
Genome-Wide Identification of the GRAS Transcription Factor Family in Sweet Orange and the Regulation of Salt Stress-Enhanced Plant Salt Tolerance in Sweet Orange by CsGRAS15 and CsGRAS27


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Access to Data and Materials
2.2. Genome-Wide Identification and Preliminary Physicochemical Characterization of the GRAS Transcription Factor Family in Sweet Orange (Citrus sinensis)
2.3. Sequence Similarity and Phylogenetic Analysis
2.4. Chromosomal Localization, Duplicate, and Covariance Analysis
2.5. Analysis of Gene Structure and Structural and Conserved Motifs
2.6. Analysis of Cis-Acting Elements
2.7. GO Function and KEGG Pathway Enrichment Analysis of GRAS Genes
2.8. Plant Material, Growing, and Salt Treatments
2.9. Transcriptome Sequencing Analysis
2.10. qRT-PCR Analysis
3. Results
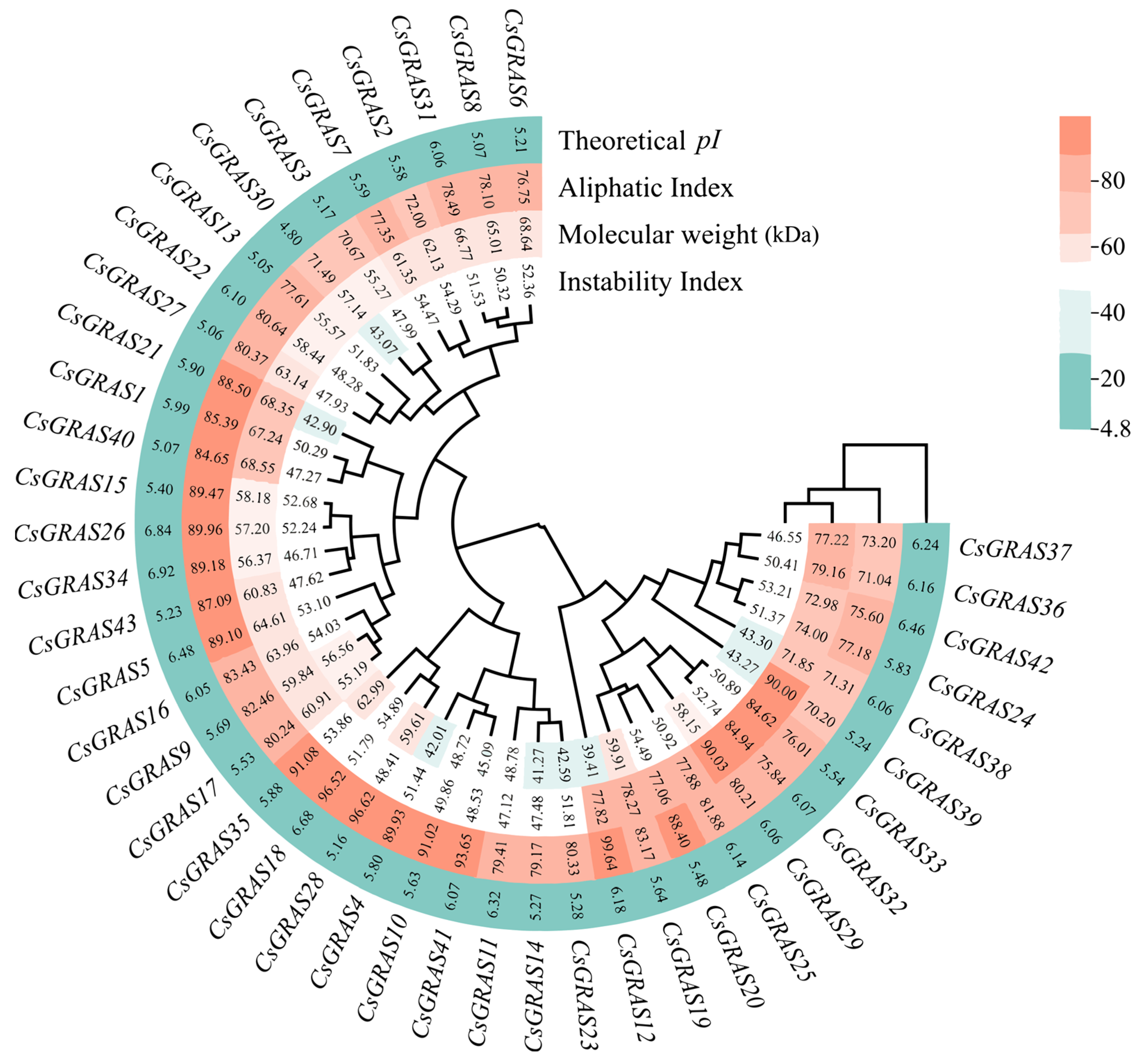
3.1. Identification and Physicochemical Characterization of the Sweet Orange GRAS Transcription Factor Family
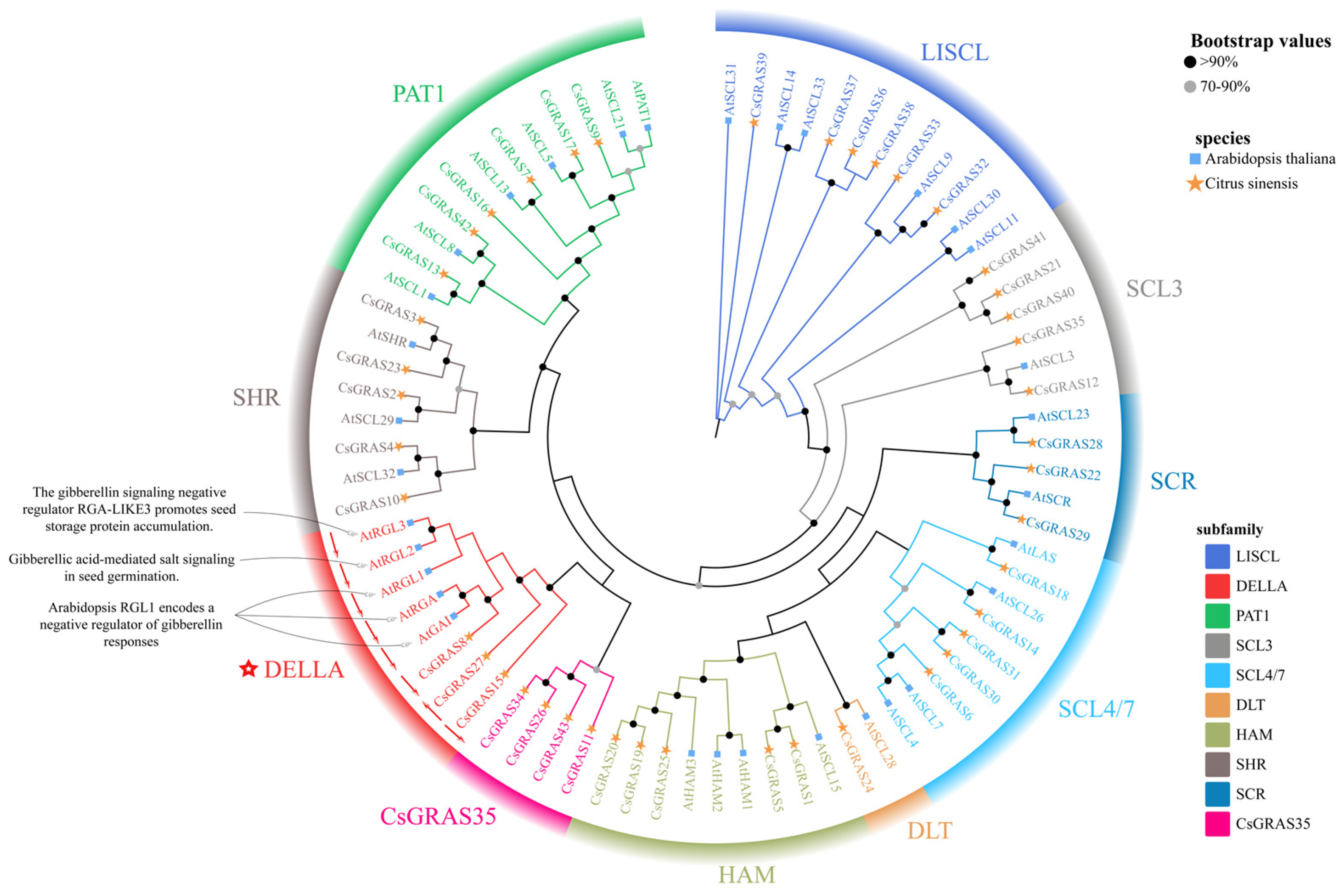
3.2. Sequence Similarity and Phylogenetic Analysis of the Sweet Orange GRAS Gene Family
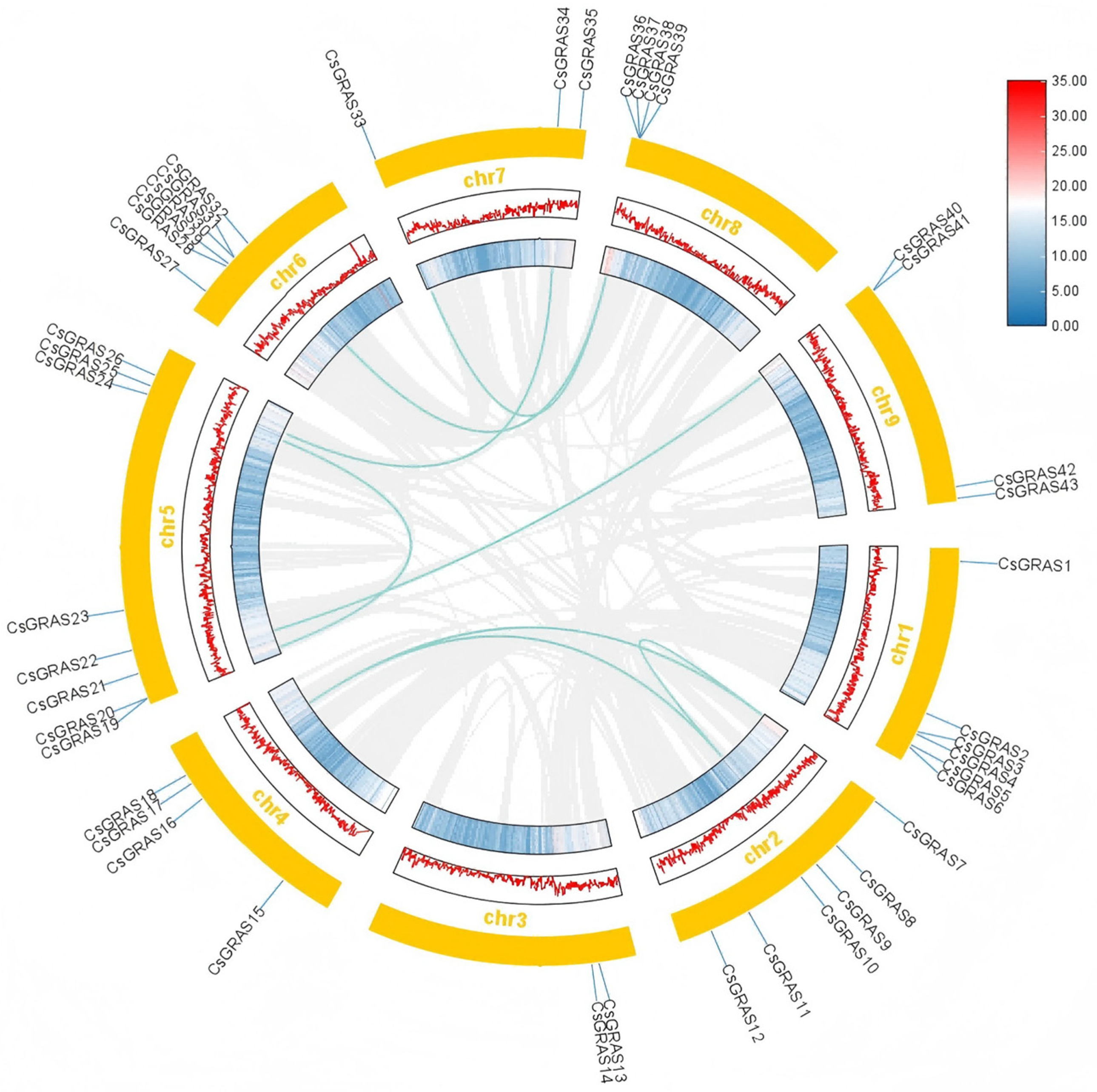
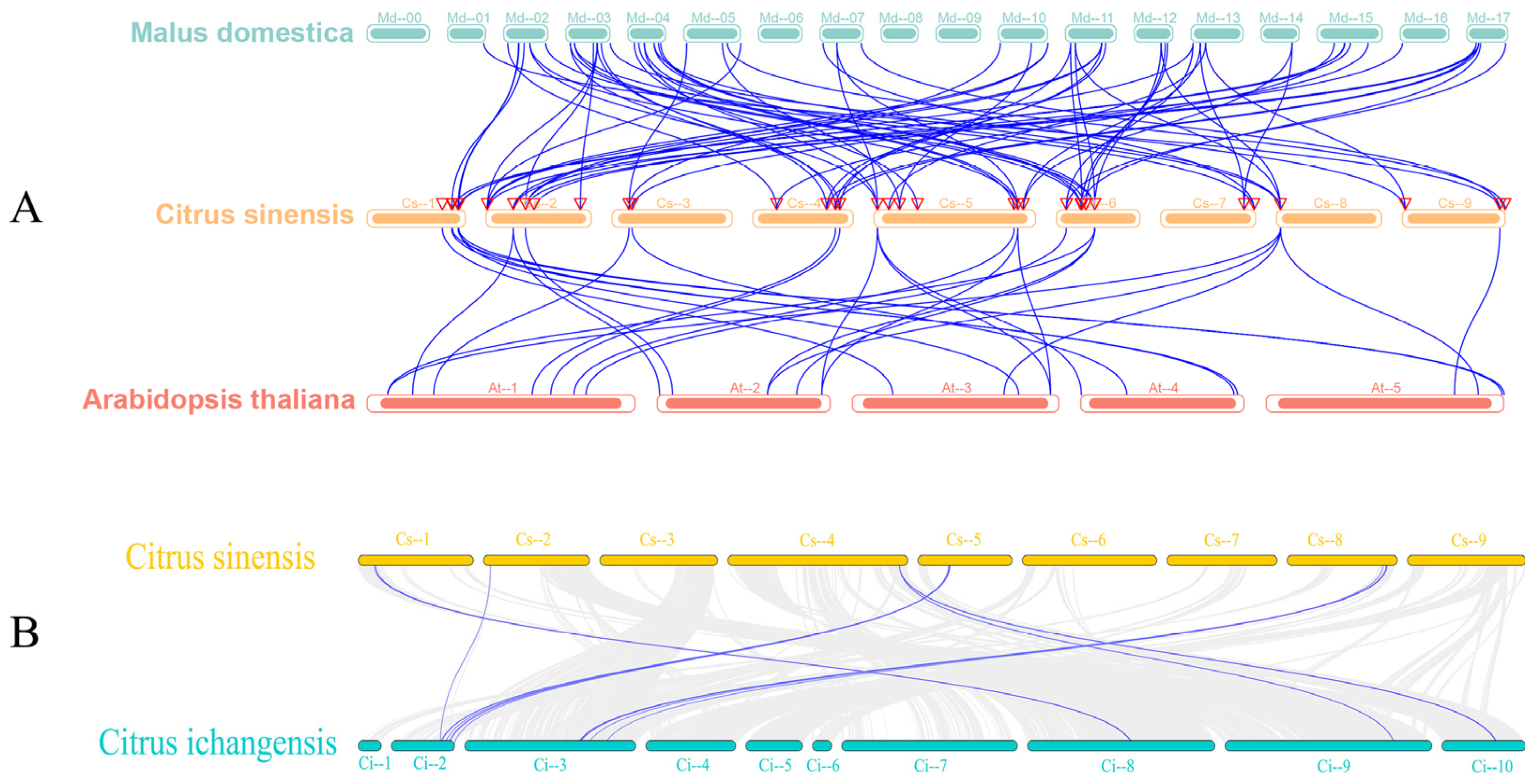
3.3. Chromosomal Localization, Gene, and Covariance Analysis of the Sweet Orange GRAS Transcription Factor Family
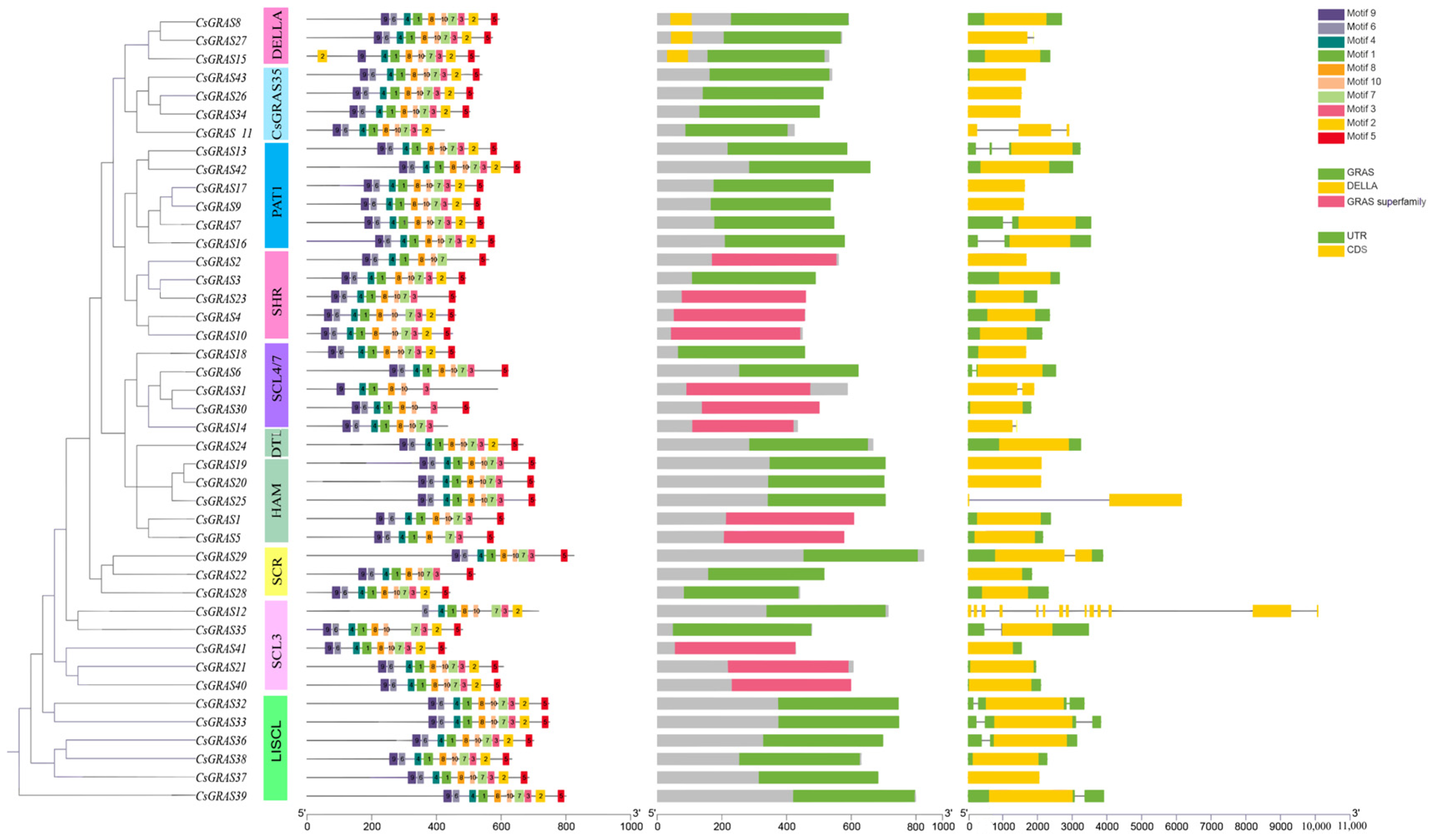
3.4. Analysis of the Gene Structure and Structural and Conserved Motifs of the Sweet Orange GRAS Transcription Factor Family
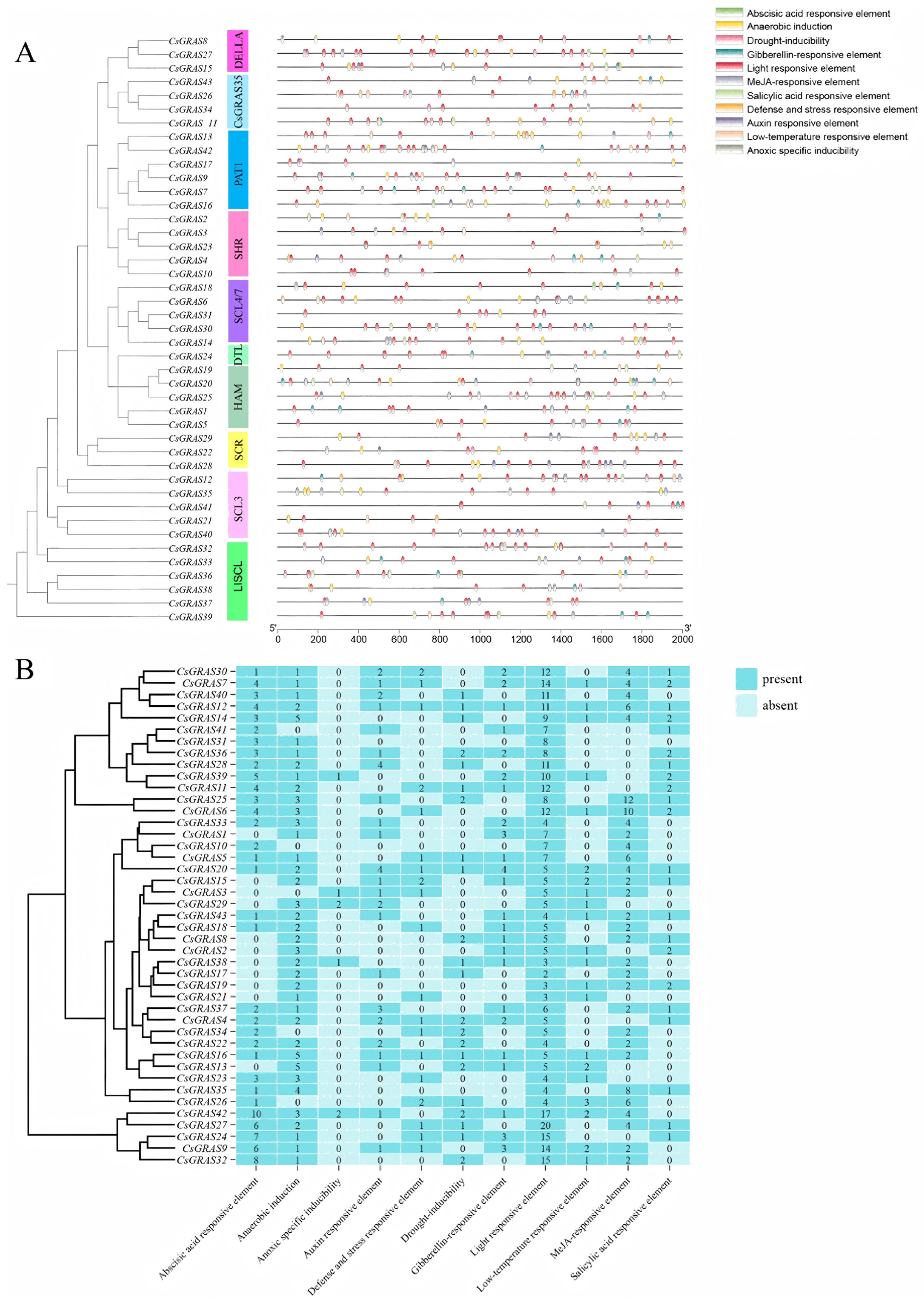
3.5. Analysis of Critical Cis-Acting Elements in the Sweet Orange GRAS Promoter
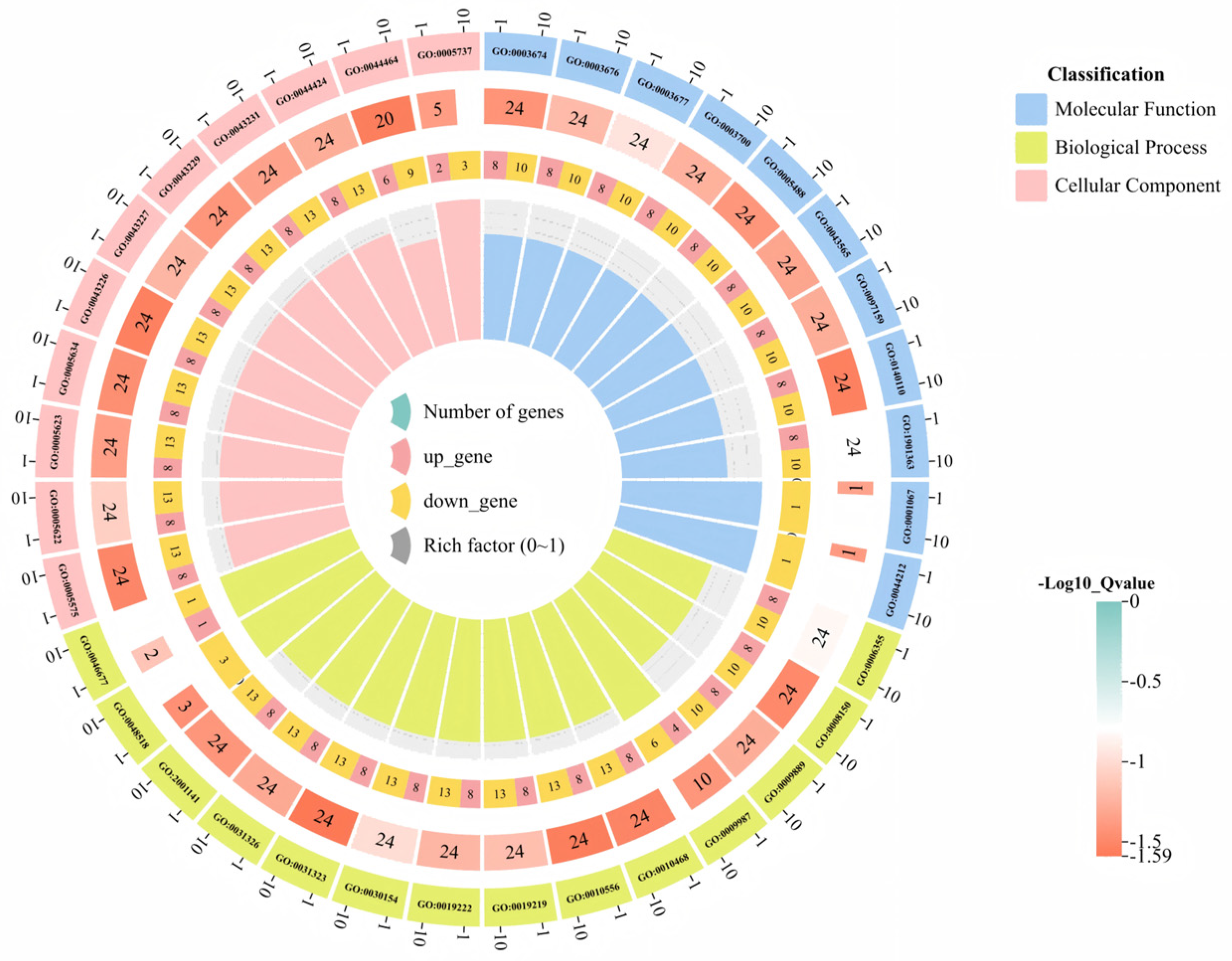
3.6. GO Functional Annotation and KEGG Enrichment Analysis of the Sweet Orange GRAS Transcription Factor Family
