011, 286, 20710–20726. [Google Scholar] [CrossRef] [PubMed]
- Bartels, T.; Choi, J.G.; Selkoe, D.J. α-synuclein occurs as a helically folded tetramer that resists aggregation. Nature 2012, 477, 107–110. [Google Scholar] [CrossRef]
- Dettmer, U.; Newman, A.J.; Luth, E.S.; Bartels, T.; Selkoe, D. In vivo cross-linking reveals principally oligomeric forms of α-synuclein and β-synuclein in neurons and non-neural cells. J. Biol. Chem. 2013, 288, 6371–6385. [Google Scholar] [CrossRef] [PubMed]
- Gould, N.; Mor, D.E.; Lightfoot, R.; Malkus, K.; Giasson, B.; Ischiropoulos, H. Evidence of native α-synuclein conformers in the human brain. J. Biol. Chem. 2014, 289, 7929–7934. [Google Scholar] [CrossRef] [PubMed]
- Kara, E.; Lewis, P.A.; Ling, H.; Proukakis, C.; Houlden, H.; Hardy, J. α-Synuclein mutations cluster around a putative protein loop. Neurosci. Lett. 2013, 546, 67–70. [Google Scholar] [CrossRef] [PubMed]
- Cremades, N.; Cohen, S.I.A.; Deas, E.; Abramov, A.Y.; Chen, A.Y.; Orte, A.; Sandal, M.; Clarke, R.W.; Dunne, P.; Aprile, F.A.; et al. Direct observation of the interconversion of normal and toxic forms of α-synuclein. Cell 2012, 149, 1048–1059. [Google Scholar] [CrossRef] [PubMed]
- Lorenzen, N.; Nielsen, S.B.; Buell, A.K.; Kaspersen, J.D.; Arosio, P.; Vad, B.S.; Paslawski, W.; Christiansen, G.; Valnickova-Hansen, Z.; Andreasen, M.; et al. The role of stable α-synuclein oligomers in the molecular events underlying amyloid formation. J. Am. Chem. Soc. 2014, 136, 3859–3868. [Google Scholar] [CrossRef] [PubMed]
- Lashuel, H.A.; Petre, B.M.; Wall, J.; Simon, M.; Nowak, R.J.; Walz, T.; Lansbury, P.T. α-Synuclein, especially the Parkinson’s disease-associated mutants, forms pore-like annular and tubular protofibrils. J. Mol. Biol. 2002, 322, 1089–1102. [Google Scholar] [CrossRef] [PubMed]
- Lindersson, E.; Beedholm, R.; Højrup, P.; Moos, T.; Gai, W.; Hendil, K.B.; Jensen, P.H. Proteasomal inhibition by alpha-synuclein filaments and oligomers. J. Biol. Chem. 2004, 279, 12924–12934. [Google Scholar] [CrossRef] [PubMed]
- Bolognesi, B.; Kumita, J.R.; Barros, T.P.; Esbjorner, E.K.; Luheshi, L.M.; Crowther, D.C.; Wilson, M.R.; Dobson, C.M.; Favrin, G.; Yerbury, J.J. ANS binding reveals common features of cytotoxic amyloid species. ACS Chem. Biol. 2010, 5, 735–740. [Google Scholar] [CrossRef] [PubMed]
- Paleologou, K.E.; Kragh, C.L.; Mann, D.M.A.; Salem, S.A.; Al-Shami, R.; Allsop, D.; Hassan, A.H.; Jensen, P.H.; El-Agnaf, O.M.A. Detection of elevated levels of soluble alpha-synuclein oligomers in post-mortem brain extracts from patients with dementia with Lewy bodies. Brain 2009, 132, 1093–1101. [Google Scholar] [CrossRef] [PubMed]
- Van Rooijen, B.D.; Claessens, M.M.A.E.; Subramaniam, V. Lipid bilayer disruption by oligomeric alpha-synuclein depends on bilayer charge and accessibility of the hydrophobic core. Biochim. Biophys. Acta 2009, 1788, 1271–1278. [Google Scholar] [CrossRef] [PubMed]
- Danzer, K.M.; Haasen, D.; Karow, A.R.; Moussaud, S.; Habeck, M.; Giese, A.; Kretzschmar, H.; Hengerer, B.; Kostka, M. Different species of α-synuclein oligomers induce calcium influx and seeding. J. Neurosci. 2007, 27, 9220–9232. [Google Scholar] [CrossRef] [PubMed]
