Effect of Metals on Kinetic Pathways of Amyloid-β Aggregation

{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
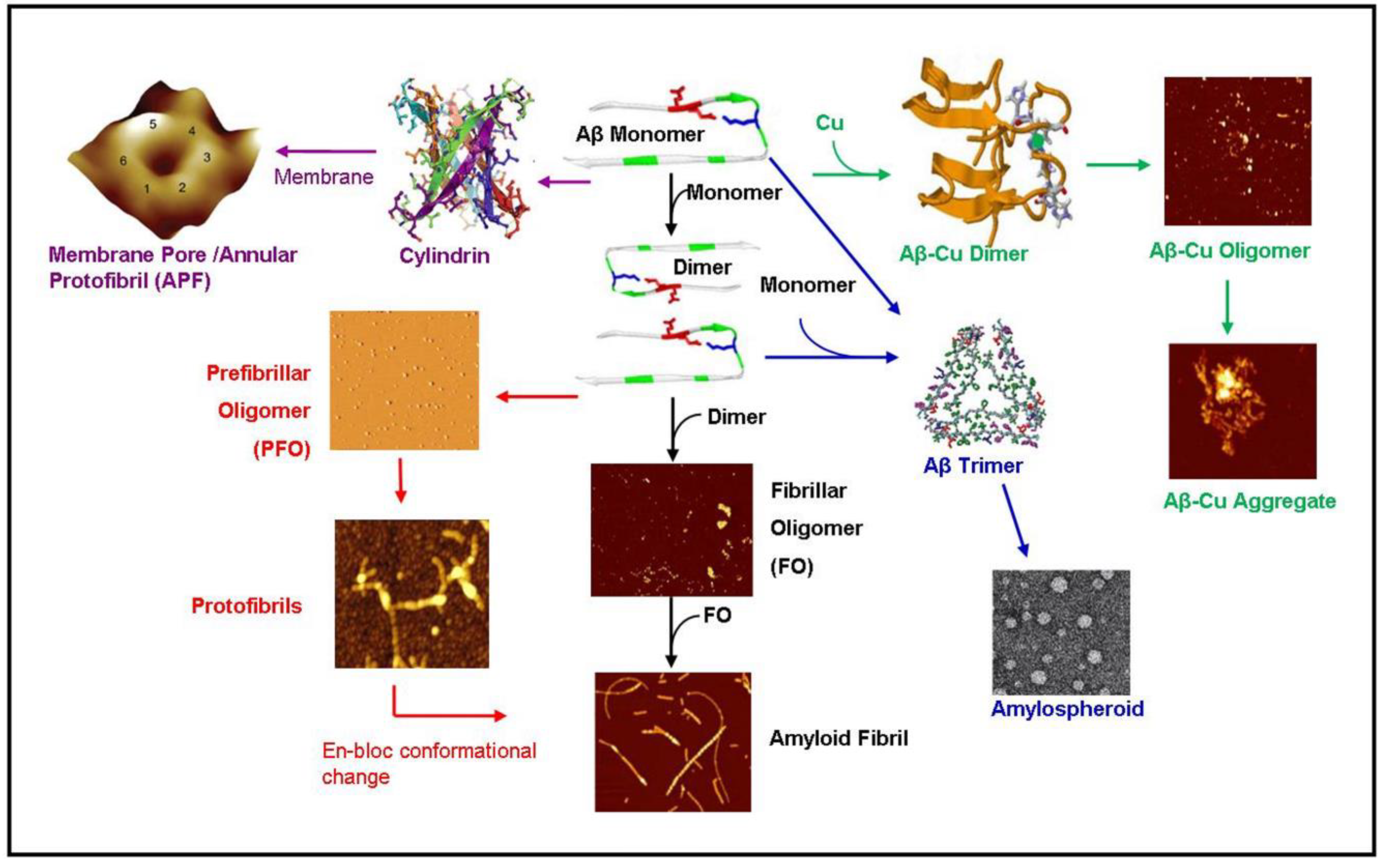
2. Fibrillar and Oligomeric Structures
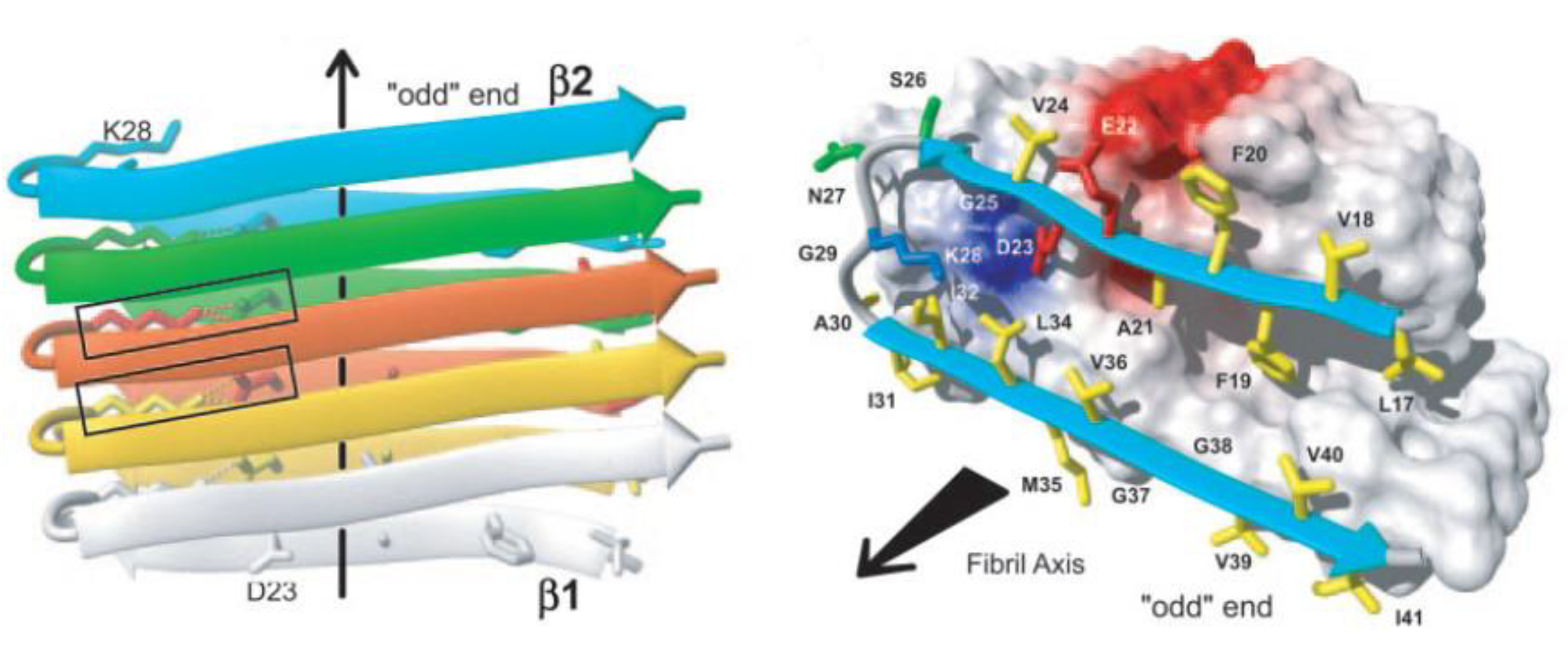
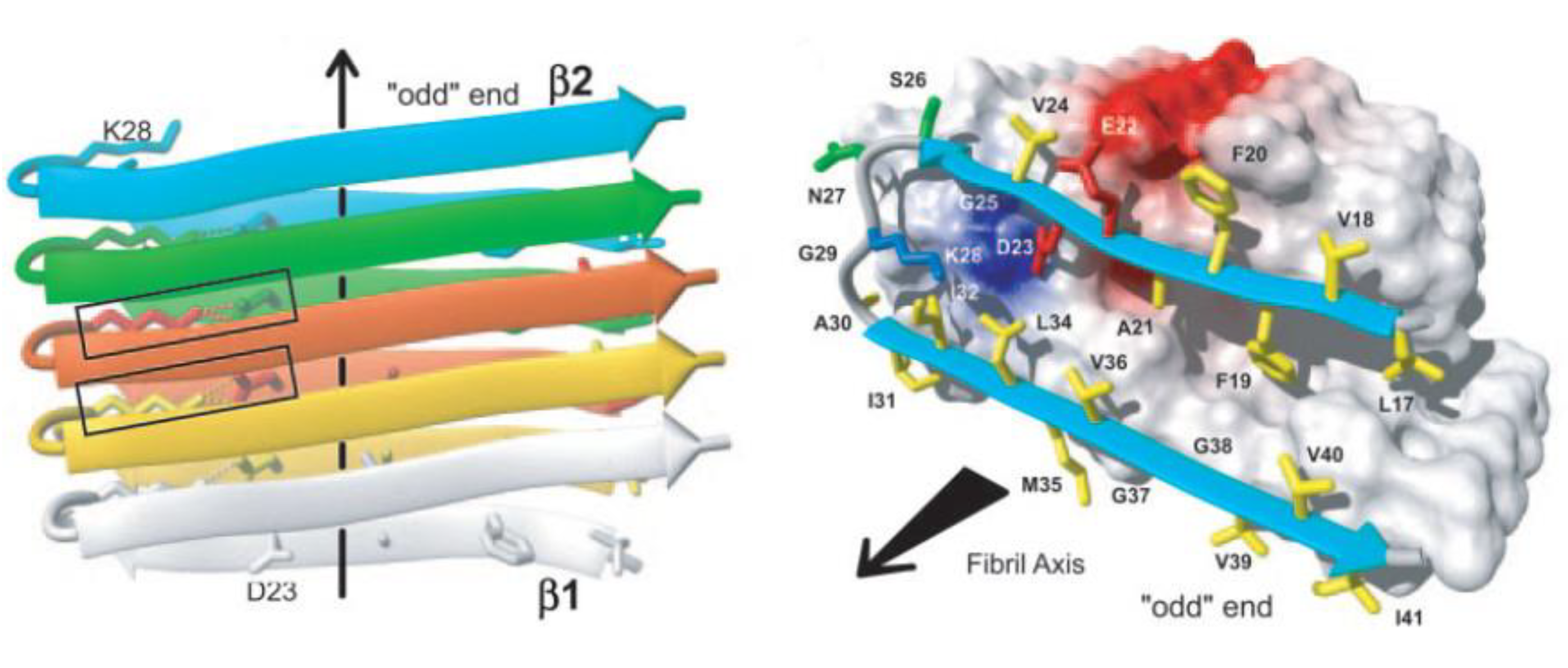
2.1. The Amyloid Fibril
2.2. The Amyloid Oligomer
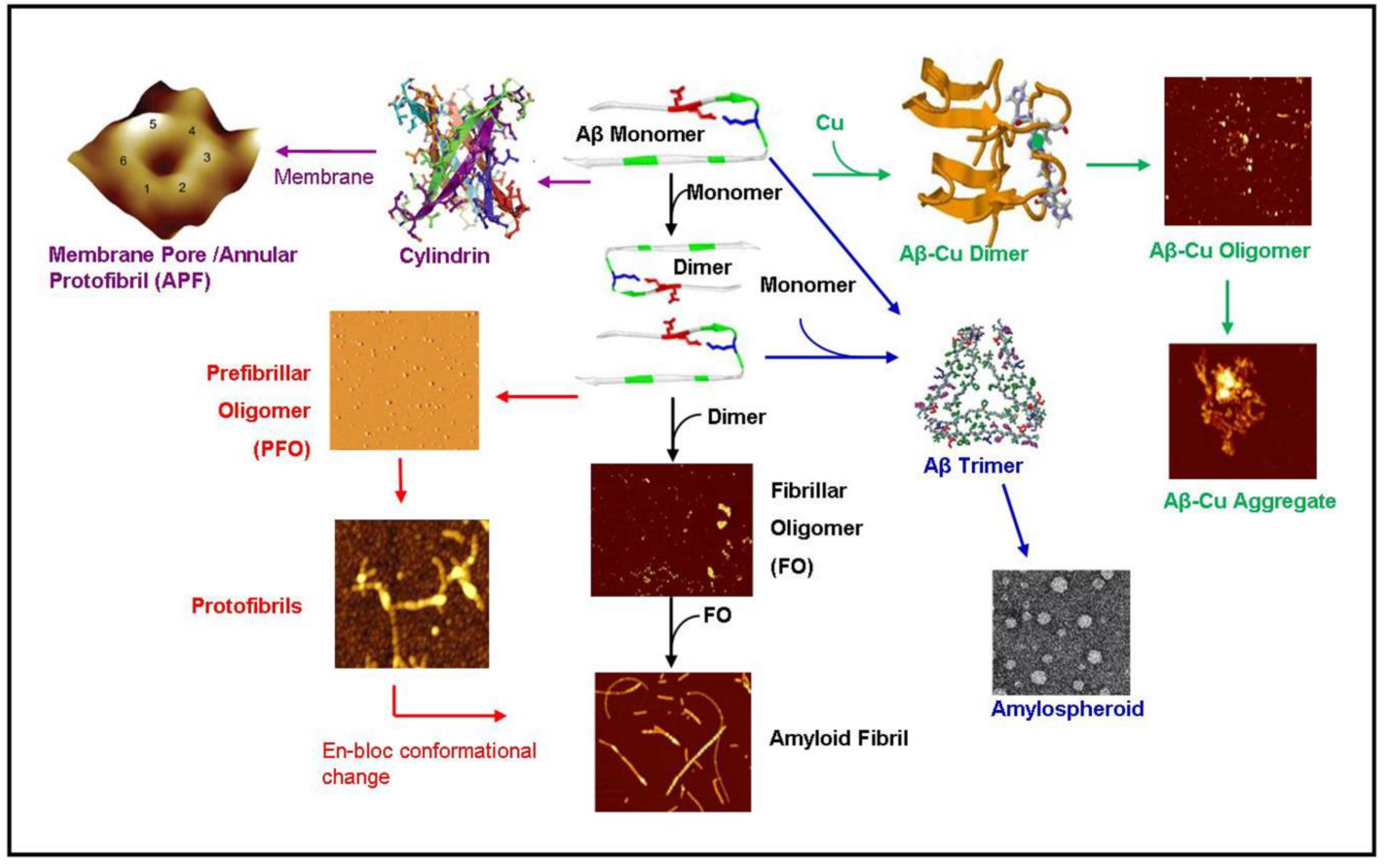
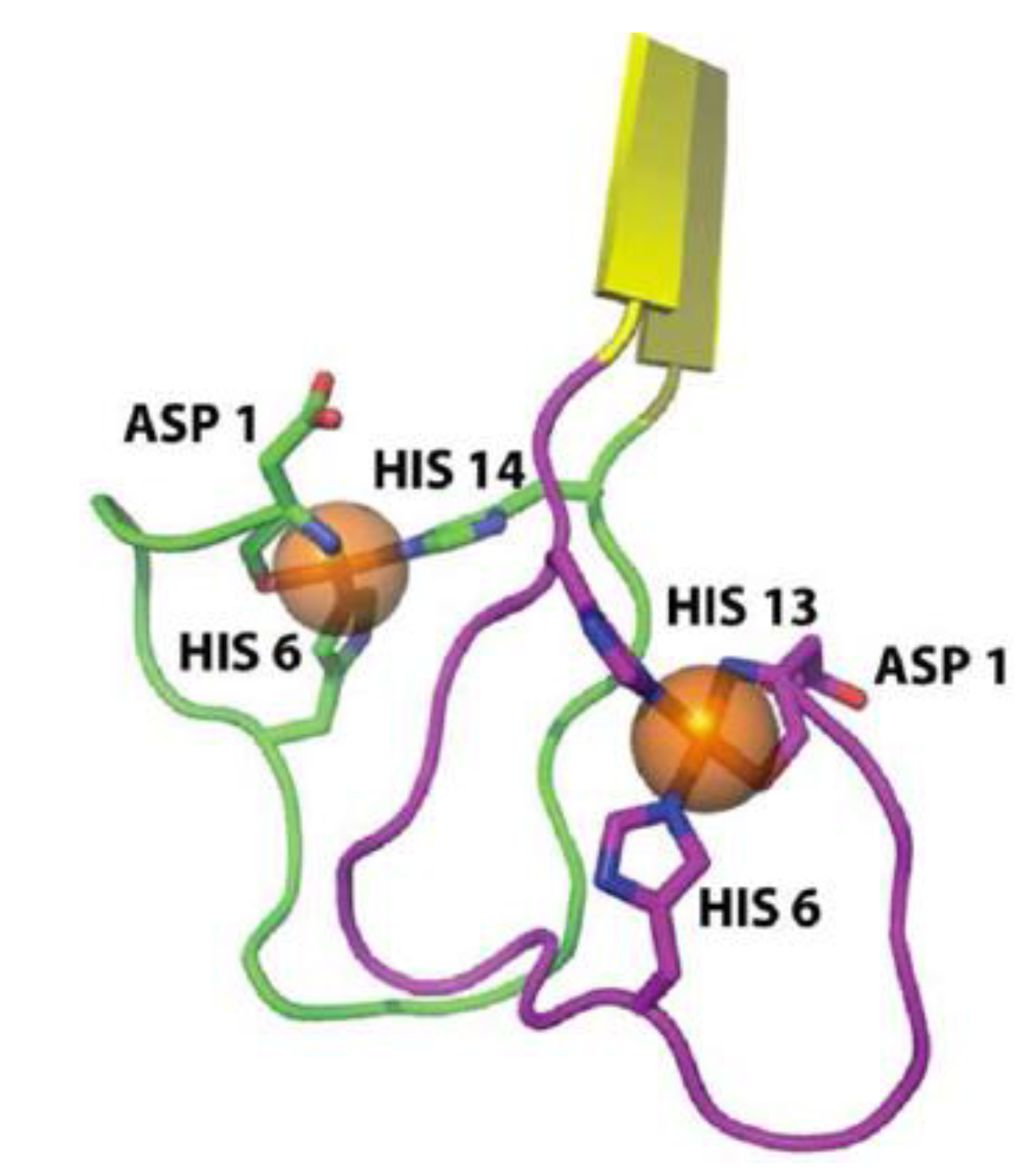
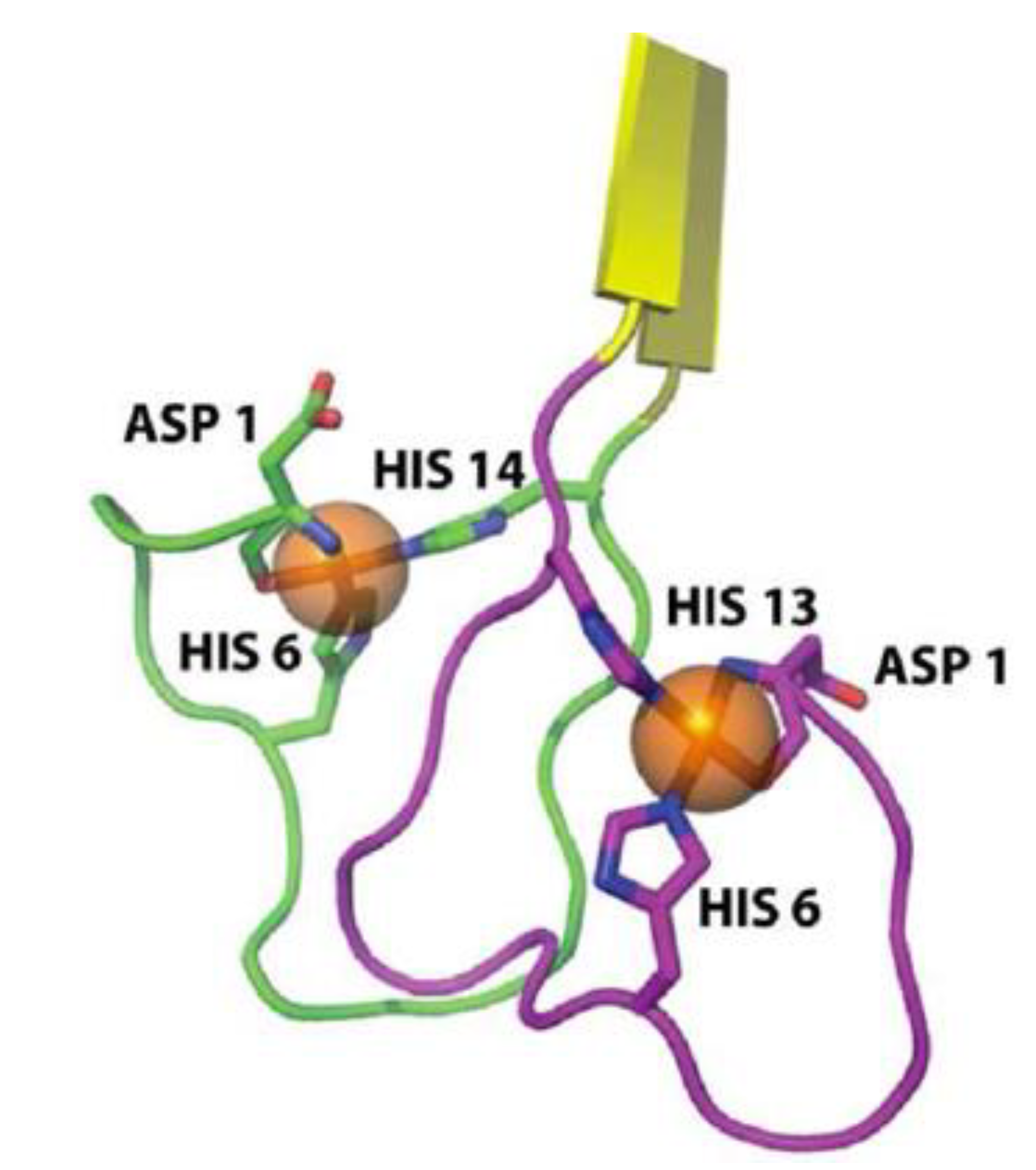
2.3. Metal-Induced Amyloid Structures
3. Aggregation Kinetics, Thermodynamics and Aggregation Pathways
3.1. In Vitro Environment
3.2. Copper Environment
3.3. Zinc Environment
3.4. Physiological Environment
4. Neurotoxicity of Metals
5. Conclusions
Acknowledgments
Conflicts of Interest
References
- Chiti, F.; Dobson, C.M. Protein misfolding, functional amyloid, and human disease. Annu. Rev. Biochem. 2006, 75, 333–366. [Google Scholar] [CrossRef]
- Necula, M.; Kayed, R.; Milton, S.; Glabe, C. Small molecule inhibitors of aggregation indicate that amyloid-β oligomerization and fibrillization pathways are independent and distinct. J. Biol. Chem. 2007, 282, 10311–10324. [Google Scholar] [CrossRef]
- Yamaguchi, T.; Yagi, H.; Goto, Y.; Matsuzaki, K.; Hoshino, M. A disulfide-linked amyloid-beta peptide dimer forms a protofibril-like oligomer through a distinct pathway from amyloid fibril formation. Biochemistry 2010, 49, 7100–7107. [Google Scholar] [CrossRef]
- Matsumura, S.; Shinoda, K.; Yamada, M.; Yokojima, S.; Inoue, M.; Ohnishi, T.; Shimada, T.; Kikuchi, K.; Masui, D.; Hashimoto, S.; et al. Two distinct amyloid-β -protein (aβ) assembly pathways leading to oligomers and fibrils identified by combined fluorescence correlation spectroscopy , morphology , and toxicity analyses. J. Biol. Chem. 2011, 286, 11555–11562. [Google Scholar] [CrossRef]
- Bieschke, J.; Herbst, M.; Wiglenda, T.; Friedrich, R.P.; Boeddrich, A.; Schiele, F.; Kleckers, D.; Lopez del Amo, J.M.; Grüning, B.; Wang, Q.; et al. Small-molecule conversion of toxic oligomers to nontoxic β-sheet-rich amyloid fibrils. Nat. Chem. Biol. 2012, 8, 93–101. [Google Scholar]
- Eskici, G.; Axelsen, P.H. Copper and oxidative stress in the pathogenesis of Alzheimer’s disease. Biochemistry 2012, 51, 6289–6311. [Google Scholar] [CrossRef]
- Curtain, C.C.; Ali, F.E.; Smith, D.G.; Bush, A.I.; Masters, C.L.; Barnham, K.J. Metal ions, pH, and cholesterol regulate the interactions of Alzheimer’s disease amyloid-beta peptide with membrane lipid. J. Biol. Chem. 2003, 278, 2977–2982. [Google Scholar]
- Bush, A.I.; Tanzi, R.E. Therapeutics for Alzheimer’s disease based on the metal hypothesis. Neurother. J. Am. Soc. Exp. Neurother. 2008, 5, 421–432. [Google Scholar] [CrossRef]
- Huang, X.; Atwood, C.S.; Moir, R.D.; Hartshorn, M.; Tanzi, R.; Bush, A. Trace metal contamination initiates the apparent auto-aggregation, amyloidosis, and oligomerization of Alzheimer’s Abeta peptides. J. Biol. Inorg. Chem. 2004, 9, 954–960. [Google Scholar] [CrossRef]
- Tõugu, V.; Karafin, A.; Palumaa, P. Binding of zinc(II) and copper(II) to the full-length Alzheimer’s amyloid-beta peptide. J. Neurochem. 2008, 104, 1249–1259. [Google Scholar] [CrossRef]