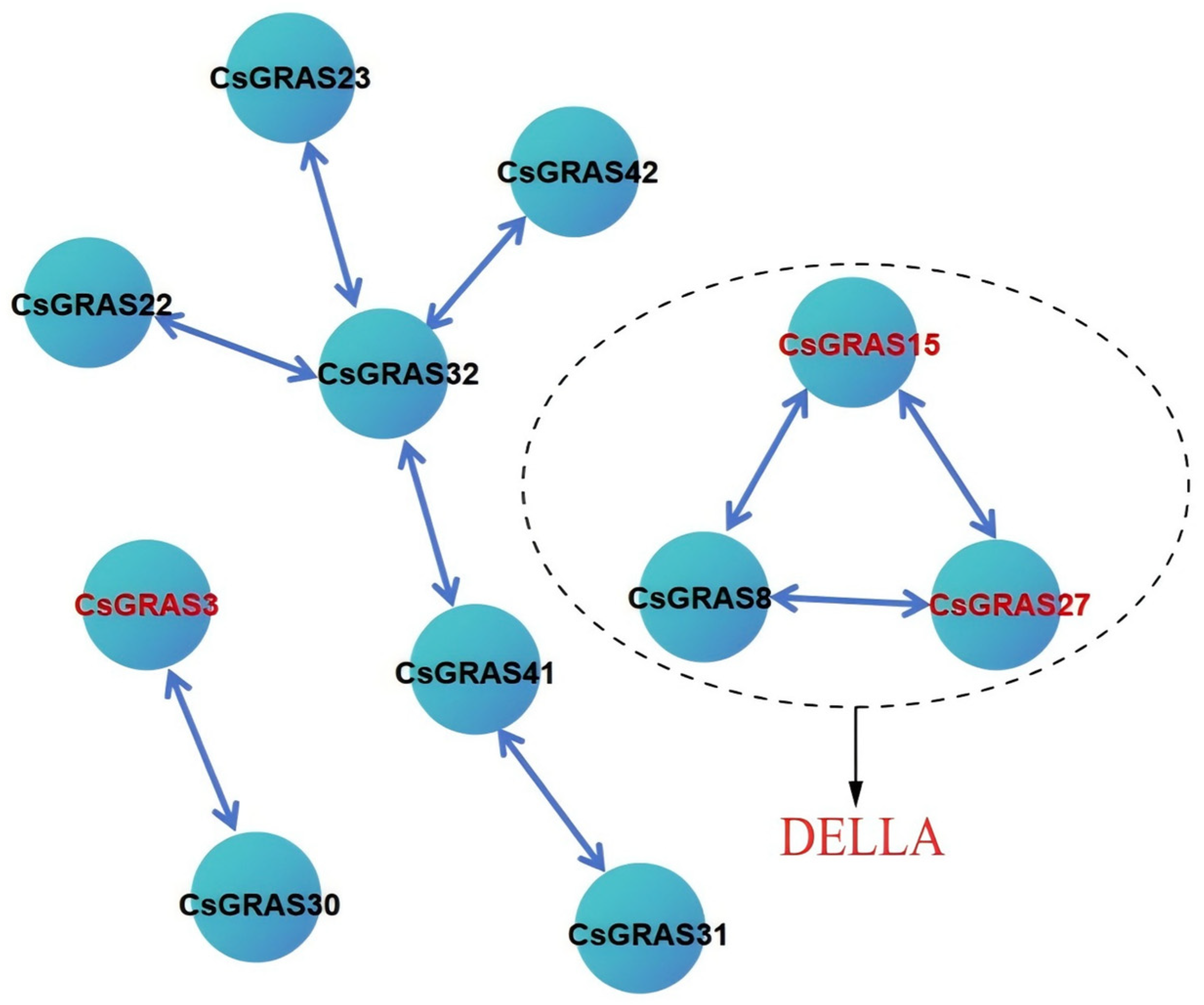
3.7. Analysis of Protein Network Interactions of the Sweet Orange GRAS Transcription Factor Family
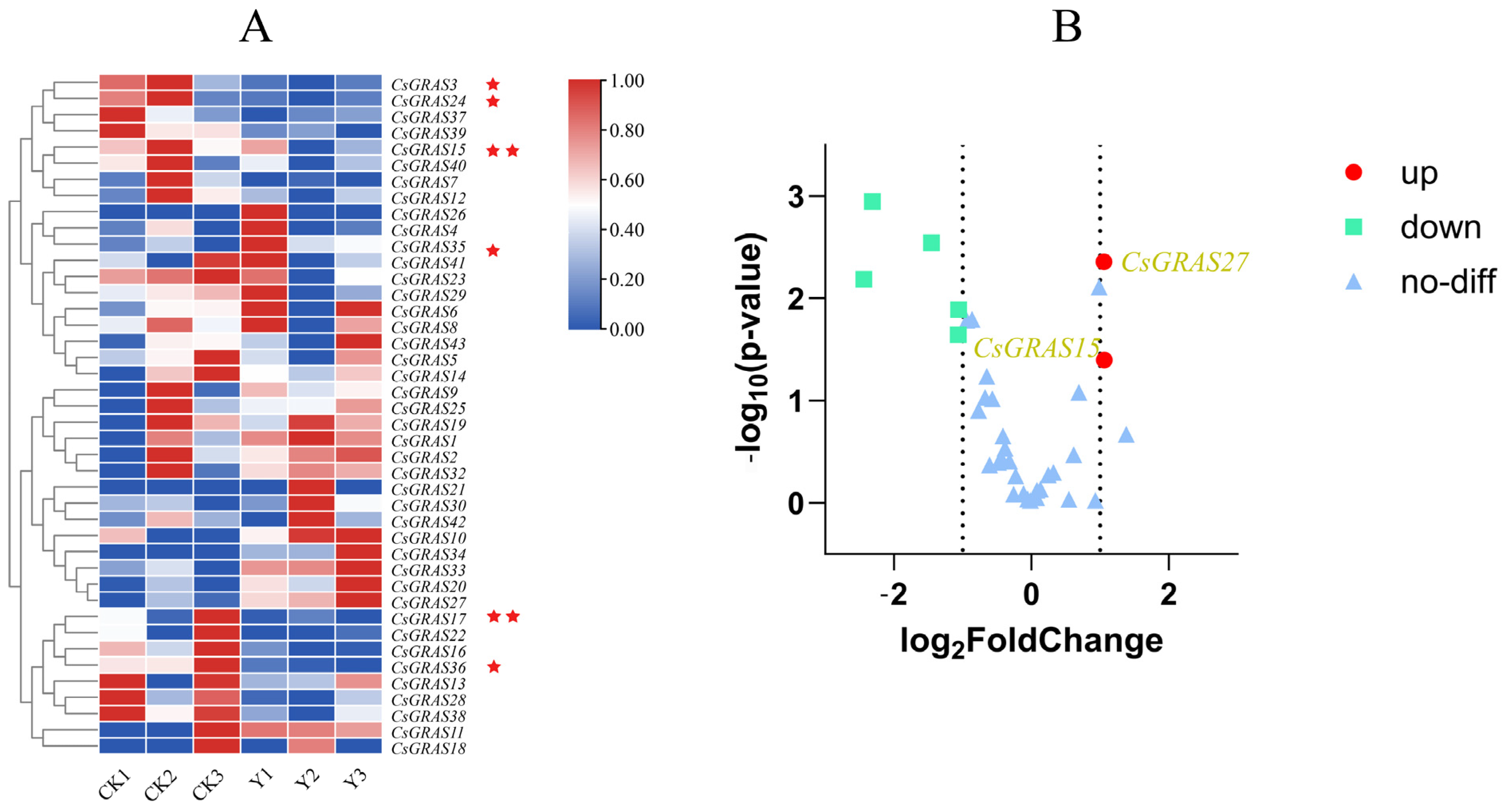
3.8. Expression Pattern Analysis of CsGRAS Transcription Factor Family Genes Under Salt Stress
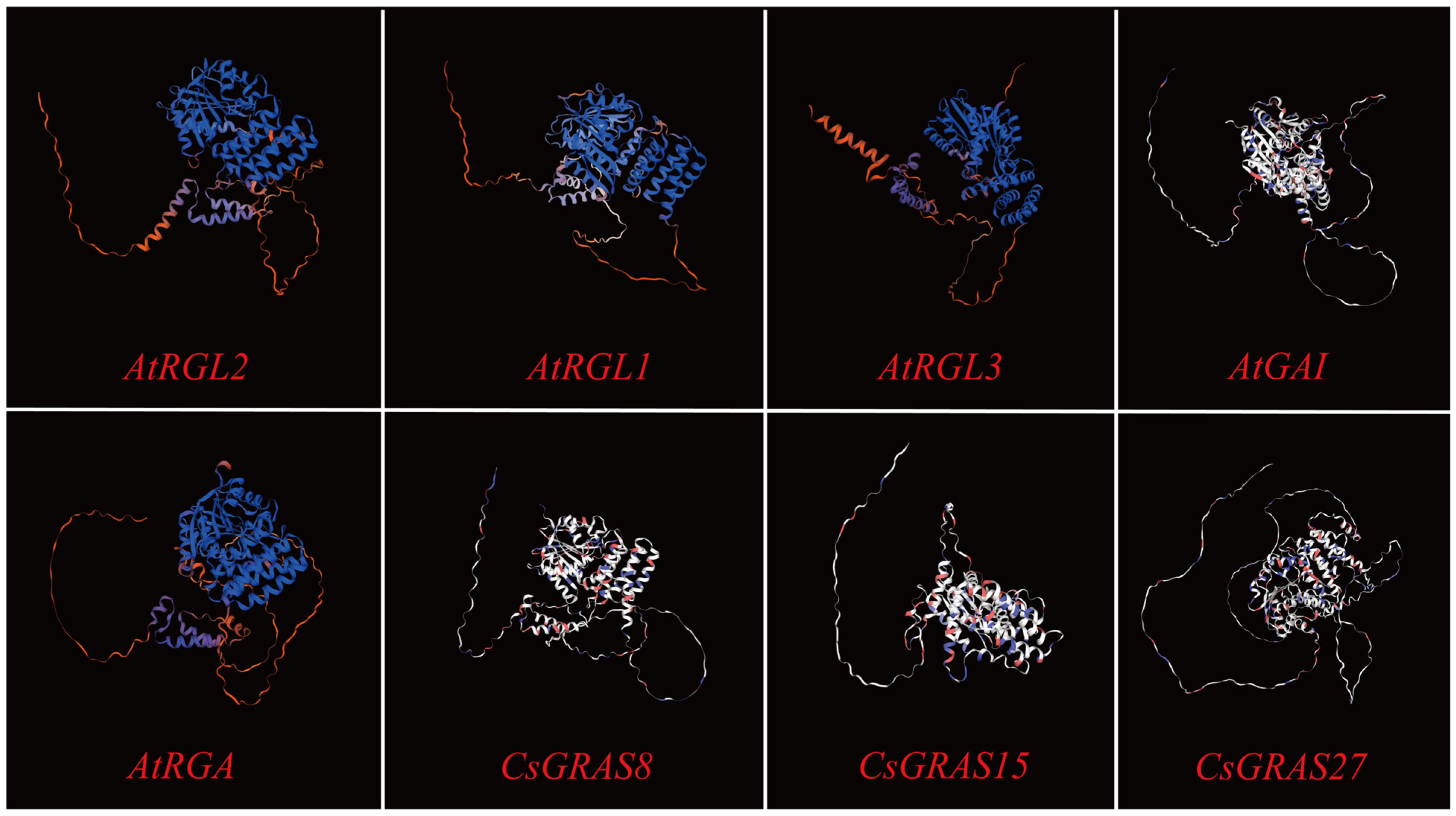
3.9. Multiple Sequence Alignment and Protein Tertiary Structure Prediction of DELLA Subfamily Genes
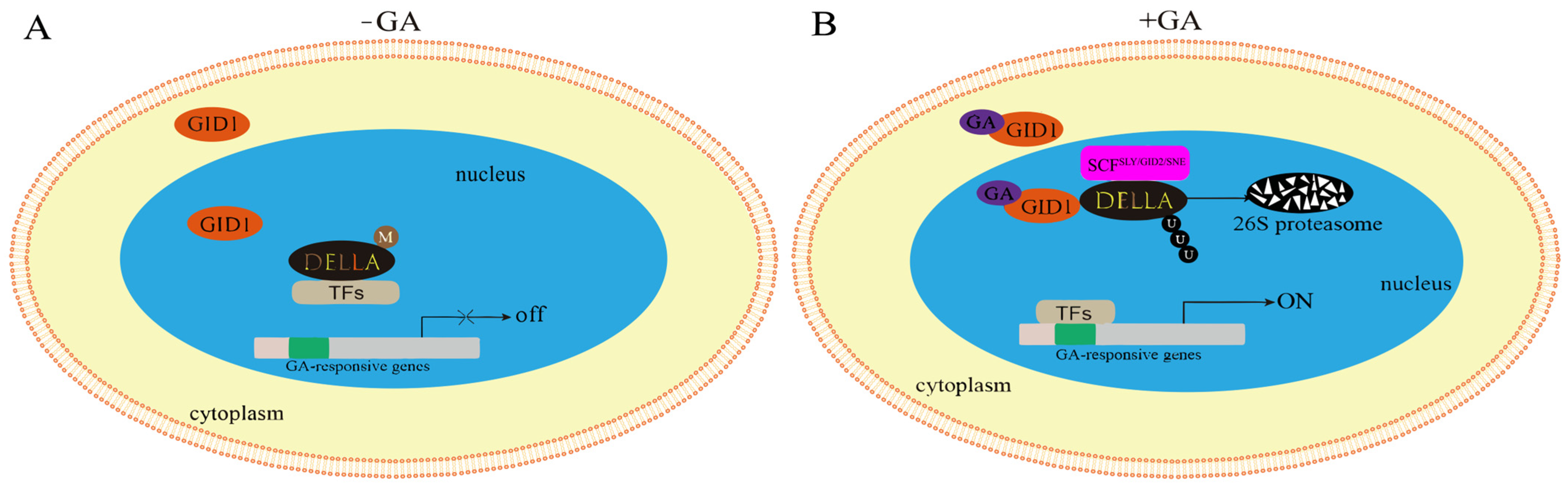
3.10. Analysis of Gibberellin Signaling Under Salt Stress
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Katz, E.; Fon, M.; Lee, Y.J.; Phinney, B.S.; Sadka, A.; Blumwald, E. The citrus fruit proteome: Insights into citrus fruit metabolism. Planta 2007, 226, 989–1005. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Ma, Y.; Qiu, Y.X.; Long, S.S.; Dai, W.S. Genome-wide characterization and expression profiling of the TGA gene family in sweet orange (Citrus sinensis) reveal CsTGA7 responses to multiple phytohormones and abiotic stresses. Front. Plant Sci. 2025, 16, 1530242. [Google Scholar] [CrossRef]
- Ahmed, F.F.; Dola, F.S.; Zohra, F.T.; Rahman, S.M.; Konak, J.N.; Sarkar, M.A.R. Genome-wide identification, classification, and characterization of lectin gene superfamily in sweet orange (Citrus sinensis L.). PLoS ONE 2023, 18, e0294233. [Google Scholar] [CrossRef]
- Wang, S.; Duan, Z.; Yan, Q.; Wu, F.; Zhou, P.; Zhang, J. Genome–Wide Identification of the GRAS Family Genes in Melilotus albus and Expression Analysis under Various Tissues and Abiotic Stresses. Int. J. Mol. Sci. 2022, 23, 7403. [Google Scholar] [CrossRef]
