
α-Synuclein-Induced Synapse Damage in Cultured Neurons Is Mediated by Cholesterol-Sensitive Activation of Cytoplasmic Phospholipase A2

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
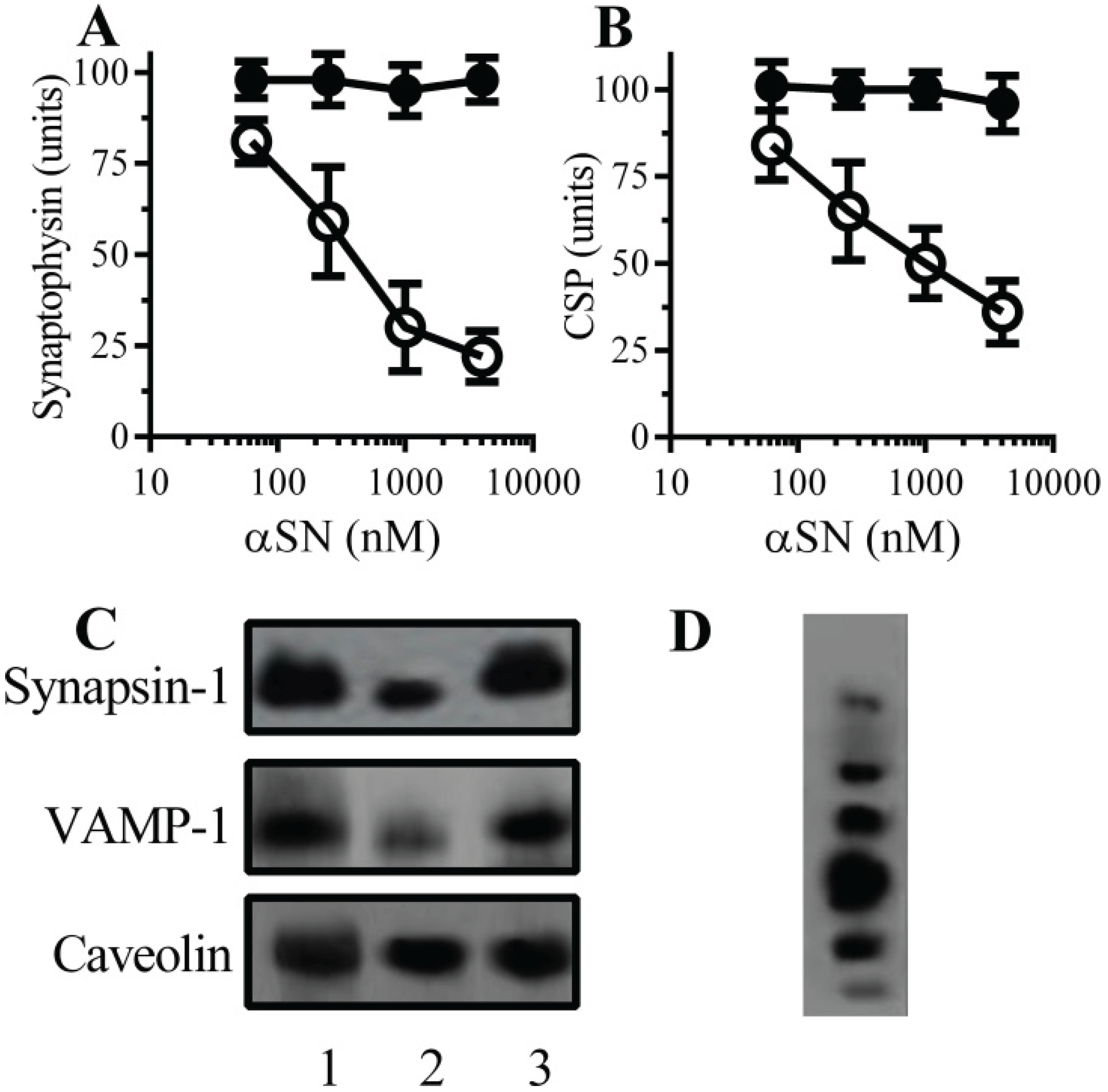
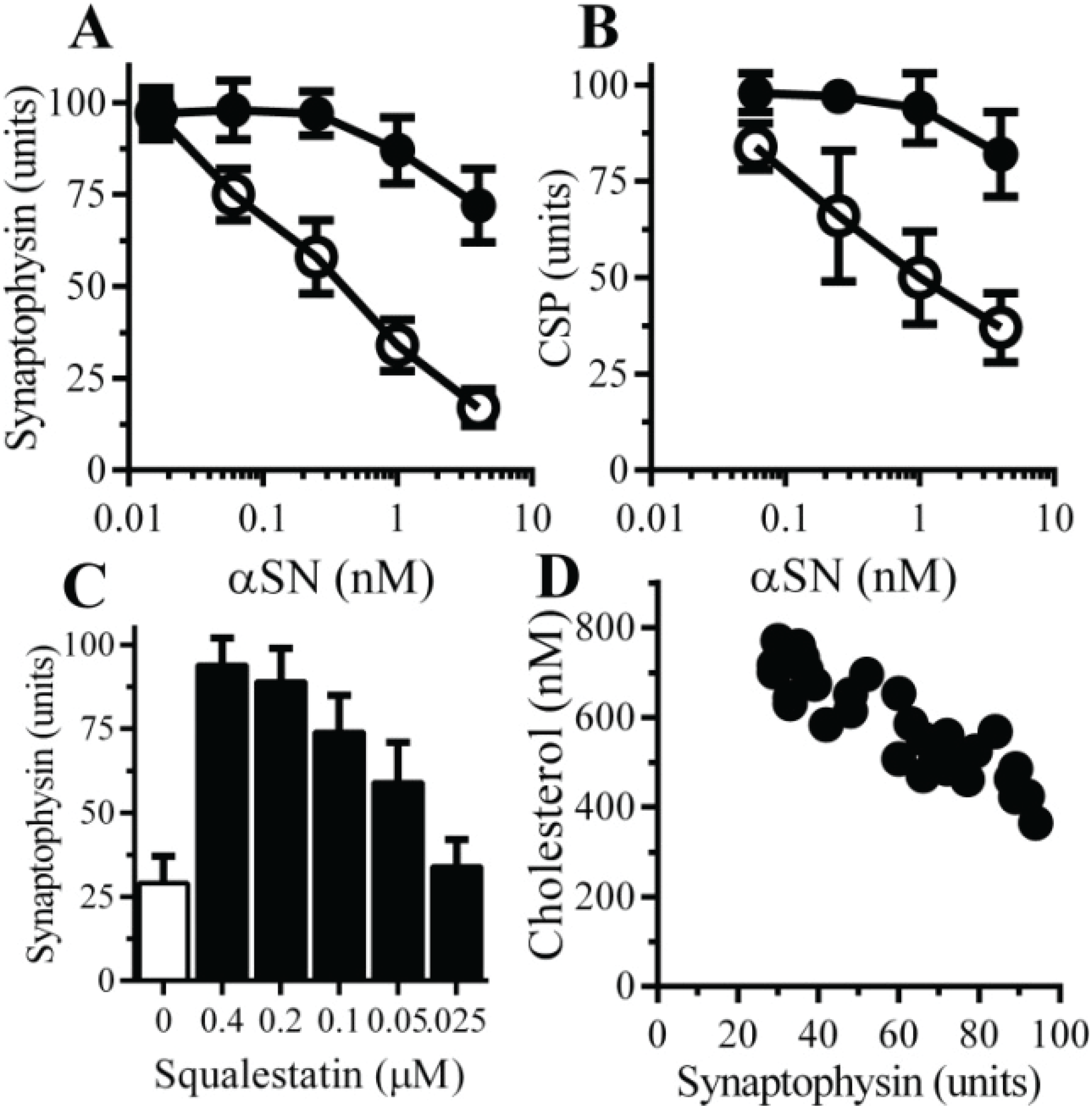
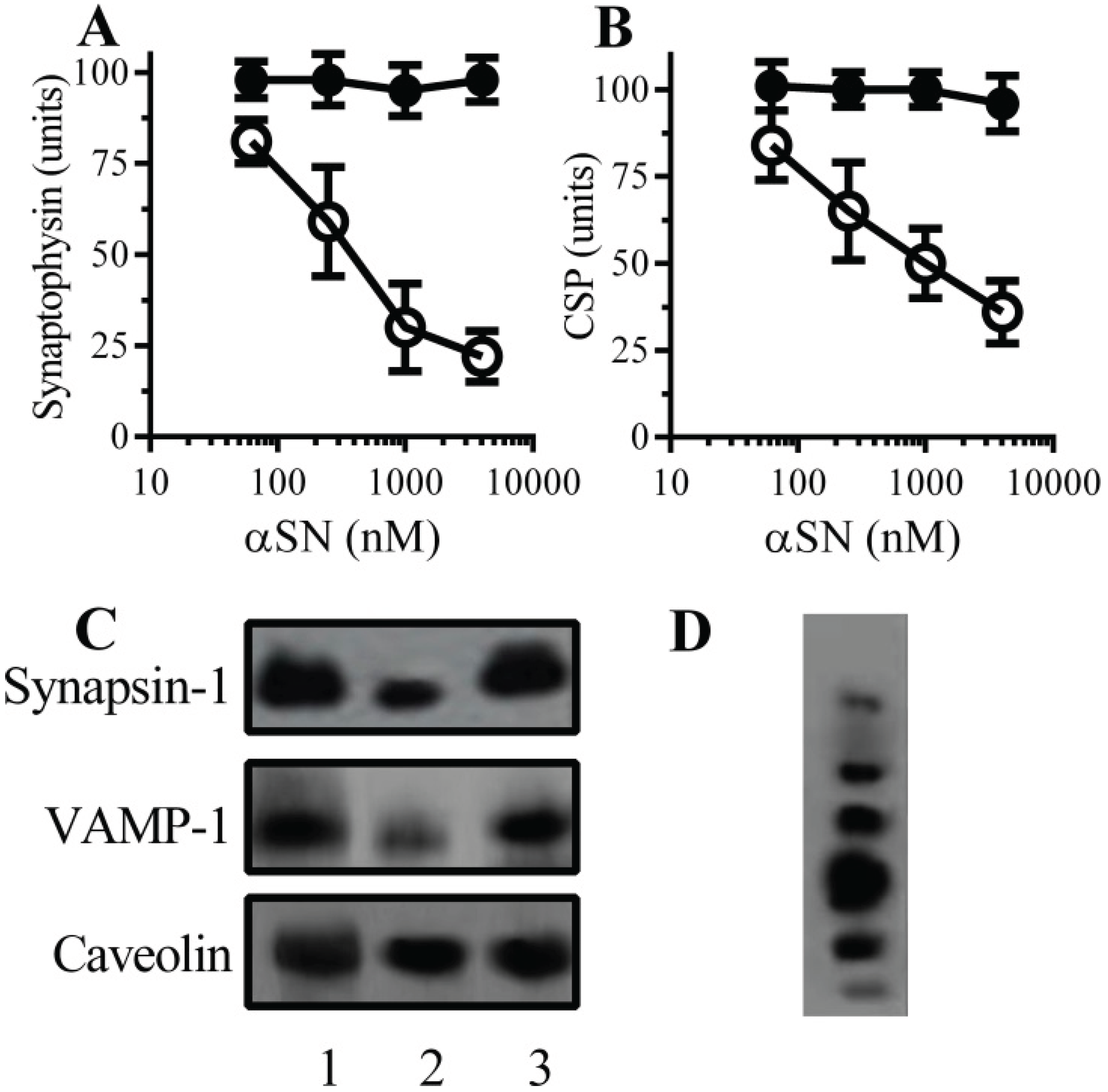
2.1. αSN Triggered the Loss of Synaptic Proteins from Cultured Neurons
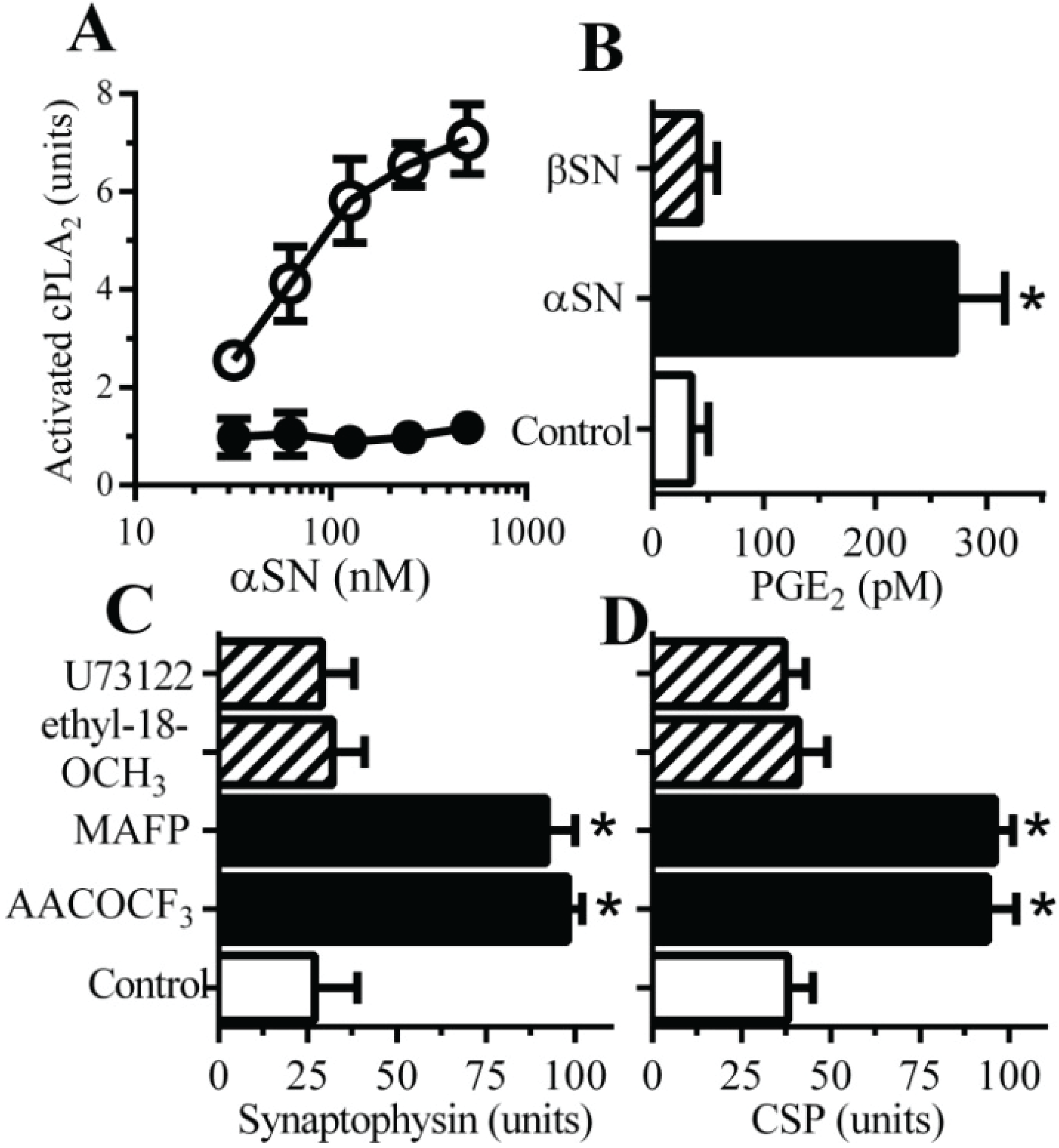
2.2. PLA2 Inhibitors Protect Neurons against αSN-Induced Synapse Damage
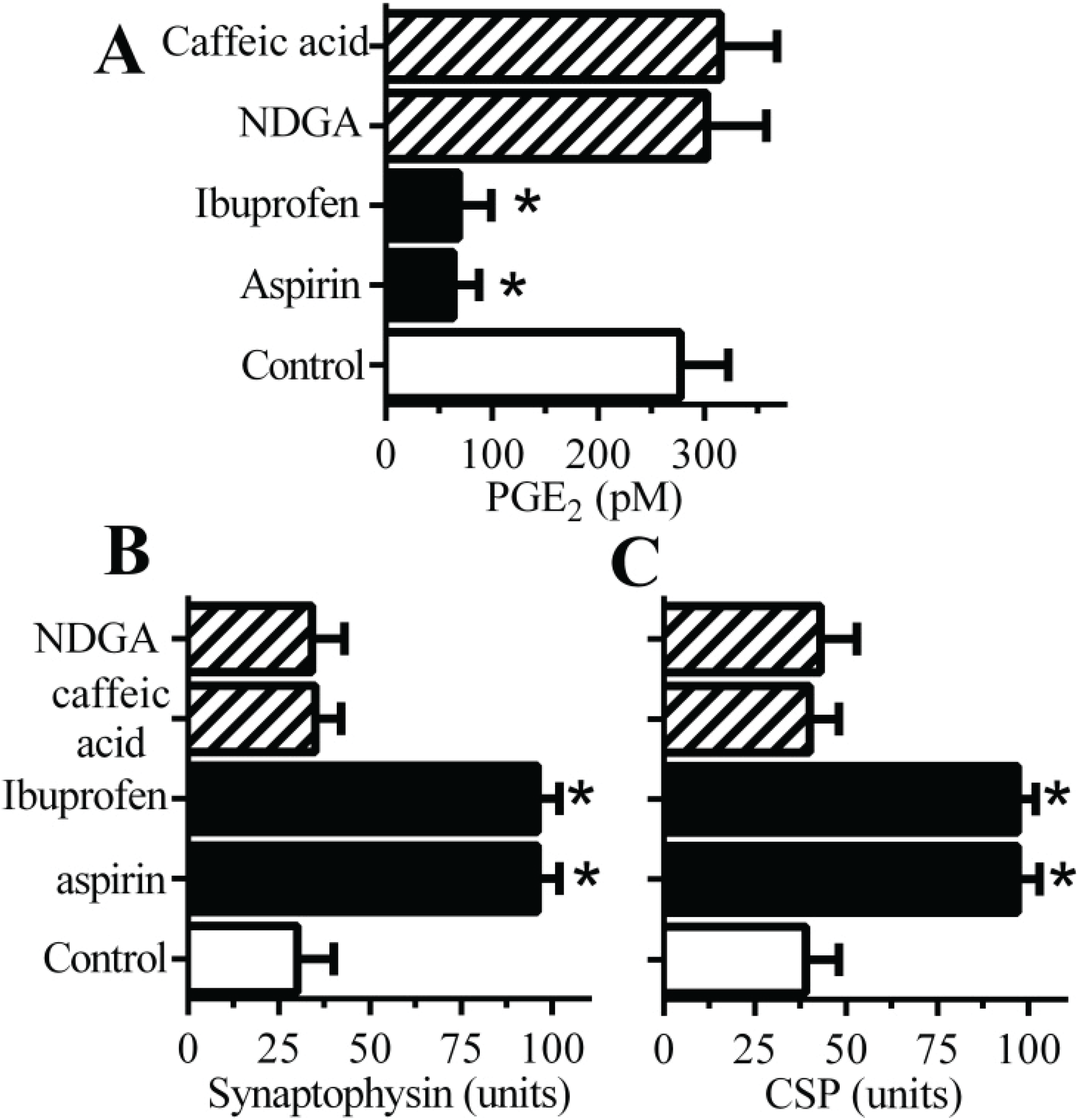
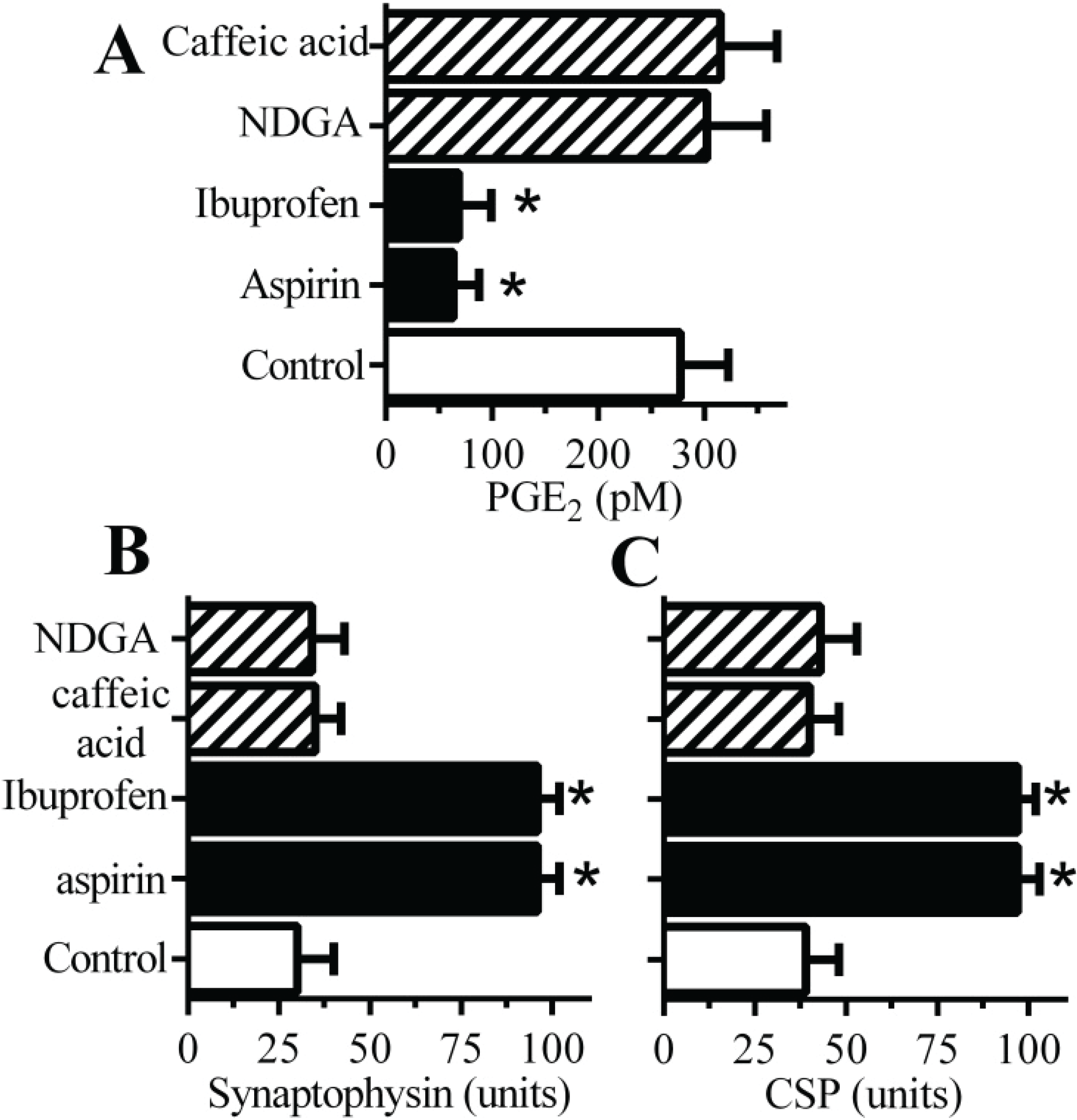
2.3. Cyclooxygenase Inhibitors Protect Neurons against αSN-Induced Synapse Damage
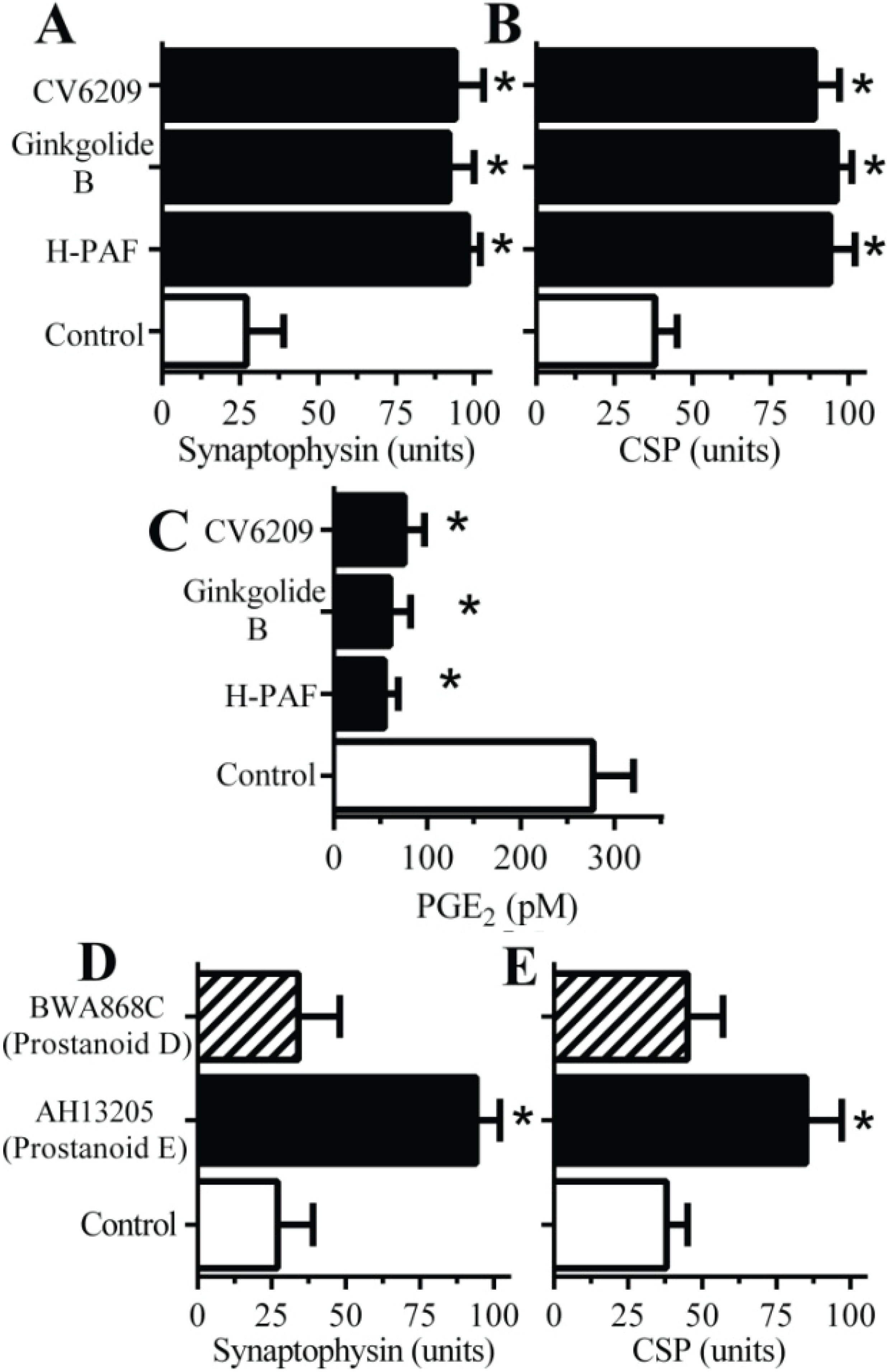
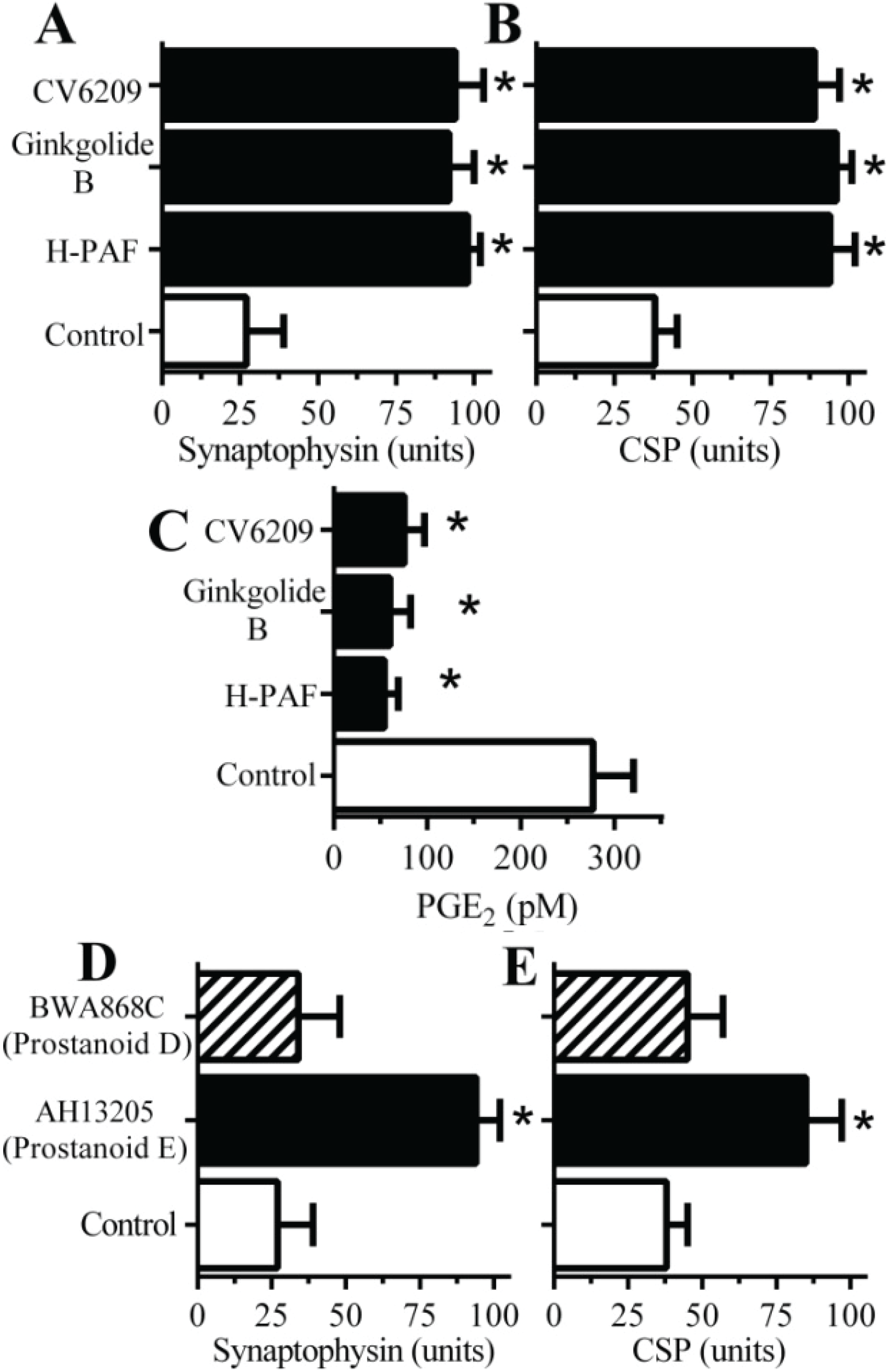
2.4. PAF Antagonists Protected against αSN-Induced Synapse Degeneration
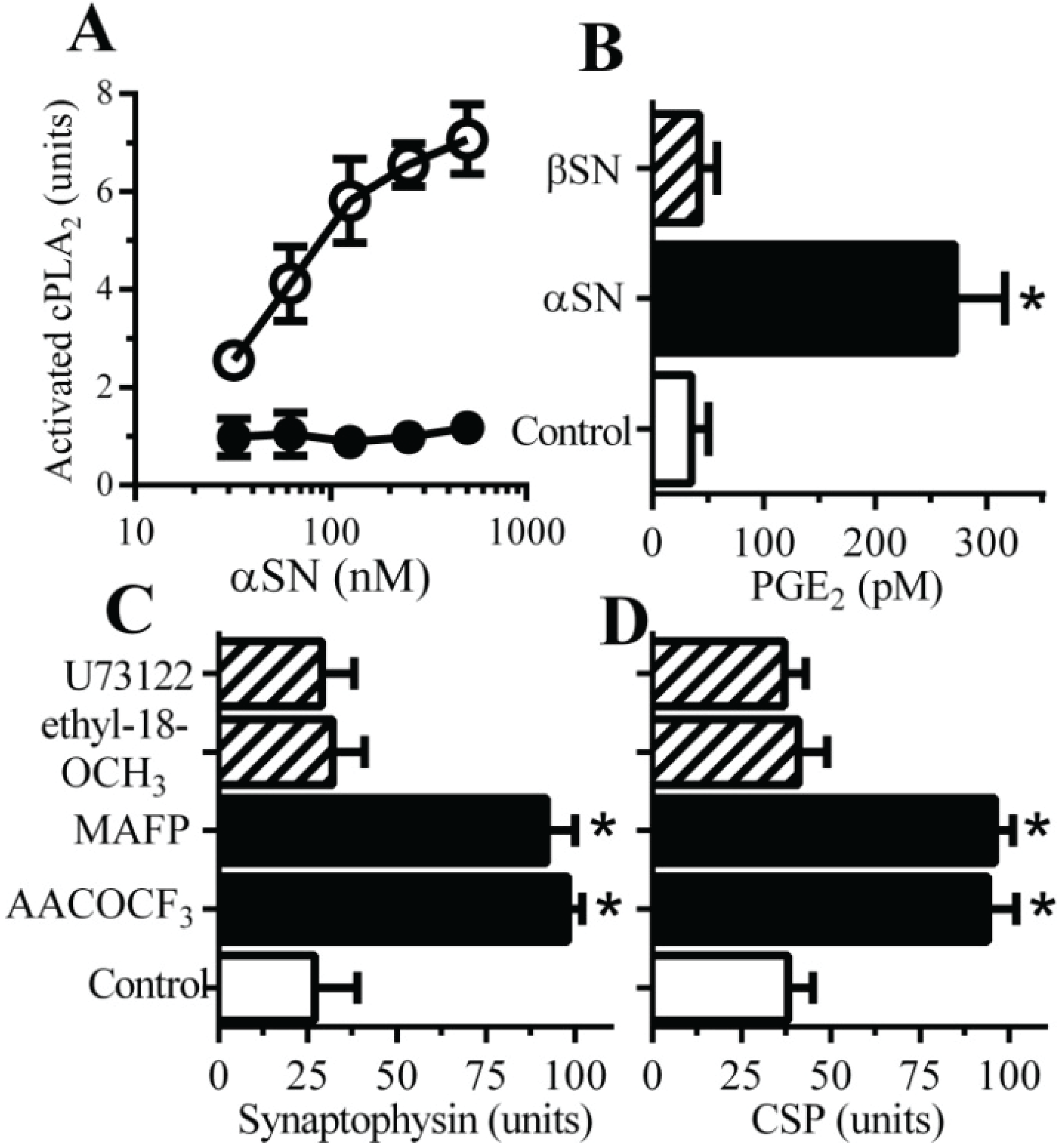
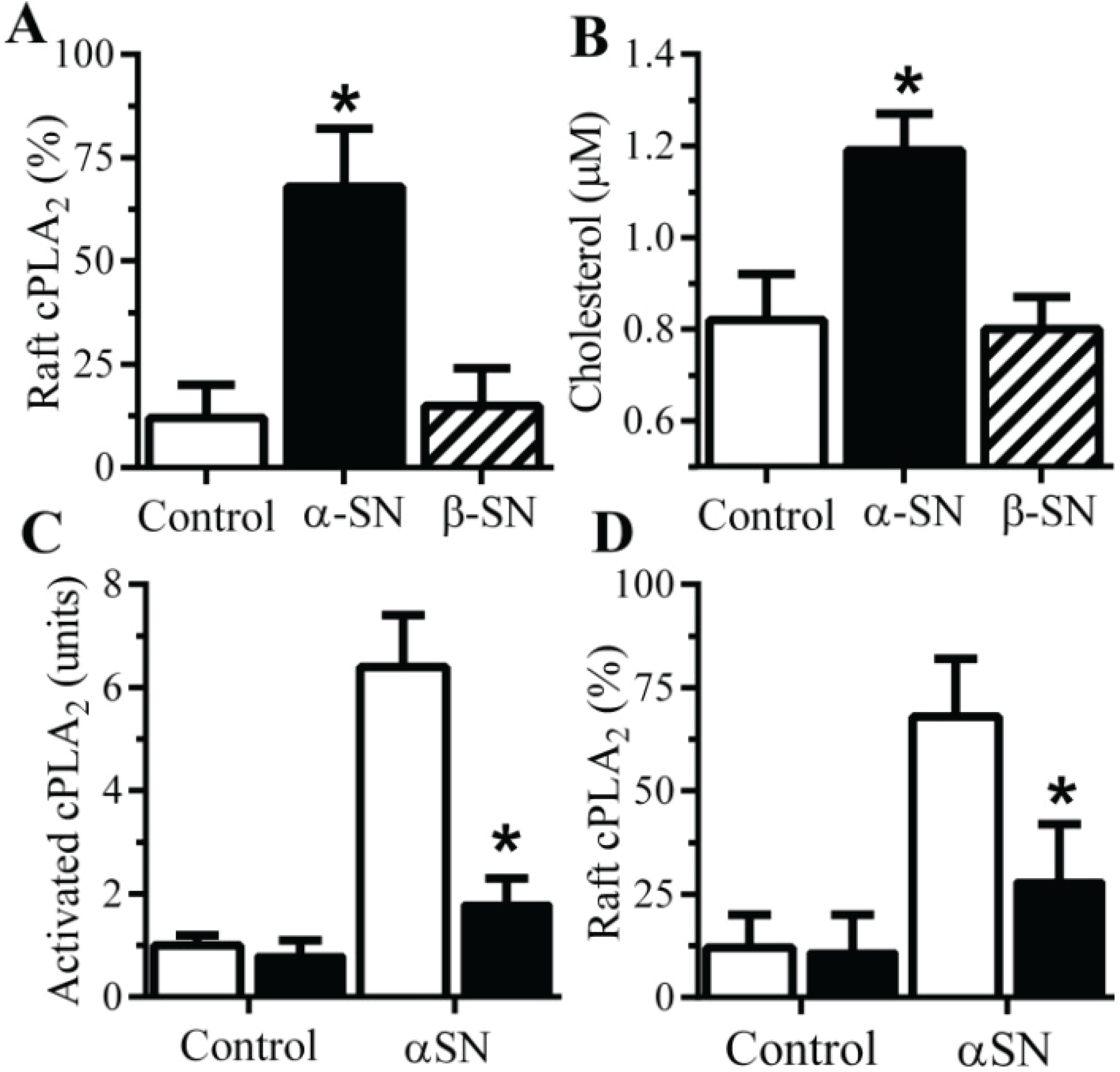
2.5. αSN-Induced Activation of cPLA2 Is Cholesterol Sensitive
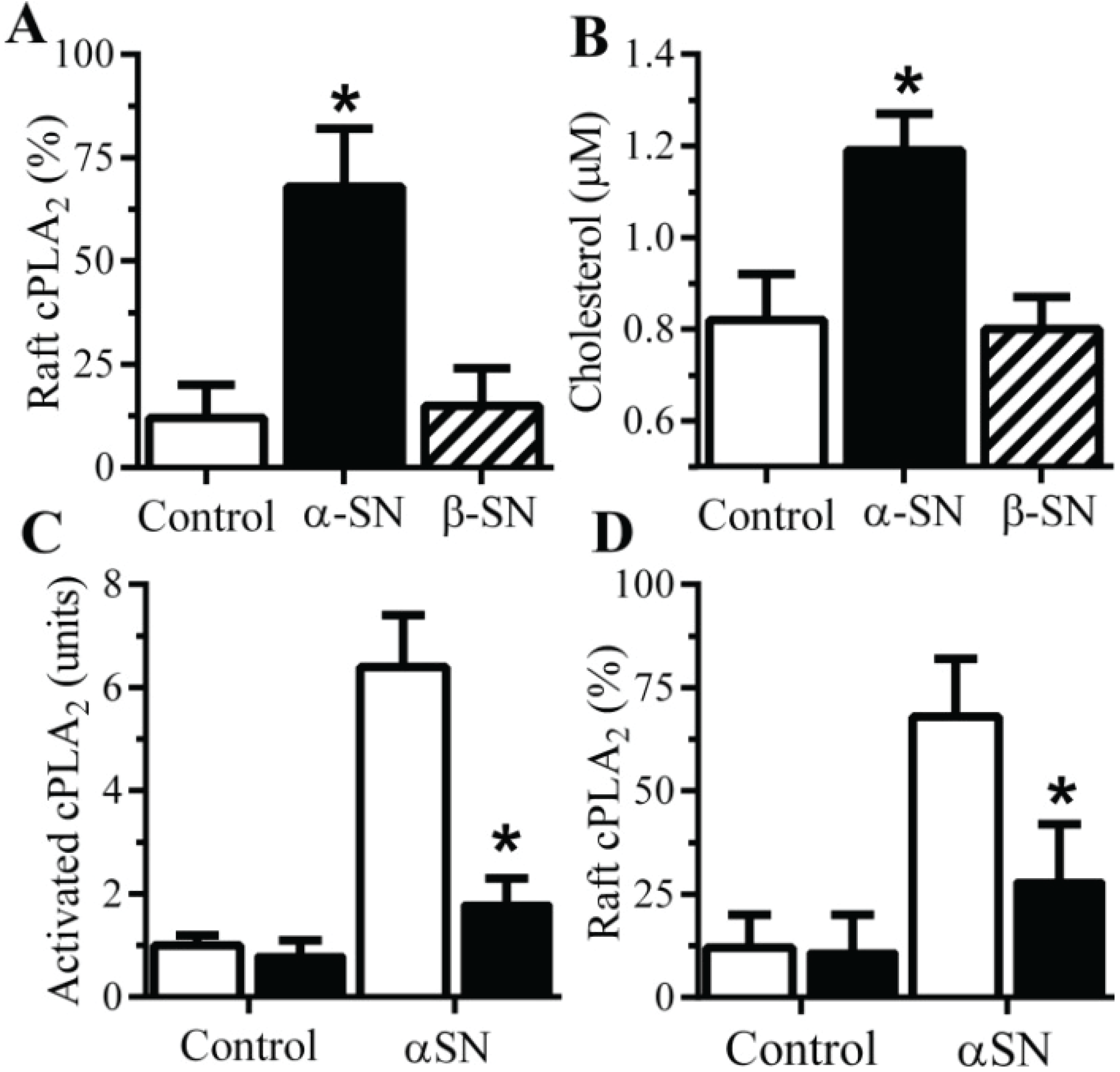
2.6. The αSN-Induced Synapse Damage Is Cholesterol Sensitive
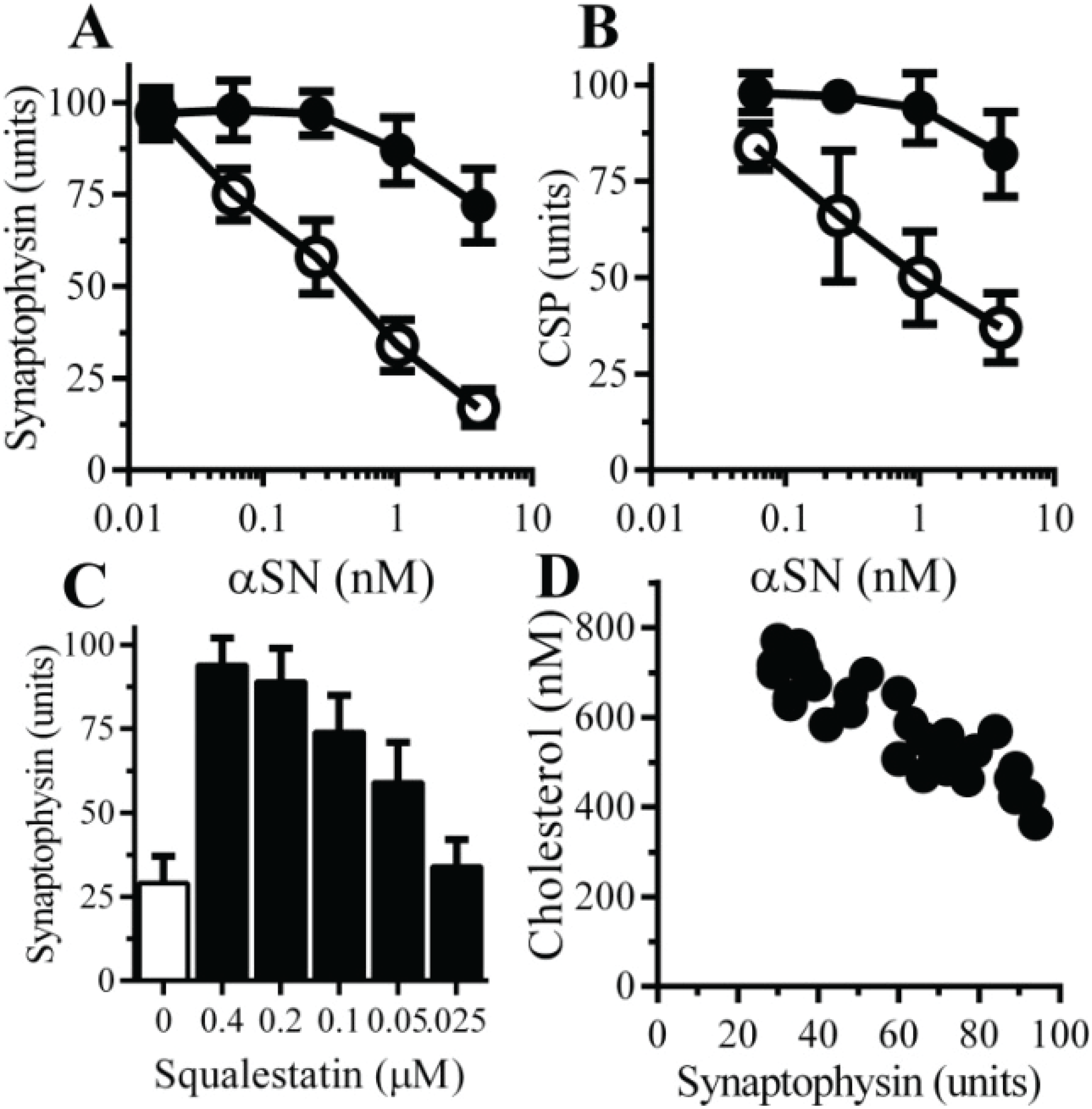
3. Discussion
4. Experimental Section
5. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Poewe, W. Non-motor symptoms in Parkinson’s disease. Eur. J. Neurol. 2008, 15, 14–20. [Google Scholar] [CrossRef] [PubMed]
- Aarsland, D.; Andersen, K.; Larsen, J.P.; Lolk, A.; Kragh-Sorensen, P. Prevalence and characteristics of dementia in Parkinson disease: An 8-year prospective study. Arch. Neurol. 2003, 60, 387–392. [Google Scholar] [CrossRef] [PubMed]
- McKeith, I.G.; Dickson, D.W.; Lowe, J.; Emre, M.; O’Brien, J.T.; Feldman, H.; Cummings, J.; Duda, J.E.; Lippa, C.; Perry, E.K.; et al. Diagnosis and management of dementia with Lewy bodies: Third report of the DLB Consortium. Neurology 2005, 65, 1863–1872. [Google Scholar] [CrossRef]