- Kostka, M.; Högen, T.; Danzer, K.M.; Levin, J.; Habeck, M.; Wirth, A.; Wagner, R.; Glabe, C.G.; Finger, S.; Heinzelmann, U.; et al. Single particle characterization of iron-induced pore-forming alpha-synuclein oligomers. J. Biol. Chem. 2008, 283, 10992–11003. [Google Scholar] [CrossRef] [PubMed]
- Choi, B.-K.; Choi, M.-G.; Kim, J.-Y.; Yang, Y.; Lai, Y.; Kweon, D.-H.; Lee, N.K.; Shin, Y.-K. Large α-synuclein oligomers inhibit neuronal SNARE-mediated vesicle docking. Proc. Natl. Acad. Sci. USA 2013, 110, 4087–4092. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Griggs, A.; Rochet, J.-C.; Stanciu, L.A. In vitro study of α-synuclein protofibrils by cryo-EM suggests a Cu2+-dependent aggregation pathway. Biophys. J. 2013, 104, 2706–2713. [Google Scholar] [CrossRef] [PubMed]
- Pham, C.L.L.; Cappai, R. The interplay between lipids and dopamine on α-synuclein oligomerization and membrane binding. Biosci. Rep. 2013, 33, 807–814. [Google Scholar] [CrossRef]
- Wright, J.A.; Wang, X.; Brown, D.R. Unique copper-induced oligomers mediate alpha-synuclein toxicity. FASEB J. 2009, 23, 2384–2393. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Moualla, D.; Wright, J.A.; Brown, D.R. Copper binding regulates intracellular alpha-synuclein localisation, aggregation and toxicity. J. Neurochem. 2010, 113, 704–714. [Google Scholar] [CrossRef] [PubMed]
- Campioni, S.; Mannini, B.; Zampagni, M.; Pensalfini, A.; Parrini, C.; Evangelisti, E.; Relini, A.; Stefani, M.; Dobson, C.M.; Cecchi, C.; et al. A causative link between the structure of aberrant protein oligomers and their toxicity. Nat. Chem. Biol. 2010, 6, 140–147. [Google Scholar] [CrossRef] [PubMed]
- Zampagni, M.; Cascella, R.; Casamenti, F.; Grossi, C.; Evangelisti, E.; Wright, D.; Becatti, M.; Liguri, G.; Mannini, B.; Campioni, S.; et al. A comparison of the biochemical modifications caused by toxic and non-toxic protein oligomers in cells. J. Cell. Mol. Med. 2011, 15, 2106–2116. [Google Scholar] [CrossRef] [PubMed]
- Olzscha, H.; Schermann, S.M.; Woerner, A.C.; Pinkert, S.; Hecht, M.H.; Tartaglia, G.G.; Vendruscolo, M.; Hayer-Hartl, M.; Hartl, F.U.; Vabulas, R.M. Amyloid-like aggregates sequester numerous metastable proteins with essential cellular functions. Cell 2011, 144, 67–78. [Google Scholar] [CrossRef] [PubMed]
- Tsigelny, I.F.; Bar-On, P.; Sharikov, Y.; Crews, L.; Hashimoto, M.; Miller, M.A.; Keller, S.H.; Platoshyn, O.; Yuan, J.X.-J.; Masliah, E. Dynamics of alpha-synuclein aggregation and inhibition of pore-like oligomer development by beta-synuclein. FEBS J. 2007, 274, 1862–1877. [Google Scholar] [CrossRef] [PubMed]
- Tsigelny, I.F.; Sharikov, Y.; Wrasidlo, W.; Gonzalez, T.; Desplats, P.A.; Spencer, B.; Masliah, E. Role of alpha-synuclein penetration into the membrane in the mechanisms of oligomer pore formation. FEBS J. 2012, 279, 1000–1013. [Google Scholar] [CrossRef] [PubMed]
