
V-ATPase and Lysosomal Energy Sensing in Periodontitis and Medicine-Related Osteonecrosis of the Jaw

Abstract
1. Introduction
2. Lysosomal Energy Sensing and Periodontitis
2.1. Rapamycin/Sirolimus
2.2. Metformin
2.3. Glucagon-like Peptide-1 (GLP-1) Agonists
2.4. Salicylate
2.5. Resveratrol
2.6. 2-Deoxy-D-Glucose
2.7. Dorsomorphin (Compound C)
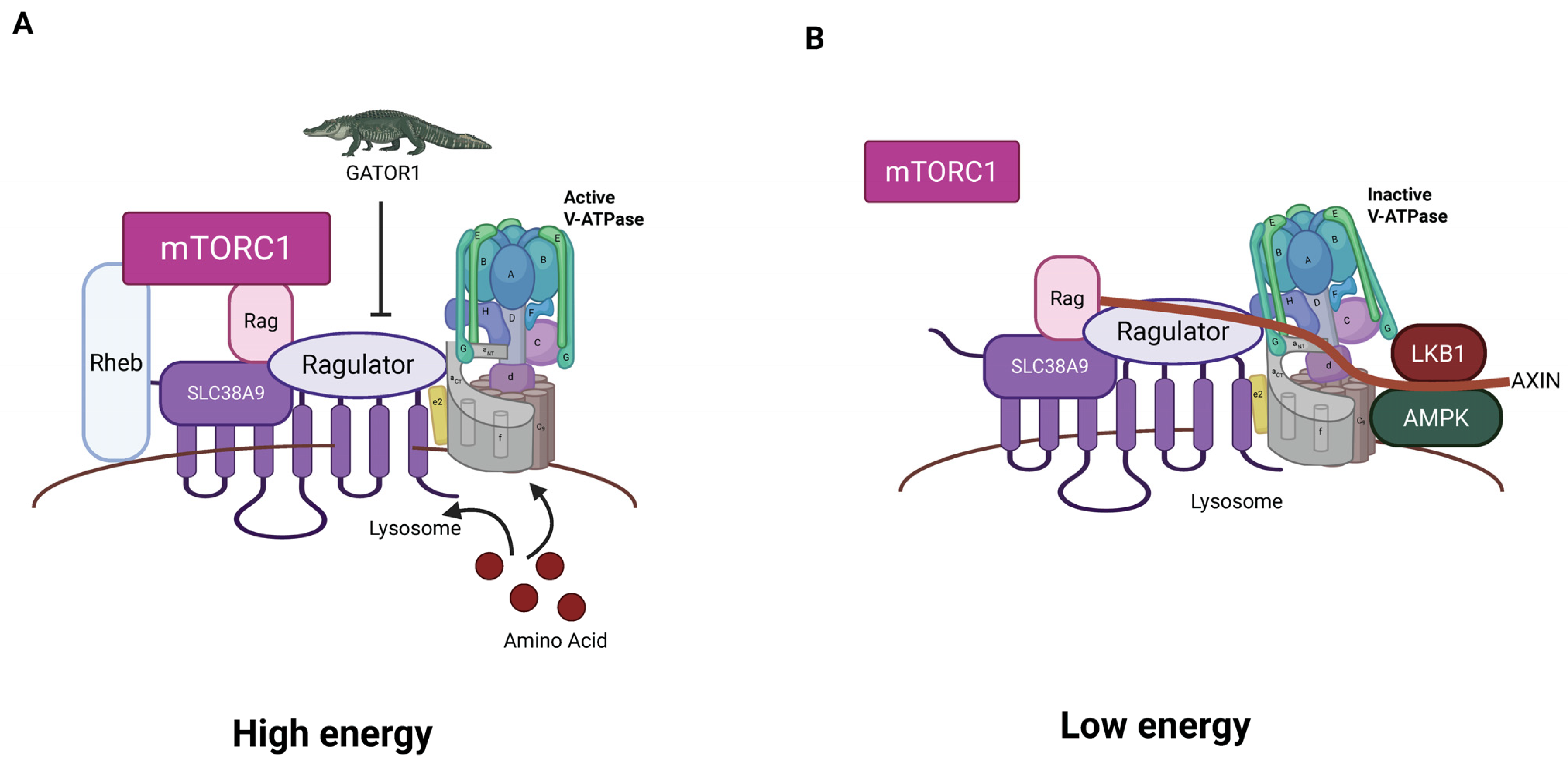
3. Key Players in Lysosomal Energy Sensing
3.1. V-ATPase

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Protein | Location | Disease |
|---|---|---|---|
| ATP6V1A | A | ubiquitous | Developmental and epileptic encephalopathies, cutis laxa, cardiac abnormalities, dysmorphic facial features, and severe hypotonia lysosome dysfunction [98,99] |
| ATP6V1B1 | B1 | kidney, epididymis | Renal tubular acidosis with deafness [100] |
| ATP6V1B2 | B2 | ubiquitous | Dominant Deafness–Onychodystrophy Syndrome (DDOD), Zimmermann–Laband Syndrome 2 (ZLS2), DOORS (Deafness, Onychodystrophy, Osteodystrophy, Intellectual Disability, and Seizures [101,102,103] |
| ATP6V1C1 | C1 | ubiquitous | DOORS syndrome (Deafness, Onychodystrophy, Osteodystrophy, Impaired Intellectual Development, And Seizures Syndrome), autosomal recessive osteopetrosis, and other lysosomal storage disorders [102] |
| ATP6V1C2 | C2 | lung, kidney | Renal tubular acidosis with deafness [104] |
| ATP6V1D | D | ubiquitous | |
| ATP6V1E1 | E1 | testis | Autosomal recessive cutis laxa type Iic [103] |
| ATP6V1E2 | E2 | ubiquitous | |
| ATP6V1F | F | ubiquitous | |
| ATP6V1G1 | G1 | ubiquitous | |
| ATP6V1G2 | G2 | neural | |
| ATP6V1G3 | G3 | kidney, epididymis | |
| ATP6V!H | H | ubiquitous | lysosome dysfunction [98,99] |
| ATP6V0a1 | a1 | ubiquitous | Developmental and epileptic encephalopathy: early-onset seizures, developmental delays, intellectual disabilities, neurodegenerative disorders bone, kidney disorders. Neurodegenerative disorders: Parkinson’s disease, Alzheimer’s disease, and other conditions affecting the brain [105,106] |
| ATP6V0a2 | a2 | ubiquitous | Autosomal recessive cutis laxa type IIA (ARCL2A) and, in some cases, wrinkly skin syndrome [107] |
| ATP6V0a3 | a3 | osteoclast (ubiquitous?) | Autosomal recessive osteopetrosis [108] |
| ATP6V0a4 | a4 | kidney, epididymis | Autosomal recessive distal renal tubular acidosis, renal tubular acidosis with deafness [109] |
| ATP6V0b | b, c″ | ubiquitous | |
| ATP6V0c | c | ubiquitous | |
| ATP6V0d1 | d1 | ubiquitous | |
| ATP6V0d2 | d2 | kidney, epididymis | |
| ATP6V0e | e | ubiquitous | |
| ATP6AP1 | AP1, AC45 | ubiquitous | Congenital Disorder of Glycosylation type, Follicular lymphoma, immunodeficiency with hepatopathy, cognitive impairment [110,111,112] |
| ATP6AP2 | AP2, Prorenin Receptor | ubiquitous | X-linked syndromic intellectual disability (Hedera type), X-linked Parkinsonism–spasticity syndrome, and congenital disorder of glycosylation type 2R autophagic liver disease [113,114] |