- Bonini, N.M.; Giasson, B.I. Snaring the function of alpha-synuclein. Cell 2005, 123, 359–361. [Google Scholar] [CrossRef] [PubMed]
- Cabin, D.E.; Shimazu, K.; Murphy, D.; Cole, N.B.; Gottschalk, W.; McIlwain, K.L.; Orrison, B.; Chen, A.; Ellis, C.E.; Paylor, R.; et al. Synaptic vesicle depletion correlates with attenuated synaptic responses to prolonged repetitive stimulation in mice lacking alpha-synuclein. J. Neurosci. 2002, 22, 8797–8807. [Google Scholar] [PubMed]
- Clayton, D.F.; George, J.M. The synucleins: A family of proteins involved in synaptic function, plasticity, neurodegeneration and disease. Trends Neurosci. 1998, 21, 249–254. [Google Scholar] [CrossRef] [PubMed]
- Kazantsev, A.G.; Kolchinsky, A.M. Central role of alpha-synuclein oligomers in neurodegeneration in Parkinson disease. Arch. Neurol. 2008, 65, 1577–1581. [Google Scholar] [CrossRef] [PubMed]
- Kramer, M.L.; Schulz-Schaeffer, W.J. Presynaptic α-synuclein aggregates, not Lewy bodies, cause neurodegeneration in dementia with Lewy bodies. J. Neurosci. 2007, 27, 1405–1410. [Google Scholar] [CrossRef] [PubMed]
- Lee, V.M.Y.; Trojanowski, J.Q. Mechanisms of Parkinson’s disease linked to pathological α-synuclein: New targets for drug discovery. Neuron 2006, 52, 33–38. [Google Scholar] [CrossRef] [PubMed]
- Desplats, P.; Lee, H.J.; Bae, E.J.; Patrick, C.; Rockenstein, E.; Crews, L.; Spencer, B.; Masliah, E.; Lee, S.J. Inclusion formation and neuronal cell death through neuron-to-neuron transmission of α-synuclein. Proc. Natl. Acad. Sci. USA 2009, 106, 13010–13015. [Google Scholar] [CrossRef] [PubMed]
- Brundin, P.; Melki, R.; Kopito, R. Prion-like transmission of protein aggregates in neurodegenerative diseases. Nat. Rev. Mol. Cell Biol. 2010, 11, 301–307. [Google Scholar] [CrossRef]