- Butterfield, S.M.; Lashuel, H.A. Amyloidogenic protein-membrane interactions: Mechanistic insight from model systems. Angew. Chem. Int. Ed. Engl. 2010, 49, 5628–5654. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, F.; Levin, J.; Kamp, F.; Kretzschmar, H.; Giese, A.; Bötzel, K. Single-channel electrophysiology reveals a distinct and uniform pore complex formed by α-synuclein oligomers in lipid membranes. PLOS ONE 2012, 7, e42545. [Google Scholar] [CrossRef] [PubMed]
- Yoshiike, Y.; Kayed, R.; Milton, S.C.; Takashima, A.; Glabe, C.G. Pore-forming proteins share structural and functional homology with amyloid oligomers. Neuromol. Med. 2007, 9, 270–275. [Google Scholar] [CrossRef]
- Kayed, R.; Sokolov, Y.; Edmonds, B.; McIntire, T.M.; Milton, S.C.; Hall, J.E.; Glabe, C.G. Permeabilization of lipid bilayers is a common conformation-dependent activity of soluble amyloid oligomers in protein misfolding diseases. J. Biol. Chem. 2004, 279, 46363–46366. [Google Scholar] [CrossRef] [PubMed]
- Sokolov, Y.; Kozak, J.A.; Kayed, R.; Chanturiya, A.; Glabe, C.; Hall, J.E. Soluble amyloid oligomers increase bilayer conductance by altering dielectric structure. J. Gen. Physiol. 2006, 128, 637–647. [Google Scholar] [CrossRef] [PubMed]
- Capone, R.; Quiroz, F.G.; Prangkio, P.; Saluja, I.; Anna, M.; Bautista, M.R.; Turner, R.S.; Yang, J.; Mayer, M. Amyloid-β-induced ion flux in artificial lipid bilayers and neuronal cells: Resolving a controversy. Neurotox. Res. 2009, 16, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Zijlstra, N.; Subramaniam, V. α-Synuclein oligomers: An amyloid pore? Insights into mechanisms of α-synuclein oligomer-lipid interactions. Mol. Neurobiol. 2013, 47, 613–621. [Google Scholar] [CrossRef] [PubMed]
- Stöckl, M.; Claessens, M.M.A.E.; Subramaniam, V. Kinetic measurements give new insights into lipid membrane permeabilization by α-synuclein oligomers. Mol. Biosyst. 2012, 8, 338–345. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, K. α-Synuclein and mitochondria: Partners in crime? Neurotherapeutics 2013, 10, 391–399. [Google Scholar] [CrossRef] [PubMed]
- Devi, L.; Raghavendran, V.; Prabhu, B.M.; Avadhani, N.G.; Anandatheerthavarada, H.K. Mitochondrial import and accumulation of α-synuclein impair complex I in human dopaminergic neuronal cultures and Parkinson disease brain. J. Biol. Chem. 2008, 283, 9089–9100. [Google Scholar] [CrossRef] [PubMed]
- Sarafian, T.A.; Ryan, C.M.; Souda, P.; Masliah, E.; Kar, U.K.; Vinters, H.V.; Mathern, G.W.; Faull, K.F.; Whitelegge, J.P.; Watson, J.B. Impairment of mitochondria in adult mouse brain overexpressing predominantly full-length, N-terminally acetylated human α-synuclein. PLOS ONE 2013, 8, e63557. [Google Scholar] [CrossRef] [PubMed]
- Luth, E.S.; Stavrovskaya, I.G.; Bartels, T.; Kristal, B.S.; Selkoe, D.J. Soluble, prefibrillar α-synuclein oligomers promote complex I-dependent, Ca2+-induced mitochondrial dysfunction. J. Biol. Chem. 2014, 289, 21490–21507. [Google Scholar] [CrossRef] [PubMed]