3.2. mTORC1
3.3. L-AMPK
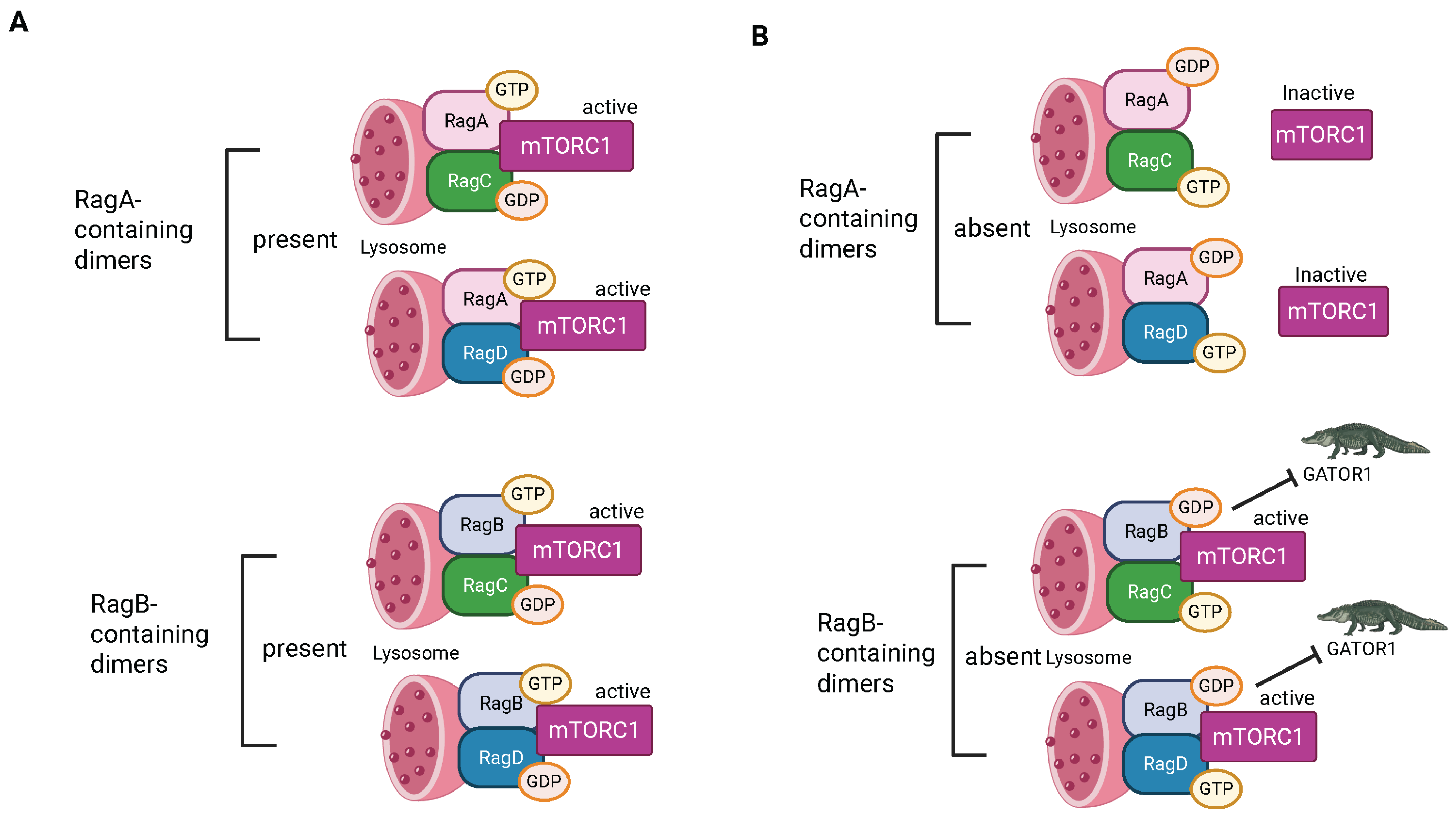
3.4. Ragulator/RAG Complex
3.5. Associations Between V-ATPase, Ragulator, L-AMPK, and mTORC1
3.6. Sugar Sensing by Aldolase
3.7. Role of V-ATPase Assembly and Disassembly
4. Energy Sensing and Periodontal Disease: How Does Inflammation Result from Energy Sensing?
4.1. RAGE Activation and AMPK
4.2. Pattern Recognition Receptors (PRRs)
5. Energy Sensing and MRONJ
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| V-ATPase | vacuolar H+-ATPase |
| L-AMPK | lysosomal AMP-activated protein kinase complex |
| mTORC1 | mammalian target of rapamycin complex 1 |
| MRONJ | medicine-related osteonecrosis of the jaw |
| RAG | Ras-related GTPase |
| AXIN | axis inhibition protein |
| GLP-1 | Glucagon-like Peptide-1 |
| FOXO | Forkhead Box O |
| 2-DG | 2-Deoxy-d-glucose |
| TDLc2 | TBC/LysM-Associated Domain Containing 2 |
| mEAK7 | Mammalian Enhancer-of-Akt-1–7 |
| RAPTOR | regulatory-associated protein of mTOR |
| MLST8 | mammalian lethal with sec-13 protein 8 |
| PRAS40 | proline-rich Akt substrate of 40 kDa |
| TTI1 | Tel2 interacting protein 1 |
| TEL2 | Telomere maintenance 2 interacting protein 2 |
| DEPTOR DEP | domain containing mTOR-interacting protein |
| RICTOR | Rapamycin-insensitive companion of mTOR |
| PROCTOR | protein observed with RICTOR |
| LKB-1 | Liver Kinase B1 |
| PEN2 | presenilin enhancer 2 |
| TSC2 | Tuberous sclerosis complex 2 |
| GEF | Guanine exchange factor |
| GAP | GTPase-activating protein |
| SLC38A9 | Solute carrier family 38 member 9 |
| GATOR | GTPase-activating protein toward Rags |
| Rheb | Ras homolog enriched in brain |
| PI-3 kinase | Phosphatidylinositol 3-kinase |
| AKT | Protein kinase B |
| LAMTOR | Late endosomal/lysosomal adaptor and MAPK and mTOR activator |
| TRPV | Transient Receptor Potential V |
| AGE | Advanced glycation end product |
| PRR | Pattern Recognition Receptors |
| TLR | Toll-like receptor |
| NOD1 | nucleotide-binding oligomerization domain-containing protein 1 |
| cGAS | cyclic GMP-AMP-synthase |
| STING | Stimulator of Interferon Genes |
| BRONJ | bisphosphonate-related osteonecrosis of the jaw |
| RANK | receptor activator of nuclear factor κ B |
References
- Glickman, I. The Relation of Experimental Diabetes to Periodontal Disease. Am. J. Orthod. 1947, 33, 703–722. [Google Scholar] [CrossRef]
- Glickman, I. The Periodontium, Pancreas and Blood Sugar Levels in Experimental Diabetes. J. Dent. Res. 1946, 25, 169. [Google Scholar] [PubMed]
- Glickman, I. The Periodontal Structures in Experimental Diabetes. N. Y. J. Dent. 1946, 16, 226–251. [Google Scholar]
- Gonzalez, A.; Hall, M.N.; Lin, S.C.; Hardie, D.G. Ampk and Tor: The Yin and Yang of Cellular Nutrient Sensing and Growth Control. Cell Metab. 2020, 31, 472–492. [Google Scholar] [CrossRef] [PubMed]
- Collins, M.P.; Forgac, M. Regulation and Function of V-Atpases in Physiology and Disease. Biochim. Biophys. Acta Biomembr. 2020, 1862, 183341. [Google Scholar] [CrossRef]
- Eaton, A.F.; Merkulova, M.; Brown, D. The H(+)-Atpase (V-Atpase): From Proton Pump to Signaling Complex in Health and Disease. Am. J. Physiol.-Cell Physiol. 2021, 320, C392–C414. [Google Scholar] [CrossRef]
- Holliday, L.S. Vacuolar H(+)-Atpases (V-Atpases) as Therapeutic Targets: A Brief Review and Recent Developments. Biotarget 2017, 1, 675430. [Google Scholar] [CrossRef]
- Abbas, Y.M.; Wu, D.; Bueler, S.A.; Robinson, C.V.; Rubinstein, J.L. Structure of V-Atpase from the Mammalian Brain. Science 2020, 367, 1240–1246. [Google Scholar] [CrossRef]
- Collins, M.P.; Forgac, M. Regulation of V-Atpase Assembly in Nutrient Sensing and Function of V-Atpases in Breast Cancer Metastasis. Front. Physiol. 2018, 9, 902. [Google Scholar] [CrossRef]
- Hardie, D.G.; Lin, S.C. Amp-Activated Protein Kinase-Not Just an Energy Sensor. F1000Research 2017, 6, 1724. [Google Scholar] [CrossRef]
- Szwed, A.; Kim, E.; Jacinto, E. Regulation and Metabolic Functions of Mtorc1 and Mtorc2. Physiol. Rev. 2021, 101, 1371–1426. [Google Scholar] [CrossRef] [PubMed]
- Bar-Peled, L.; Sabatini, D.M. Regulation of Mtorc1 by Amino Acids. Trends Cell Biol. 2014, 24, 400–406. [Google Scholar] [CrossRef] [PubMed]
- Tsujimoto, K.; Takamatsu, H.; Kumanogoh, A. The Ragulator Complex: Delving Its Multifunctional Impact on Metabolism and Beyond. Inflamm. Regen. 2023, 43, 28. [Google Scholar] [CrossRef]
- Qiu, L.; Sun, Y.; Ning, H.; Chen, G.; Zhao, W.; Gao, Y. The Scaffold Protein Axin1: Gene Ontology, Signal Network, and Physiological Function. Cell Commun. Signal. 2024, 22, 77. [Google Scholar] [CrossRef]
- Condon, K.J.; Sabatini, D.M. Nutrient Regulation of Mtorc1 at a Glance. J. Cell Sci. 2019, 132, jcs222570. [Google Scholar] [CrossRef] [PubMed]
- Saxton, R.A.; Sabatini, D.M. Mtor Signaling in Growth, Metabolism, and Disease. Cell 2017, 168, 960–976. [Google Scholar] [CrossRef]
- Lama-Sherpa, T.D.; Jeong, M.H.; Jewell, J.L. Regulation of Mtorc1 by the Rag Gtpases. Biochem. Soc. Trans. 2023, 51, 655–664. [Google Scholar] [CrossRef]
- Jewell, J.L. Rag-Ulating Mtorc1 with Amino Acids. Nat. Rev. Mol. Cell Biol. 2021, 22, 587. [Google Scholar] [CrossRef]
- Lamming, D.W. Inhibition of the Mechanistic Target of Rapamycin (Mtor)-Rapamycin and Beyond. Cold Spring Harb. Perspect. Med. 2016, 6, a25924. [Google Scholar] [CrossRef]
- An, J.Y.; Quarles, E.K.; Mekvanich, S.; Kang, A.; Liu, A.; Santos, D.; Miller, R.A.; Rabinovitch, P.S.; Cox, T.C.; Kaeberlein, M. Rapamycin Treatment Attenuates Age-Associated Periodontitis in Mice. Geroscience 2017, 39, 457–463. [Google Scholar] [CrossRef]
- Li, X.; Chang, B.; Wang, B.; Bu, W.; Zhao, L.; Liu, J.; Meng, L.; Wang, L.; Xin, Y.; Wang, D.; et al. Rapamycin Promotes Osteogenesis under Inflammatory Conditions. Mol. Med. Rep. 2017, 16, 8923–8929. [Google Scholar] [CrossRef] [PubMed]
- Feng, C.; Liu, Y.; Zhang, B.Y.; Zhang, H.; Shan, F.Y.; Li, T.Q.; Zhao, Z.N.; Wang, X.X.; Zhang, X.Y. Rapamycin Inhibits Osteoclastogenesis and Prevents Lps-Induced Alveolar Bone Loss by Oxidative Stress Suppression. ACS Omega 2023, 8, 20739–20754. [Google Scholar] [CrossRef] [PubMed]
- Hudson, J.; Kaeberlein, T.; Mahal, A.; Wong, N.; Ghorbanifarajzadeh, M.; Radella, F.; Isman, A.; Nyquist, A.; Zalzala, S.; Haddad, G.; et al. Evaluation of Off-Label Rapamycin Use on Oral Health. Geroscience 2024, 46, 4135–4146. [Google Scholar] [CrossRef]
- King, R.; Tanna, N.; Patel, V. Medication-Related Osteonecrosis of the Jaw Unrelated to Bisphosphonates and Denosumab—A Review. Oral Surg. Oral Med. Oral Pathol. Oral Radiol. 2019, 127, 289–299. [Google Scholar] [CrossRef]
- Kim, J.; You, Y.J. Regulation of Organelle Function by Metformin. IUBMB Life 2017, 69, 459–469. [Google Scholar] [CrossRef] [PubMed]
- Foretz, M.; Guigas, B.; Viollet, B. Metformin: Update on Mechanisms of Action and Repurposing Potential. Nat. Rev. Endocrinol. 2023, 19, 460–476. [Google Scholar] [CrossRef]
- Isop, L.M.; Neculau, A.E.; Necula, R.D.; Kakucs, C.; Moga, M.A.; Dima, L. Metformin: The Winding Path from Understanding Its Molecular Mechanisms to Proving Therapeutic Benefits in Neurodegenerative Disorders. Pharmaceuticals 2023, 16, 1714. [Google Scholar] [CrossRef]
- Ma, T.; Tian, X.; Zhang, B.; Li, M.; Wang, Y.; Yang, C.; Wu, J.; Wei, X.; Qu, Q.; Yu, Y.; et al. Low-Dose Metformin Targets the Lysosomal Ampk Pathway through Pen2. Nature 2022, 603, 159–165. [Google Scholar] [CrossRef]
- Neves, V.C.M.; Okajima, L.S.; Elbahtety, E.; Joseph, S.; Daly, J.; Menon, A.; Fan, D.; Volkyte, A.; Mainas, G.; Fung, K.; et al. Repurposing Metformin for Periodontal Disease Management as a Form of Oral-Systemic Preventive Medicine. J. Transl. Med. 2023, 21, 655. [Google Scholar] [CrossRef]
- Silva, F.F.V.E.; Chauca-Bajana, L.; Caponio, V.C.A.; Cueva, K.A.S.; Velasquez-Ron, B.; Padin-Iruegas, M.E.; Almeida, L.L.; Lorenzo-Pouso, A.I.; Suarez-Penaranda, J.M.; Perez-Sayans, M. Regeneration of Periodontal Intrabony Defects Using Platelet-Rich Fibrin (Prf): A Systematic Review and Network Meta-Analysis. Odontology 2024, 112, 1047–1068. [Google Scholar] [CrossRef]
