

The Advance of Single-Cell RNA Sequencing Applications in Ocular Physiology and Disease Research

Abstract
1. Introduction
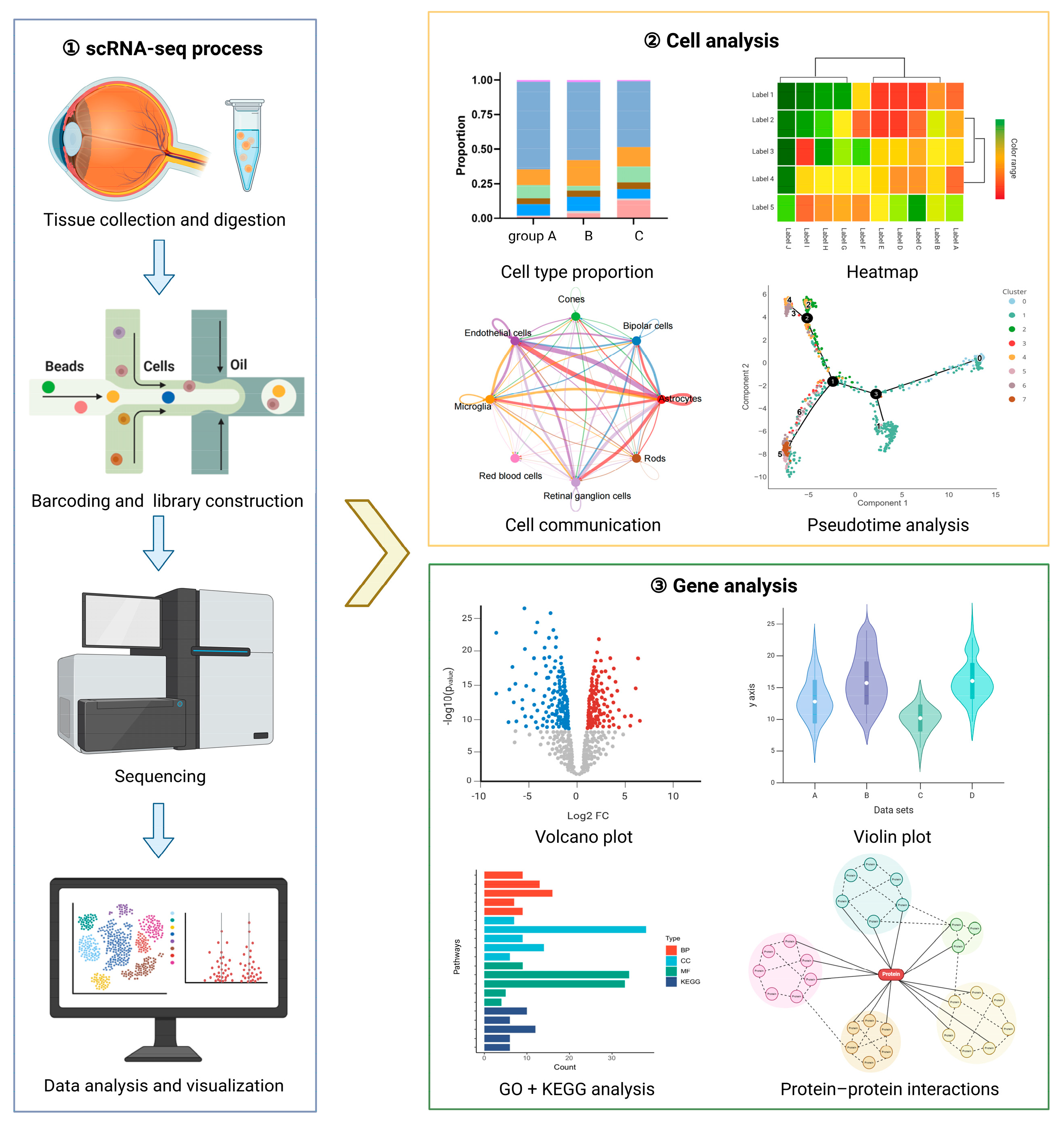
2. scRNA-Seq Technology
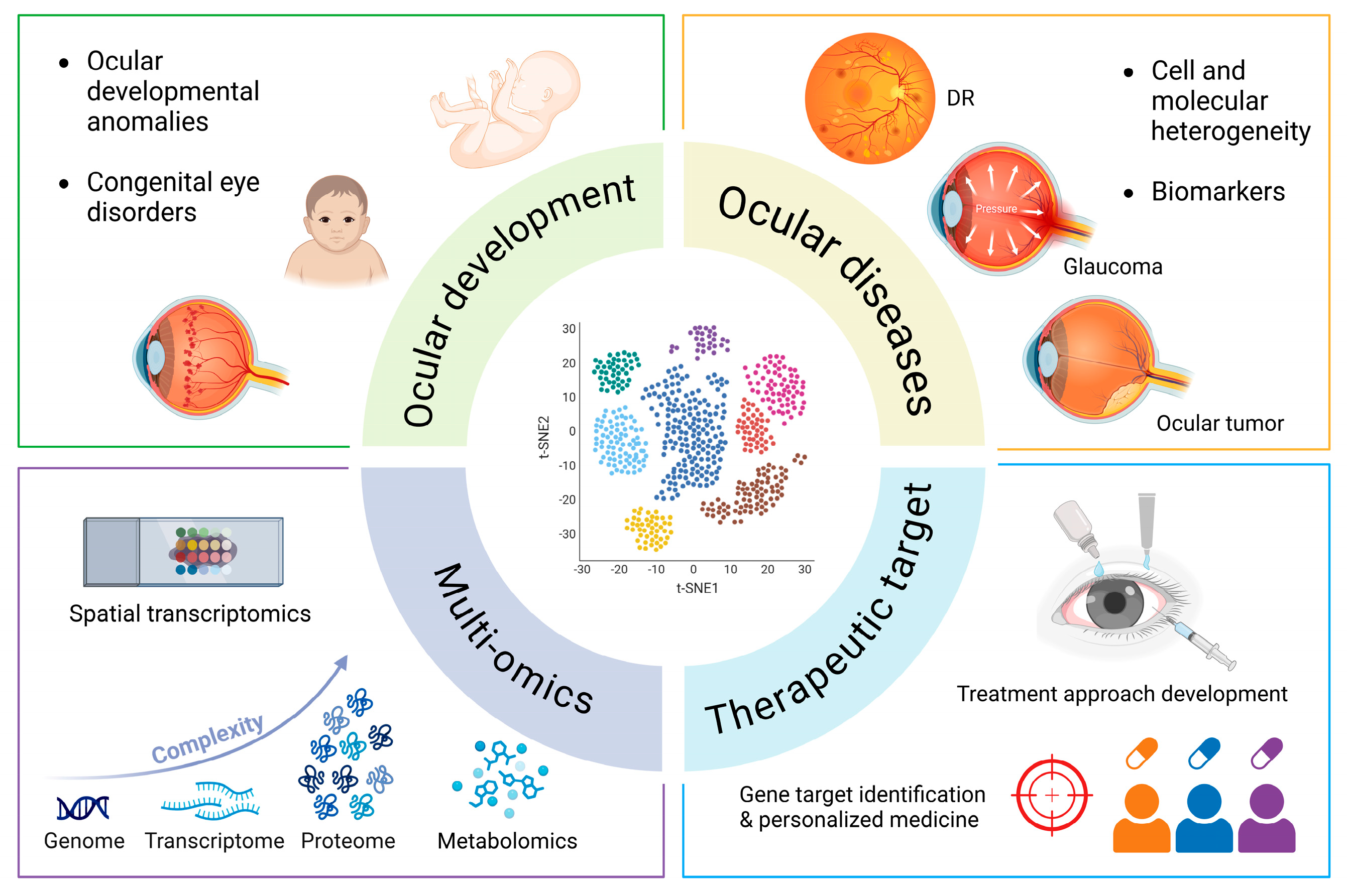
3. Ocular Tissue Development
4. Application of scRNA-Seq in Ocular Diseases
4.1. Myopia
4.2. Ocular Surface and Corneal Diseases
4.3. Glaucoma
4.4. Uveitis
4.5. Retinal Diseases

{kind=link}
{kind=link}
{kind=link}
| Disease | Sample | Cell Number | Method | Key Findings | Reference |
|---|---|---|---|---|---|
| Glaucoma | Trabecular meshwork and neighboring tissues from 8 human eyes from 4 donors | 8758 | scRNA-seq | Schlemm’s canal exhibits a unique combination of lymphatic and vascular gene expression. Mapped key glaucoma-related genes to specific cell clusters, revealing their roles in IOP regulation. | Patel et al. [40] (2020) |
| SECs from C57BL/6J and 129/Sj mouse strains | ~4500 SECs (bulk RNA-seq); 903 single SECs (scRNA-seq and snRNA-seq) | bulk RNA-seq, scRNA-seq, snRNA-seq | Schlemm’s canal cells have a lymphatic-biased identity and identified key marker genes. Characterized interactions between SECs and trabecular meshwork cells, providing insights into the regulation of aqueous humor outflow and IOP. | Balasubramanian et al. [41] (2024) | |
| Anterior segment issues from 7 human eyes, cynomolgus macaque, rhesus macaque, pig, and mouse | 24,023 (human); 5158 (rhesus macaque), 9155 (cynomolgus macaque), 6709 (pig), 5067 (mouse) | scRNA-seq | Generated a cell atlas of aqueous humor outflow pathways in humans and four model species. Exhibited conservation and differences in cell types and gene expression across species, providing insights into glaucoma pathogenesis. | Van et al. [43] (2020) | |
| Retinas from WT mice and IGFBPL1 KO mice | 3500 (scRNA-seq) | scRNA-seq combined with bulk RNA-seq | IGFBPL1 deficiency leads to microglial activation and progressive neurodegeneration in the retina and brain, while IGFBPL1 resets pro-inflammatory microglia to a homeostatic state via IGF1R signaling. | Pan et al. [49] (2023) | |
| Ciliary body and contiguous tissues from adult mice with ocular hypertension models | Not specified | scRNA-seq | CRISPR-CasRx-mediated disruption of Aqp1/Adrb2/Rock1/Rock2 genes reduces IOP and RGC damage in mice. | Yao et al. [51] (2024) | |
| Uveitis | Cervical draining lymph node cells from NC, EAU control, and EAU mice | 47,048 | scRNA-seq | Hif1α identified as a potential participant in autoimmune uveitis pathogenesis by regulating Th-17, Th1, and regulatory T cells. | Zhu et al. [52] (2023) |
| Retina from 1 healthy mouse and 2 EAU mice for scRNA-seq; RPE from 3 healthy mice and 2 EAU mice for bulk RNA-seq | 11,516 (scRNA-seq) | scRNA-seq combined with bulk RNA-seq | During EAU, interactions exist between Müller glia and T cell/natural killer cell subsets. RPE cells exhibited an epithelial-to-mesenchymal transition signature during EAU. | Quinn et al. [53] (2024) | |
| Aqueous humor from 3 BD patients and 3 VKHD patients | 47,048 | scRNA-seq combined with scTCR-seq | BD uveitis shows significant myeloid cell infiltration and CD8+ T cell clonality with cytotoxic phenotype, while VKHD uveitis is dominated by CD4+ T cells with Th1-like phenotype. | Kang et al. [54] (2023) | |
| 3 healthy adult mice, 6 EAU model mice (3 untreated, 3 treated with CsA) | 41,349 | scRNA-seq | CsA reversed EAU-associated changes in immune cell composition and gene expression, and reduced the differentiation and immunoglobulin secretion of plasma B cells, and restored the balance between pathogenic T cells and Tregs. | Duan et al. [55] (2022) | |
| 3 healthy adult mice, 6 EAU model mice (3 untreated, 3 treated with MMF) | 41,349 | scRNA-seq | MMF reduced the differentiation tendency from naïve to effector phenotypes and downregulated pathogenic cytokine production in Th1 and Th17 cells. MMF also inhibited B-cell immunoglobulin production and antigen processing and presentation. | Wang et al. [56] (2023) | |
| 6 healthy adult mice, 12 EAU model mice (6 untreated, 6 treated with DMF) | 41,349 | scRNA-seq | DMF treatment effectively ameliorated EAU symptoms by reversing the Teff/Treg imbalance and inhibiting the ocular infiltration of Teff cells. DMF downregulated PIM1 and CXCR4 expression, which are critical for T cell differentiation and migration. DMF also reduced the proportion of plasma cells by inhibiting PIM1 expression in B cells. | Zhu et al. [57] (2024) | |