- Esteves, A.R.; Gozes, I.; Cardoso, S.M. The rescue of microtubule-dependent traffic recovers mitochondrial function in Parkinson’s disease. Biochim. Biophys. Acta 2014, 1842, 7–21. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Wang, T.; Pei, Z.; Miller, D.S.; Wu, X.; Block, M.L.; Wilson, B.; Zhang, W.; Zhou, Y.; Hong, J.-S.; et al. Aggregated alpha-synuclein activates microglia: A process leading to disease progression in Parkinson’s disease. FASEB J. 2005, 19, 533–542. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Jin, J.; Davis, J.; Zhou, Y.; Wang, Y.; Liu, J.; Lockhart, P.J.; Zhang, J. Oligomeric alpha-synuclein inhibits tubulin polymerization. Biochem. Biophys. Res. Commun. 2007, 356, 548–553. [Google Scholar] [CrossRef] [PubMed]
- Prots, I.; Veber, V.; Brey, S.; Campioni, S.; Buder, K.; Riek, R.; Böhm, K.J.; Winner, B. α-Synuclein oligomers impair neuronal microtubule-kinesin interplay. J. Biol. Chem. 2013, 288, 21742–21754. [Google Scholar] [CrossRef] [PubMed]
- Junn, E.; Mouradian, M.M. Human alpha-synuclein over-expression increases intracellular reactive oxygen species levels and susceptibility to dopamine. Neurosci. Lett. 2002, 320, 146–150. [Google Scholar] [CrossRef] [PubMed]
- Choi, D.-H.; Cristóvão, A.C.; Guhathakurta, S.; Lee, J.; Joh, T.H.; Beal, M.F.; Kim, Y.-S. NADPH oxidase 1-mediated oxidative stress leads to dopamine neuron death in Parkinson’s disease. Antioxid. Redox Signal. 2012, 16, 1033–1045. [Google Scholar] [CrossRef] [PubMed]
- Béraud, D.; Hathaway, H.A.; Trecki, J.; Chasovskikh, S.; Johnson, D.A.; Johnson, J.A.; Federoff, H.J.; Shimoji, M.; Mhyre, T.R.; Maguire-Zeiss, K.A. Microglial activation and antioxidant responses induced by the Parkinson’s disease protein α-synuclein. J. Neuroimmune Pharmacol. 2013, 8, 94–117. [Google Scholar] [CrossRef] [PubMed]
- Stefanova, N.; Fellner, L.; Reindl, M.; Masliah, E.; Poewe, W.; Wenning, G.K. Toll-like receptor 4 promotes α-synuclein clearance and survival of nigral dopaminergic neurons. Am. J. Pathol. 2011, 179, 954–963. [Google Scholar] [CrossRef] [PubMed]
- Kim, C.; Ho, D.-H.; Suk, J.-E.; You, S.; Michael, S.; Kang, J.; Joong Lee, S.; Masliah, E.; Hwang, D.; Lee, H.-J.; et al. Neuron-released oligomeric α-synuclein is an endogenous agonist of TLR2 for paracrine activation of microglia. Nat. Commun. 2013. [Google Scholar] [CrossRef]
- Hoozemans, J.J.M.; van Haastert, E.S.; Nijholt, D.A.T.; Rozemuller, A.J.M.; Scheper, W. Activation of the unfolded protein response is an early event in Alzheimer’s and Parkinson’s disease. Neurodegener. Dis. 2012, 10, 212–215. [Google Scholar] [CrossRef] [PubMed]
- Colla, E.; Jensen, P.H.; Pletnikova, O.; Troncoso, J.C.; Glabe, C.; Lee, M.K. Accumulation of toxic α-synuclein oligomer within endoplasmic reticulum occurs in α-synucleinopathy in vivo. J. Neurosci. 2012, 32, 3301–3305. [Google Scholar] [CrossRef] [PubMed]
- Colla, E.; Coune, P.; Liu, Y.; Pletnikova, O.; Troncoso, J.C.; Schneider, B.L.; Lee, M.K. Endoplasmic reticulum stress is important for the manifestations of α-synucleinopathy in vivo. J. Neurosci. 2012, 32, 3306–3320. [Google Scholar] [CrossRef] [PubMed]
- Su, L.J.; Auluck, P.K.; Outeiro, T.F.; Yeger-Lotem, E.; Kritzer, J.A.; Tardiff, D.F.; Strathearn, K.E.; Liu, F.; Cao, S.; Hamamichi, S.; et al. Compounds from an unbiased chemical screen reverse both ER-to-Golgi trafficking defects and mitochondrial dysfunction in Parkinson’s disease models. Dis. Model. Mech. 2010, 3, 194–208. [Google Scholar] [CrossRef] [PubMed]