- Rauk, A. The chemistry of Alzheimer’s disease. Chem. Soc. Rev. 2009, 38, 2698–2715. [Google Scholar] [CrossRef]
- Azimi, S.; Rauk, A. On the involvement of copper binding to the N-terminus of the amyloid Beta Peptide of Alzheimer’s disease: A computational study on model systems. Int. J. Alzheimers. Dis. 2011, 2011, 539762. [Google Scholar]
- Hane, F.; Tran, G.; Attwood, S.J.; Leonenko, Z. Cu 2+ affects amyloid-β (1–42) aggregation by increasing peptide-peptide binding forces. PLoS One 2013, 8, e59005. [Google Scholar]
- Innocenti, M.; Salvietti, E.; Guidotti, M.; Casini, A.; Bellandi, S.; Foresti, M.L.; Gabbiani, C.; Pozzi, A.; Zatta, P.; Messori, L. Trace copper(II) or zinc(II) ions drastically modify the aggregation behavior of amyloid-beta1–42: An AFM study. J. Alzheimer’s Dis. 2010, 19, 1323–1329. [Google Scholar]
- Bolognin, S.; Messori, L.; Drago, D.; Gabbiani, C.; Cendron, L.; Zatta, P. Aluminum, copper, iron and zinc differentially alter amyloid-Aβ(1–42) aggregation and toxicity. Int. J. Biochem. Cell Biol. 2011, 43, 877–885. [Google Scholar] [CrossRef]
- Mobley, D.L.; Cox, D.L.; Singh, R.R.P.; Maddox, M.W.; Longo, M.L. Modeling amyloid beta-peptide insertion into lipid bilayers. Biophys. J. 2004, 86, 3585–3597. [Google Scholar] [CrossRef]
- Singh, I.; Sagare, A.P.; Coma, M.; Perlmutter, D.; Gelein, R.; Bell, R.D.; Deane, R.J.; Zhong, E.; Parisi, M.; Ciszewski, J.; et al. Low levels of copper disrupt brain amyloid-β homeostasis by altering its production and clearance. Proc. Natl. Acad. Sci. USA 2013, 110, 14771–14776. [Google Scholar] [CrossRef]
- Markesbury, W.; Lovell, M. Damage to lipids, proteins, DNA and RNA in mild cognative impairment. Arch. Neurol. 2007, 64, 954–956. [Google Scholar] [CrossRef]
- Nelson, R.; Eisenberg, D. Recent atomic models of amyloid fibril structure. Curr. Opin. Struct. Biol. 2006, 16, 260–265. [Google Scholar] [CrossRef]
- Nelson, R.; Sawaya, M.R.; Balbirnie, M.; Madsen, A.; Riekel, C.; Grothe, R.; Eisenberg, D. Structure of the cross-β spine of amyloid-like fibrils. Nature 2005, 435, 773–778. [Google Scholar] [CrossRef]
- Luhrs, T.; Rotter, C.; Adrian, M.; Riek-Loher, D.; Bohrmann, B.; Dobeli, H. 3D structure of Alzheimer’s amyloid-beta(1-42) fibrils. Proc. Natl. Acad. Sci. USA 2005, 102, 17342–17347. [Google Scholar] [CrossRef]
- Kirchner, D.; Abraham, C.; Selkoe, D. X-ray diffraction from intraneuronal paired helical filaments and extraneuronal amyloid fibers in Alzheimer disease indicates cross-beta conformation. Proc. Natl. Acad. Sci. USA 1986, 83, 503–507. [Google Scholar] [CrossRef]
- Makin, O.S.; Atkins, E.; Sikorski, P.; Johansson, J.; Serpell, L. Molecular basis for amyloid fibril formation and stability. Proc. Natl. Acad. Sci. USA 2005, 102, 315–320. [Google Scholar] [CrossRef]
- Buchete, N.; Tycko, R.; Hummer, G. Molecular dynamics simulations of Alzheimer’s beta-amyloid protofilaments. J. Mol. Biol. 2005, 353, 804–821. [Google Scholar] [CrossRef]
- Ma, B.; Nussinov, R. Stabilities and conformations of Alzheimer’s β-amyloid peptide oligomers (Aβ16–22, Aβ16–35, and Aβ10–35): Sequence effects. Proc. Natl. Acad. Sci. USA 2002, 99, 14126–14131. [Google Scholar] [CrossRef]