| CDLNs from 9 normal mice, 9 EAU model mice (6 untreated, 3 treated with GMSCs) | 71,000 | scRNA-seq | GMSC treatment alleviated EAU symptoms, reduced retinal immune cell infiltration, and restored Th17/Treg balance by regulating Th17/Treg-related genes and suppressing pro-inflammatory Th17 cell formation. | Gao et al. [58] (2023) | |
| 2 WT C57BL/6J mice and 2 EAU model mice | 20,448 | scRNA-seq | EAU immune response is primarily driven by Th1 cells, and CCR5-overexpressing MSCs enhance homing capacity and improve immunomodulatory outcomes in preventing EAU. | Yuan et al. [59] (2024) | |
| Retina diseases | 4 healthy human PBMCs, 4 DME human PBMCs | 57,650 | scRNA-seq | The presence of innate immune dysregulation in the peripheral blood of DME patients with T2D, and pro-inflammatory CD14+ monocytes predominated in promoting inflammation. | Ma et al. [65] (2021) |
| 10 retinas from 5 diabetic mice, 10 retinas from 5 WT mice | 31,256 | scRNA-seq | Identified two microglial subpopulations and three EC populations in retinal cells of diabetic retinopathy. Found CSF1/CSF1R crosstalk dysregulation associated with PDR. | Ben et al. [66] (2024) | |
| 5 rat retinal samples (2 normal SD rats, 3 DR rats) | 35,910 | scRNA-seq | Constructed a communication network among ECs, pericytes, and two Müller cell subtypes in the early stage of DR. | Wang et al. [67] (2022) | |
| 4 fibrovascular membranes from PDR patients | 4044 | scRNA-seq | Identified a subset of macrophages expressing proangiogenic cytokines and a pericyte-myofibroblast transdifferentiating subcluster. | Corano et al. [68] (2023) | |
| 129 human donor eyes (106 control and 23 AMD patients) for bulk RNA-seq; 5 control donor eyes for snRNA-seq | 100,055 nuclei (snRNA-seq) | snRNA-seq combined with bulk RNA-seq | Identified 15 putative causal genes for AMD, with some highly expressed in the RPE. | Orozco et al. [69] (2020) | |
| 3 human donor eyes (2 controls and 1 AMD) | 4335 | scRNA-seq | Discovered three novel cell markers: DNASE1L3 for ECs, ABCB5 for melanocytes, and SLC39A12 for RPE cells. Constructed cell-specific TF regulatory loops, highlighting key TFs in AMD pathogenesis. | Wang et al. [70] (2023) | |
| Retina from 2 adult humans for scRNA-seq, retina from 15 adult humans with and without AMD for bulk RNA-seq | 92,385 (scRNA-seq) | scRNA-seq combined with bulk RNA-seq | Identified 9772 and 1214 differentially expressed genes in the macula and periphery for advanced AMD vs. control comparison. Enrichment in complement and coagulation, antigen presentation, tissue remodeling signaling pathways. | Lyu et al. [71] (2021) | |
| RPE/choroid from 3 human donors (2 normal eyes and 1 neovascular AMD eye) for scRNA-seq; RPE/choroid from 96 normal human donors for bulk RNA-seq | 4766 (scRNA-seq) | scRNA-seq combined with bulk RNA-seq | Identified VEGF-, BMP-, and tenascin-mediated pathways as strong intercellular communication pathways related to aging and senescence. AMD samples showed higher senescence scores than normal cells | Dhirachaikulpanich et al. [72] (2022) | |
| Mouse retinas from the OIR model and control mice | 76,164 | scRNA-seq (BD Rhapsody platform) | Pericyte sub-population 2, highly expressing Col1a1, was vulnerable to pathological angiogenesis, and Col1a1 expression was upregulated in the aqueous humor of patients with PDR or ROP. | Xia et al. [73] (2023) |
4.6. Ocular Tumor
5. Summary and Prospect
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| scRNA-seq | Single-cell RNA sequencing |
| UMI | Unique molecular identifier |
| PCR | Polymerase Chain Reaction |
| snRNA-seq | Single-nucleus RNA sequencing |
| LPCs | Limbal progenitor cells |
| ROPA | Retinoic Acid-Related Orphan Receptor Alpha |
| CECs | Corneal epithelial cells |
| ATAC-seq | Assay for transposase-accessible chromatin sequencing |
| LECs | Lens epithelial cells |
| LFCs | Lens fiber cells |
| RGCs | Retinal ganglion cells |
| RPCs | Retinal progenitor cells |
| TACs | Transit-amplifying cells |
| DED | Dry eye disease |