- Castillo-Carranza, D.L.; Zhang, Y.; Guerrero-Muñoz, M.J.; Kayed, R.; Rincon-Limas, D.E.; Fernandez-Funez, P. Differential activation of the ER stress factor XBP1 by oligomeric assemblies. Neurochem. Res. 2012, 37, 1707–1717. [Google Scholar] [CrossRef] [PubMed]
- Thayanidhi, N.; Helm, J.R.; Nycz, D.C.; Bentley, M.; Liang, Y.; Hay, J.C. α-Synuclein delays endoplasmic reticulum (ER)-to-Golgi transport in mammalian cells by antagonizing ER/Golgi SNAREs. Mol. Biol. Cell 2010, 21, 1850–1863. [Google Scholar] [CrossRef] [PubMed]
- Cooper, A.A.; Gitler, A.D.; Cashikar, A.; Haynes, C.M.; Kathryn, J.; Bhullar, B.; Liu, K.; Xu, K.; Strathearn, K.E.; Cao, S.; et al. α-Synuclein blocks ER-Golgi traffic and Rab1 rescues neuron loss in Parkinson’s models. Science 2006, 313, 324–328. [Google Scholar] [CrossRef] [PubMed]
- Xilouri, M.; Brekk, O.R.; Stefanis, L. α-Synuclein and protein degradation systems: A reciprocal relationship. Mol. Neurobiol. 2013, 47, 537–551. [Google Scholar] [CrossRef] [PubMed]
- Emmanouilidou, E.; Stefanis, L.; Vekrellis, K. Cell-produced α-synuclein oligomers are targeted to, and impair, the 26S proteasome. Neurobiol. Aging 2010, 31, 953–968. [Google Scholar] [CrossRef] [PubMed]
- Snyder, H.; Mensah, K.; Theisler, C.; Lee, J.; Matouschek, A.; Wolozin, B. Aggregated and monomeric alpha-synuclein bind to the S6’ proteasomal protein and inhibit proteasomal function. J. Biol. Chem. 2003, 278, 11753–11759. [Google Scholar] [CrossRef] [PubMed]
- Xilouri, M.; Vogiatzi, T.; Vekrellis, K.; Park, D.; Stefanis, L. Abberant alpha-synuclein confers toxicity to neurons in part through inhibition of chaperone-mediated autophagy. PLOS ONE 2009, 4, e5515. [Google Scholar] [CrossRef] [PubMed]
- Cuervo, A.M.; Stefanis, L.; Fredenburg, R.; Lansbury, P.T.; Sulzer, D. Impaired degradation of mutant alpha-synuclein by chaperone-mediated autophagy. Science 2004, 305, 1292–1295. [Google Scholar] [CrossRef] [PubMed]
- Martinez-vicente, M.; Talloczy, Z.; Kaushik, S.; Massey, A.C.; Mazzulli, J.; Mosharov, E.V.; Hodara, R.; Fredenburg, R.; Wu, D.; Follenzi, A.; et al. Dopamine-modified α-synuclein blocks chaperone-mediated autophagy. J. Clin. Investig. 2008, 118, 777–788. [Google Scholar] [PubMed]
- Celej, M.S.; Sarroukh, R.; Goormaghtigh, E.; Fidelio, G.D.; Ruysschaert, J.-M.; Raussens, V. Toxic prefibrillar α-synuclein amyloid oligomers adopt a distinctive antiparallel β-sheet structure. Biochem. J. 2012, 443, 719–726. [Google Scholar] [CrossRef] [PubMed]
- Pieri, L.; Madiona, K.; Bousset, L.; Melki, R. Fibrillar α-synuclein and huntingtin exon 1 assemblies are toxic to the cells. Biophys. J. 2012, 102, 2894–2905. [Google Scholar] [CrossRef] [PubMed]
- Desplats, P.; Lee, H.; Bae, E.; Patrick, C.; Rockenstein, E.; Crews, L.; Spencer, B.; Masliah, E.; Lee, S. Inclusion formation and neuronal cell death through neuron-to-neuron transmission of α-synuclein. PNAS 2009, 106, 13010–13015. [Google Scholar] [CrossRef] [PubMed]