- Rauk, A. Why is the amyloid beta peptide of Alzheimer’s disease neurotoxic? Dalt. Trans. 2008, 1273–1282. [Google Scholar] [CrossRef]
- Baumketner, A.; Bernstein, S.; Wyttenbach, T.; Lazo, N.; Teplow, D.; Bowers, M.; Shea, J. Structure of the 21–30 fragment of amyloid beta-protein. Protein Sci. 2006, 15, 1239–1947. [Google Scholar] [CrossRef]
- Harper, J.; Lieber, C.; Lansbury, P. Atomic force microscopic imaging of seeded fibril formation and fibril branching by the Alzheimer’s disease amyloid-β protein. Chem. Biol. 1997, 4, 951–959. [Google Scholar] [CrossRef]
- Goldsbury, C.; Kistler, J.; Aebi, U.; Arvinte, T.; Cooper, G.J.S. Watching amyloid fibrils grow by time-lapse atomic force microscopy. J. Mol. Biol. 1999, 285, 33–39. [Google Scholar] [CrossRef]
- Antzutkin, O.N.; Balbach, J.J.; Leapman, R.D.; Rizzo, N.W.; Reed, J.; Tycko, R. Multiple quantum solid-state NMR indicates a parallel, not antiparallel , organization of β-sheets in Alzheimer’s β-amyloid fibrils. Proc. Natl. Acad. Sci. USA 2000, 97, 13045–13050. [Google Scholar]
- Balbach, J.J.; Petkova, A.T.; Oyler, N.A.; Antzutkin, O.N.; Gordon, D.J.; Meredith, S.C.; Tycko, R. Supramolecular structure in full-length Alzheimer’s β-amyloid fibrils: Evidence for a parallel β-sheet organization from solid-state nuclear magnetic resonance. Biophys. J. 2002, 83, 1205–1216. [Google Scholar] [CrossRef]
- Qiang, W.; Yau, W.; Luo, Y.; Mattson, M.P.; Tycko, R. Antiparallel β-sheet architecture in Iowa-mutant β-amyloid fibrils. Proc. Natl. Acad. Sci. USA 2012, 109, 4443–4448. [Google Scholar]
- Kayed, R.; Head, E.; Thompson, J.; McIntire, T.; Milton, S.; Cotman, C.; Glabe, C. Common structure of soluble amyloid oligomers implies common mechanism of pathogenesis. Science 2003, 300, 486–489. [Google Scholar] [CrossRef]
- Bucciantini, M.; Giannoni, E.; Chiti, F.; Baroni, F.; Formigli, L. Inherent toxicity of aggregates implies a common mechanism for protein misfolding diseases. Nature 2002, 416, 507–511. [Google Scholar] [CrossRef]
- Soreghan, B.; Kosmoski, J.; Glabe, C. Surfactant properties of Alzheimer’s A beta peptides and the mechanism of amyloid aggregation. J. Biol. Chem. 1994, 18, 28551–28554. [Google Scholar]
- Harper, J.D.; Lansbury, P.T. Models of amyloid seeding in Alzheimer’s disease and scrapie: Mechanistic truths and physiological consequences of the time-dependent solubility of amyloid proteins. Annu. Rev. Biochem. 1997, 66, 385–407. [Google Scholar] [CrossRef]
- Walsh, D.; Hartley, D.; Kusumoto, Y.; Fezoui, Y.; Condron, M.; Lomakin, A.; Benedek, G.; Selkoe, D.; Teplow, D. Amyloid β Protein Fibrillogenesis. J. Biol. Chem. 1999, 274, 25945–25952. [Google Scholar] [CrossRef]
- Stroud, J.C.; Liu, C.; Teng, P.K.; Eisenberg, D. Toxic fibrillar oligomers of amyloid-β have cross-β structure. Proc. Natl. Acad. Sci. USA 2012, 109, 7717–7722. [Google Scholar] [CrossRef]
- Lambert, M.P.; Barlow, K.; Chromy, B.; Edwards, C.; Freed, R.; Liosatos, M.; Morgan, T.E.; Rozovsky, I.; Trommer, B.; Viola, K.L.; et al. Diffusible, nonfibrillar ligands derived from Abeta1–42 are potent central nervous system neurotoxins. Proc. Natl. Acad. Sci. USA 1998, 95, 6448–6453. [Google Scholar] [CrossRef]
