
PRMT5 Identified as a Viable Target for Combination Therapy in Preclinical Models of Pancreatic Cancer
 , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
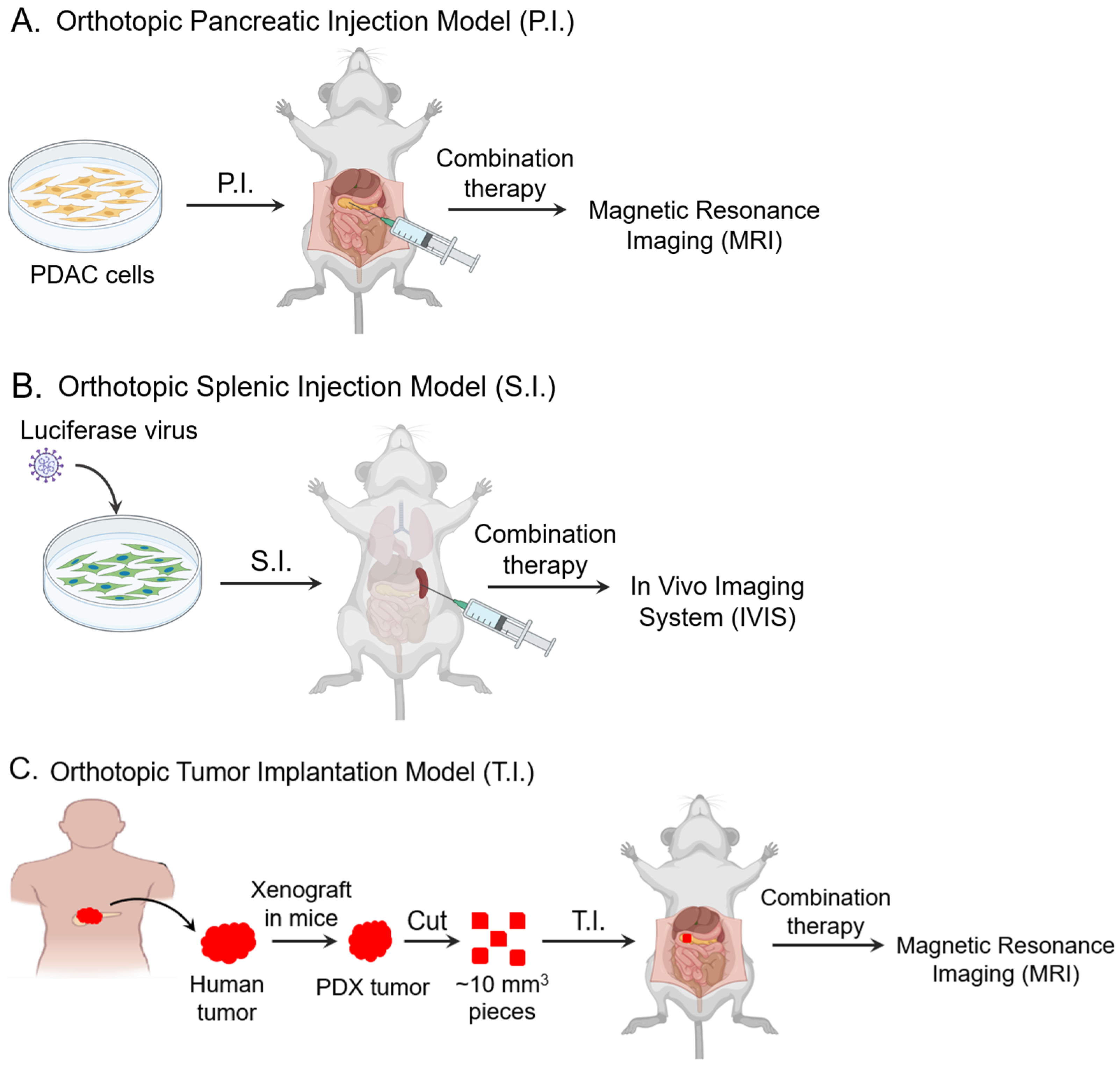
2.1. Cell Lines and Patient-Derived Xenografts
2.2. PRMT5 KO in the Orthotopic Pancreatic Injection (P.I.) Model
2.3. PRMT5 KO in the Metastatic Splenic Injection (S.I.) Model
2.4. PRMT5 Pharmacologic Inhibition in the PDX Orthotopic Tumor Implantation (T.I.) Model
2.5. Immunohistochemistry
2.6. Statistical Analysis
3. Results
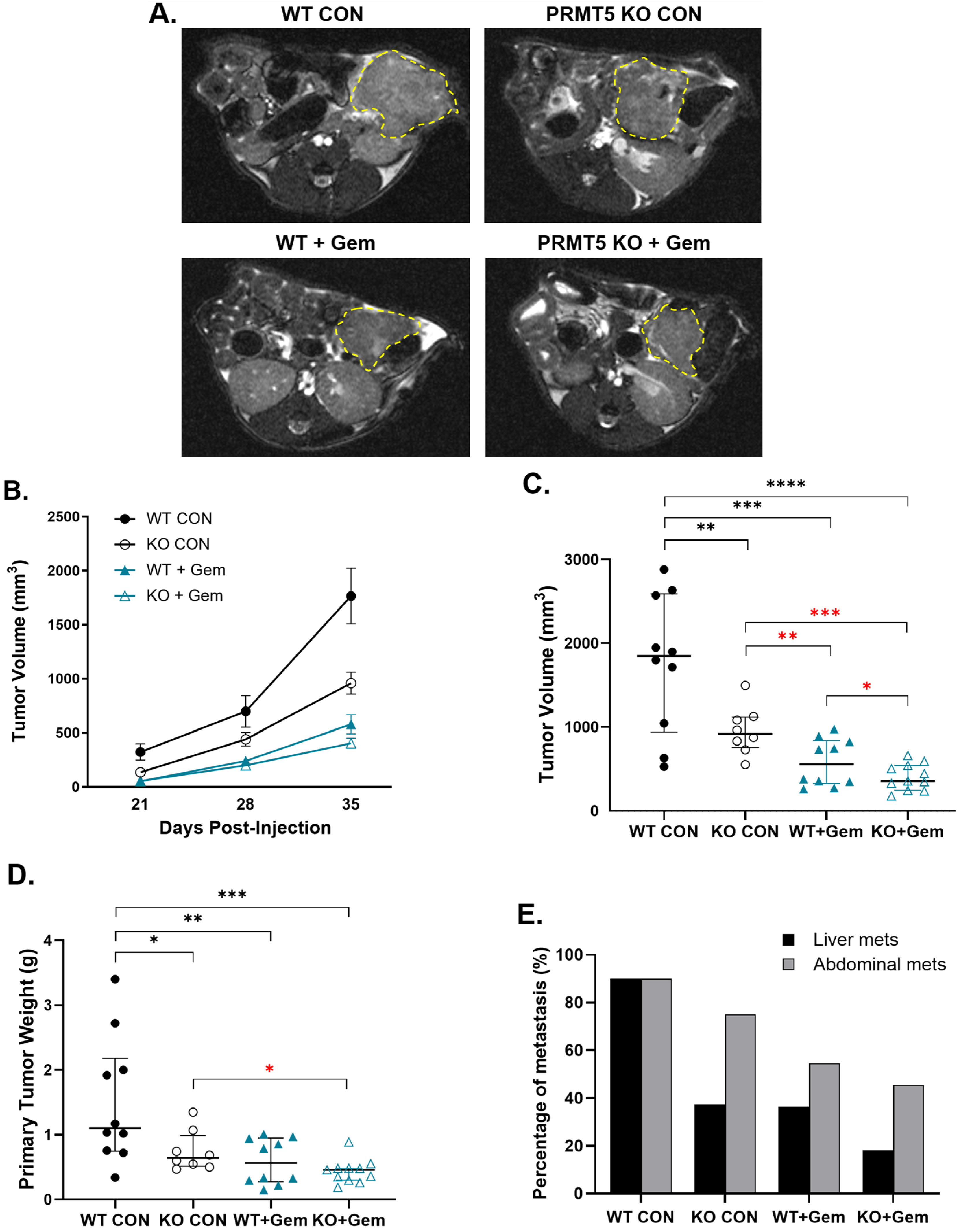
3.1. PRMT5 Depletion Decreases Pancreatic Tumor Growth in Vivo
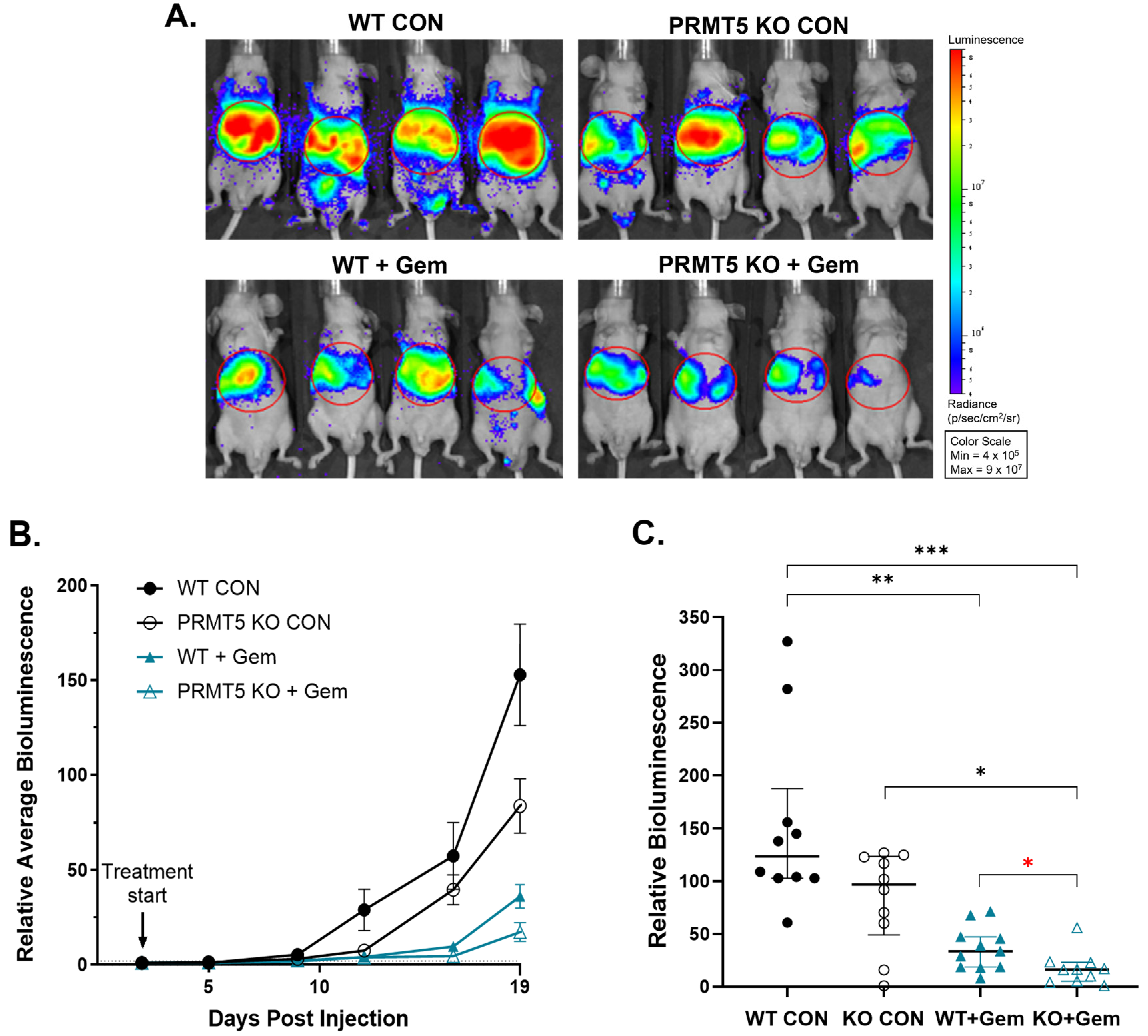
3.2. PRMT5 Depletion Attenuates Pancreatic Tumor Metastasis in Vivo
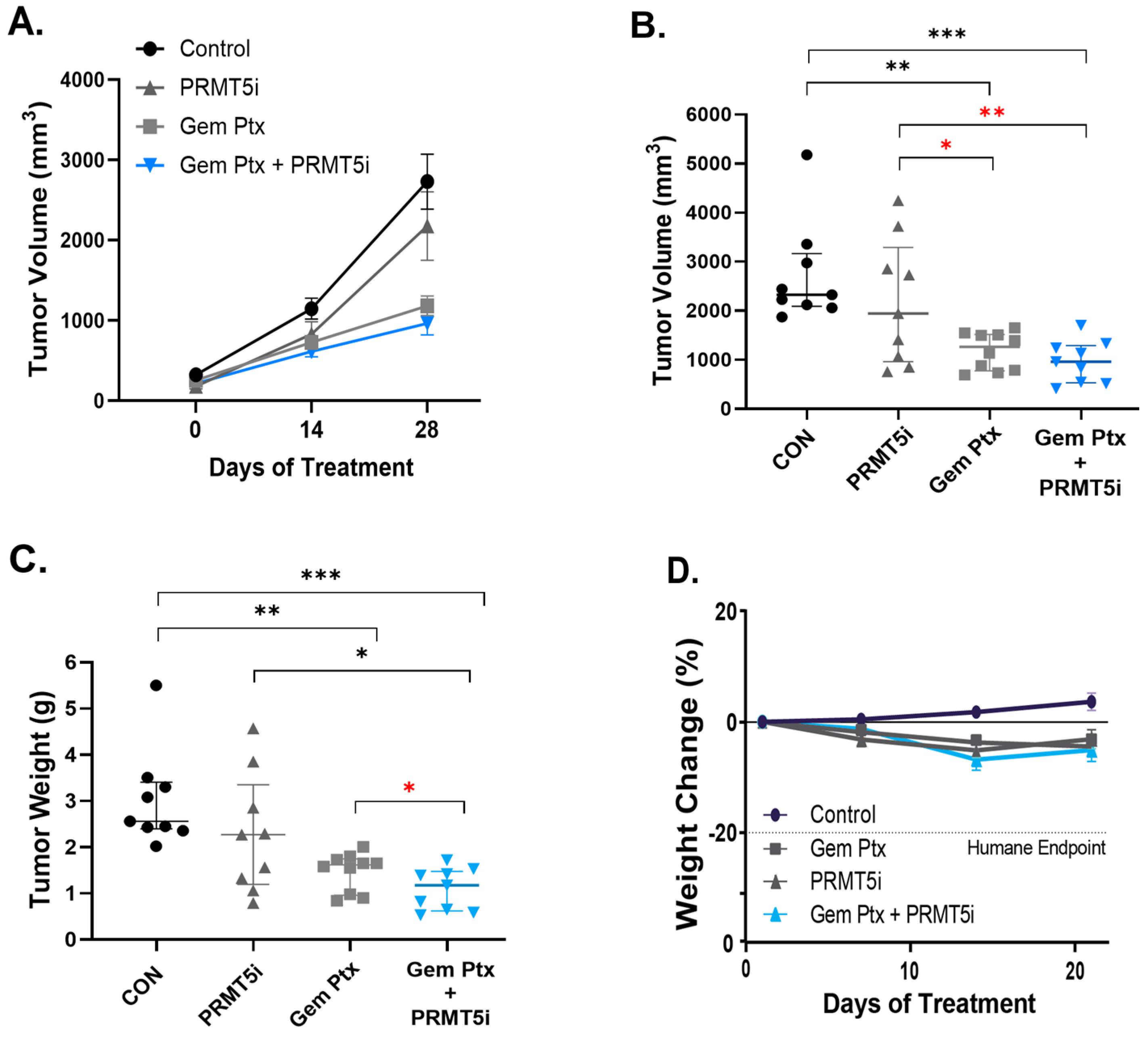
3.3. Combination Therapy of Pharmacologic PRMT5 Inhibition with Chemotherapeutics Synergistically Inhibits PDAC Tumor In Vivo
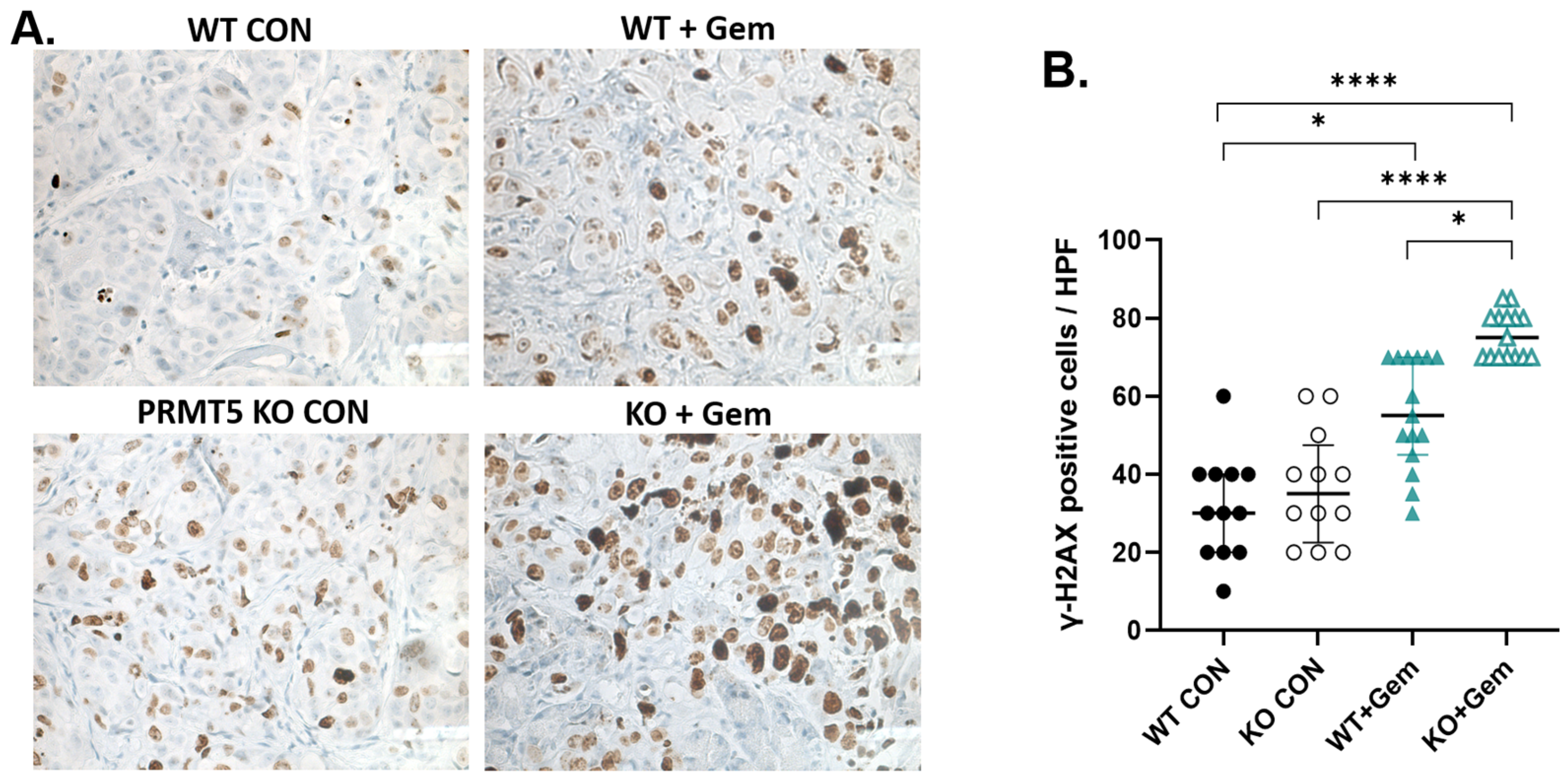
3.4. PRMT5 Depletion Increases DNA Damage in Pancreatic Tumors
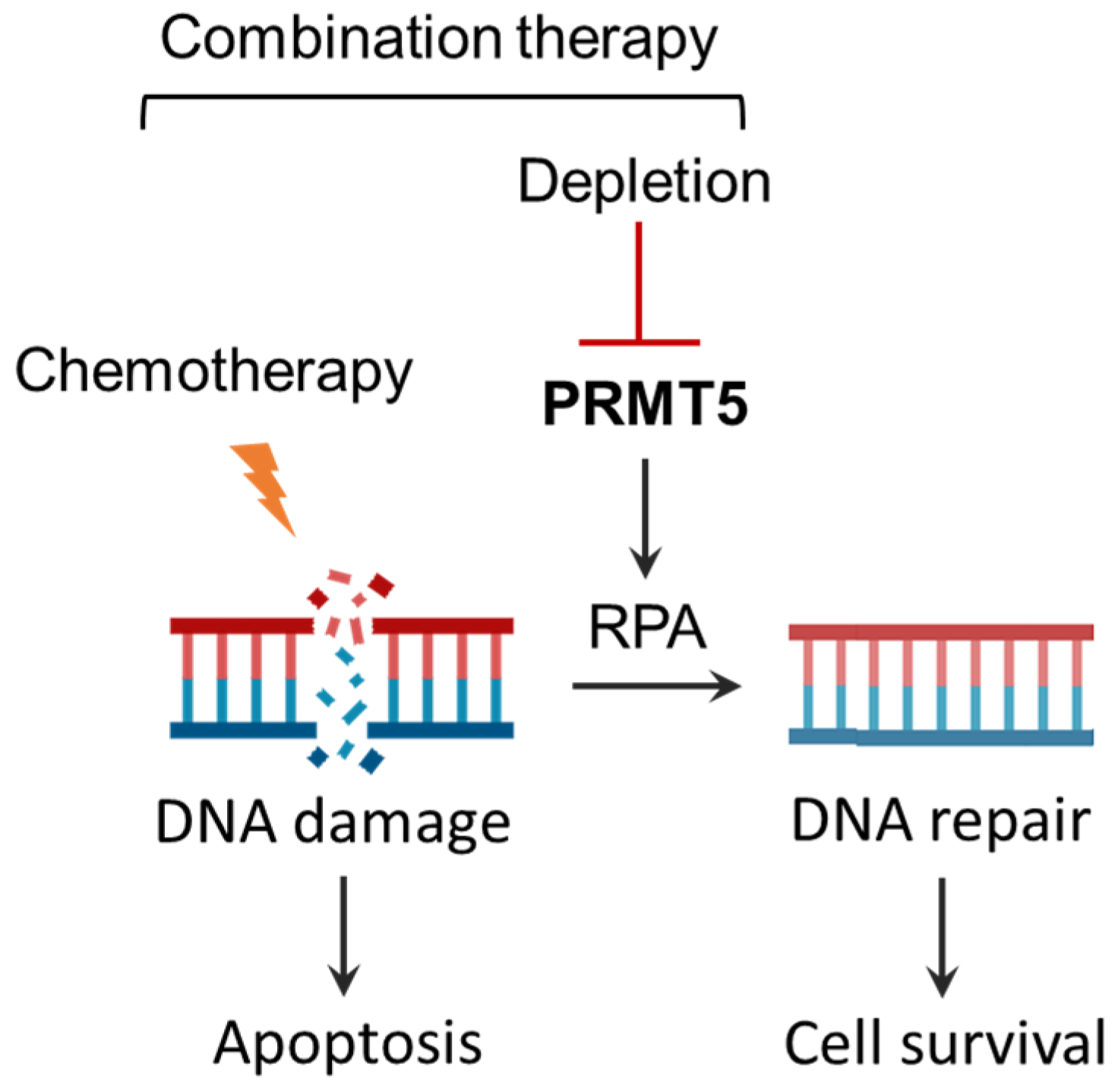
3.5. Summary of Effects of Combination Therapy of PRMT5 Inhibition with Chemotherapeutics
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| PRMT5 | Protein arginine methyltransferase 5 |
| PDAC | Pancreatic ductal adenocarcinoma |
| PDX | Patient-derived xenograft |
| MRI | Volumetric magnetic resonance imaging |
| I.P. | Intraperitoneal injection |
| S.Q. | Subcutaneous injection |
| P.O. | Per oral administration |
| P.I. | Pancreatic injection |
| S.I. | Splenic injection |
| T.I. | Pancreatic implant of tumor |
| IVIS | In Vivo Imaging Systems |
| Gem | Gemcitabine |
| Ptx | Paclitaxel |
| PRMT5i | PRMT5 inhibitor |
| IHC | Immunohistochemistry |