- Hansen, C.; Angot, E.; Bergström, A.; Steiner, J.A.; Pieri, L.; Paul, G.; Outeiro, T.F.; Melki, R.; Kallunki, P.; Fog, K. α-Synuclein propagates from mouse brain to grafted dopaminergic neurons and seeds aggregation in cultured human cells. J. Clin. Investig. 2011, 121, 715–725. [Google Scholar] [CrossRef] [PubMed]
- Volpicelli-Daley, L.A.; Luk, K.C.; Patel, T.P.; Tanik, S.A.; Dawn, M.; Stieber, A.; Meany, D.F.; Trojanowski, J.Q.; Lee, V.M. Exogenous α-synuclein fibrils induce Lewy Body pathology leading to synaptic dysfunction and neuron death. Neuron 2012, 72, 57–71. [Google Scholar] [CrossRef]
- Tanik, S.A.; Schultheiss, C.E.; Volpicelli-Daley, L.A.; Brunden, K.R.; Lee, V.M.Y. Lewy body-like α-synuclein aggregates resist degradation and impair macroautophagy. J. Biol. Chem. 2013, 288, 15194–15210. [Google Scholar] [CrossRef] [PubMed]
- Jan, A.; Adolfsson, O.; Allaman, I.; Buccarello, A.-L.; Magistretti, P.J.; Pfeifer, A.; Muhs, A.; Lashuel, H.A. Abeta42 neurotoxicity is mediated by ongoing nucleated polymerization process rather than by discrete Abeta42 species. J. Biol. Chem. 2011, 286, 8585–8596. [Google Scholar] [CrossRef] [PubMed]
- Wan, O.W.; Chung, K.K.K. The role of alpha-synuclein oligomerization and aggregation in cellular and animal models of Parkinson’s disease. PLOS ONE 2012, 7, e38545. [Google Scholar] [CrossRef] [PubMed]
- Cherny, R.A.; Culvenor, J.G.; Bottomley, S.P.; Masters, C.L. Dopamine promotes α-synuclein aggregation into SDS-resistant soluble oligomers via a distinct folding pathway. FASEB J. 2005, 19, 1377–1379. [Google Scholar] [PubMed]
- Sparr, E.; Engel, M.F.M.; Sakharov, D.V.; Sprong, M.; Jacobs, J.; de Kruijff, B.; Höppener, J.W.M.; Killian, J.A. Islet amyloid polypeptide-induced membrane leakage involves uptake of lipids by forming amyloid fibers. FEBS Lett. 2004, 577, 117–120. [Google Scholar] [CrossRef] [PubMed]
- Gai, W.; Yuan, H.; Li, X.; Power, J.; Blumbergs, P.; Jensen, P. In situ and in vitro study of colocalization and segregation of alpha-synuclein, ubiquitin, and lipids in Lewy bodies. Exp. Neurol. 2000, 166, 324–333. [Google Scholar] [CrossRef] [PubMed]
- Reynolds, N.P.; Soragni, A.; Rabe, M.; Verdes, D.; Liverani, E.; Handschin, S.; Riek, R.; Seeger, S. Mechanism of membrane interaction and disruption by α-synuclein. J. Am. Chem. Soc. 2011, 133, 19366–19375. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.-H.; Hong, C.-S.; Lee, S.; Yang, J.-E.; Park, Y.I.; Lee, D.; Hyeon, T.; Jung, S.; Paik, S.R. Radiating amyloid fibril formation on the surface of lipid membranes through unit-assembly of oligomeric species of α-synuclein. PLOS ONE 2012, 7, e47580. [Google Scholar] [CrossRef] [PubMed]
- Chaudhary, H.; Stefanovic, A.N.D.; Subramaniam, V.; Claessens, M.M.A.E. Membrane interactions and fibrillization of α-synuclein play an essential role in membrane disruption. FEBS Lett. 2014, 588, 4457–4463. [Google Scholar] [CrossRef] [PubMed]
- Plotegher, N.; Greggio, E.; Bisaglia, M.; Bubacco, L. Biophysical groundwork as a hinge to unravel the biology of α-synuclein aggregation and toxicity. Q. Rev. Biophys. 2014, 47, 1–48. [Google Scholar] [CrossRef] [PubMed]