- Glabe, C.G. Structural classification of toxic amyloid oligomers. J. Biol. Chem. 2008, 283, 29639–29643. [Google Scholar] [CrossRef]
- Yu, X.; Zheng, J. Polymorphic structures of Alzheimer’s β-amyloid globulomers. PLoS One 2011, 6, e20575. [Google Scholar]
- Kayed, R.; Pensalfini, A.; Margol, L.; Sokolov, Y.; Sarsoza, F.; Head, E.; Hall, J.; Glabe, C. Annular protofibrils are a structurally and functionally distinct type of amyloid oligomer. J. Biol. Chem. 2009, 284, 4230–4237. [Google Scholar]
- Bernstein, S.; Dupuis, N.; Lazo, N.; Wyttenbach, T.; Condron, M.; Bitan, G.; Teplow, D.; Shea, J.; Ruotolo, B.; Robinson, C.; et al. Amyloid-β protein oligomerization and the importance of tetramers and dodecamers in the aetiology of Alzheimer’s disease. Nat.Chem. 2009, 1, 326–331. [Google Scholar] [CrossRef]
- Quist, A.; Doudevski, I.; Lin, H.; Azimova, R.; Ng, D.; Frangione, B.; Kagan, B.; Ghiso, J. Amyloid ion channels: A common structural link for protein-misfolding disease. Proc. Natl. Acad. Sci. USA 2005, 102, 10427–10432. [Google Scholar]
- Lin, H.A.I.; Bhatia, R.; Lal, R. Amyloid-beta protein forms ion channels: Implications form Alzeimer’s diease pathophysiology. FASEB J. 2001, 15, 2433–2444. [Google Scholar] [CrossRef]
- Connelly, L.; Jang, H.; Arce, F.T.; Capone, R.; Kotler, S.A.; Ramachandran, S.; Kagan, B.L.; Nussinov, R.; Lal, R. Atomic force microscopy and MD simulations reveal pore-like structures of all-D-enantiomer of Alzheimer’s β-Amyloid peptide: Relevance to the ion channel mechanism of AD pathology. Phys. Chem. B 2012, 116, 1728–1735. [Google Scholar]
- Benilova, I.; Karran, E.; de Strooper, B. The toxic Aβ oligomer and Alzheimer’s disease: An emperor in need of clothes. Nature 2012, 15, 349–357. [Google Scholar]
- Gu, L.; Liu, C.; Guo, Z. Structural insights into Aβ42 oligomers using site-directed spin labeling. J. Biol. Chem. 2013, 228, 18673–18683. [Google Scholar]
- Laganowsky, A.; Liu, C.; Sawaya, M.R.; Whitelegge, J.P.; Park, J.; Zhao, M.; Pensalfini, A.; Soriaga, A.B.; Landau, M.; Teng, P.K.; et al. Atomic view of a toxic amyloid small oligomer. Science 2012, 335, 1228–1231. [Google Scholar] [CrossRef]
- Lal, R.; Lin, H.; Quist, A.P. Amyloid beta ion channel: 3D structure and relevance to amyloid channel paradigm. Biochim. Biophys. Acta 2007, 1768, 1966–1975. [Google Scholar]
- Lal, R.; Ramachandran, S.; Arnsdorf, M.F. Multidimensional atomic force microscopy: A versatile novel technology for nanopharmacology research. AAPS J. 2010, 12, 716–728. [Google Scholar] [CrossRef]
- Jang, H.; Arce, F.T.; Ramachandran, S.; Capone, R.; Lal, R.; Nussinov, R. β-barrel topology of Alzheimer’S β-amyloid ion channels. J. Mol. Biol. 2010, 404, 917–934. [Google Scholar] [CrossRef]
- Moores, B.; Drolle, E.; Attwood, S.J.; Simons, J.; Leonenko, Z. Effect of surfaces on amyloid fibril formation. PLoS One 2011, 6, e25954. [Google Scholar]