- Elsheery, N.I.; Helaly, M.N.; El-Hoseiny, H.M.; Alam-Eldein, S.M. Zinc Oxide and Silicone Nanoparticles to Improve the Resistance Mechanism and Annual Productivity of Salt-Stressed Mango Trees. Agronomy 2020, 10, 558. [Google Scholar] [CrossRef]
- Saiki, S.T.; Ishida, A.; Yoshimura, K.; Yazaki, K. Physiological mechanisms of drought-induced tree die-off in relation to carbon, hydraulic and respiratory stress in a drought-tolerant woody plant. Sci. Rep. 2017, 7, 2995. [Google Scholar] [CrossRef]
- van Zelm, E.; Zhang, Y.; Testerink, C. Salt Tolerance Mechanisms of Plants. Annu. Rev. Plant Biol. 2020, 71, 403–433. [Google Scholar] [CrossRef] [PubMed]
- Rasool, S.; Ahmad, A.; Siddiqi, T.O.; Ahmad, P. Changes in growth, lipid peroxidation and some key antioxidant enzymes in chickpea genotypes under salt stress. Acta Physiol. Plant. 2013, 4, 35. [Google Scholar] [CrossRef]
- Salladini, E.; Jørgensen, M.L.M.; Theisen, F.F.; Skriver, K. Intrinsic Disorder in Plant Transcription Factor Systems: Functional Implications. Int. J. Mol. Sci. 2020, 21, 9755. [Google Scholar] [CrossRef]
- Negi, Y.; Kumar, K. Cloning, homology modelling and expression analysis of Oryza sativa WNK gene family. Int. J. Biol. Macromol. 2023, 229, 994–1008. [Google Scholar] [CrossRef]
- Mizoi, J.; Shinozaki, K.; Yamaguchi-Shinozaki, K. AP2/ERF family transcription factors in plant abiotic stress responses. Biochim. Biophys. Acta (BBA)-Gene Regul. Mech. 2012, 1819, 86–96. [Google Scholar] [CrossRef]
- Liu, C.; Mao, B.; Ou, S.; Wang, W.; Liu, L.; Wu, Y.; Chu, C.; Wang, X. OsbZIP71, a bZIP transcription factor, confers salinity and drought tolerance in rice. Plant Mol. Biol. 2013, 84, 19–36. [Google Scholar] [CrossRef]