| γ-H2AX | Phosphorylated gamma-H2AX |
| HPF | High-power fields |
References
- Rahib, L.; Smith, B.D.; Aizenberg, R.; Rosenzweig, A.B.; Fleshman, J.M.; Matrisian, L.M. Projecting cancer incidence and deaths to 2030: The unexpected burden of thyroid, liver, and pancreas cancers in the United States. Cancer Res. 2014, 74, 2913–2921. [Google Scholar] [CrossRef]
- Siegel, R.L.; Kratzer, T.B.; Giaquinto, A.N.; Sung, H.; Jemal, A. Cancer statistics, 2025. CA A Cancer J. Clin. 2025, 75, 10–45. [Google Scholar] [CrossRef]
- Covarrubias-Zambrano, O.; Agarwal, D.; Kalubowilage, M.; Ehsan, S.; Yapa, A.S.; Covarrubias, J.; Kasi, A.; Natarajan, B.; Bossmann, S.H. Protease activity-based nanobiosensors for early detection of pancreatic cancer. Med. Res. Arch. 2024, 12, 5632. [Google Scholar] [CrossRef]
- Tempero, M.A.; Malafa, M.P.; Al-Hawary, M.; Asbun, H.; Bain, A.; Behrman, S.W.; Binder, E.; Cardin, D.B.; Cha, C.; Chiorean, E.G. Pancreatic Adenocarcinoma, Version 2.2017, NCCN Clinical Practice Guidelines in Oncology. J. Natl. Compr. Cancer Netw. 2017, 15, 1028–1061. [Google Scholar] [CrossRef] [PubMed]
- Kamisawa, T.; Wood, L.D.; Itoi, T.; Takaori, K. Pancreatic cancer. Lancet 2016, 388, 73–85. [Google Scholar] [CrossRef] [PubMed]
- Spadi, R.; Brusa, F.; Ponzetti, A.; Chiappino, I.; Birocco, N.; Ciuffreda, L.; Satolli, M.A. Current therapeutic strategies for advanced pancreatic cancer: A review for clinicians. World J. Clin. Oncol. 2016, 7, 27–43. [Google Scholar] [CrossRef] [PubMed]
- Szlachta, K.; Kuscu, C.; Tufan, T.; Adair, S.J.; Shang, S.; Michaels, A.D.; Mullen, M.G.; Fischer, N.L.; Yang, J.; Liu, L.; et al. CRISPR knockout screening identifies combinatorial drug targets in pancreatic cancer and models cellular drug response. Nat. Commun. 2018, 9, 4275. [Google Scholar] [CrossRef]