- Miron, R.J.; Moraschini, V.; Estrin, N.E.; Shibli, J.A.; Cosgarea, R.; Jepsen, K.; Jervoe-Storm, P.M.; Sculean, A.; Jepsen, S. Periodontal Regeneration Using Platelet-Rich Fibrin. Furcation Defects: A Systematic Review with Meta-Analysis. Periodontology 2000, 2024; Online ahead of print. [Google Scholar]
- Neves, V.C.M.; Savchenko, V.; Daly, J.; Sharpe, P. Periodontal Ageing and Its Management Via Pharmacological Glucose Modulation. Front. Dent. Med. 2024, 5, 1415960. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Song, J.H.; Kim, J.W.; Kwon, S.H.; Piao, X.; Oh, S.H.; Park, S.G.; Kim, S.H.; Ryu, J.H.; Kim, O.S.; et al. Metformin Reverses Periodontal Destruction Caused by Experimental Periodontitis by Inhibiting Interleukin-1β Activity. J. Periodontol. 2025; Online ahead of print. [Google Scholar]
- Liu, F.; Han, R.; Nie, S.; Cao, Y.; Zhang, X.; Gao, F.; Wang, Z.; Xing, L.; Ouyang, Z.; Sui, L.; et al. Metformin Rejuvenates Nap1l2-Impaired Immunomodulation of Bone Marrow Mesenchymal Stem Cells Via Metabolic Reprogramming. Cell Prolif. 2024, 57, e13612. [Google Scholar] [CrossRef] [PubMed]
- Jia, L.; Xiong, Y.; Zhang, W.; Ma, X.; Xu, X. Metformin Promotes Osteogenic Differentiation and Protects against Oxidative Stress-Induced Damage in Periodontal Ligament Stem Cells Via Activation of the Akt/Nrf2 Signaling Pathway. Exp. Cell Res. 2020, 386, 111717. [Google Scholar] [CrossRef]
- Xiang, M.; Liu, Y.; Guo, Q.; Liao, C.; Xiao, L.; Xiang, M.; Guan, X.; Liu, J. Metformin Enhances the Therapeutic Effects of Extracellular Vesicles Derived from Human Periodontal Ligament Stem Cells on Periodontitis. Sci. Rep. 2024, 14, 19940. [Google Scholar] [CrossRef]
- Ren, C.; Hao, X.; Wang, L.; Hu, Y.; Meng, L.; Zheng, S.; Ren, F.; Bu, W.; Wang, H.; Li, D.; et al. Metformin Carbon Dots for Promoting Periodontal Bone Regeneration Via Activation of Erk/Ampk Pathway. Adv. Health Mater. 2021, 10, e2100196. [Google Scholar] [CrossRef] [PubMed]
- Tseng, C.H. Metformin and Risk of Gingival/Periodontal Diseases in Diabetes Patients: A Retrospective Cohort Study. Front. Endocrinol. 2022, 13, 1036885. [Google Scholar] [CrossRef] [PubMed]
- Park, S.H.; Kang, M.A.; Moon, Y.J.; Jang, K.Y.; Kim, J.R. Metformin Coordinates Osteoblast/Osteoclast Differentiation Associated with Ischemic Osteonecrosis. Aging 2020, 12, 4727–4741. [Google Scholar] [CrossRef]
- Nakagawa, T.; Tsuka, S.; Aonuma, F.; Nodai, T.; Munemasa, T.; Tamura, A.; Mukaibo, T.; Kondo, Y.; Masaki, C.; Hosokawa, R. Effects of Metformin on the Prevention of Bisphosphonate-Related Osteonecrosis of the Jaw-Like Lesions in Rats. J. Prosthodont. Res. 2021, 65, 219–224. [Google Scholar] [CrossRef]
- Zhuang, J.; Zu, J.; Zhou, C.; Sun, Y.; Kong, P.; Jing, Y. Bioinformatic Data Mining for Candidate Drugs Affecting Risk of Bisphosphonate-Related Osteonecrosis of the Jaw (Bronj) in Cancer Patients. Dis. Markers 2022, 2022, 3348480. [Google Scholar] [CrossRef]
- Drucker, D.J. Mechanisms of Action and Therapeutic Application of Glucagon-Like Peptide-1. Cell Metab. 2018, 27, 740–756. [Google Scholar] [CrossRef]
- Muller, T.D.; Finan, B.; Bloom, S.R.; D’Alessio, D.; Drucker, D.J.; Flatt, P.R.; Fritsche, A.; Gribble, F.; Grill, H.J.; Habener, J.F.; et al. Glucagon-Like Peptide 1 (Glp-1). Mol. Metab. 2019, 30, 72–130. [Google Scholar] [CrossRef] [PubMed]
- Joy, S.V.; Rodgers, P.T.; Scates, A.C. Incretin Mimetics as Emerging Treatments for Type 2 Diabetes. Ann. Pharmacother. 2005, 39, 110–118. [Google Scholar] [CrossRef] [PubMed]
- Nauck, M.A.; Meier, J.J. Glucagon-Like Peptide 1 and Its Derivatives in the Treatment of Diabetes. Regul. Pept. 2005, 128, 135–148. [Google Scholar] [CrossRef]
- Lu, W.; Zhou, Z.; Jiang, N.; Han, J. An Updated Patent Review of Glp-1 Receptor Agonists (2020-Present). Expert. Opin. Ther. Pat. 2023, 33, 597–612. [Google Scholar] [CrossRef]
- Birajdar, S.V.; Mazahir, F.; Alam, M.I.; Kumar, A.; Yadav, A.K. Repurposing and Clinical Attributes of Antidiabetic Drugs for the Treatment of Neurodegenerative Disorders. Eur. J. Pharmacol. 2023, 961, 176117. [Google Scholar] [CrossRef]
- Sawada, N.; Adachi, K.; Nakamura, N.; Miyabe, M.; Ito, M.; Kobayashi, S.; Miyajima, S.I.; Suzuki, Y.; Kikuchi, T.; Mizutani, M.; et al. Glucagon-Like Peptide-1 Receptor Agonist Liraglutide Ameliorates the Development of Periodontitis. J. Diabetes Res. 2020, 2020, 8843310. [Google Scholar] [CrossRef]
- Wang, Z.; Wang, X.; Zhang, L.; Wang, B.; Xu, B.; Zhang, J. Glp-1 Inhibits Pkcbeta2 Phosphorylation to Improve the Osteogenic Differentiation Potential of Hpdlscs in the Age Microenvironment. J. Diabetes Complicat. 2020, 34, 107495. [Google Scholar] [CrossRef]
- Gheonea, T.C.; Surlin, P.; Nicolae, F.M.; Gheorghe, D.N.; Popescu, D.M.; Rogoveanu, I. Dipeptidyl-Peptidase-4 and Glucagon-Like-Peptide-1, a Link in the Connection between Periodontitis and Diabetes Mellitus-What Do We Know So Far?—A Scoping Review. J. Clin. Med. 2024, 13, 903. [Google Scholar] [CrossRef] [PubMed]
- Solini, A.; Suvan, J.; Santini, E.; Gennai, S.; Seghieri, M.; Masi, S.; Petrini, M.; D’Aiuto, F.; Graziani, F. Periodontitis Affects Glucoregulatory Hormones in Severely Obese Individuals. Int. J. Obes. 2019, 43, 1125–1129. [Google Scholar] [CrossRef]
- Mohamed, H.G.; Idris, S.B.; Mustafa, M.; Ahmed, M.F.; Astrom, A.N.; Mustafa, K.; Ibrahim, S.O. Impact of Chronic Periodontitis on Levels of Glucoregulatory Biomarkers in Gingival Crevicular Fluid of Adults with and without Type 2 Diabetes. PLoS ONE 2015, 10, e0127660. [Google Scholar] [CrossRef]
- Suvan, J.; Masi, S.; Harrington, Z.; Santini, E.; Raggi, F.; D’Aiuto, F.; Solini, A. Effect of Treatment of Periodontitis on Incretin Axis in Obese and Nonobese Individuals: A Cohort Study. J. Clin. Endocrinol. Metab. 2021, 106, e74–e82. [Google Scholar] [CrossRef] [PubMed]
- Ohara-Nemoto, Y.; Nakasato, M.; Shimoyama, Y.; Baba, T.T.; Kobayakawa, T.; Ono, T.; Yaegashi, T.; Kimura, S.; Nemoto, T.K. Degradation of Incretins and Modulation of Blood Glucose Levels by Periodontopathic Bacterial Dipeptidyl Peptidase 4. Infect. Immun. 2017, 85, e00277-17. [Google Scholar] [CrossRef]
- Ahmad, P.; Estrin, N.; Farshidfar, N.; Zhang, Y.; Miron, R.J. Glucagon-Like Peptide 1 Receptor Agonists (Glp-1ras) Improve Periodontal and Peri-Implant Health in Type 2 Diabetes Mellitus. J. Periodontal. Res. 2025; Online ahead of print. [Google Scholar]
- Hawley, S.A.; Fullerton, M.D.; Ross, F.A.; Schertzer, J.D.; Chevtzoff, C.; Walker, K.J.; Peggie, M.W.; Zibrova, D.; Green, K.A.; Mustard, K.J.; et al. The Ancient Drug Salicylate Directly Activates Amp-Activated Protein Kinase. Science 2012, 336, 918–922. [Google Scholar] [CrossRef]
- Shi, Y.; Zhang, R.; Da, N.; Wang, Y.; Yang, J.; Li, B.; He, X. Aspirin Loaded Extracellular Vesicles Inhibit Inflammation of Macrophages Via Switching Metabolic Phenotype in Periodontitis. Biochem. Biophys. Res. Commun. 2023, 667, 25–33. [Google Scholar] [CrossRef] [PubMed]
- Wu, L.; Luo, Z.; Liu, Y.; Jia, L.; Jiang, Y.; Du, J.; Guo, L.; Bai, Y.; Liu, Y. Aspirin Inhibits Rankl-Induced Osteoclast Differentiation in Dendritic Cells by Suppressing Nf-Kappab and Nfatc1 Activation. Stem Cell Res. Ther. 2019, 10, 375. [Google Scholar] [CrossRef]
- Hasan, F.; Ikram, R.; Simjee, S.U.; Iftakhar, K.; Asadullah, K.; Usman, M. The Effects of Aspirin Gel and Mouthwash on Levels of Salivary Biomarkers Pge2, Tnf-Alpha and Nitric Oxide in Patients with Periodontal Diseases. Pak. J. Pharm. Sci. 2019, 32, 2019–2023. [Google Scholar] [PubMed]
- Rogina, B.; Tissenbaum, H.A. Sirt1, Resveratrol and Aging. Front. Genet. 2024, 15, 1393181. [Google Scholar] [CrossRef]
- Zhou, D.D.; Cheng, J.; Li, J.; Wu, S.X.; Xiong, R.G.; Huang, S.Y.; Cheung, P.C.; Li, H.B. Resveratrol and Its Analogues: Anti-Ageing Effects and Underlying Mechanisms. Subcell Biochem. 2024, 107, 183–203. [Google Scholar]
- Zhu, L.; Yang, M.; Fan, L.; Yan, Q.; Zhang, L.; Mu, P.; Lu, F. Interaction between Resveratrol and Sirt1: Role in Neurodegenerative Diseases. Naunyn Schmiedebergs Arch. Pharmacol. 2025, 398, 89–101. [Google Scholar] [CrossRef]