- Erpen, L.; Devi, H.S.; Grosser, J.W.; Dutt, M. Potential use of the DREB/ERF, MYB, NAC and WRKY transcription factors to improve abiotic and biotic stress in transgenic plants. Plant Cell Tissue Organ Cult. (PCTOC) 2017, 132, 1–25. [Google Scholar] [CrossRef]
- Pysh, L.D.; Wysocka-Diller, J.W.; Camilleri, C.; Bouchez, D.; Benfey, P.N. The GRAS gene family in Arabidopsis: Sequence characterization and basic expression analysis of the SCARECROW-LIKE genes. Plant J. 2002, 18, 111–119. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.; Iyer, L.M.; Aravind, L. Bacterial GRAS domain proteins throw new light on gibberellic acid response mechanisms. Bioinformatics 2012, 28, 2407–2411. [Google Scholar] [CrossRef]
- Itoh, H.; Ueguchi, T.M.; Sato, Y.; Ashikari, M.; Matsuoka, M. The Gibberellin Signaling Pathway Is Regulated by the Appearance and Disappearance of SLENDER RICE1 in Nuclei. Plant Cell 2002, 14, 57–70. [Google Scholar] [CrossRef]
- Cenci, A.; Rouard, M. Evolutionary Analyses of GRAS Transcription Factors in Angiosperms. Front. Plant Sci. 2017, 8, 273. [Google Scholar] [CrossRef] [PubMed]
- Sun, X.; Jones William, T.; Rikkerink Erik, H.A. GRAS proteins: The versatile roles of intrinsically disordered proteins in plant signalling. Biochem. J. 2012, 442, 1–12. [Google Scholar] [CrossRef]
- Waseem, M.; Nkurikiyimfura, O.; Niyitanga, S.; Jakada, B.H.; Shaheen, I.; Aslam, M.M. GRAS transcription factors emerging regulator in plants growth, development, and multiple stresses. Mol. Biol. Rep. 2022, 49, 9673–9685. [Google Scholar] [CrossRef]
- Neves, C.; Ribeiro, B.; Amaro, R.; Expósito, J.; Grimplet, J.; Fortes, A.M. Network of GRAS transcription factors in plant development, fruit ripening and stress responses. Hortic. Res. 2023, 10, uhad220. [Google Scholar] [CrossRef]