- Wei, X.; Yang, J.; Adair, S.J.; Ozturk, H.; Kuscu, C.; Lee, K.Y.; Kane, W.J.; O’Hara, P.E.; Liu, D.; Demirlenk, Y.M.; et al. Targeted CRISPR screening identifies PRMT5 as synthetic lethality combinatorial target with gemcitabine in pancreatic cancer cells. Proc. Natl. Acad. Sci. USA 2020, 117, 28068–28079. [Google Scholar] [CrossRef]
- Xiao, W.; Chen, X.; Liu, L.; Shu, Y.; Zhang, M.; Zhong, Y. Role of protein arginine methyltransferase 5 in human cancers. Biomed. Pharmacother. 2019, 114, 108790. [Google Scholar] [CrossRef]
- Karkhanis, V.; Hu, Y.-J.; Baiocchi, R.A.; Imbalzano, A.N.; Sif, S. Versatility of PRMT5-induced methylation in growth control and development. Trends Biochem. Sci. 2011, 36, 633–641. [Google Scholar] [CrossRef]
- Huang, L.; Zhang, X.O.; Rozen, E.J.; Sun, X.; Sallis, B.; Verdejo-Torres, O.; Wigglesworth, K.; Moon, D.; Huang, T.; Cavaretta, J.P.; et al. PRMT5 activates AKT via methylation to promote tumor metastasis. Nat. Commun. 2022, 13, 3955. [Google Scholar] [CrossRef] [PubMed]
- Qin, Y.; Hu, Q.; Xu, J.; Ji, S.; Dai, W.; Liu, W.; Xu, W.; Sun, Q.; Zhang, Z.; Ni, Q.; et al. PRMT5 enhances tumorigenicity and glycolysis in pancreatic cancer via the FBW7/cMyc axis. Cell Commun. Signal. 2019, 17, 30. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.; Ronai, Z.A. PRMT5 function and targeting in cancer. Cell Stress 2020, 4, 199–215. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Hu, W.; Yuan, Y. Protein Arginine Methyltransferase 5 (PRMT5) as an Anticancer Target and Its Inhibitor Discovery. J. Med. Chem. 2018, 61, 9429–9441. [Google Scholar] [CrossRef]