| FECD | Fuchs’ endothelial corneal dystrophy |
| Th1 | T helper 1 |
| Treg | Regulatory T |
| TM | Trabecular meshwork |
| SC | Schlemm’s canal |
| IOP | Intraocular pressure |
| CDLNs | Cervical draining lymph nodes |
| EAU | Experimental autoimmune uveitis |
| Hif1α | Hypoxia-inducible factor 1 alpha |
| DEG | Differentially expressed gene |
| RPE | Retinal pigment epithelium |
| BD | Behcet’s disease |
| VKHD | Vogt–Koyanagi–Harada disease |
| CsA | Cyclosporine A |
| MMF | Mycophenolate mofetil |
| DMF | Dimethyl fumarate |
| MSCs | Mesenchymal stem cells |
| GMSCs | Gingiva-derived mesenchymal stem cells |
| DR | Diabetic retinopathy |
| PDR | Proliferative DR |
| AEBP1 | Adipocyte enhancer-binding protein 1 |
| AMD | Age-related macular degeneration |
| GWAS | Genome-wide association studies |
| eQTL | Expression quantitative trait locus |
| GEO | Gene Expression Omnibus |
| OIR | Oxygen-induced retinopathy |
| ROP | Retinopathy of prematurity |
| CoM | Conjunctival melanoma |
| CAFs | Cancer-associated fibroblasts |
| UM | Uveal melanoma |
| scATAC-seq | Single-cell assay for transposase-accessible chromatin sequencing |
References
- Zong, Y.; Xiao, S.; Lei, D.; Li, H. Discoveries in Retina Physiology and Disease Biology Using Single-Cell RNA Sequencing. Front. Biosci. 2023, 28, 247. [Google Scholar] [CrossRef]
- Shiau, F.; Ruzycki, P.A.; Clark, B.S. A single-cell guide to retinal development: Cell fate decisions of multipotent retinal progenitors in scRNA-seq. Dev. Biol. 2021, 478, 41–58. [Google Scholar] [CrossRef]
- Li, X.; Wang, C.Y. From bulk, single-cell to spatial RNA sequencing. Int. J. Oral. Sci. 2021, 13, 36. [Google Scholar] [CrossRef]
- Rossin, E.J.; Sobrin, L.; Kim, L.A. Single-cell RNA sequencing: An overview for the ophthalmologist. Semin. Ophthalmol. 2021, 36, 191–197. [Google Scholar] [CrossRef]
- Tan, Y.; Huang, J.; Li, D.; Zou, C.; Liu, D.; Qin, B. Single-cell RNA sequencing in dissecting microenvironment of age-related macular degeneration: Challenges and perspectives. Ageing Res. Rev. 2023, 90, 102030. [Google Scholar] [CrossRef] [PubMed]
- Macosko Evan, Z.; Basu, A.; Satija, R.; Nemesh, J.; Shekhar, K.; Goldman, M.; Tirosh, I.; Bialas Allison, R.; Kamitaki, N.; Martersteck Emily, M.; et al. Highly Parallel Genome-wide Expression Profiling of Individual Cells Using Nanoliter Droplets. Cell 2015, 161, 1202–1214. [Google Scholar] [CrossRef] [PubMed]
- Voigt, A.P.; Mullin, N.K.; Stone, E.M.; Tucker, B.A.; Scheetz, T.E.; Mullins, R.F. Single-cell RNA sequencing in vision research: Insights into human retinal health and disease. Prog. Retin. Eye Res. 2021, 83, 100934. [Google Scholar] [CrossRef]
- Voigt, A.P.; Whitmore, S.S.; Lessing, N.D.; DeLuca, A.P.; Tucker, B.A.; Stone, E.M.; Mullins, R.F.; Scheetz, T.E. Spectacle: An interactive resource for ocular single-cell RNA sequencing data analysis. Exp. Eye Res. 2020, 200, 108204. [Google Scholar] [CrossRef] [PubMed]
- Lake, B.B.; Ai, R.; Kaeser, G.E.; Salathia, N.S.; Yung, Y.C.; Liu, R.; Wildberg, A.; Gao, D.; Fung, H.-L.; Chen, S.; et al. Neuronal subtypes and diversity revealed by single-nucleus RNA sequencing of the human brain. Science 2016, 352, 1586–1590. [Google Scholar] [CrossRef]
- Li, M.; Guo, H.; Wang, B.; Han, Z.; Wu, S.; Liu, J.; Huang, H.; Zhu, J.; An, F.; Lin, Z.; et al. The single-cell transcriptomic atlas and RORA-mediated 3D epigenomic remodeling in driving corneal epithelial differentiation. Nat. Commun. 2024, 15, 256. [Google Scholar] [CrossRef]
- Collin, J.; Queen, R.; Zerti, D.; Bojic, S.; Dorgau, B.; Moyse, N.; Molina, M.M.; Yang, C.; Dey, S.; Reynolds, G.; et al. A single cell atlas of human cornea that defines its development, limbal progenitor cells and their interactions with the immune cells. Ocul. Surf. 2021, 21, 279–298. [Google Scholar] [CrossRef]
- Swarup, A.; Phansalkar, R.; Morri, M.; Agarwal, A.; Subramaniam, V.; Li, B.; Wu, A.Y. Single-cell transcriptomic analysis of corneal organoids during development. Stem Cell Rep. 2023, 18, 2482–2497. [Google Scholar] [CrossRef] [PubMed]
- Zhu, M.C.; Hu, W.; Lin, L.; Yang, Q.W.; Zhang, L.; Xu, J.L.; Xu, Y.T.; Liu, J.S.; Zhang, M.D.; Tong, X.Y.; et al. Single-cell RNA sequencing reveals new subtypes of lens superficial tissue in humans. Cell Prolif. 2023, 56, e13477. [Google Scholar] [CrossRef]