- AlHayani, D.A.; Kubaev, A.; Uthirapathy, S.; Mandaliya, V.; Ballal, S.; Kalia, R.; Arya, R.; Gabble, B.C.; Alasheqi, M.Q.; Kadhim, A.J. Insights into the Therapeutic Potential of Sirt1-Modifying Compounds for Alzheimer’s Disease: A Focus on Molecular Mechanisms. J. Mol. Neurosci. 2025, 75, 29. [Google Scholar] [CrossRef]
- Nikniaz, S.; Vaziri, F.; Mansouri, R. Impact of Resveratrol Supplementation on Clinical Parameters and Inflammatory Markers in Patients with Chronic Periodontitis: A Randomized Clinical Trail. BMC Oral. Health 2023, 23, 177. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.N.; Han, L.; Iyer, S.; de Cabo, R.; Zhao, H.; O’Brien, C.A.; Manolagas, S.C.; Almeida, M. Sirtuin1 Suppresses Osteoclastogenesis by Deacetylating Foxos. Mol. Endocrinol. 2015, 29, 1498–1509. [Google Scholar] [CrossRef]
- Qu, Q.; Chen, Y.; Wang, Y.; Wang, W.; Long, S.; Yang, H.Y.; Wu, J.; Li, M.; Tian, X.; Wei, X.; et al. Lithocholic Acid Binds Tulp3 to Activate Sirtuins and Ampk to Slow down Ageing. Nature, 2024; Online ahead of print. [Google Scholar]
- Blair, H.C.; Teitelbaum, S.L.; Ghiselli, R.; Gluck, S. Osteoclastic Bone Resorption by a Polarized Vacuolar Proton Pump. Science 1989, 245, 855–857. [Google Scholar] [CrossRef]
- Huang, C.Y.; Chen, S.H.; Lin, T.; Liao, Y.W.; Chang, Y.C.; Chen, C.C.; Yu, C.C.; Chen, C.J. Resveratrol Attenuates Advanced Glycation End Product-Induced Senescence and Inflammation in Human Gingival Fibroblasts. J. Dent. Sci. 2024, 19, 580–586. [Google Scholar] [CrossRef]
- Shi, J.; Zhang, Y.; Zhang, X.; Chen, R.; Wei, J.; Hou, J.; Wang, B.; Lai, H.; Huang, Y. Remodeling Immune Microenvironment in Periodontitis Using Resveratrol Liposomes as an Antibiotic-Free Therapeutic Strategy. J. Nanobiotechnol. 2021, 19, 429. [Google Scholar] [CrossRef]
- Jiang, K.; Li, J.; Jiang, L.; Li, H.; Lei, L. Pink1-Mediated Mitophagy Reduced Inflammatory Responses to Porphyromonas Gingivalis in Macrophages. Oral Dis. 2023, 29, 3665–3676. [Google Scholar] [CrossRef] [PubMed]
- Hashim, N.T.; Babiker, R.; Chaitanya, N.; Mohammed, R.; Priya, S.P.; Padmanabhan, V.; Ahmed, A.; Dasnadi, S.P.; Islam, M.S.; Gismalla, B.G.; et al. New Insights in Natural Bioactive Compounds for Periodontal Disease: Advanced Molecular Mechanisms and Therapeutic Potential. Molecules 2025, 30, 807. [Google Scholar] [CrossRef]
- Yang, G.; Collins, J.M.; Rafiee, R.; Singh, S.; Langaee, T.; McDonough, C.W.; Holliday, L.S.; Wang, D.; Lamba, J.K.; Kim, Y.S.; et al. Sirt1 Gene Snp Rs932658 Is Associated with Medication-Related Osteonecrosis of the Jaw. J. Bone Miner. Res. 2021, 36, 347–356. [Google Scholar] [CrossRef] [PubMed]
- Bojtor, B.; Vaszilko, M.; Armos, R.; Tobias, B.; Podani, J.; Szentpeteri, S.; Balla, B.; Lengyel, B.; Piko, H.; Illes, A.; et al. Analysis of Sirt1 Gene Snps and Clinical Characteristics in Medication-Related Osteonecrosis of the Jaw. Int. J. Mol. Sci. 2024, 25, 3646. [Google Scholar] [CrossRef]
- Cui, Y.; Zhang, W.; Yang, P.; Zhu, S.; Luo, S.; Li, M. Menaquinone-4 Prevents Medication-Related Osteonecrosis of the Jaw through the Sirt1 Signaling-Mediated Inhibition of Cellular Metabolic Stresses-Induced Osteoblast Apoptosis. Free Radic. Biol. Med. 2023, 206, 33–49. [Google Scholar] [CrossRef]
- Su, W.; Li, J.; Jiang, L.; Lei, L.; Li, H. Hexokinase 2-Mediated Glycolysis Supports Inflammatory Responses to Porphyromonas Gingivalis in Gingival Fibroblasts. BMC Oral. Health 2023, 23, 103. [Google Scholar] [CrossRef] [PubMed]
- He, Y.; Wang, Y.; Jia, X.; Li, Y.; Yang, Y.; Pan, L.; Zhao, R.; Han, Y.; Wang, F.; Guan, X.; et al. Glycolytic Reprogramming Controls Periodontitis-Associated Macrophage Pyroptosis Via Ampk/Sirt1/Nf-Kappab Signaling Pathway. Int. Immunopharmacol. 2023, 119, 110192. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Li, F.; Zhu, C.; Zhang, C.; Le, Y.; Li, Z.; Wan, Q. The Glycolytic Enzyme Pkm2 Regulates Inflammatory Osteoclastogenesis by Modulating Stat3 Phosphorylation. J. Biol. Chem. 2025, 301, 108389. [Google Scholar] [CrossRef]
- Lee, Y.S.; Kim, Y.S.; Lee, S.Y.; Kim, G.H.; Kim, B.J.; Lee, S.H.; Lee, K.U.; Kim, G.S.; Kim, S.W.; Koh, J.M. Amp Kinase Acts as a Negative Regulator of Rankl in the Differentiation of Osteoclasts. Bone 2010, 47, 926–937. [Google Scholar] [CrossRef]
- Wang, H.; Rubinstein, J.L. Cryoem of V-Atpases: Assembly, Disassembly, and Inhibition. Curr. Opin. Struct. Biol. 2023, 80, 102592. [Google Scholar] [CrossRef]
- Wang, R.; Long, T.; Hassan, A.; Wang, J.; Sun, Y.; Xie, X.S.; Li, X. Cryo-Em Structures of Intact V-Atpase from Bovine Brain. Nat. Commun. 2020, 11, 3921. [Google Scholar] [CrossRef]
- Cotter, K.; Stransky, L.; McGuire, C.; Forgac, M. Recent Insights into the Structure, Regulation, and Function of the V-Atpases. Trends Biochem. Sci. 2015, 40, 611–622. [Google Scholar] [CrossRef] [PubMed]
- Feng, R.; Liu, F.; Li, R.; Zhou, Z.; Lin, Z.; Lin, S.; Deng, S.; Li, Y.; Nong, B.; Xia, Y.; et al. The Rapid Proximity Labeling System Phastid Identifies Atp6ap1 as an Unconventional Gef for Rheb. Cell Res. 2024, 34, 355–369. [Google Scholar] [CrossRef]
- Li, S.; Ouyang, X.; Su, B. Atp6ap1 Was Phast-Id’ed as a Long-Sought Gef for Rheb. Cell Res. 2024, 34, 397–398. [Google Scholar] [CrossRef]
- Chu, A.; Zirngibl, R.A.; Manolson, M.F. The V-Atpase A3 Subunit: Structure, Function and Therapeutic Potential of an Essential Biomolecule in Osteoclastic Bone Resorption. Int. J. Mol. Sci. 2021, 22, 6934. [Google Scholar] [CrossRef]
- Nakanishi-Matsui, M.; Matsumoto, N. V-Atpase A3 Subunit in Secretory Lysosome Trafficking in Osteoclasts. Biol. Pharm. Bull. 2022, 45, 1426–1431. [Google Scholar] [CrossRef] [PubMed]
- Susani, L.; Pangrazio, A.; Sobacchi, C.; Taranta, A.; Mortier, G.; Savarirayan, R.; Villa, A.; Orchard, P.; Vezzoni, P.; Albertini, A.; et al. Tcirg1-Dependent Recessive Osteopetrosis: Mutation Analysis, Functional Identification of the Splicing Defects, and in Vitro Rescue by U1 Snrna. Hum. Mutat. 2004, 24, 225–235. [Google Scholar] [CrossRef]
- Frattini, A.; Orchard, P.J.; Sobacchi, C.; Giliani, S.; Abinun, M.; Mattsson, J.P.; Keeling, D.J.; Andersson, A.K.; Wallbrandt, P.; Zecca, L.; et al. Defects in Tcirg1 Subunit of the Vacuolar Proton Pump Are Responsible for a Subset of Human Autosomal Recessive Osteopetrosis. Nat. Genet. 2000, 25, 343–346. [Google Scholar] [CrossRef] [PubMed]
- Shadur, B.; Zaidman, I.; NaserEddin, A.; Lokshin, E.; Hussein, F.; Oron, H.C.; Avni, B.; Grisariu, S.; Stepensky, P. Successful Hematopoietic Stem Cell Transplantation for Osteopetrosis Using Reduced Intensity Conditioning. Pediatr. Blood Cancer 2018, 65, e27010. [Google Scholar] [CrossRef]
- Kantaputra, P.N.; Thawanaphong, S.; Issarangporn, W.; Klangsinsirikul, P.; Ohazama, A.; Sharpe, P.; Supanchart, C. Long-Term Survival in Infantile Malignant Autosomal Recessive Osteopetrosis Secondary to Homozygous P.Arg526gln Mutation in Clcn7. Am. J. Med. Genet. A 2012, 158A, 909–916. [Google Scholar] [CrossRef]
- Even-Or, E.; Stepensky, P. How We Approach Malignant Infantile Osteopetrosis. Pediatr. Blood Cancer 2021, 68, e28841. [Google Scholar] [CrossRef]
- Oot, R.A.; Wilkens, S. Human V-Atpase Function Is Positively and Negatively Regulated by Tldc Proteins. Structure 2024, 32, 989–1000.e6. [Google Scholar] [CrossRef] [PubMed]
- Eaton, A.F.; Danielson, E.C.; Tu, L.J.; Brown, D.; Merkulova, M. Knockout of the V-Atpase Interacting Protein Tldc2 in B-Type Kidney Intercalated Cells Impairs Urine Alkalinization. Am. J. Physiol. Ren. Physiol. 2025, 328, F890–F906. [Google Scholar] [CrossRef]
- Tan, Y.Z.; Abbas, Y.M.; Wu, J.Z.; Wu, D.; Keon, K.A.; Hesketh, G.G.; Bueler, S.A.; Gingras, A.C.; Robinson, C.V.; Grinstein, S.; et al. Cryoem of Endogenous Mammalian V-Atpase Interacting with the Tldc Protein Meak-7. Life Sci. Alliance 2022, 5, e202201527. [Google Scholar] [CrossRef]
- Heitman, J.; Movva, N.R.; Hall, M.N. Targets for Cell Cycle Arrest by the Immunosuppressant Rapamycin in Yeast. Science 1991, 253, 905–909. [Google Scholar] [CrossRef]