- Lee, M.H.; Kim, B.; Song, S.K.; Heo, J.O.; Yu, N.I.; Lee, S.A.; Kim, M.; Kim, D.G.; Sohn, S.O.; Lim, C.E.; et al. Large-scale analysis of the GRAS gene family in Arabidopsis thaliana. Plant Mol. Biol. 2008, 67, 659–670. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Zhao, Y.; Wu, Y. Expression and Purification of the GRAS Domain of Os-SCL7 from Rice for Structural Studies. Bio-Protocol 2017, 7, e2122. [Google Scholar] [CrossRef]
- Liu, X.; Widmer, A. Genome-wide Comparative Analysis of the GRAS Gene Family in Populus, Arabidopsis and Rice. Plant Mol. Biol. Rep. 2014, 32, 1129–1145. [Google Scholar] [CrossRef]
- Abarca, D.; Pizarro, A.; Hernández, I.; Sánchez, C.; Solana, S.P.; del Amo, A.; Carneros, E.; Díaz-Sala, C. The GRAS gene family in pine: Transcript expression patterns associated with the maturation-related decline of competence to form adventitious roots. BMC Plant Biol. 2014, 14, 354. [Google Scholar] [CrossRef] [PubMed]
- Grimplet, J.; Agudelo-Romero, P.; Teixeira, R.T.; Martinez-Zapater, J.M.; Fortes, A.M. Structural and Functional Analysis of the GRAS Gene Family in Grapevine Indicates a Role of GRAS Proteins in the Control of Development and Stress Responses. Front. Plant Sci. 2016, 7, 353. [Google Scholar] [CrossRef]
- Wang, L.; Ding, X.; Gao, Y.; Yang, S. Genome-wide identification and characterization of GRAS genes in soybean (Glycine max). BMC Plant Biol. 2020, 20, 415. [Google Scholar] [CrossRef]
- Pilkington, S.M.; Crowhurst, R.; Hilario, E. A manually annotated Actinidia chinensis var. chinensis (kiwifruit) genome highlights the challenges associated with draft genomes and gene prediction in plants. BMC Genom. 2018, 19, 257. [Google Scholar] [CrossRef]