- Zhang, J.; Roberts, J.M.; Chang, F.; Schwakopf, J.; Vetter, M.L. Jarid2 promotes temporal progression of retinal progenitors via repression of Foxp1. Cell Rep. 2023, 42, 112237. [Google Scholar] [CrossRef]
- Biswas, S.; Shahriar, S.; Bachay, G.; Arvanitis, P.; Jamoul, D.; Brunken, W.J.; Agalliu, D. Glutamatergic neuronal activity regulates angiogenesis and blood-retinal barrier maturation via Norrin/beta-catenin signaling. bioRxiv 2024. [Google Scholar] [CrossRef]
- Brown, N.L.; Patel, S.; Brzezinski, J.; Glaser, T. Math5 is required for retinal ganglion cell and optic nerve formation. Development 2001, 128, 2497–2508. [Google Scholar] [CrossRef]
- Mehta, K.; Daghsni, M.; Raeisossadati, R.; Xu, Z.; Davis, E.; Naidich, A.; Wang, B.; Tao, S.; Pi, S.; Chen, W.; et al. A cis-regulatory module underlies retinal ganglion cell genesis and axonogenesis. Cell Rep. 2024, 43, 114291. [Google Scholar] [CrossRef]
- Lu, Y.; Shiau, F.; Yi, W.; Lu, S.; Wu, Q.; Pearson, J.D.; Kallman, A.; Zhong, S.; Hoang, T.; Zuo, Z.; et al. Single-Cell Analysis of Human Retina Identifies Evolutionarily Conserved and Species-Specific Mechanisms Controlling Development. Dev. Cell 2020, 53, 473–491 e9. [Google Scholar] [CrossRef] [PubMed]
- Mullin, N.K.; Bohrer, L.R.; Voigt, A.P.; Lozano, L.P.; Wright, A.T.; Bonilha, V.L.; Mullins, R.F.; Stone, E.M.; Tucker, B.A. NR2E3 loss disrupts photoreceptor cell maturation and fate in human organoid models of retinal development. J. Clin. Investig. 2024, 134, e173892. [Google Scholar] [CrossRef]
- Xiao, Y.; Mao, X.; Hu, X.; Yuan, S.; Chen, X.; Dai, W.; Zhang, S.; Li, Y.; Chen, M.; Mao, P.; et al. Single-Cell Transcriptomic Profiling of Human Retinal Organoids Revealed a Role of IGF1-PHLDA1 Axis in Photoreceptor Precursor Specification. Investig. Ophthalmol. Vis. Sci. 2022, 63, 9. [Google Scholar] [CrossRef]
- Dorgau, B.; Collin, J.; Rozanska, A.; Zerti, D.; Unsworth, A.; Crosier, M.; Hussain, R.; Coxhead, J.; Dhanaseelan, T.; Patel, A.; et al. Single-cell analyses reveal transient retinal progenitor cells in the ciliary margin of developing human retina. Nat. Commun. 2024, 15, 3567. [Google Scholar] [CrossRef]
- Dorgau, B.; Collin, J.; Rozanska, A.; Boczonadi, V.; Moya-Molina, M.; Unsworth, A.; Hussain, R.; Coxhead, J.; Dhanaseelan, T.; Armstrong, L.; et al. Deciphering the spatiotemporal transcriptional and chromatin accessibility of human retinal organoid development at the single-cell level. iScience 2024, 27, 109397. [Google Scholar] [CrossRef]
- Wahle, P.; Brancati, G.; Harmel, C.; He, Z.; Gut, G.; Del Castillo, J.S.; Xavier da Silveira Dos Santos, A.; Yu, Q.; Noser, P.; Fleck, J.S.; et al. Multimodal spatiotemporal phenotyping of human retinal organoid development. Nat. Biotechnol. 2023, 41, 1765–1775. [Google Scholar] [CrossRef] [PubMed]
- Murgiano, L.; Becker, D.; Spector, C.; Carlin, K.; Santana, E.; Niggel, J.K.; Jagannathan, V.; Leeb, T.; Pearce-Kelling, S.; Aguirre, G.D.; et al. CCDC66 frameshift variant associated with a new form of early-onset progressive retinal atrophy in Portuguese Water Dogs. Sci. Rep. 2020, 10, 21162. [Google Scholar] [CrossRef]
- Gerding, W.M.; Schreiber, S.; Schulte-Middelmann, T.; de Castro Marques, A.; Atorf, J.; Akkad, D.A.; Dekomien, G.; Kremers, J.; Dermietzel, R.; Gal, A.; et al. Ccdc66 null mutation causes retinal degeneration and dysfunction. Hum. Mol. Genet. 2011, 20, 3620–3631. [Google Scholar] [CrossRef]
- Chen, X.; Tong, P.; Jiang, Y.; Cheng, Z.; Zang, L.; Yang, Z.; Lan, W.; Xia, K.; Hu, Z.; Tian, Q. CCDC66 mutations are associated with high myopia through affected cell mitosis. J. Med. Genet. 2024, 61, 262–269. [Google Scholar] [CrossRef]
- Yuan, J.; Zhuang, Y.Y.; Liu, X.; Zhang, Y.; Li, K.; Chen, Z.J.; Li, D.; Chen, H.; Liang, J.; Yao, Y.; et al. Exome-wide association study identifies KDELR3 mutations in extreme myopia. Nat. Commun. 2024, 15, 6703. [Google Scholar] [CrossRef] [PubMed]
- Yao, Y.; Chen, Z.; Wu, Q.; Lu, Y.; Zhou, X.; Zhu, X. Single-cell RNA sequencing of retina revealed novel transcriptional landscape in high myopia and underlying cell-type-specific mechanisms. Med. Comm. 2023, 4, e372. [Google Scholar] [CrossRef] [PubMed]