- Braak, H.; del Tredici, K.; Rub, U.; de Vos, R.A.; Jansen Steur, E.N.; Braak, E. Staging of brain pathology related to sporadic Parkinson’s disease. Neurobiol. Aging 2003, 24, 197–211. [Google Scholar] [CrossRef] [PubMed]
- Galvin, J.E.; Uryu, K.; Lee, V.M.; Trojanowski, J.Q. Axon pathology in Parkinson’s disease and Lewy body dementia hippocampus contains alpha-, beta-, and gamma-synuclein. Proc. Natl. Acad. Sci. USA 1999, 96, 13450–13455. [Google Scholar] [CrossRef] [PubMed]
- Chung, C.Y.; Koprich, J.B.; Siddiqi, H.; Isacson, O. Dynamic changes in presynaptic and axonal transport proteins combined with striatal neuroinflammation precede dopaminergic neuronal loss in a rat model of AAV alpha-synucleinopathy. J. Neurosci. 2009, 29, 3365–3373. [Google Scholar] [CrossRef] [PubMed]
- Bate, C.; Gentleman, S.; Williams, A. α-synuclein induced synapse damage is enhanced by amyloid-b1–42. Mol. Neurodegener. 2010, 5. [Google Scholar] [CrossRef]
- Bate, C.; Tayebi, M.; Williams, A. Phospholipase A2 inhibitors protect against prion and Ab mediated synapse degeneration. Mol. Neurodegener. 2010. [Google Scholar] [CrossRef]
- Sze, C.I.; Troncoso, J.C.; Kawas, C.; Mouton, P.; Price, D.L.; Martin, L.J. Loss of the presynaptic vesicle protein synaptophysin in hippocampus correlates with cognitive decline in Alzheimer disease. J. Neuropath Exp. Neurol. 1997, 56, 933–944. [Google Scholar] [CrossRef] [PubMed]
- Terry, R.D.; Masliah, E.; Salmon, D.P.; Butters, N.; deTeresa, R.; Hill, R.; Hansen, L.A.; Katzman, R. Physical basis of cognitive alterations in Alzheimer’s disease: Synapse loss is the major correlate of cognitive impairment. Ann. Neurol. 1991, 30, 572–580. [Google Scholar] [CrossRef] [PubMed]
- Bate, C.; Williams, A. Amyloid-β-induced synapse damage is mediated via cross-linkage of the cellular prion protein. J. Biol. Chem. 2011, 286, 37955–37963. [Google Scholar] [CrossRef] [PubMed]