- Heitman, J.; Koller, A.; Kunz, J.; Henriquez, R.; Schmidt, A.; Movva, N.R.; Hall, M.N. The Immunosuppressant Fk506 Inhibits Amino Acid Import in Saccharomyces Cerevisiae. Mol. Cell Biol. 1993, 13, 5010–5019. [Google Scholar] [PubMed]
- Arriola Apelo, S.I.; Lamming, D.W. Rapamycin: An Inhibitor of Aging Emerges from the Soil of Easter Island. J. Gerontol. A Biol. Sci. Med. Sci. 2016, 71, 841–849. [Google Scholar] [CrossRef] [PubMed]
- Sabatini, D.M.; Erdjument-Bromage, H.; Lui, M.; Tempst, P.; Snyder, S.H. Raft1: A Mammalian Protein That Binds to Fkbp12 in a Rapamycin-Dependent Fashion and Is Homologous to Yeast Tors. Cell 1994, 78, 35–43. [Google Scholar] [CrossRef]
- Sirchia, F.; Taietti, I.; Donesana, M.; Bassanese, F.; Clemente, A.M.; Barbato, E.; Orsini, A.; Ferretti, A.; Marseglia, G.L.; Savasta, S.; et al. Expanding the Spectrum of Autosomal Dominant ATP6V1A-Related Disease: Case Report and Literature Review. Genes 2024, 15, 1219. [Google Scholar] [CrossRef]
- Li, B.; Song, L.; Liu, X.; Ji, J.-J.; He, Y.-Y.; Zhang, D.-M.; Xu, J.; Sun, H.; Shi, Z.; Wang, J.; et al. ATP6V1A variants are associated with childhood epilepsy with favorable outcome. Seizure Eur. J. Epilepsy 2024, 116, 81–86. [Google Scholar] [CrossRef]
- Stover, E.H.; Borthwick, K.J.; Bavalia, C.; Eady, N.; Fritz, D.M.; Rungroj, N.; Giersch, A.B.; Morton, C.C.; Axon, P.R.; Akil, I.; et al. Novel Atp6v1b1 and Atp6v0a4 Mutations in Autosomal Recessive Distal Renal Tubular Acidosis with New Evidence for Hearing Loss. J. Med. Genet. 2002, 39, 796–803. [Google Scholar] [CrossRef] [PubMed]
- Wang, F.; Yang, Y.; Klionsky, D.J.; Malek, S.N. Mutations in V-Atpase in Follicular Lymphoma Activate Autophagic Flux Creating a Targetable Dependency. Autophagy 2023, 19, 716–719. [Google Scholar] [CrossRef]
- Carpentieri, G.; Cecchetti, S.; Bocchinfuso, G.; Radio, F.C.; Leoni, C.; Onesimo, R.; Calligari, P.; Pietrantoni, A.; Ciolfi, A.; Ferilli, M.; et al. Dominantly Acting Variants In atp6v1c1 and atp6v1b2 Cause a Multisystem Phenotypic Spectrum by Altering Lysosomal and/or Autophagosome Function. Hum. Genet. Genom. Adv. 2024, 5, 100349. [Google Scholar] [CrossRef]
- Van Damme, T.; Gardeitchik, T.; Mohamed, M.; Guerrero-Castillo, S.; Freisinger, P.; Guillemyn, B.; Kariminejad, A.; Dalloyaux, D.; van Kraaij, S.; Lefeber, D.J.; et al. Mutations in Atp6v1e1 or Atp6v1a Cause Autosomal-Recessive Cutis Laxa. Am. J. Hum. Genet. 2017, 100, 216–227. [Google Scholar] [CrossRef]
- Corniere, N.; Eladari, D. Identification of Atp6v1c2 as a Novel Candidate Gene for Distal Tubular Acidosis. Kidney Int. 2020, 97, 452–455. [Google Scholar] [CrossRef]
- Aoto, K.; Kato, M.; Akita, T.; Nakashima, M.; Mutoh, H.; Akasaka, N.; Tohyama, J.; Nomura, Y.; Hoshino, K.; Ago, Y.; et al. Atp6v0a1 Encoding the A1-Subunit of the V0 Domain of Vacuolar H(+)-Atpases Is Essential for Brain Development in Humans and Mice. Nat. Commun. 2021, 12, 2107. [Google Scholar] [CrossRef]
- Bott, L.C.; Forouhan, M.; Lieto, M.; Sala, A.J.; Ellerington, R.; Johnson, J.O.; Speciale, A.A.; Criscuolo, C.; Filla, A.; Chitayat, D.; et al. Variants in Atp6v0a1 Cause Progressive Myoclonus Epilepsy and Developmental and Epileptic Encephalopathy. Brain Commun. 2021, 3, fcab245. [Google Scholar] [CrossRef] [PubMed]
- Hucthagowder, V.; Morava, E.; Kornak, U.; Lefeber, D.J.; Fischer, B.; Dimopoulou, A.; Aldinger, A.; Choi, J.; Davis, E.C.; Abuelo, D.N.; et al. Loss-of-Function Mutations in Atp6v0a2 Impair Vesicular Trafficking, Tropoelastin Secretion and Cell Survival. Hum. Mol. Genet. 2009, 18, 2149–2165. [Google Scholar] [CrossRef] [PubMed]
- Scimeca, J.C.; Quincey, D.; Parrinello, H.; Romatet, D.; Grosgeorge, J.; Gaudray, P.; Philip, N.; Fischer, A.; Carle, G.F. Novel Mutations in the Tcirg1 Gene Encoding the A3 Subunit of the Vacuolar Proton Pump in Patients Affected by Infantile Malignant Osteopetrosis. Hum. Mutat. 2003, 21, 151–157. [Google Scholar] [CrossRef]
- Smith, A.N.; Skaug, J.; Choate, K.A.; Nayir, A.; Bakkaloglu, A.; Ozen, S.; Hulton, S.A.; Sanjad, S.A.; Al-Sabban, E.A.; Lifton, R.P.; et al. Mutations in Atp6n1b, Encoding a New Kidney Vacuolar Proton Pump 116-Kd Subunit, Cause Recessive Distal Renal Tubular Acidosis with Preserved Hearing. Nat. Genet. 2000, 26, 71–75. [Google Scholar] [CrossRef]
- Morales-Romero, B.; Munoz-Pujol, G.; Artuch, R.; Garcia-Cazorla, A.; O’Callaghan, M.; Sykut-Cegielska, J.; Campistol, J.; Moreno-Lozano, P.J.; Oud, M.M.; Wevers, R.A.; et al. Genome and Rna Sequencing Were Essential to Reveal Cryptic Intronic Variants Associated to Defective Atp6ap1 Mrna Processing. Mol. Genet. Metab. 2024, 142, 108511. [Google Scholar] [CrossRef] [PubMed]
- Dimitrov, B.; Himmelreich, N.; Ederveen, A.L.H.; Luchtenborg, C.; Okun, J.G.; Breuer, M.; Hutter, A.M.; Carl, M.; Guglielmi, L.; Hellwig, A.; et al. Cutis Laxa, Exocrine Pancreatic Insufficiency and Altered Cellular Metabolomics as Additional Symptoms in a New Patient with Atp6ap1-Cdg. Mol. Genet. Metab. 2018, 123, 364–374. [Google Scholar] [CrossRef]
- Jansen, E.J.; Timal, S.; Ryan, M.; Ashikov, A.; van Scherpenzeel, M.; Graham, L.A.; Mandel, H.; Hoischen, A.; Iancu, T.C.; Raymond, K.; et al. Atp6ap1 Deficiency Causes an Immunodeficiency with Hepatopathy, Cognitive Impairment and Abnormal Protein Glycosylation. Nat. Commun. 2016, 7, 11600. [Google Scholar] [CrossRef]
- Hirose, T.; Cabrera-Socorro, A.; Chitayat, D.; Lemonnier, T.; Feraud, O.; Cifuentes-Diaz, C.; Gervasi, N.; Mombereau, C.; Ghosh, T.; Stoica, L.; et al. Atp6ap2 Variant Impairs Cns Development and Neuronal Survival to Cause Fulminant Neurodegeneration. J. Clin. Investig. 2019, 129, 2145–2162. [Google Scholar] [CrossRef]
- Cannata Serio, M.; Rujano, M.A.; Simons, M. Mutations in Atp6ap2 Cause Autophagic Liver Disease in Humans. Autophagy 2018, 14, 1088–1089. [Google Scholar] [CrossRef]
- Lorenz, M.C.; Heitman, J. Tor Mutations Confer Rapamycin Resistance by Preventing Interaction with Fkbp12-Rapamycin. J. Biol. Chem. 1995, 270, 27531–27537. [Google Scholar] [CrossRef]
- Alarcon, C.M.; Cardenas, M.E.; Heitman, J. Mammalian Raft1 Kinase Domain Provides Rapamycin-Sensitive Tor Function in Yeast. Genes. Dev. 1996, 10, 279–288. [Google Scholar] [CrossRef]
- Laplante, M.; Sabatini, D.M. Regulation of Mtorc1 and Its Impact on Gene Expression at a Glance. J. Cell Sci. 2013, 126, 1713–1719. [Google Scholar] [CrossRef] [PubMed]
- Sabatini, D.M. Mtor and Cancer: Insights into a Complex Relationship. Nat. Rev. Cancer 2006, 6, 729–734. [Google Scholar] [CrossRef] [PubMed]
- Almeida, L.O.; Silva, L.C.; Emerick, C.; Santos, J.A.D.; Castilho, R.M.; Squarize, C.H. Head and Neck Cancer Stem Cell Maintenance Relies on Mtor Signaling, Specifically Involving the Mechanistic Target of Rapamycin Complexes 1 and 2 (Mtorc1 and Mtorc2). Arch. Oral. Biol. 2024, 157, 105840. [Google Scholar] [CrossRef]
- Efeyan, A.; Zoncu, R.; Sabatini, D.M. Amino Acids and Mtorc1: From Lysosomes to Disease. Trends Mol. Med. 2012, 18, 524–533. [Google Scholar] [CrossRef] [PubMed]
- Xu, W.; Chen, H.; Xiao, H. Mtorc2: A Neglected Player in Aging Regulation. J. Cell Physiol. 2024, 239, e31363. [Google Scholar] [CrossRef]
- Ragupathi, A.; Kim, C.; Jacinto, E. The Mtorc2 Signaling Network: Targets and Cross-Talks. Biochem. J. 2024, 481, 45–91. [Google Scholar] [CrossRef]
- Sarbassov, D.D.; Ali, S.M.; Kim, D.H.; Guertin, D.A.; Latek, R.R.; Erdjument-Bromage, H.; Tempst, P.; Sabatini, D.M. Rictor, a Novel Binding Partner of Mtor, Defines a Rapamycin-Insensitive and Raptor-Independent Pathway That Regulates the Cytoskeleton. Curr. Biol. 2004, 14, 1296–1302. [Google Scholar] [CrossRef]
- Zong, Y.; Zhang, C.S.; Li, M.; Wang, W.; Wang, Z.; Hawley, S.A.; Ma, T.; Feng, J.W.; Tian, X.; Qi, Q.; et al. Hierarchical Activation of Compartmentalized Pools of Ampk Depends on Severity of Nutrient or Energy Stress. Cell Res. 2019, 29, 460–473. [Google Scholar] [CrossRef]