- Yao, Y.; Wei, L.; Chen, Z.; Li, H.; Qi, J.; Wu, Q.; Zhou, X.; Lu, Y.; Zhu, X. Single-cell RNA sequencing: Inhibited Notch2 signalling underlying the increased lens fibre cells differentiation in high myopia. Cell Prolif. 2023, 56, e13412. [Google Scholar] [CrossRef]
- Hao, J.; Xie, Y.; Wei, H.; Yang, Z.; Zhang, R.; Ma, Z.; Zhang, M.; Du, X.; Yin, X.; Liu, J.; et al. Electroacupuncture Slows Experimental Myopia Progression by Improving Retinal Mitochondrial Function: A Study Based on Single-Cell RNA Sequencing. Adv. Biol. 2024, 8, e2400269. [Google Scholar] [CrossRef]
- Ligocki, A.J.; Fury, W.; Gutierrez, C.; Adler, C.; Yang, T.; Ni, M.; Bai, Y.; Wei, Y.; Lehmann, G.L.; Romano, C. Molecular characteristics and spatial distribution of adult human corneal cell subtypes. Sci. Rep. 2021, 11, 16323. [Google Scholar] [CrossRef]
- Li, J.M.; Kim, S.; Zhang, Y.; Bian, F.; Hu, J.; Lu, R.; Pflugfelder, S.C.; Chen, R.; Li, D.Q. Single-Cell Transcriptomics Identifies a Unique Entity and Signature Markers of Transit-Amplifying Cells in Human Corneal Limbus. Investig. Ophthalmol. Vis. Sci. 2021, 62, 36. [Google Scholar] [CrossRef]
- Li, Y.; Jeong, J.; Song, W. Molecular Characteristics and Distribution of Adult Human Corneal Immune Cell Types. Front. Immunol. 2022, 13, 798346. [Google Scholar] [CrossRef]
- Zhang, X.; Han, P.; Qiu, J.; Huang, F.; Luo, Q.; Cheng, J.; Shan, K.; Yang, Y.; Zhang, C. Single-cell RNA sequencing reveals the complex cellular niche of pterygium. Ocul. Surf. 2024, 32, 91–103. [Google Scholar] [CrossRef]
- Liu, Z.; Xie, H.; Li, L.; Jiang, D.; Qian, Y.; Zhu, X.; Dai, M.; Li, Y.; Wei, R.; Luo, Z.; et al. Single-cell landscape reveals the epithelial cell-centric pro-inflammatory immune microenvironment in dry eye development. Mucosal Immunol. 2024, 17, 491–507. [Google Scholar] [CrossRef]
- Niu, X.; Xu, M.; Zhu, J.; Zhang, S.; Yang, Y. Identification of the immune-associated characteristics and predictive biomarkers of keratoconus based on single-cell RNA-sequencing and bulk RNA-sequencing. Front. Immunol. 2023, 14, 1220646. [Google Scholar] [CrossRef]
- Buffault, J.; Reboussin, É.; Blond, F.; Guillonneau, X.; Bastelica, P.; Kessal, K.; Akkurt Arslan, M.; Melik-Parsadaniantz, S.; Réaux-le Goazigo, A.; Labbé, A.; et al. RNA-seq transcriptomic profiling of TGF-β2-exposed human trabecular meshwork explants: Advancing insights beyond conventional cell culture models. Exp. Cell Res. 2024, 442, 114220. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, M.; Sato, T.; Tsugeno, Y.; Umetsu, A.; Suzuki, S.; Furuhashi, M.; Ida, Y.; Hikage, F.; Ohguro, H. Human Trabecular Meshwork (HTM) Cells Treated with TGF-β2 or Dexamethasone Respond to Compression Stress in Different Manners. Biomedicines 2022, 10, 1338. [Google Scholar] [CrossRef]
- Carnes, M.U.; Allingham, R.R.; Ashley-Koch, A.; Hauser, M.A. Transcriptome analysis of adult and fetal trabecular meshwork, cornea, and ciliary body tissues by RNA sequencing. Exp. Eye Res. 2018, 167, 91–99. [Google Scholar] [CrossRef] [PubMed]
- Patel, G.; Fury, W.; Yang, H.; Gomez-Caraballo, M.; Bai, Y.; Yang, T.; Adler, C.; Wei, Y.; Ni, M.; Schmitt, H.; et al. Molecular taxonomy of human ocular outflow tissues defined by single-cell transcriptomics. Proc. Natl. Acad. Sci. USA 2020, 117, 12856–12867. [Google Scholar] [CrossRef] [PubMed]
- Balasubramanian, R.; Kizhatil, K.; Li, T.; Tolman, N.; Bhandari, A.; Clark, G.; Bupp-Chickering, V.; Kelly, R.A.; Zhou, S.; Peregrin, J.; et al. Transcriptomic profiling of Schlemm’s canal cells reveals a lymphatic-biased identity and three major cell states. eLife 2024, 13, RP96459. [Google Scholar] [CrossRef]
- Jia, X.; Wu, J.; Chen, X.; Hou, S.; Li, Y.; Zhao, L.; Zhu, Y.; Li, Z.; Deng, C.; Su, W.; et al. Cell atlas of trabecular meshwork in glaucomatous non-human primates and DEGs related to tissue contract based on single-cell transcriptomics. iScience 2023, 26, 108024. [Google Scholar] [CrossRef]
- van Zyl, T.; Yan, W.; McAdams, A.; Peng, Y.R.; Shekhar, K.; Regev, A.; Juric, D.; Sanes, J.R. Cell atlas of aqueous humor outflow pathways in eyes of humans and four model species provides insight into glaucoma pathogenesis. Proc. Natl. Acad. Sci. USA 2020, 117, 10339–10349. [Google Scholar] [CrossRef] [PubMed]