- Francescangeli, E.; Domanska-Janik, K.; Goracci, G. Relative contribution of the de novo and remodelling pathways to the synthesis of platelet-activating factor in brain areas and during ischemia. J. Lipid Mediat. Cell Signal. 1996, 14, 89–98. [Google Scholar] [CrossRef] [PubMed]
- Bate, C.; Tayebi, M.; Williams, A. Ginkgolides protect against amyloid-β1–42-mediated synapse damage in vitro. Mol. Neurodegener. 2008. [Google Scholar] [CrossRef]
- Teixeira, M.M.; al Rashed, S.; Rossi, A.G.; Hellewell, P.G. Characterization of the prostanoid receptors mediating inhibition of PAF-induced aggregation of guinea-pig eosinophils. Br. J. Pharmacol. 1997, 121, 77–82. [Google Scholar] [CrossRef] [PubMed]
- Sugimoto, Y.; Narumiya, S. Prostaglandin E receptors. J. Biol. Chem. 2007, 282, 11613–11617. [Google Scholar] [CrossRef] [PubMed]
- Nalefski, E.A.; Sultzman, L.A.; Martin, D.M.; Kriz, R.W.; Towler, P.S.; Knopf, J.L.; Clark, J.D. Delineation of two functionally distinct domains of cytosolic phospholipase A2, a regulatory Ca2+-dependent lipid-binding domain and a Ca2+-independent catalytic domain. J. Biol. Chem. 1994, 269, 18239–18249. [Google Scholar] [PubMed]
- Crick, D.C.; Suders, J.; Kluthe, C.M.; Andres, D.A.; Waechter, C.J. Selective inhibition of cholesterol biosynthesis in brain cells by squalestatin 1. J. Neurochem. 1995, 65, 1365–1373. [Google Scholar] [CrossRef] [PubMed]
- Bate, C.; Williams, A. Squalestatin protects neurons and reduces the activation of cytoplasmic phospholipase A2 by Aβ1–42. Neuropharmacology 2007, 53, 222–231. [Google Scholar] [CrossRef] [PubMed]
- Farooqui, A.A.; Horrocks, L.A. Phospholipase A2-generated lipid mediators in the brain: The good, the bad, and the ugly. Neuroscientist 2006, 12, 245–260. [Google Scholar] [CrossRef] [PubMed]