- Yue, J.; Liu, J.; Tang, W.; Wu, Y.Q.; Tang, X.; Li, W.; Yang, Y.; Wang, L.; Huang, S.; Fang, C.; et al. Kiwifruit Genome Database (KGD): A comprehensive resource for kiwifruit genomics. Hortic. Res. 2020, 7, 117. [Google Scholar] [CrossRef]
- Madeira, F.; Park, Y.m.; Lee, J.; Buso, N.; Gur, T.; Madhusoodanan, N.; Basutkar, P.; Tivey, A.R.N.; Potter, S.C.; Finn, R.D.; et al. The EMBL-EBI search and sequence analysis tools APIs in 2019. Nucleic Acids Res. 2019, 47, W636–W641. [Google Scholar] [CrossRef]
- Yin, T.; Han, P.; Xi, D.; Yu, W.; Zhu, L.; Du, C.; Yang, N.; Liu, X.; Zhang, H. Genome-wide identification, characterization, and expression profile of NBS-LRR gene family in sweet orange (Citrus sinensis). Gene 2023, 854, 147117. [Google Scholar] [CrossRef]
- Chen, C.; Chen, H.; Zhang, Y.; Thomas, H.R.; Frank, M.H.; He, Y.; Xia, R. TBtools: An Integrative Toolkit Developed for Interactive Analyses of Big Biological Data. Mol. Plant 2020, 13, 1194–1202. [Google Scholar] [CrossRef] [PubMed]
- Xi, D.X.; Yin, T.; Han, P.C.; Yang, X.Y.; Zhang, M.J.; Du, C.J.; Zhang, H.Y.; Liu, X.Z. Genome-Wide Identification of Sweet Orange WRKY Transcription Factors and Analysis of Their Expression in Response to Infection by. Curr. Issues Mol. Biol. 2023, 45, 1250–1271. [Google Scholar] [CrossRef]
- Gao, X.; Gao, Z.; Zhang, M.; Qiao, H.; Jiang, S.; Zhang, W.; Xiong, Y.; Jin, S.; Fu, H. Identifying Relationships between Glutathione S-Transferase-2 Single Nucleotide Polymorphisms and Hypoxia Tolerance and Growth Traits in Macrobrachium nipponense. Animals 2024, 14, 666. [Google Scholar] [CrossRef]
- Tian, C.; Wan, P.; Sun, S.; Li, J.; Chen, M. Genome-Wide Analysis of the GRAS Gene Family in Rice and Arabidopsis. Plant Mol. Biol. 2004, 54, 519–532. [Google Scholar] [CrossRef] [PubMed]