- González-Loyola, A.; Bovay, E.; Kim, J.; Lozano, T.W.; Sabine, A.; Renevey, F.; Arroz-Madeira, S.; Rapin, A.; Wypych, T.P.; Rota, G.; et al. FOXC2 controls adult lymphatic endothelial specialization, function, and gut lymphatic barrier preventing multiorgan failure. Sci. Adv. 2021, 7, eabf4335. [Google Scholar] [CrossRef] [PubMed]
- Medina-Trillo, C.; Aroca-Aguilar, J.D.; Ferre-Fernández, J.J.; Alexandre-Moreno, S.; Morales, L.; Méndez-Hernández, C.D.; García-Feijoo, J.; Escribano, J. Role of FOXC2 and PITX2 rare variants associated with mild functional alterations as modifier factors in congenital glaucoma. PLoS ONE 2019, 14, e0211029. [Google Scholar] [CrossRef] [PubMed]
- Ujiie, N.; Norden, P.R.; Fang, R.; Beckmann, L.; Cai, Z.; Kweon, J.; Liu, T.; Tan, C.; Kuhn, M.S.; Stamer, W.D.; et al. Differential roles of FOXC2 in the trabecular meshwork and Schlemm’s canal in glaucomatous pathology. Life Sci. Alliance 2023, 6, e202201721. [Google Scholar] [CrossRef]
- Thomson, B.R.; Liu, P.; Onay, T.; Du, J.; Tompson, S.W.; Misener, S.; Purohit, R.R.; Young, T.L.; Jin, J.; Quaggin, S.E. Cellular crosstalk regulates the aqueous humor outflow pathway and provides new targets for glaucoma therapies. Nat. Commun. 2021, 12, 6072. [Google Scholar] [CrossRef]
- Ashok, A.; Singh, N.; Chaudhary, S.; Bellamkonda, V.; Kritikos, A.E.; Wise, A.S.; Rana, N.; McDonald, D.; Ayyagari, R. Retinal Degeneration and Alzheimer’s Disease: An Evolving Link. Int. J. Mol. Sci. 2020, 21, 7290. [Google Scholar] [CrossRef]
- Pan, L.; Cho, K.S.; Wei, X.; Xu, F.; Lennikov, A.; Hu, G.; Tang, J.; Guo, S.; Chen, J.; Kriukov, E.; et al. IGFBPL1 is a master driver of microglia homeostasis and resolution of neuroinflammation in glaucoma and brain tauopathy. Cell Rep. 2023, 42, 112889. [Google Scholar] [CrossRef]
- Wang, S.; Tong, S.; Jin, X.; Li, N.; Dang, P.; Sui, Y.; Liu, Y.; Wang, D. Single-cell RNA sequencing analysis of the retina under acute high intraocular pressure. Neural Regen. Res. 2024, 19, 2522–2531. [Google Scholar] [CrossRef]
- Yao, M.; Zeng, Z.; Li, S.; Zou, Z.; Chen, Z.; Chen, X.; Gao, Q.; Zhao, G.; Chen, A.; Li, Z.; et al. CRISPR-CasRx-mediated disruption of Aqp1/Adrb2/Rock1/Rock2 genes reduces intraocular pressure and retinal ganglion cell damage in mice. Nat. Commun. 2024, 15, 6395. [Google Scholar] [CrossRef]
- Zhu, L.; Li, H.; Wang, R.; Li, Z.; Zhao, S.; Peng, X.; Su, W. Identification of Hif1α as a Potential Participant in Autoimmune Uveitis Pathogenesis Using Single-Cell Transcriptome Analysis. Investig. Ophthalmol. Vis. Sci. 2023, 64, 24. [Google Scholar] [CrossRef]
- Quinn, J.; Salman, A.; Paluch, C.; Jackson-Wood, M.; McClements, M.E.; Luo, J.; Davis, S.J.; Cornall, R.J.; MacLaren, R.E.; Dendrou, C.A.; et al. Single-cell transcriptomic analysis of retinal immune regulation and blood-retinal barrier function during experimental autoimmune uveitis. Sci. Rep. 2024, 14, 20033. [Google Scholar] [CrossRef]
- Kang, H.; Sun, H.; Yang, Y.; Tuong, Z.K.; Shu, M.; Wei, Y.; Zhang, Y.; Yu, D.; Tao, Y. Autoimmune uveitis in Behcet’s disease and Vogt-Koyanagi-Harada disease differ in tissue immune infiltration and T cell clonality. Clin. Transl. Immunol. 2023, 12, e1461. [Google Scholar] [CrossRef]
- Duan, R.; Xie, L.; Li, H.; Wang, R.; Liu, X.; Tao, T.; Yang, S.; Gao, Y.; Lin, X.; Su, W. Insights gained from Single-Cell analysis of immune cells on Cyclosporine A treatment in autoimmune uveitis. Biochem. Pharmacol. 2022, 202, 115116. [Google Scholar] [CrossRef]
- Wang, R.; Zhu, L.; Li, H.; Peng, X.; Zhao, S.; Su, W. Single-Cell transcriptomes of immune cells provide insights into the therapeutic effects of mycophenolate mofetil on autoimmune uveitis. Int. Immunopharmacol. 2023, 119, 110223. [Google Scholar] [CrossRef] [PubMed]
- Zhu, L.; Li, H.; Peng, X.; Li, Z.; Zhao, S.; Wu, D.; Chen, J.; Li, S.; Jia, R.; Li, Z.; et al. Beneficial mechanisms of dimethyl fumarate in autoimmune uveitis: Insights from single-cell RNA sequencing. J. Neuroinflammation 2024, 21, 112. [Google Scholar] [CrossRef] [PubMed]
- Gao, Y.; Duan, R.; Li, H.; Jiang, L.; Tao, T.; Liu, X.; Zhu, L.; Li, Z.; Chen, B.; Zheng, S.; et al. Single-cell analysis of immune cells on gingiva-derived mesenchymal stem cells in experimental autoimmune uveitis. iScience 2023, 26, 106729. [Google Scholar] [CrossRef] [PubMed]