- Kryukov, G.V.; Wilson, F.H.; Ruth, J.R.; Paulk, J.; Tsherniak, A.; Marlow, S.E.; Vazquez, F.; Weir, B.A.; Fitzgerald, M.E.; Tanaka, M.; et al. MTAP deletion confers enhanced dependency on the PRMT5 arginine methyltransferase in cancer cells. Science 2016, 351, 1214–1218. [Google Scholar] [CrossRef]
- Hamard, P.J.; Santiago, G.E.; Liu, F.; Karl, D.L.; Martinez, C.; Man, N.; Mookhtiar, A.K.; Duffort, S.; Greenblatt, S.; Verdun, R.E.; et al. PRMT5 Regulates DNA Repair by Controlling the Alternative Splicing of Histone-Modifying Enzymes. Cell Rep. 2018, 24, 2643–2657. [Google Scholar] [CrossRef]
- Carter, J.; Hulse, M.; Sivakumar, M.; Burtell, J.; Thodima, V.; Wang, M.; Agarwal, A.; Vykuntam, K.; Spruance, J.; Bhagwat, N.; et al. PRMT5 Inhibitors Regulate DNA Damage Repair Pathways in Cancer Cells and Improve Response to PARP Inhibition and Chemotherapies. Cancer Res. Commun. 2023, 3, 2233–2243. [Google Scholar] [CrossRef]
- Bhandari, K.; Ding, W.Q. Protein Arginine Methyltransferases in Pancreatic Ductal Adenocarcinoma: New Molecular Targets for Therapy. Int. J. Mol. Sci. 2024, 25, 3958. [Google Scholar] [CrossRef]
- Feustel, K.; Falchook, G.S. Protein arginine methyltransferase 5 (PRMT5) inhibitors in oncology clinical trials: A review. J. Immunother. Precis. Oncol. 2022, 5, 58–67. [Google Scholar] [CrossRef]
- Vieito, M.; Moreno, V.; Spreafico, A.; Brana, I.; Wang, J.S.; Preis, M.; Hernández, T.; Genta, S.; Hansen, A.R.; Doger, B.; et al. Phase 1 Study of JNJ-64619178, a Protein Arginine Methyltransferase 5 Inhibitor, in Advanced Solid Tumors. Clin. Cancer Res. 2023, 29, 3592–3602. [Google Scholar] [CrossRef]