- Hane, F.; Drolle, E.; Gaikwad, R.; Faught, E.; Leonenko, Z. Amyloid-β aggregation on model lipid membranes: An atomic force microscopy study. J. Alzheimer’s Dis. 2011, 26, 485–494. [Google Scholar]
- Drolle, E.; Gaikwad, R.M.; Leonenko, Z. Nanoscale electrostatic domains in cholesterol-laden lipid membranes create a target for amyloid binding. Biophys. J. 2012, 103, L27–L29. [Google Scholar] [CrossRef]
- Bokvist, M.; Lindström, F.; Watts, A.; Gröbner, G. Two types of Alzheimer’s β-amyloid (1–40) peptide membrane interactions: Aggregation preventing transmembrane anchoring vs. accelerated surface fibril formation. J. Mol. Biol. 2004, 335, 1039–1049. [Google Scholar] [CrossRef]
- Lee, W.; Lee, H.; Woo Lee, S.; Sung Yoon, D.; Eom, K.; Kwon, T. Mapping the surface charge distribution of amyloid fibril. Appl. Phys. Lett. 2012, 101, 043703–043704. [Google Scholar] [CrossRef]
- Burke, K.A.; Yates, E.A.; Legleiter, J.; Montie, H.L.; Jefferson, T. Biophysical insights into how surfaces, including lipid membranes, modulate protein aggregation related to neurodegeneration. Front. Neurol. 2013, 4, 1–17. [Google Scholar]
- Burke, K.; Yates, E.; Legleiter, J. Amyloid-forming proteins alter the local mechanical properties of lipid membranes. Biochemistry 2013, 52, 808–817. [Google Scholar] [CrossRef]
- Yip, C.M.; Darabie, A.A.; McLaurin, J. Abeta42-peptide assembly on lipid bilayers. J. Mol. Biol. 2002, 318, 97–107. [Google Scholar] [CrossRef]
- Jang, H.; Teran, F.; Ramachandran, S.; Capone, R.; Azimova, R. Truncated β-amyloid peptide channels provide an alternative mechanism for Alzheimer ’ s disease and down syndrome. Proc. Natl. Acad. Sci. USA 2010, 107, 2–7. [Google Scholar] [CrossRef]
- Wu, J.W.; Breydo, L.; Isas, J.M.; Lee, J.; Kuznetsov, Y.G.; Langen, R.; Glabe, C. Fibrillar oligomers nucleate the oligomerization of monomeric amyloid beta but do not seed fibril formation. J. Biol. Chem. 2010, 285, 6071–6079. [Google Scholar]
- Lee, J.; Culyba, E.; Powers, E.; Kelly, J. Amyloid-b forms fibrils by nucleated conformational conversion of oligomers. Nat. Chem. Biol. 2011, 7, 602–609. [Google Scholar] [CrossRef]
- Nilsberth, C.; Westlind-Danielsson, A.; Eckman, C.; Condron, M.; Axelman, K.; Forsell, C.; Stenh, C.; Luthman, J.; Teplow, D.; Younkin, S.; et al. The “Arctic” APP mutation (E693G) causes Alzheimer’s disease by enhanced Aβ protofibril formation. Nat. Neurosci. 2001, 4, 887–893. [Google Scholar] [CrossRef]
- Hoshi, M.; Sato, M.; Matsumoto, S.; Noguchi, A.; Yasutake, K.; Yoshida, N.; Sato, K. Spherical aggregates of beta-amyloid (amylospheroid) show high neurotoxicity and activate tau protein kinase I/glycogen synthase kinase-3beta. Proc. Natl. Acad. Sci. USA 2003, 100, 6370–6375. [Google Scholar] [CrossRef]
- Paravastu, A.K.; Leapman, R.D.; Yau, W.; Tycko, R. Molecular structural basis for polymorphism in Alzheimer’ s beta-amyloid fibrils. Proc. Natl. Acad. Sci. USA 2008, 105, 18349–18354. [Google Scholar]
- Pedersen, J.T.; Østergaard, J.; Rozlosnik, N.; Gammelgaard, B.; Heegaard, N.H.H. Cu(II) mediates kinetically distinct, non-amyloidogenic aggregation of amyloid-beta peptides. J. Biol. Chem. 2011, 286, 26952–26963. [Google Scholar] [CrossRef]