- Emmanouilidou, E.; Melachroinou, K.; Roumeliotis, T.; Garbis, S.D.; Ntzouni, M.; Margaritis, L.H.; Stefanis, L.; Vekrellis, K. Cell-produced alpha-synuclein is secreted in a calcium-dependent manner by exosomes and impacts neuronal survival. J. Neurosci. 2010, 30, 6838–6851. [Google Scholar] [CrossRef] [PubMed]
- Winner, B.; Jappelli, R.; Maji, S.K.; Desplats, P.A.; Boyer, L.; Aigner, S. In vivo demonstration that α-synuclein oligomers are toxic. PNAS 2011, 108, 4194–4199. [Google Scholar] [CrossRef] [PubMed]
- Rockenstein, E.; Nuber, S.; Overk, C.R.; Ubhi, K.; Mante, M.; Patrick, C.; Adame, A.; Trejo-Morales, M.; Gerez, J.; Picotti, P.; et al. Accumulation of oligomer-prone α-synuclein exacerbates synaptic and neuronal degeneration in vivo. Brain 2014, 137, 1496–1513. [Google Scholar] [CrossRef] [PubMed]
- Karpinar, D.P.; Balija, M.B.G.; Kügler, S.; Opazo, F.; Rezaei-Ghaleh, N.; Wender, N.; Kim, H.-Y.; Taschenberger, G.; Falkenburger, B.H.; Heise, H.; et al. Pre-fibrillar alpha-synuclein variants with impaired beta-structure increase neurotoxicity in Parkinson’s disease models. EMBO J. 2009, 28, 3256–3268. [Google Scholar] [CrossRef] [PubMed]
- Taschenberger, G.; Garrido, M.; Tereshchenko, Y.; Bähr, M.; Zweckstetter, M.; Kügler, S. Aggregation of αSynuclein promotes progressive in vivo neurotoxicity in adult rat dopaminergic neurons. Acta Neuropathol. 2012, 123, 671–683. [Google Scholar] [CrossRef] [PubMed]
- Ardah, M.T.; Paleologou, K.E.; Lv, G.; Abul Khair, S.B.; Kazim, A.S.; Minhas, S.T.; Al-Tel, T.H.; Al-Hayani, A.A.; Haque, M.E.; Eliezer, D.; et al. Structure activity relationship of phenolic acid inhibitors of α-synuclein fibril formation and toxicity. Front. Aging Neurosci. 2014. [Google Scholar] [CrossRef]
- Bieschke, J.; Russ, J.; Friedrich, R.P.; Ehrnhoefer, D.E.; Wobst, H.; Neugebauer, K.; Wanker, E.E. EGCG remodels mature alpha-synuclein and amyloid-beta fibrils and reduces cellular toxicity. Proc. Natl. Acad. Sci. USA 2010, 107, 7710–7715. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Gillardon, F.; Hengerer, B.; Berlinicke, C. Baicalein reduces E46K α-synuclein aggregation in vitro and protects cells against E46K α-synuclein toxicity in cell models of familiar Parkinsonism. J. Neurochem. 2011, 114, 419–429. [Google Scholar]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Roberts, H.L.; Brown, D.R. Seeking a Mechanism for the Toxicity of Oligomeric α-Synuclein. Biomolecules 2015, 5, 282-305. https://doi.org/10.3390/biom5020282
Roberts HL, Brown DR. Seeking a Mechanism for the Toxicity of Oligomeric α-Synuclein. Biomolecules. 2015; 5(2):282-305. https://doi.org/10.3390/biom5020282
Chicago/Turabian StyleRoberts, Hazel L., and David R. Brown. 2015. "Seeking a Mechanism for the Toxicity of Oligomeric α-Synuclein" Biomolecules 5, no. 2: 282-305. https://doi.org/10.3390/biom5020282
APA StyleRoberts, H. L., & Brown, D. R. (2015). Seeking a Mechanism for the Toxicity of Oligomeric α-Synuclein. Biomolecules, 5(2), 282-305. https://doi.org/10.3390/biom5020282