- Engstrom, L.D.; Aranda, R.; Waters, L.; Moya, K.; Bowcut, V.; Vegar, L.; Trinh, D.; Hebbert, A.; Smith, C.R.; Kulyk, S.; et al. MRTX1719 Is an MTA-Cooperative PRMT5 Inhibitor That Exhibits Synthetic Lethality in Preclinical Models and Patients with MTAP-Deleted Cancer. Cancer Discov. 2023, 13, 2412–2431. [Google Scholar] [CrossRef] [PubMed]
- Rodon, J.; Rodriguez, E.; Maitland, M.L.; Tsai, F.Y.; Socinski, M.A.; Berlin, J.D.; Thomas, J.S.; Al Baghdadi, T.; Wang, I.M.; Guo, C.; et al. A phase I study to evaluate the safety, pharmacokinetics, and pharmacodynamics of PF-06939999 (PRMT5 inhibitor) in patients with selected advanced or metastatic tumors with high incidence of splicing factor gene mutations. ESMO Open 2024, 9, 102961. [Google Scholar] [CrossRef] [PubMed]
- Rodon, J.; Prenen, H.; Sacher, A.; Villalona-Calero, M.; Penel, N.; El Helali, A.; Rottey, S.; Yamamoto, N.; Ghiringhelli, F.; Goebeler ME; et al. First-in-human study of AMG 193, an MTA-cooperative PRMT5 inhibitor, in patients with MTAP-deleted solid tumors: Results from phase I dose exploration. Ann. Oncol. 2024, 35, 1138–1147. [Google Scholar] [CrossRef] [PubMed]
- Herreros-Villanueva, M.; Hijona, E.; Cosme, A.; Bujanda, L. Mouse models of pancreatic cancer. World J. Gastroenterol. 2012, 18, 1286–1294. [Google Scholar] [CrossRef]
- Stokes, J.B.; Adair, S.J.; Slack-Davis, J.K.; Walters, D.M.; Tilghman, R.W.; Hershey, E.D.; Lowrey, B.; Thomas, K.S.; Bouton, A.H.; Hwang, R.F.; et al. Inhibition of focal adhesion kinase by PF-562,271 inhibits the growth and metastasis of pancreatic cancer concomitant with altering the tumor microenvironment. Mol. Cancer Ther. 2011, 10, 2135–2145. [Google Scholar] [CrossRef]
- Walters, D.M.; Stokes, J.B.; Adair, S.J.; Stelow, E.B.; Borgman, C.A.; Lowrey, B.T.; Xin, W.; Blais, E.M.; Lee, J.K.; Papin, J.A.; et al. Clinical, molecular and genetic validation of a murine orthotopic xenograft model of pancreatic adenocarcinoma using fresh human specimens. PLoS ONE 2013, 8, e77065. [Google Scholar] [CrossRef]