- You, J.; Wang, Y.; Zhang, Y.; Dossa, K.; Li, D.; Zhou, R.; Wang, L.; Zhang, X. Genome-wide identification and expression analyses of genes involved in raffinose accumulation in sesame. Sci. Rep. 2018, 8, 4331. [Google Scholar] [CrossRef] [PubMed]
- Wierzbicki, A.T.; Blevins, T.; Swiezewski, S. Long Noncoding RNAs in Plants. Annu. Rev. Plant Biol. 2021, 72, 245–271. [Google Scholar] [CrossRef]
- Marchler-Bauer, A.; Lu, S.; Anderson, J.B.; Chitsaz, F.; Derbyshire, M.K.; DeWeese-Scott, C.; Fong, J.H.; Geer, L.Y.; Geer, R.C.; Gonzales, N.R.; et al. CDD: A Conserved Domain Database for the functional annotation of proteins. Nucleic Acids Res. 2010, 39, D225–D229. [Google Scholar] [CrossRef] [PubMed]
- Lescot, M.; Déhais, P.; Thijs, G.; Marchal, K.; Moreau, Y.; Van de Peer, Y.; Rouzé, P.; Rombauts, S. PlantCARE, a database of plant cis-acting regulatory elements and a portal to tools for in silico analysis of promoter sequences. Nucleic Acids Res. 2002, 30, 325–327. [Google Scholar] [CrossRef]
- Cantalapiedra, C.P.; Hernández-Plaza, A.; Letunic, I.; Bork, P.; Huerta-Cepas, J.; Tamura, K. eggNOG-mapper v2: Functional Annotation, Orthology Assignments, and Domain Prediction at the Metagenomic Scale. Mol. Biol. Evol. 2021, 38, 5825–5829. [Google Scholar] [CrossRef]
- Zi, Y.; Zhang, M.; Yang, X.; Zhao, K.; Yin, T.; Wen, K.; Li, X.; Liu, X.; Zhang, H. Identification of the sweet orange (Citrus sinensis) bHLH gene family and the role of CsbHLH55 and CsbHLH87 in regulating salt stress. Plant Genome 2024, 17, e20502. [Google Scholar] [CrossRef]
- Benjamini, Y.; Yekutieli, D. The control of the false discovery rate in multiple testing under dependency. Ann. Stat. 2001, 29, 1165–1188. [Google Scholar] [CrossRef]