- Minicozzi, V.; Stellato, F.; Comai, M.; Dalla Serra, M.; Potrich, C.; Meyer-Klaucke, W.; Morante, S. Identifying the minimal copper- and zinc-binding site sequence in amyloid-beta peptides. J. Biol. Chem. 2008, 283, 10784–10792. [Google Scholar]
- Faller, P.; Hureau, C. Bioinorganic chemistry of copper and zinc ions coordinated to amyloid-b peptide. Dalt. Trans. 2009, 7, 1080–1094. [Google Scholar] [CrossRef]
- Sarell, C.J.; Wilkinson, S.R.; Viles, J.H. Substoichiometric levels of Cu2+ ions accelerate the kinetics of fiber formation and promote cell toxicity of amyloid-beta from Alzheimer disease. J. Biol. Chem. 2010, 285, 41533–41540. [Google Scholar] [CrossRef]
- Gunderson, W.A.; Hernández-Guzmán, J.; Karr, J.W.; Sun, L.; Szalai, V.A.; Warncke, K. Local structure and global patterning of Cu2+ binding in fibrillar Amyloid-β [Aβ(1−40)] protein. J. Am. Chem. Soc. 2012, 134, 18330–18337. [Google Scholar]
- Necula, M.; Breydo, L.; Milton, S.; Kayed, R.; van der Veer, W.; Tone, P.; Glabe, C. Methylene blue inhibits amyloid Abeta oligomerization by promoting fibrillization. Biochemistry 2007, 46, 8850–8860. [Google Scholar] [CrossRef]
- Bellesia, G.; Shea, J. Diversity of kinetic pathways in amyloid fibril formation Diversity of kinetic pathways in amyloid fibril formation. J. Chem. Phys. 2009, 131, 111102. [Google Scholar] [CrossRef]
- Bellesia, G.; Shea, J. Effect of β-sheet propensity on peptide aggregation effect of β-sheet propensity on peptide aggregation. J. Chem. Phys. 2009, 130, 145103. [Google Scholar] [CrossRef]
- Esler, W.; Stimson, E.; Ghilardi, J.; Vinters, H.; Lee, J.; Mantyh, P.; Maggio, J. In Vitro growth of Alzheimer’s disease B-amyloid plaques displays first-order kinetics. Biochemistry 1996, 35, 749–757. [Google Scholar]
- Ladiwala, A.R.; Litt, J.; Kane, R.; Aucoin, D.; Smith, S.; Ranjan, S.; Davis, J.; van Nostrand, W.E.; Tessier, P.M. Conformational differences between two Amyloid-β oligomers of similar size and dissimilar toxicity. J. Biol. Chem. 2012, 287, 24765–24773. [Google Scholar] [CrossRef]
- Faller, P.; Hureau, C.; Berthoumieu, O. Role of metal ions in the self-assembly of the Alzheimer’s Amyloid-β peptide. Inorg. Chem. 2013, 52, 12193–12206. [Google Scholar]
- Viles, J.H. Metal ions and amyloid fiber formation in neurodegenerative diseases. Copper, zinc and iron in Alzheimer’s, Parkinson’s and prion diseases. Coord. Chem. Rev. 2012, 256, 2271–2284. [Google Scholar] [CrossRef]
- Bonda, D.; Lee, H.; Blair, J.; Zhu, X.; Perry, G.; Smith, M. Role of metal dyshomeostasis in Alzheimer’s disease. Metallomics 2011, 3, 267–270. [Google Scholar]
- Ha, C.; Ryu, J.; Park, C.B. Metal ions differentially influence the aggregation and deposition of Alzheimer’s beta-Amyloid on a solid template. Biochemistry 2007, 46, 6118–6125. [Google Scholar]
- Bush, A.; Pettingell, W.; Multhaup, G.; Paradis, M.; Vonsattel, J.; Gusella, J.; Beyreuther, K.; Masters, C.; Tanzi, R. Rapid induction of Alzheimer A beta amyloid formation by zinc. Science 1994, 265, 1464–1467. [Google Scholar]