- Lindberg, J.M.; Newhook, T.E.; Adair, S.J.; Walters, D.M.; Kim, A.J.; Stelow, E.B.; Parsons, J.T.; Bauer, T.W. Co-treatment with panitumumab and trastuzumab augments response to the MEK inhibitor trametinib in a patient-derived xenograft model of pancreatic cancer. Neoplasia 2014, 16, 562–571. [Google Scholar] [CrossRef]
- NRC. Update of the Guide for the Care and Use of Laboratory Animals. In Guide for the Care and Use of Laboratory Animals, 8th ed.; National Academies Press: Washington, DC, USA, 2011. [Google Scholar]
- Newhook, T.E.; Lindberg, J.M.; Adair, S.J.; Kim, A.J.; Stelow, E.B.; Rahma, O.E.; Parsons, J.T.; Bauer, T.W. Adjuvant Trametinib Delays the Outgrowth of Occult Pancreatic Cancer in a Mouse Model of Patient-Derived Liver Metastasis. Ann. Surg. Oncol. 2016, 23, 1993–2000. [Google Scholar] [CrossRef]
- Michaels, A.D.; Newhook, T.E.; Adair, S.J.; Morioka, S.; Goudreau, B.J.; Nagdas, S.; Mullen, M.G.; Persily, J.B.; Bullock, T.N.; Slingluff Jr, C.L.; et al. CD47 Blockade as an Adjuvant Immunotherapy for Resectable Pancreatic Cancer. Clin. Cancer Res. 2018, 24, 1415–1425. [Google Scholar] [CrossRef]
- Rajendran, S.; Salwa, S.; Gao, X.; Tabirca, S.; O’Hanlon, D.; O’Sullivan, G.C.; Tangney, M. Murine bioluminescent hepatic tumour model. J. Vis. Exp. 2010, 41, 1977. [Google Scholar]
- Edinger, M.; Cao, Y.A.; Hornig, Y.S.; Jenkins, D.E.; Verneris, M.R.; Bachmann, M.H.; Negrin, R.S.; Contag, C.H. Advancing animal models of neoplasia through in vivo bioluminescence imaging. Eur. J. Cancer. 2002, 38, 2128–2136. [Google Scholar] [CrossRef]