- Yuan, F.; Zhang, R.; Li, J.; Lei, Q.; Wang, S.; Jiang, F.; Guo, Y.; Xiang, M. CCR5-overexpressing mesenchymal stem cells protect against experimental autoimmune uveitis: Insights from single-cell transcriptome analysis. J. Neuroinflammation 2024, 21, 136. [Google Scholar] [CrossRef]
- Lukowski, S.W.; Lo, C.Y.; Sharov, A.A.; Nguyen, Q.; Fang, L.; Hung, S.S.; Zhu, L.; Zhang, T.; Grunert, U.; Nguyen, T.; et al. A single-cell transcriptome atlas of the adult human retina. EMBO J. 2019, 38, e100811. [Google Scholar] [CrossRef]
- Liang, Q.; Dharmat, R.; Owen, L.; Shakoor, A.; Li, Y.; Kim, S.; Vitale, A.; Kim, I.; Morgan, D.; Liang, S.; et al. Single-nuclei RNA-seq on human retinal tissue provides improved transcriptome profiling. Nat. Commun. 2019, 10, 5743. [Google Scholar] [CrossRef]
- Yamagata, M.; Yan, W.; Sanes, J.R. A cell atlas of the chick retina based on single-cell transcriptomics. eLife 2021, 10, e63907. [Google Scholar] [CrossRef] [PubMed]
- Teo, Z.L.; Tham, Y.C.; Yu, M.; Chee, M.L.; Rim, T.H.; Cheung, N.; Bikbov, M.M.; Wang, Y.X.; Tang, Y.; Lu, Y.; et al. Global Prevalence of Diabetic Retinopathy and Projection of Burden through 2045: Systematic Review and Meta-analysis. Ophthalmology 2021, 128, 1580–1591. [Google Scholar] [CrossRef]
- Van Hove, I.; De Groef, L.; Boeckx, B.; Modave, E.; Hu, T.-T.; Beets, K.; Etienne, I.; Van Bergen, T.; Lambrechts, D.; Moons, L.; et al. Single-cell transcriptome analysis of the Akimba mouse retina reveals cell-type-specific insights into the pathobiology of diabetic retinopathy. Diabetologia 2020, 63, 2235–2248. [Google Scholar] [CrossRef]
- Ma, P.; Zhang, P.; Chen, S.; Shi, W.; Ye, J.; Chen, S.; Ju, R.; Liu, B.; Zheng, Y.; Liu, Y. Immune Cell Landscape of Patients With Diabetic Macular Edema by Single-Cell RNA Analysis. Front. Pharmacol. 2021, 12, 754933. [Google Scholar] [CrossRef]
- Ben, S.; Ma, Y.; Bai, Y.; Zhang, Q.; Zhao, Y.; Xia, J.; Yao, M. Microglia-endothelial cross-talk regulates diabetes-induced retinal vascular dysfunction through remodeling inflammatory microenvironment. iScience 2024, 27, 109145. [Google Scholar] [CrossRef]
- Wang, Y.; Yang, X.; Li, Q.; Zhang, Y.; Chen, L.; Hong, L.; Xie, Z.; Yang, S.; Deng, X.; Cao, M.; et al. Single-cell RNA sequencing reveals the Muller subtypes and inner blood-retinal barrier regulatory network in early diabetic retinopathy. Front. Mol. Neurosci. 2022, 15, 1048634. [Google Scholar] [CrossRef]
- Corano Scheri, K.; Lavine, J.A.; Tedeschi, T.; Thomson, B.R.; Fawzi, A.A. Single-cell transcriptomics analysis of proliferative diabetic retinopathy fibrovascular membranes reveals AEBP1 as fibrogenesis modulator. JCI Insight 2023, 8, e172062. [Google Scholar] [CrossRef] [PubMed]
- Orozco, L.D.; Chen, H.H.; Cox, C.; Katschke, K.J., Jr.; Arceo, R.; Espiritu, C.; Caplazi, P.; Nghiem, S.S.; Chen, Y.J.; Modrusan, Z.; et al. Integration of eQTL and a Single-Cell Atlas in the Human Eye Identifies Causal Genes for Age-Related Macular Degeneration. Cell Rep. 2020, 30, 1246–1259.e6. [Google Scholar] [CrossRef]
- Wang, W.; Lin, P.; Wang, S.; Zhang, G.; Chen, C.; Lu, X.; Zhuang, Y.; Su, J.; Wang, H.; Xu, L. In-depth mining of single-cell transcriptome reveals the key immune-regulated loops in age-related macular degeneration. Front. Mol. Neurosci. 2023, 16, 1173123. [Google Scholar] [CrossRef] [PubMed]
- Lyu, Y.; Zauhar, R.; Dana, N.; Strang, C.E.; Hu, J.; Wang, K.; Liu, S.; Pan, N.; Gamlin, P.; Kimble, J.A.; et al. Implication of specific retinal cell-type involvement and gene expression changes in AMD progression using integrative analysis of single-cell and bulk RNA-seq profiling. Sci. Rep. 2021, 11, 15612. [Google Scholar] [CrossRef]
- Dhirachaikulpanich, D.; Lagger, C.; Chatsirisupachai, K.; de Magalhães, J.P.; Paraoan, L. Intercellular communication analysis of the human retinal pigment epithelial and choroidal cells predicts pathways associated with aging, cellular senescence and age-related macular degeneration. Front. Aging Neurosci. 2022, 14, 1016293. [Google Scholar] [CrossRef]
- Xia, M.; Jiao, L.; Wang, X.H.; Tong, M.; Yao, M.D.; Li, X.M.; Yao, J.; Li, D.; Zhao, P.Q.; Yan, B. Single-cell RNA sequencing reveals a unique pericyte type associated with capillary dysfunction. Theranostics 2023, 13, 2515–2530. [Google Scholar] [CrossRef]