- Farooqui, A.A.; Ong, W.Y.; Horrocks, L.A. Inhibitors of brain phospholipase A2 activity: Their neuropharmacological effects and therapeutic importance for the treatment of neurologic disorders. Phamacol. Rev. 2006, 58, 591–620. [Google Scholar]
- Malaplate-Armand, C.; Florent-Bechard, S.; Youssef, I.; Koziel, V.; Sponne, I.; Kriem, B.; Leininger-Muller, B.; Olivier, J.L.; Oster, T.; Pillot, T. Soluble oligomers of amyloid-β peptide induce neuronal apoptosis by activating a cPLA2-dependent sphingomyelinase-ceramide pathway. Neurobiol. Dis. 2006, 23, 178–189. [Google Scholar] [CrossRef] [PubMed]
- Kriem, B.; Sponne, I.; Fifre, A.; Malaplate-Armand, C.; Lozac’h-Pillot, K.; Koziel, V.; Yen-Potin, F.T.; Bihain, B.; Oster, T.; Olivier, J.L.; et al. Cytosolic phospholipase A2 mediates neuronal apoptosis induced by soluble oligomers of the amyloid-β peptide. FASEB J. 2005, 19, 85–87. [Google Scholar]
- Klivenyi, P.; Beal, M.F.; Ferrante, R.J.; Andreassen, O.A.; Wermer, M.; Chin, M.R.; Bonventre, J.V. Mice deficient in group IV cytosolic phospholipase A2 are resistant to MPTP neurotoxicity. J. Neurochem. 1998, 71, 2634–2637. [Google Scholar] [CrossRef] [PubMed]
- Sanchez-Mejia, R.O.; Newman, J.W.; Toh, S.; Yu, G.Q.; Zhou, Y.; Halabisky, B.; Cisse, M.; Scearce-Levie, K.; Cheng, I.H.; Gan, L.; et al. Phospholipase A2 reduction ameliorates cognitive deficits in a mouse model of Alzheimer’s disease. Nat. Neurosci. 2008, 11, 1311–1318. [Google Scholar] [CrossRef] [PubMed]
- Sun, G.Y.; Xu, J.; Jensen, M.D.; Simonyi, A. Phospholipase A2 in the central nervous system: Implications for neurodegenerative diseases. J. Lipid Res. 2004, 45, 205–213. [Google Scholar] [CrossRef] [PubMed]