- Lin, S.C.; Hardie, D.G. Ampk: Sensing Glucose as Well as Cellular Energy Status. Cell Metab. 2018, 27, 299–313. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.S.; Hawley, S.A.; Zong, Y.; Li, M.; Wang, Z.; Gray, A.; Ma, T.; Cui, J.; Feng, J.W.; Zhu, M.; et al. Fructose-1,6-Bisphosphate and Aldolase Mediate Glucose Sensing by Ampk. Nature 2017, 548, 112–116. [Google Scholar] [CrossRef]
- Lu, M.; Holliday, L.S.; Zhang, L.; Dunn, W.A., Jr.; Gluck, S.L. Interaction between Aldolase and Vacuolar H+-Atpase: Evidence for Direct Coupling of Glycolysis to the Atp-Hydrolyzing Proton Pump. J. Biol. Chem. 2001, 276, 30407–30413. [Google Scholar] [CrossRef] [PubMed]
- Kelly, D.M.; O’Hara, P.V.; Kelleher, E.M.; Casserly, L.F. Osteopetrosis with Renal Tubular Acidosis and Cerebral Calcification. Kidney Int. 2018, 93, 1020. [Google Scholar] [CrossRef] [PubMed]
- Whyte, M.P. Carbonic Anhydrase Ii Deficiency. Bone 2023, 169, 116684. [Google Scholar] [CrossRef]
- Lu, M.; Ammar, D.; Ives, H.; Albrecht, F.; Gluck, S.L. Physical Interaction between Aldolase and Vacuolar H+-Atpase Is Essential for the Assembly and Activity of the Proton Pump. J. Biol. Chem. 2007, 282, 24495–24503. [Google Scholar] [CrossRef]
- Lu, M.; Sautin, Y.Y.; Holliday, L.S.; Gluck, S.L. The Glycolytic Enzyme Aldolase Mediates Assembly, Expression, and Activity of Vacuolar H+-Atpase. J. Biol. Chem. 2004, 279, 8732–8739. [Google Scholar] [CrossRef]
- Van Nostrand, J.L.; Hellberg, K.; Luo, E.C.; Van Nostrand, E.L.; Dayn, A.; Yu, J.; Shokhirev, M.N.; Dayn, Y.; Yeo, G.W.; Shaw, R.J. Ampk Regulation of Raptor and Tsc2 Mediate Metformin Effects on Transcriptional Control of Anabolism and Inflammation. Genes. Dev. 2020, 34, 1330–1344. [Google Scholar] [CrossRef]
- Wang, S.; Li, H.; Yuan, M.; Fan, H.; Cai, Z. Role of Ampk in Autophagy. Front. Physiol. 2022, 13, 1015500. [Google Scholar] [CrossRef]
- Herzig, S.; Shaw, R.J. Ampk: Guardian of Metabolism and Mitochondrial Homeostasis. Nat. Rev. Mol. Cell Biol. 2018, 19, 121–135. [Google Scholar] [CrossRef]
- Valenstein, M.L.; Lalgudi, P.V.; Gu, X.; Kedir, J.F.; Taylor, M.S.; Chivukula, R.R.; Sabatini, D.M. Rag-Ragulator Is the Central Organizer of the Physical Architecture of the Mtorc1 Nutrient-Sensing Pathway. Proc. Natl. Acad. Sci. USA 2024, 121, e2322755121. [Google Scholar] [CrossRef] [PubMed]
- Mu, Z.; Wang, L.; Deng, W.; Wang, J.; Wu, G. Structural Insight into the Ragulator Complex Which Anchors Mtorc1 to the Lysosomal Membrane. Cell Discov. 2017, 3, 17049. [Google Scholar] [CrossRef]
- Shen, K.; Sabatini, D.M. Ragulator and Slc38a9 Activate the Rag Gtpases through Noncanonical Gef Mechanisms. Proc. Natl. Acad. Sci. USA 2018, 115, 9545–9550. [Google Scholar] [CrossRef] [PubMed]
- Rogala, K.B.; Gu, X.; Kedir, J.F.; Abu-Remaileh, M.; Bianchi, L.F.; Bottino, A.M.S.; Dueholm, R.; Niehaus, A.; Overwijn, D.; Fils, A.P.; et al. Structural Basis for the Docking of Mtorc1 on the Lysosomal Surface. Science 2019, 366, 468–475. [Google Scholar] [CrossRef] [PubMed]
- Cui, Z.; Napolitano, G.; de Araujo, M.E.G.; Esposito, A.; Monfregola, J.; Huber, L.A.; Ballabio, A.; Hurley, J.H. Structure of the Lysosomal Mtorc1-Tfeb-Rag-Ragulator Megacomplex. Nature 2023, 614, 572–579. [Google Scholar] [CrossRef]
- Zoncu, R.; Efeyan, A.; Sabatini, D.M. Mtor: From Growth Signal Integration to Cancer, Diabetes and Ageing. Nat. Rev. Mol. Cell Biol. 2011, 12, 21–35. [Google Scholar] [CrossRef]
- Hees, J.T.; Harbauer, A.B. Metabolic Regulation of Mitochondrial Protein Biogenesis from a Neuronal Perspective. Biomolecules 2022, 12, 1595. [Google Scholar] [CrossRef]
- Yang, H.; Yu, Z.; Chen, X.; Li, J.; Li, N.; Cheng, J.; Gao, N.; Yuan, H.X.; Ye, D.; Guan, K.L.; et al. Structural Insights into Tsc Complex Assembly and Gap Activity on Rheb. Nat. Commun. 2021, 12, 339. [Google Scholar] [CrossRef]
- Sutera, P.; Kim, J.; Kumar, R.; Deek, R.A.; Stephenson, R.; Mayer, T.; Saraiya, B.; Ghodoussipour, S.; Jang, T.; Golombos, D.; et al. Pik3/Akt/Mtor Pathway Alterations in Metastatic Castration-Sensitive Prostate Cancer. Prostate 2024, 84, 1301–1308. [Google Scholar] [CrossRef]
- Coupland, C.E.; Karimi, R.; Bueler, S.A.; Liang, Y.; Courbon, G.M.; Di Trani, J.M.; Wong, C.J.; Saghian, R.; Youn, J.Y.; Wang, L.Y.; et al. High-Resolution Electron Cryomicroscopy of V-Atpase in Native Synaptic Vesicles. Science 2024, 385, 168–174. [Google Scholar] [CrossRef]
- Zhang, C.S.; Jiang, B.; Li, M.; Zhu, M.; Peng, Y.; Zhang, Y.L.; Wu, Y.Q.; Li, T.Y.; Liang, Y.; Lu, Z.; et al. The Lysosomal V-Atpase-Ragulator Complex Is a Common Activator for Ampk and Mtorc1, Acting as a Switch between Catabolism and Anabolism. Cell Metab. 2014, 20, 526–540. [Google Scholar] [CrossRef] [PubMed]
- Jansen, E.J.; van Bakel, N.H.; Loohuis, N.F.O.; Hafmans, T.G.; Arentsen, T.; Coenen, A.J.; Scheenen, W.J.; Martens, G.J. Identification of Domains within the V-Atpase Accessory Subunit Ac45 Involved in V-Atpase Transport and Ca2+-Dependent Exocytosis. J. Biol. Chem. 2012, 287, 27537–27546. [Google Scholar] [CrossRef] [PubMed]
- Ratto, E.; Chowdhury, S.R.; Siefert, N.S.; Schneider, M.; Wittmann, M.; Helm, D.; Palm, W. Direct Control of Lysosomal Catabolic Activity by Mtorc1 through Regulation of V-Atpase Assembly. Nat. Commun. 2022, 13, 4848. [Google Scholar] [CrossRef]
- Wolfe, M.S. Presenilin, Gamma-Secretase, and the Search for Pathogenic Triggers of Alzheimer’s Disease. Biochemistry 2025, 64, 1662–1672. [Google Scholar] [CrossRef] [PubMed]
- Son, S.M.; Shin, H.J.; Byun, J.; Kook, S.Y.; Moon, M.; Chang, Y.J.; Mook-Jung, I. Metformin Facilitates Amyloid-β Generation by β- and Gamma-Secretases via Autophagy Activation. J. Alzheimer’s Dis. 2016, 51, 1197–1208. [Google Scholar] [CrossRef]
- Alzamora, R.; Al-Bataineh, M.M.; Liu, W.; Gong, F.; Li, H.; Thali, R.F.; Joho-Auchli, Y.; Brunisholz, R.A.; Satlin, L.M.; Neumann, D.; et al. Amp-Activated Protein Kinase Regulates the Vacuolar H+-Atpase Via Direct Phosphorylation of the a Subunit (Atp6v1a) in the Kidney. Am. J. Physiol. Ren. Physiol. 2013, 305, F943–F956. [Google Scholar] [CrossRef]
- Li, J.; Knudsen, J.R.; Henriquez-Olguin, C.; Li, Z.; Birk, J.B.; Persson, K.W.; Hellsten, Y.; Offergeld, A.; Jarassier, W.; Le Grand, F.; et al. Axin1 Knockout Does Not Alter Ampk/Mtorc1 Regulation and Glucose Metabolism in Mouse Skeletal Muscle. J. Physiol. 2021, 599, 3081–3100. [Google Scholar] [CrossRef]
- Chen, S.H.; Bubb, M.R.; Yarmola, E.G.; Zuo, J.; Jiang, J.; Lee, B.S.; Lu, M.; Gluck, S.L.; Hurst, I.R.; Holliday, L.S. Vacuolar H+-Atpase Binding to Microfilaments: Regulation in Response to Phosphatidylinositol 3-Kinase Activity and Detailed Characterization of the Actin-Binding Site in Subunit B. J. Biol. Chem. 2004, 279, 7988–7998. [Google Scholar] [CrossRef]
- Lee, B.S.; Gluck, S.L.; Holliday, L.S. Interaction between Vacuolar H(+)-Atpase and Microfilaments during Osteoclast Activation. J. Biol. Chem. 1999, 274, 29164–29171. [Google Scholar] [CrossRef]
- Holliday, L.S.; Lu, M.; Lee, B.S.; Nelson, R.D.; Solivan, S.; Zhang, L.; Gluck, S.L. The Amino-Terminal Domain of the B Subunit of Vacuolar H+-Atpase Contains a Filamentous Actin Binding Site. J. Biol. Chem. 2000, 275, 32331–32337. [Google Scholar] [CrossRef]
- Zuo, J.; Jiang, J.; Chen, S.H.; Vergara, S.; Gong, Y.; Xue, J.; Huang, H.; Kaku, M.; Holliday, L.S. Actin Binding Activity of Subunit B of Vacuolar H+-Atpase Is Involved in Its Targeting to Ruffled Membranes of Osteoclasts. J. Bone Min. Res. 2006, 21, 714–721. [Google Scholar] [CrossRef] [PubMed]
- Vitavska, O.; Merzendorfer, H.; Wieczorek, H. The V-Atpase Subunit C Binds to Polymeric F-Actin as Well as to Monomeric G-Actin and Induces Cross-Linking of Actin Filaments. J. Biol. Chem. 2005, 280, 1070–1076. [Google Scholar] [CrossRef]