- Lin, H.; Luengo, J.I. Nucleoside protein arginine methyltransferase 5 (PRMT5) inhibitors. Bioorg. Med. Chem. Lett. 2019, 29, 1264–1269. [Google Scholar] [CrossRef]
- Mah, L.J.; El-Osta, A.; Karagiannis, T.C. γH2AX: A sensitive molecular marker of DNA damage and repair. Leukemia 2010, 24, 679–686. [Google Scholar] [CrossRef]
- Rahmanian, N.; Shokrzadeh, M.; Eskandani, M. Recent advances in γH2AX biomarker-based genotoxicity assays: A marker of DNA damage and repair. DNA Repair 2021, 108, 103243. [Google Scholar] [CrossRef] [PubMed]
- Kola, I.; Landis, J. Can the pharmaceutical industry reduce attrition rates? Nat. Rev. Drug Discov. 2004, 3, 711–716. [Google Scholar] [CrossRef] [PubMed]
- He, M.; Henderson, M.; Muth, S.; Murphy, A.; Zheng, L. Preclinical mouse models for immunotherapeutic and non-immunotherapeutic drug development for pancreatic ductal adenocarcinoma. Ann. Pancreat. Cancer. 2020, 3, 7. [Google Scholar] [CrossRef]
- Ho, W.J.; Jaffee, E.M.; Zheng, L. The tumour microenvironment in pancreatic cancer—Clinical challenges and opportunities. Nat. Rev. Clin. Oncol. 2020, 17, 527–540. [Google Scholar] [CrossRef] [PubMed]
- Stathis, A.; Moore, M.J. Advanced pancreatic carcinoma: Current treatment and future challenges. Nat. Rev. Clin. Oncol. 2010, 7, 163–172. [Google Scholar] [CrossRef]
- Medicine USNLo. Study to Evaluate the Safety, Tolerability & Efficacy of TNG462 in Combination in PDAC & NSCLC Patients. Available online: https://clinicaltrials.gov/ct2/show/NCT06922591 (accessed on 8 May 2025).
- Medicine USNLo. Safety and Tolerability of TNG456 Alone and in Combination with Abemaciclib in Patients with Solid Tumors with MTAP Loss. Available online: https://clinicaltrials.gov/ct2/show/NCT06810544 (accessed on 8 May 2025).