- Bellizzi, M.J.; Lu, S.M.; Masliah, E.; Gelbard, H.A. Synaptic activity becomes excitotoxic in neurons exposed to elevated levels of platelet-activating factor. J. Clin. Investig. 2005, 115, 3185–3192. [Google Scholar] [CrossRef] [PubMed]
- Rojas, P.; Serrano-García, N.; Mares-Sámano, J.J.; Medina-Campos, O.N.; Pedraza-Chaverri, J.; Ögren, S.O. EGb761 protects against nigrostriatal dopaminergic neurotoxicity in 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine-induced Parkinsonism in mice: Role of oxidative stress. Eur. J. Neurosci. 2008, 28, 41–50. [Google Scholar] [CrossRef]
- Rojas, P.; Montes, P.; Rojas, C.; Serrano-García, N.; Rojas-Castañeda, J.C. Effect of a phytopharmaceutical medicine, Ginko biloba extract 761, in an animal model of Parkinson’s disease: Therapeutic perspectives. Nutrition 2012, 28, 1081–1088. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; Bazan, N.G. Endogenous PGE2 regulates membrane excitability and synaptic transmission in hippocampal CA1 pyramidal neurons. J. Neurphysiol. 2005, 93, 929–941. [Google Scholar] [CrossRef]
- Maxfield, F.R.; Tabas, I. Role of cholesterol and lipid organization in disease. Nature 2005, 438, 612–621. [Google Scholar] [CrossRef] [PubMed]
- Dolga, A.M.; Culmsee, C.; de Lau, L.; Winter, Y.; Oertel, W.H.; Luiten, P.G.M.; Eisel, U.L.M. Statins—Increasing or reducing the risk of Parkinson’s disease? Exp. Neurol. 2011, 228, 1–4. [Google Scholar] [CrossRef] [PubMed]
- Hu, G.; Antikainen, R.; Jousilahti, P.; Kivipelto, M.; Tuomilehto, J. Total cholesterol and the risk of Parkinson disease. Neurology 2008, 70, 1972–1979. [Google Scholar] [CrossRef] [PubMed]
- Koob, A.O.; Ubhi, K.; Paulsson, J.F.; Kelly, J.; Rockenstein, E.; Mante, M.; Adame, A.; Masliah, E. Lovastatin ameliorates α-synuclein accumulation and oxidation in transgenic mouse models of α-synucleinopathies. Exp. Neurol. 2010, 221, 267–274. [Google Scholar] [CrossRef] [PubMed]