- Mantyh, P.; Ghilardi, J.; Rogers, S.; DeMaster, E.; Allen, C.; Stimson, E.; Maggio, J. Aluminum, iron, and zinc ions promote aggregation of physiological concentrations of B-Amyloid peptide. J. Neurochem. 1993, 61, 1171–1174. [Google Scholar]
- Garai, K.; Sahoo, B.; Kaushalya, S.; Desai, R.; Maiti, S. Zinc lowers Amyloid-beta toxicity by selectively precipitating aggregation. Biochemistry 2007, 46, 10655–10663. [Google Scholar]
- Bogden, J.; Troiano, R.; Joselow, M. Copper, zinc, magnesium, and calcium in plasma and cerebrospinal fluid of patients with neurological diseases. Clin. Chem. 1977, 23, 485–489. [Google Scholar]
- Oe, T.; Ackermann, B.; Inoue, K.; Berna, M.; Garner, C.; Gelfanova, V. Quantitative analysis of Amyloid β peptides in cerebrospinal fluid of Alzheimer’s disease patients by immunoaffinity purification and stable isotope dilution liquid chromatography/negative electrospray ionization tandem mass. Rapid Commun. Mass Spectrom. 2006, 20, 3723–3735. [Google Scholar]
- Cirrito, J.; May, P.; O’Dell, M.; Taylor, J.; Parsadanian, M.; Cramer, K. In vivo assessment of brain interstitial fluid with microdialysis reveals plaque-associated changes in Amyloid-β metabolism and half-life. Neuroscience 2003, 23, 8844–8853. [Google Scholar]
- Kardos, J.; Kovacs, I.; Hajos, F.; Kalman, M.; Simonyi, M. Nerve-endings from rat-brain tissue release copper upon depolarization: A possible role in regulating neuronal excitability. Neurosci. Lett. 1989, 103, 139–144. [Google Scholar]
- Smith, D.; Cappai, R.; Barnham, K. The redox chemistry of the Alzheimer’s disease amyloid beta peptide. Biochim. Biophys. Acta 2007, 1768, 1976–1990. [Google Scholar]
- Perczel, A.; Hudaky, P.; Palfi, V. Dead-end street of protein folding: Thermodynamic rationale of amyloid fibril formation. J. Am. Chem. Soc. 2007, 129, 14959–14965. [Google Scholar]
- He, X.; Giurleo, J.T.; Talaga, D.S. Role of small oligomers on the amyloidogenic aggregation free-energy landscape. J. Mol. Biol. 2010, 395, 134–154. [Google Scholar]
- Barz, B.; Urbanc, B. Dimer formation enhances structural differences explicit-solvent molecular dynamics study. PLoS One 2012, 7, e34345. [Google Scholar]
- Han, D.; Wang, H.; Yang, P. Molecular modeling of zinc and copper binding with Alzheimer’s amyloid beta-peptide. Biometals 2008, 21, 189–196. [Google Scholar]
- Stefani, M. Progress in Neurobiology Structural features and cytotoxicity of amyloid oligomers: Implications in Alzheimer’s disease and other diseases with amyloid deposits. Prog. Neurobiol. 2012, 99, 226–245. [Google Scholar]
- Tõugu, V.; Tiiman, A.; Palumaa, P. Interactions of Zn(II) and Cu(II) ions with Alzheimer’s amyloid-beta peptide. Metal ion binding, contribution to fibrillization and toxicity. Metallomics 2011, 3, 250–261. [Google Scholar]
- Faller, P. Copper and zinc binding to amyloid-beta: Coordination, dynamics, aggregation, reactivity and metal-ion transfer. Chembiochem 2009, 10, 2837–2845. [Google Scholar]
- Sengupta, P.; Garai, K.; Sahoo, B.; Shi, Y.; Callaway, D.; Maiti, S. The amyloid beta peptide (Abeta(1–40)) is thermodynamically soluble at physiological concentrations. Biochemistry 2003, 42, 10506–10513. [Google Scholar]