- Shi, H.; Tian, H.; Zhu, T.; Liao, Q.; Liu, C.; Yuan, P.; Li, Y.; Yang, J.; Zong, C.; Jia, S.; et al. Single-cell sequencing depicts tumor architecture and empowers clinical decision in metastatic conjunctival melanoma. Cell Discov. 2024, 10, 63. [Google Scholar] [CrossRef]
- Liao, Q.; Shi, H.; Yang, J.; Ge, S.; Jia, R.; Song, X.; Chai, P.; Jia, R. FTO elicits tumor neovascularization in cancer-associated fibroblasts through eliminating m(6)A modifications of multiple pro-angiogenic factors. Cancer Lett. 2024, 592, 216911. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Tang, Z.; Wan, Q.; Huang, W.; Li, X.; Huang, X.; Zheng, S.; Lu, C.; Wu, J.; Li, Z.; et al. Machine learning and single-cell RNA sequencing reveal relationship between intratumor CD8(+) T cells and uveal melanoma metastasis. Cancer Cell Int. 2024, 24, 359. [Google Scholar] [CrossRef] [PubMed]
- Li, K.; Huang, J.; Tan, Y.; Sun, J.; Zhou, M. Single-cell and bulk transcriptome analysis reveals tumor cell heterogeneity and underlying molecular program in uveal melanoma. J. Transl. Med. 2024, 22, 1020. [Google Scholar] [CrossRef] [PubMed]
- Li, K.; Sun, L.; Wang, Y.; Cen, Y.; Zhao, J.; Liao, Q.; Wu, W.; Sun, J.; Zhou, M. Single-cell characterization of macrophages in uveal melanoma uncovers transcriptionally heterogeneous subsets conferring poor prognosis and aggressive behavior. Exp. Mol. Med. 2023, 55, 2433–2444. [Google Scholar] [CrossRef]
- Li, J.; Cao, D.; Jiang, L.; Zheng, Y.; Shao, S.; Zhuang, A.; Xiang, D. ITGB2-ICAM1 axis promotes liver metastasis in BAP1-mutated uveal melanoma with retained hypoxia and ECM signatures. Cell. Oncol. 2024, 47, 951–965. [Google Scholar] [CrossRef]
- Liu, Y.; Hu, W.; Xie, Y.; Tang, J.; Ma, H.; Li, J.; Nie, J.; Wang, Y.; Gao, Y.; Cheng, C.; et al. Single-cell transcriptomics enable the characterization of local extension in retinoblastoma. Commun. Biol. 2024, 7, 11. [Google Scholar] [CrossRef]
- Cuadrado-Vilanova, M.; Liu, J.; Paco, S.; Aschero, R.; Burgueno, V.; Sirab, N.; Pascual-Pasto, G.; Correa, G.; Balaguer-Lluna, L.; Castillo-Ecija, H.; et al. Identification of immunosuppressive factors in retinoblastoma cell secretomes and aqueous humor from patients. J. Pathol. 2022, 257, 327–339. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Li, Y.; Han, Y.; Feng, Y.; Zhou, M.; Zong, C.; He, X.; Jia, R.; Xu, X.; Fan, J. Single-cell transcriptome profiling reveals intratumoural heterogeneity and malignant progression in retinoblastoma. Cell Death Dis. 2021, 12, 1100. [Google Scholar] [CrossRef] [PubMed]
- Tran, H.T.N.; Ang, K.S.; Chevrier, M.; Zhang, X.; Lee, N.Y.S.; Goh, M.; Chen, J. A benchmark of batch-effect correction methods for single-cell RNA sequencing data. Genome Biol. 2020, 21, 12. [Google Scholar] [CrossRef] [PubMed]
- Leek, J.T.; Scharpf, R.B.; Bravo, H.C.; Simcha, D.; Langmead, B.; Johnson, W.E.; Geman, D.; Baggerly, K.; Irizarry, R.A. Tackling the widespread and critical impact of batch effects in high-throughput data. Nat. Rev. Genet. 2010, 11, 733–739. [Google Scholar] [CrossRef]
- Wang, S.K.; Nair, S.; Li, R.; Kraft, K.; Pampari, A.; Patel, A.; Kang, J.B.; Luong, C.; Kundaje, A.; Chang, H.Y. Single-cell multiome of the human retina and deep learning nominate causal variants in complex eye diseases. Cell Genom. 2022, 2, 100164. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cheng, Y.; Gu, S.; Lu, X.; Pei, C. The Advance of Single-Cell RNA Sequencing Applications in Ocular Physiology and Disease Research. Biomolecules 2025, 15, 1120. https://doi.org/10.3390/biom15081120
Cheng Y, Gu S, Lu X, Pei C. The Advance of Single-Cell RNA Sequencing Applications in Ocular Physiology and Disease Research. Biomolecules. 2025; 15(8):1120. https://doi.org/10.3390/biom15081120
Chicago/Turabian StyleCheng, Ying, Sihan Gu, Xueqing Lu, and Cheng Pei. 2025. "The Advance of Single-Cell RNA Sequencing Applications in Ocular Physiology and Disease Research" Biomolecules 15, no. 8: 1120. https://doi.org/10.3390/biom15081120
APA StyleCheng, Y., Gu, S., Lu, X., & Pei, C. (2025). The Advance of Single-Cell RNA Sequencing Applications in Ocular Physiology and Disease Research. Biomolecules, 15(8), 1120. https://doi.org/10.3390/biom15081120