- Wahner, A.D.; Bronstein, J.M.; Bordelon, Y.M.; Ritz, B. Statin use and the risk of Parkinson disease. Neurology 2008, 70, 1418–1422. [Google Scholar] [CrossRef] [PubMed]
- Wolozin, B.; Kellman, W.; Ruosseau, P.; Celesia, G.G.; Siegel, G. Decreased prevalence of alzheimer disease associated with 3-hydroxy-3-methyglutaryl coenzyme A reductase inhibitors. Arch. Neurol. 2000, 57, 1439–1443. [Google Scholar] [CrossRef] [PubMed]
- Samii, A.; Carleton, B.C.; Etminan, M. Statin use and the risk of Parkinson disease: A nested case control study. J. Clin. Neurosci. 2008, 15, 1272–1273. [Google Scholar] [CrossRef] [PubMed]
- Becker, C.; Meier, C.R. Statins and the risk of Parkinson disease: An update on the controversy. Expert Opin. Drug Saf. 2009, 8, 261–271. [Google Scholar] [CrossRef] [PubMed]
- Baxter, A.; Fitzgerald, B.J.; Hutson, J.L.; McCarthy, A.D.; Motteram, J.M.; Ross, B.C.; Sapra, M.; Snowden, M.A.; Watson, N.S.; Williams, R.J. Squalestatin 1, a potent inhibitor of squalene synthase, which lowers serum cholesterol in vivo. J. Biol. Chem. 1992, 267, 11705–11708. [Google Scholar] [PubMed]
- Pike, L.J. Lipid rafts: Heterogeneity on the high seas. Biochem. J. 2004, 378, 281–292. [Google Scholar] [CrossRef] [PubMed]
- Simons, K.; Toomre, D. Lipid rafts and signal transduction. Nat. Rev. Mol. Cell Biol. 2000, 1, 31–39. [Google Scholar] [CrossRef] [PubMed]
- Graziani, A.; Bricko, V.; Carmignani, M.; Graier, W.F.; Groschner, K. Cholesterol- and caveolin-rich membrane domains are essential for phospholipase A2-dependent EDHF formation. Cardiovasc. Res. 2004, 64, 234–242. [Google Scholar] [CrossRef] [PubMed]
- Bate, C.; Tayebi, M.; Williams, A. Sequestration of free cholesterol in cell membranes by prions correlates with cytoplasmic phospholipase A2 activation. BMC Biol. 2008. [Google Scholar] [CrossRef]