- Kowalik-Jankowska, L.; Ruta, M.; Wisniewska, K.; Lankiewicz, L. Coordination abilities of the 1–16 and 1–28 fragments of beta-amyloid peptide towards copper(II) ions: A combined potentiometric and spectroscopic study. J. Inorg. Biochem. 2003, 95, 270–282. [Google Scholar]
- Hatcher, J.; Hong, L.; Bush, W.; Carducci, T.; Simon, J. Quantification of the binding constant of copper(II) to the amyloid-beta peptide. J. Phys. Chem. B 2008, 112, 8160–8164. [Google Scholar]
- Sarell, C.; Syme, C.; Rigby, S.; Viles, J. Copper(II) binding to amyloid-beta fibrils of Alzheimer’s disease reveals a picomolar affinity: Stoichiometry and coordination geometry are independent of Abeta oligomeric form. Biochemistry 2009, 48, 4388–4402. [Google Scholar]
- Doraiswamy, P.; Finefrock, A. Metals in our minds: Therapeutic implications for neurodegenerative disorders. Lancet Neurol. 2004, 3, 431–434. [Google Scholar]
- Pearl, R. The Rate of Living: Being an Account of Some experimental Studies on the Biology of Life Duration; AA Knopf: New York, NY, USA, 1928. [Google Scholar]
- Smith, M.; Numomura, A.; Zhu, X.; Takeda, A.; Perry, G. Metabolic, metallic, and mitotic sources of oxidative stress in Alzheimer disease. Antioxid. Redox Signal 2000, 2, 413–420. [Google Scholar]
- Markesbury, W. Oxidative stress hypothesis in Alzheimer’s disease. Free Radic. Biol. Med. 1997, 23, 134–147. [Google Scholar]
- Liochev, S. The mechanism of “Fenton-like” reactions and their importance for biological systems. A biologist’s view. Met. Ions Biol. Syst. 1999, 36, 1–36. [Google Scholar]
- Paoletti, P.; Vergnano, A.; Barbour, B.; Casado, M. Zinc at glutamatergic synapses. Neuroscience 2009, 158, 126. [Google Scholar]
- Frederickson, C.J.; Koh, Y.; Bush, A.I. The neurobiology of zinc in health and disease. Nat. Rev. Neurosci. 2005, 6, 449. [Google Scholar]
- Cuajunco, M.; Goldstein, L.; Numomura, A.; Smith, M.; Lim, J.; Atwood, C. Evidence that the beta-amyloid plaques of Alzheimer’s disease represent the redox silencing and entombment of abeta by zinc. J. Biol. Chem. 2000, 275, 19439–19442. [Google Scholar]
- Lovell, M.A.; Xie, C.; Markesbery, W. Protection against amyloid beta peptide toxicity by zinc. Brain Res. 1999, 823, 88–95. [Google Scholar]
- Moreira, P.; Pereira, C.; Santos, M.S.; Oliveira, C. Effect of zinc ions on the cytotoxicity induced by the amyloid beta-peptide. Antioxid. Redox Signal. 2000, 2, 317–325. [Google Scholar]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Hane, F.; Leonenko, Z. Effect of Metals on Kinetic Pathways of Amyloid-β Aggregation. Biomolecules 2014, 4, 101-116. https://doi.org/10.3390/biom4010101
Hane F, Leonenko Z. Effect of Metals on Kinetic Pathways of Amyloid-β Aggregation. Biomolecules. 2014; 4(1):101-116. https://doi.org/10.3390/biom4010101
Chicago/Turabian StyleHane, Francis, and Zoya Leonenko. 2014. "Effect of Metals on Kinetic Pathways of Amyloid-β Aggregation" Biomolecules 4, no. 1: 101-116. https://doi.org/10.3390/biom4010101
APA StyleHane, F., & Leonenko, Z. (2014). Effect of Metals on Kinetic Pathways of Amyloid-β Aggregation. Biomolecules, 4(1), 101-116. https://doi.org/10.3390/biom4010101