- Vitavska, O.; Wieczorek, H.; Merzendorfer, H. A Novel Role for Subunit C in Mediating Binding of the H+-V-Atpase to the Actin Cytoskeleton. J. Biol. Chem. 2003, 278, 18499–18505. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Zhang, C.S.; Feng, J.W.; Wei, X.; Zhang, C.; Xie, C.; Wu, Y.; Hawley, S.A.; Atrih, A.; Lamont, D.J.; et al. Aldolase Is a Sensor for Both Low and High Glucose, Linking to Ampk and Mtorc1. Cell Res. 2021, 31, 478–481. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Zhang, C.S.; Zong, Y.; Feng, J.W.; Ma, T.; Hu, M.; Lin, Z.; Li, X.; Xie, C.; Wu, Y.; et al. Transient Receptor Potential V Channels Are Essential for Glucose Sensing by Aldolase and Ampk. Cell Metab. 2019, 30, 508–524.e12. [Google Scholar] [CrossRef]
- Kane, P.M. Targeting Reversible Disassembly as a Mechanism of Controlling V-Atpase Activity. Curr. Protein Pept. Sci. 2012, 13, 117–123. [Google Scholar] [CrossRef]
- Parra, K.J.; Kane, P.M. Reversible Association between the V1 and V0 Domains of Yeast Vacuolar H+-Atpase Is an Unconventional Glucose-Induced Effect. Mol. Cell Biol. 1998, 18, 7064–7074. [Google Scholar] [CrossRef]
- Kane, P.M.; Smardon, A.M. Assembly and Regulation of the Yeast Vacuolar H+-Atpase. J. Bioenerg. Biomembr. 2003, 35, 313–321. [Google Scholar] [CrossRef]
- Jaskolka, M.C.; Kane, P.M. Interaction between the Yeast Rave Complex and Vph1-Containing V(O) Sectors Is a Central Glucose-Sensitive Interaction Required for V-Atpase Reassembly. J. Biol. Chem. 2020, 295, 2259–2269. [Google Scholar] [CrossRef]
- Oot, R.A.; Kane, P.M.; Berry, E.A.; Wilkens, S. Crystal Structure of Yeast V1-Atpase in the Autoinhibited State. EMBO J. 2016, 35, 1694–1706. [Google Scholar] [CrossRef]
- Kishikawa, J.I.; Nakanishi, A.; Furuta, A.; Kato, T.; Namba, K.; Tamakoshi, M.; Mitsuoka, K.; Yokoyama, K. Mechanical Inhibition of Isolated V(O) from V/a-Atpase for Proton Conductance. Elife 2020, 9, e56862. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Tarsio, M.; Kane, P.M.; Rubinstein, J.L. Structure of Yeast Rave Bound to a Partial V(1) Complex. Proc. Natl. Acad. Sci. USA 2024, 121, e2414511121. [Google Scholar] [CrossRef]
- Jaskolka, M.C.; Winkley, S.R.; Kane, P.M. Rave and Rabconnectin-3 Complexes as Signal Dependent Regulators of Organelle Acidification. Front. Cell Dev. Biol. 2021, 9, 698190. [Google Scholar] [CrossRef] [PubMed]
- Kane, P.M.; Tarsio, M.; Liu, J. Early Steps in Assembly of the Yeast Vacuolar H+-Atpase. J. Biol. Chem. 1999, 274, 17275–17283. [Google Scholar] [CrossRef] [PubMed]
- Jorgensen, P.; Nishikawa, J.L.; Breitkreutz, B.J.; Tyers, M. Systematic Identification of Pathways That Couple Cell Growth and Division in Yeast. Science 2002, 297, 395–400. [Google Scholar] [CrossRef]
- Luby-Phelps, K. Cytoarchitecture and Physical Properties of Cytoplasm: Volume, Viscosity, Diffusion, Intracellular Surface Area. Int. Rev. Cytol. 2000, 192, 189–221. [Google Scholar]
- Esser, N.; Legrand-Poels, S.; Piette, J.; Scheen, A.J.; Paquot, N. Inflammation as a Link between Obesity, Metabolic Syndrome and Type 2 Diabetes. Diabetes Res. Clin. Pract. 2014, 105, 141–150. [Google Scholar] [CrossRef]
- Deng, T.; Lyon, C.J.; Bergin, S.; Caligiuri, M.A.; Hsueh, W.A. Obesity, Inflammation, and Cancer. Annu. Rev. Pathol. 2016, 11, 421–449. [Google Scholar] [CrossRef]
- Khalid, M.; Petroianu, G.; Adem, A. Advanced Glycation End Products and Diabetes Mellitus: Mechanisms and Perspectives. Biomolecules 2022, 12, 542. [Google Scholar] [CrossRef]
- Liu, Y.; Li, J.; Han, Y.; Chen, Y.; Liu, L.; Lang, J.; Yang, C.; Luo, H.; Ning, J. Advanced Glycation End-Products Suppress Autophagy by Ampk/Mtor Signaling Pathway to Promote Vascular Calcification. Mol. Cell Biochem. 2020, 471, 91–100. [Google Scholar] [CrossRef]
- Baek, C.H.; Kim, H.; Moon, S.Y.; Yang, W.S. Ampk Boosts Adam10 Shedding Activity in Human Aortic Endothelial Cells by Promoting Rab14-Dependent Adam10 Cell Surface Translocation. Biochem. Biophys. Res. Commun. 2023, 675, 54–60. [Google Scholar] [CrossRef]
- Beisswenger, P.J.; Howell, S.K.; Touchette, A.D.; Lal, S.; Szwergold, B.S. Metformin Reduces Systemic Methylglyoxal Levels in Type 2 Diabetes. Diabetes 1999, 48, 198–202. [Google Scholar] [CrossRef]
- Araujo, A.A.; Pereira, A.; Medeiros, C.; Brito, G.A.C.; Leitao, R.F.C.; Araujo, L.S.; Guedes, P.M.M.; Hiyari, S.; Pirih, F.Q.; Junior, R.F.A. Effects of Metformin on Inflammation, Oxidative Stress, and Bone Loss in a Rat Model of Periodontitis. PLoS ONE 2017, 12, e0183506. [Google Scholar] [CrossRef]
- Song, B.; Zhang, Y.L.; Chen, L.J.; Zhou, T.; Huang, W.K.; Zhou, X.; Shao, L.Q. The Role of Toll-Like Receptors in Periodontitis. Oral. Dis. 2017, 23, 168–180. [Google Scholar] [CrossRef] [PubMed]
- Kittaka, M.; Yoshimoto, T.; Levitan, M.E.; Urata, R.; Choi, R.B.; Teno, Y.; Xie, Y.; Kitase, Y.; Prideaux, M.; Dallas, S.L.; et al. Osteocyte Rankl Drives Bone Resorption in Mouse Ligature-Induced Periodontitis. J. Bone Min. Res. 2023, 38, 1521–1540. [Google Scholar] [CrossRef] [PubMed]
- Portes, J.; Bullon, B.; Quiles, J.L.; Battino, M.; Bullon, P. Diabetes Mellitus and Periodontitis Share Intracellular Disorders as the Main Meeting Point. Cells 2021, 10, 2411. [Google Scholar] [CrossRef] [PubMed]
- Sanchez-Garrido, J.; Shenoy, A.R. Regulation and Repurposing of Nutrient Sensing and Autophagy in Innate Immunity. Autophagy 2021, 17, 1571–1591. [Google Scholar] [CrossRef]
- Trindade, B.C.; Chen, G.Y. Nod1 and Nod2 in Inflammatory and Infectious Diseases. Immunol. Rev. 2020, 297, 139–161. [Google Scholar] [CrossRef]
- Singhrao, S.K.; Harding, A.; Poole, S.; Kesavalu, L.; Crean, S. Porphyromonas Gingivalis Periodontal Infection and Its Putative Links with Alzheimer’s Disease. Mediators Inflamm 2015, 2015, 137357. [Google Scholar] [CrossRef]
- Kim, Y.; Li, C.; Gu, C.; Fang, Y.; Tycksen, E.; Puri, A.; Pietka, T.A.; Sivapackiam, J.; Kidd, K.; Park, S.J.; et al. Manf Stimulates Autophagy and Restores Mitochondrial Homeostasis to Treat Autosomal Dominant Tubulointerstitial Kidney Disease in Mice. Nat. Commun. 2023, 14, 6493. [Google Scholar] [CrossRef]
- Bi, R.; Yang, Y.; Liao, H.; Ji, G.; Ma, Y.; Cai, L.; Li, J.; Yang, J.; Sun, M.; Liang, J.; et al. Porphyromonas Gingivalis Induces an Inflammatory Response Via the Cgas-Sting Signaling Pathway in a Periodontitis Mouse Model. Front. Microbiol. 2023, 14, 1183415. [Google Scholar] [CrossRef] [PubMed]
- Ruggiero, S.L.; Mehrotra, B.; Rosenberg, T.J.; Engroff, S.L. Osteonecrosis of the Jaws Associated with the Use of Bisphosphonates: A Review of 63 Cases. J. Oral Maxillofac. Surg. 2004, 62, 527–534. [Google Scholar] [CrossRef] [PubMed]
- Nagy, V.; Penninger, J.M. The Rankl-Rank Story. Gerontology 2015, 61, 534–542. [Google Scholar] [CrossRef] [PubMed]
- Gogakos, A.I.; Anastasilakis, A.D. Current and Emerging Bone Resorption Inhibitors for the Treatment of Osteoporosis. Expert. Opin. Pharmacother. 2025, 26, 265–278. [Google Scholar] [CrossRef]
- Drake, M.T.; Clarke, B.L.; Khosla, S. Bisphosphonates: Mechanism of Action and Role in Clinical Practice. Mayo Clin. Proc. 2008, 83, 1032–1045. [Google Scholar] [CrossRef]
- Coxon, F.P.; Thompson, K.; Roelofs, A.J.; Ebetino, F.H.; Rogers, M.J. Visualizing Mineral Binding and Uptake of Bisphosphonate by Osteoclasts and Non-Resorbing Cells. Bone 2008, 42, 848–860. [Google Scholar] [CrossRef]
- Dong, L.; Jiang, L.; Xu, Z.; Zhang, X. Denosumab, Teriparatide and Bisphosphonates for Glucocorticoid-Induced Osteoporosis: A Bayesian Network Meta-Analysis. Front. Pharmacol. 2024, 15, 1336075. [Google Scholar] [CrossRef]
- Son, S.; Oh, M.Y.; Yoo, B.R.; Park, H.B. Comparison of the Efficacy of Zoledronate and Denosumab in Patients with Acute Osteoporotic Vertebral Compression Fractures: A Randomized Controlled Trial. J. Clin. Med. 2024, 13, 2040. [Google Scholar] [CrossRef]
- Tang, Y.; Jin, Z.; Lu, Y.; Chen, L.; Lv, S.; Xu, T.; Tong, P.; Chen, G. Comparing the Efficacy of Antiosteoporotic Drugs in Preventing Periprosthetic Bone Loss Following Total Hip Arthroplasty: A Systematic Review and Bayesian Network Meta-Analysis. Orthop. Surg. 2024, 16, 2344–2354. [Google Scholar] [CrossRef]
- Yang, G.; Williams, R.; Wang, L.; Farhadfar, N.; Chen, Y.; Loiacono, A.T.; Bian, J.; Holliday, L.S.; Katz, J.; Gong, Y. Medication-Related Osteonecrosis of the Jaw in Cancer Patients: Result from the Oneflorida Clinical Research Consortium. J. Bone Min. Res. 2022, 37, 2466–2471. [Google Scholar] [CrossRef]