- Thais, M.E.; Carqueja, C.L.; Santos, T.G.; Silva, R.V.; Stroeh, E.; Machado, R.S.; Wahlheim, D.O.; Bianchin, M.M.; Sakamoto, A.C.; Brentani, R.R.; et al. Synaptosomal glutamate release and uptake in mice lacking the cellular prion protein. Brain Res. 2006, 1075, 13–19. [Google Scholar] [CrossRef] [PubMed]
- London, E.; Brown, D.A. Insolubility of lipids in Triton X-100: Physical origin and relationship to sphingolipid/cholesterol membrane domains (rafts). Biochim. Biophys. Acta 2000, 1508, 182–195. [Google Scholar] [CrossRef] [PubMed]
- Bate, C.; Williams, A. Monoacylated cellular prion protein modifies cell membranes, inhibits cell signaling and reduces prion formation. J. Biol. Chem. 2011, 286, 8752–8758. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bate, C.; Williams, A. α-Synuclein-Induced Synapse Damage in Cultured Neurons Is Mediated by Cholesterol-Sensitive Activation of Cytoplasmic Phospholipase A2. Biomolecules 2015, 5, 178-193. https://doi.org/10.3390/biom5010178
Bate C, Williams A. α-Synuclein-Induced Synapse Damage in Cultured Neurons Is Mediated by Cholesterol-Sensitive Activation of Cytoplasmic Phospholipase A2. Biomolecules. 2015; 5(1):178-193. https://doi.org/10.3390/biom5010178
Chicago/Turabian StyleBate, Clive, and Alun Williams. 2015. "α-Synuclein-Induced Synapse Damage in Cultured Neurons Is Mediated by Cholesterol-Sensitive Activation of Cytoplasmic Phospholipase A2" Biomolecules 5, no. 1: 178-193. https://doi.org/10.3390/biom5010178
APA StyleBate, C., & Williams, A. (2015). α-Synuclein-Induced Synapse Damage in Cultured Neurons Is Mediated by Cholesterol-Sensitive Activation of Cytoplasmic Phospholipase A2. Biomolecules, 5(1), 178-193. https://doi.org/10.3390/biom5010178