- Pawar, J.S.; Al-Amin, M.Y.; Hu, C.D. JNJ-64619178 radiosensitizes and suppresses fractionated ionizing radiation-induced neuroendocrine differentiation (NED) in prostate cancer. Front. Oncol. 2023, 13, 1126482. [Google Scholar] [CrossRef]
- Brehmer, D.; Beke, L.; Wu, T.; Millar, H.J.; Moy, C.; Sun, W.; Mannens, G.; Pande, V.; Boeckx, A.; van Heerde, E.; et al. Discovery and Pharmacological Characterization of JNJ-64619178, a Novel Small-Molecule Inhibitor of PRMT5 with Potent Antitumor Activity. Mol. Cancer Ther. 2021, 20, 2317–2328. [Google Scholar] [CrossRef]
- Soeta, T.; Sugisawa, N.; Yamamura, A.; Tanaka, N.; Imoto, H.; Tsuchiya, T.; Aizawa, T.; Okamoto, K.; Kawamura, M.; Saijo, F.; et al. MRTX1719, an MTA-cooperative PRMT5 Inhibitor, Induces Cell Cycle Arrest and Synergizes with Oxaliplatin and Gemcitabine for Enhanced Anticancer Effects. Anticancer Res. 2024, 44, 5231–5240. [Google Scholar] [CrossRef] [PubMed]
- Ayoub, N.M. Editorial: Novel Combination Therapies for the Treatment of Solid Cancers. Front. Oncol. 2021, 11, 708943. [Google Scholar] [CrossRef] [PubMed]
- Bayat Mokhtari, R.; Homayouni, T.S.; Baluch, N.; Morgatskaya, E.; Kumar, S.; Das, B.; Yeger, H. Combination therapy in combating cancer. Oncotarget 2017, 8, 38022–38043. [Google Scholar] [CrossRef] [PubMed]
- Toledo, L.; Neelsen, K.J.; Lukas, J. Replication Catastrophe: When a Checkpoint Fails because of Exhaustion. Mol. Cell. 2017, 66, 735–749. [Google Scholar] [CrossRef]
- Dahai, Y.; Sanyuan, S.; Hong, L.; Di, Z.; Chong, Z. A Relationship Between Replication Protein A and Occurrence and Prognosis of Esophageal Carcinoma. Cell Biochem. Biophys. 2013, 67, 175–180. [Google Scholar] [CrossRef] [PubMed]
- Givalos, N.; Gakiopoulou, H.; Skliri, M.; Bousboukea, K.; Konstantinidou, A.E.; Korkolopoulou, P.; Lelouda, M.; Kouraklis, G.; Patsouris, E.; Karatzas, G. Replication protein A is an independent prognostic indicator with potential therapeutic implications in colon cancer. Mod. Pathol. 2007, 20, 159–166. [Google Scholar] [CrossRef]
- Glanzer, J.G.; Liu, S.; Wang, L.; Mosel, A.; Peng, A.; Oakley, G.G. RPA inhibition increases replication stress and suppresses tumor growth. Cancer Res. 2014, 74, 5165–5172. [Google Scholar] [CrossRef]
- Zou, Y.; Liu, Y.; Wu, X.; Shell, S.M. Functions of human replication protein A (RPA): From DNA replication to DNA damage and stress responses. J. Cell. Physiol. 2006, 208, 267–273. [Google Scholar] [CrossRef]
- Mirzoeva, O.K.; Petrini, J.H. DNA damage-dependent nuclear dynamics of the Mre11 complex. Mol. Cell. Biol. 2001, 21, 281–288. [Google Scholar] [CrossRef]
- Bedford, M.T.; Clarke, S.G. Protein Arginine Methylation in Mammals: Who, What, and Why. Mol. Cell 2009, 33, 1–13. [Google Scholar] [CrossRef]
- Yu, Z.; Vogel, G.; Coulombe, Y.; Dubeau, D.; Spehalski, E.; Hébert, J.; Ferguson, D.O.; Masson, J.Y.; Richard, S. The MRE11 GAR motif regulates DNA double-strand break processing and ATR activation. Cell Res. 2012, 22, 305–320. [Google Scholar] [CrossRef] [PubMed]
- Clarke, T.L.; Sanchez-Bailon, M.P.; Chiang, K.; Reynolds, J.J.; Herrero-Ruiz, J.; Bandeiras, T.M.; Matias, P.M.; Maslen, S.L.; Skehel, J.M.; Stewart, G.S.; et al. PRMT5-Dependent Methylation of the TIP60 Coactivator RUVBL1 Is a Key Regulator of Homologous Recombination. Mol. Cell 2017, 65, 900–916.e7. [Google Scholar] [CrossRef] [PubMed]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wei, X.; Kane, W.J.; Adair, S.J.; Nagdas, S.; Liu, D.; Bauer, T.W. PRMT5 Identified as a Viable Target for Combination Therapy in Preclinical Models of Pancreatic Cancer. Biomolecules 2025, 15, 948. https://doi.org/10.3390/biom15070948
Wei X, Kane WJ, Adair SJ, Nagdas S, Liu D, Bauer TW. PRMT5 Identified as a Viable Target for Combination Therapy in Preclinical Models of Pancreatic Cancer. Biomolecules. 2025; 15(7):948. https://doi.org/10.3390/biom15070948
Chicago/Turabian StyleWei, Xiaolong, William J. Kane, Sara J. Adair, Sarbajeet Nagdas, Denis Liu, and Todd W. Bauer. 2025. "PRMT5 Identified as a Viable Target for Combination Therapy in Preclinical Models of Pancreatic Cancer" Biomolecules 15, no. 7: 948. https://doi.org/10.3390/biom15070948
APA StyleWei, X., Kane, W. J., Adair, S. J., Nagdas, S., Liu, D., & Bauer, T. W. (2025). PRMT5 Identified as a Viable Target for Combination Therapy in Preclinical Models of Pancreatic Cancer. Biomolecules, 15(7), 948. https://doi.org/10.3390/biom15070948