- Yang, G.; Singh, S.; McDonough, C.W.; Lamba, J.K.; Hamadeh, I.; Holliday, L.S.; Wang, D.; Katz, J.; Lakatos, P.A.; Balla, B.; et al. Genome-Wide Association Study Identified Chromosome 8 Locus Associated with Medication-Related Osteonecrosis of the Jaw. Clin. Pharmacol. Ther. 2021, 110, 1558–1569. [Google Scholar] [CrossRef] [PubMed]
- Otsuru, M.; Fujiki, Y.; Soutome, S.; Nakamura, N.; Miyoshi, T.; Naruse, T.; Ohnuma, M.; Hotokezaka, Y.; Rokutanda, S.; Umeda, M. Risk Factors for Dental Findings of the Development of Medication-Related Osteonecrosis of the Jaw: Investigation of 3734 Teeth in Cancer Patients Receiving High Dose Antiresorptive Agents. J. Dent. Sci. 2024, 19, 203–210. [Google Scholar] [CrossRef]
- Castillo, E.J.; Messer, J.G.; Abraham, A.M.; Jiron, J.M.; Alekseyenko, A.V.; Israel, R.; Thomas, S.; Gonzalez-Perez, G.M.; Croft, S.; Gohel, A.; et al. Preventing or Controlling Periodontitis Reduces the Occurrence of Osteonecrosis of the Jaw (Onj) in Rice Rats (Oryzomys Palustris). Bone 2021, 145, 115866. [Google Scholar] [CrossRef] [PubMed]
- Bone, H.G.; Wagman, R.B.; Brandi, M.L.; Brown, J.P.; Chapurlat, R.; Cummings, S.R.; Czerwinski, E.; Fahrleitner-Pammer, A.; Kendler, D.L.; Lippuner, K.; et al. 10 Years of Denosumab Treatment in Postmenopausal Women with Osteoporosis: Results from the Phase 3 Randomised Freedom Trial and Open-Label Extension. Lancet Diabetes Endocrinol. 2017, 5, 513–523. [Google Scholar] [CrossRef]
- Narayan, N.; Vadde, T.; Sandesara, M.; Divity, S.; Mamytova, A.; Tagaev, T. Efficacy and Safety of Efpeglenatide in Patients with Type 2 Diabetes and Obesity: A Systematic Review. Cureus 2025, 17, e77089. [Google Scholar] [CrossRef] [PubMed]
- Bushi, G.; Khatib, M.N.; Rohilla, S.; Singh, M.P.; Uniyal, N.; Ballal, S.; Bansal, P.; Bhopte, K.; Gupta, M.; Gaidhane, A.M.; et al. Association of Glp-1 Receptor Agonists with Risk of Suicidal Ideation and Behaviour: A Systematic Review and Meta-Analysis. Diabetes Metab. Res. Rev. 2025, 41, e70037. [Google Scholar] [CrossRef]
- Bednarczyk, M.; Dabrowska-Szeja, N.; Letowski, D.; Dziegielewska-Gesiak, S.; Waniczek, D.; Muc-Wierzgon, M. Relationship between Dietary Nutrient Intake and Autophagy-Related Genes in Obese Humans: A Narrative Review. Nutrients 2024, 16, 4003. [Google Scholar] [CrossRef]
- Wan, K.; Jin, Y.; Fan, R.; Xu, Q.; Li, X.; Yan, H.; Wang, R. Exploring Molecular Mechanisms of Exercise on Metabolic Syndrome: A Bibliometric and Visualization Study Using Citespace. Front. Endocrinol. 2024, 15, 1408466. [Google Scholar] [CrossRef]
- Lin, J.; Zhang, X.; Sun, Y.; Xu, H.; Li, N.; Wang, Y.; Tian, X.; Zhao, C.; Wang, B.; Zhu, B.; et al. Exercise Ameliorates Muscular Excessive Mitochondrial Fission, Insulin Resistance and Inflammation in Diabetic Rats Via Irisin/Ampk Activation. Sci. Rep. 2024, 14, 10658. [Google Scholar] [CrossRef]
- Kolnes, K.J.; Nilsen, E.T.F.; Brufladt, S.; Meadows, A.M.; Jeppesen, P.B.; Skattebo, O.; Johansen, E.I.; Birk, J.B.; Hojlund, K.; Hingst, J.; et al. Effects of Seven Days’ Fasting on Physical Performance and Metabolic Adaptation during Exercise in Humans. Nat. Commun. 2025, 16, 122. [Google Scholar] [CrossRef]
- Qu, Q.; Chen, Y.; Wang, Y.; Long, S.; Wang, W.; Yang, H.Y.; Li, M.; Tian, X.; Wei, X.; Liu, Y.H.; et al. Lithocholic Acid Phenocopies Anti-Ageing Effects of Calorie Restriction. Nature, 2024; Online ahead of print. [Google Scholar]
- Yuliyanasari, N.; Rejeki, P.S.; Hidayati, H.B.; Subsomwong, P.; Miftahussurur, M. The Effect of Intermittent Fasting on Preventing Obesity-Related Early Aging from a Molecular and Cellular Perspective. J. Med. Life 2024, 17, 261–272. [Google Scholar] [PubMed]
- Paoli, A.; Tinsley, G.M.; Mattson, M.P.; De Vivo, I.; Dhawan, R.; Moro, T. Common and Divergent Molecular Mechanisms of Fasting and Ketogenic Diets. Trends Endocrinol. Metab. 2024, 35, 125–141. [Google Scholar] [CrossRef] [PubMed]
- Ma, J.; Zhang, H.; Lv, Y.; Gao, M.; Gai, Z.; Liu, Y. Clinical and Genetic Characteristics of Two Cases with Developmental and Epileptic Encephalopathy 93 Caused by Novel ATP6V1A Mutations and Literature Review. Hum. Mutat. 2024, 2024, 4678670. [Google Scholar] [CrossRef]
- Mucha, B.E.; Banka, S.; Ajeawung, N.F.; Molidperee, S.; Chen, G.G.; Koenig, M.K.; Adejumo, R.B.; Till, M.; Harbord, M.; Perrier, R.; et al. A New Microdeletion Syndrome Involving Tbc1d24, Atp6v0c, and Pdpk1 Causes Epilepsy, Microcephaly, and Developmental Delay. Genet. Med. 2019, 21, 1058–1064. [Google Scholar] [CrossRef] [PubMed]
- Mattison, K.A.; Tossing, G.; Mulroe, F.; Simmons, C.; Butler, K.M.; Schreiber, A.; Alsadah, A.; Neilson, D.E.; Naess, K.; Wedell, A.; et al. Atp6v0c Variants Impair V-Atpase Function Causing a Neurodevelopmental Disorder Often Associated with Epilepsy. Brain 2023, 146, 1357–1372. [Google Scholar] [CrossRef]
- Rujano, M.A.; Serio, M.C.; Panasyuk, G.; Peanne, R.; Reunert, J.; Rymen, D.; Hauser, V.; Park, J.H.; Freisinger, P.; Souche, E.; et al. Mutations in the X-Linked Atp6ap2 Cause a Glycosylation Disorder with Autophagic Defects. J. Exp. Med. 2017, 214, 3707–3729. [Google Scholar] [CrossRef]
- Knüppel, H. Dental Power: How We Can Live Longer and Better and What Role Teeth Play as a Reflection of Health; BoD–Books on Demand: Hamburg, Germany, 2024. [Google Scholar]






| Elements of V1 subdomain of V-ATPase | ||||||||||
| ATP6V1A | ATP6V1B2 | ATP6V1C1 | ATP6V1D | ATP6V1E1 | ATP6V1F | ATP6V1G1 | ATP6V1H | |||
| Homo sapiens (mammal) | 100 | 100 | 100 | 100 | 100 | 100 | 100 | 100 | ||
| Mus musculis (mammal) | 98 | 100 | 100 | 98 | 99 | 98 | 95 | 99 | ||
| Drosphila melanogaster (insect) | 83 | 91 | 65 | 72 | 63 | 72 | 50 | 53 | ||
| Arabidopsis thaliana (land plant) | 69 | 91 | 40 | 54 | 41 | 50 | 39 | 28 | ||
| Saccharomyces cerevisiae (budding yeast) | 67 | 81 | 39 | 51 | 34 | 53 | 38 | 31 | ||
| Elements of V0 subdomain of V-ATPase | ||||||||||
| ATP6V0a1 | ATP6V0a2 | ATP6V0a3 | ATP6V0a4 | ATP6V0b | ATP6V0c | ATP6v0d1 | ATP6V0e1 | ATP6AP1 | ATP6AP2 | |
| Homo sapiens (mammal) | 100 | 100/57 | 100/53 | 100/61 | 100 | 100 | 100 | 100 | 100 | 100 |
| Mus musculis (mammal) | 96/a1 | 92/a2 | 84/a3 | 86/a4 | 99 | 91 | 100 | 100 | 86 | 94 |
| Drosphila melanogaster (insect) | 62/a1 | 45/a2 | 43/a1 | 54/a1 | 64 | 78 | 81 | 41 | 29 | 26 |
| Arabidopsis thaliana (land plant) | 43/a3 | 38/a1 | 41/a3 | 42/a2 | 56 | 62 | 51 | 46 | ||
| Saccharomyces cerevisiae (budding yeast) | 42/vph1 | 37/vph1 | 36/vph1 | 38/vph1 | 34 | 63 | 46 | 46 | ||
| Elements of mTORC1 and L-AMPK | ||||||||||
| RAPTOR | Tor | LST8 | SIN1 | RICTOR | Rheb | TSC | AMPK-α | AMPK-β | AMPK-γ | |
| Homo sapiens (mammal) | 100 | 100 | 100 | 100 | 100 | 100 | 100 | 100 | 100 | 100 |
| Mus musculis (mammal) | 93 | 99 | 98 | 97 | 95 | 99 | 87 | 99 | 97 | 97 |
| Drosphila melanogaster (insect) | 32 | 54 | 49 | 29 | 32 | 64 | 32 | 46 | 62 | 66 |
| Arabidopsis thaliana (land plant) | 41 | 42 | 45 | 53 | 38 | 30 | ||||
| Saccharomyces cerevisiae (budding yeast) | 25 | 44/42 | 47 | 32 | 25 | 37 | 41 | 31 | 37 | |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yang, X.; Holliday, L.S. V-ATPase and Lysosomal Energy Sensing in Periodontitis and Medicine-Related Osteonecrosis of the Jaw. Biomolecules 2025, 15, 997. https://doi.org/10.3390/biom15070997
Yang X, Holliday LS. V-ATPase and Lysosomal Energy Sensing in Periodontitis and Medicine-Related Osteonecrosis of the Jaw. Biomolecules. 2025; 15(7):997. https://doi.org/10.3390/biom15070997
Chicago/Turabian StyleYang, Xianrui, and Lexie Shannon Holliday. 2025. "V-ATPase and Lysosomal Energy Sensing in Periodontitis and Medicine-Related Osteonecrosis of the Jaw" Biomolecules 15, no. 7: 997. https://doi.org/10.3390/biom15070997
APA StyleYang, X., & Holliday, L. S. (2025). V-ATPase and Lysosomal Energy Sensing in Periodontitis and Medicine-Related Osteonecrosis of the Jaw. Biomolecules, 15(7), 997. https://doi.org/10.3390/biom15070997