- Kim, S.G.; Park, C.M. Gibberellic acid-mediated salt signaling in seed germination. Plant Signal. Behav. 2014, 3, 877–879. [Google Scholar] [CrossRef]
- Hu, Y.; Zhou, L.; Yang, Y.; Zhang, W.; Chen, Z.; Li, X.; Qian, Q.; Kong, F.; Li, Y.; Liu, X.; et al. The gibberellin signaling negative regulator RGA-LIKE3 promotes seed storage protein accumulation. Plant Physiol. 2021, 185, 1697–1707. [Google Scholar] [CrossRef]
- Wen, C.K.; Chang, C. Arabidopsis RGL1 Encodes a Negative Regulator of Gibberellin Responses. Plant Cell 2002, 14, 87–100. [Google Scholar] [CrossRef]
- Wang, Y.; Tang, H.; DeBarry, J.D.; Tan, X.; Li, J.; Wang, X.; Lee, T.h.; Jin, H.; Marler, B.; Guo, H.; et al. MCScanX: A toolkit for detection and evolutionary analysis of gene synteny and collinearity. Nucleic Acids Res. 2012, 40, e49. [Google Scholar] [CrossRef] [PubMed]
- Seo, M.-H.; Kim, P.M. The present and the future of motif-mediated protein–protein interactions. Curr. Opin. Struct. Biol. 2018, 50, 162–170. [Google Scholar] [CrossRef]
- Schönberger, B.; Schaal, C.; Schäfer, R.; Voß, B. RNA interactomics: Recent advances and remaining challenges. F1000Research 2018, 7, F1000. [Google Scholar] [CrossRef] [PubMed]
- Park, J.; Nguyen, K.T.; Park, E.; Jeon, J.-S.; Choi, G. DELLA Proteins and Their Interacting RING Finger Proteins Repress Gibberellin Responses by Binding to the Promoters of a Subset of Gibberellin-Responsive Genes in Arabidopsis. Plant Cell 2013, 25, 927–943. [Google Scholar] [CrossRef]
- Kanehisa, M.; Goto, S. KEGG: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 2000, 28, 27–30. [Google Scholar] [CrossRef]
- Sun, T.P. The Molecular Mechanism and Evolution of the GA–GID1–DELLA Signaling Module in Plants. Curr. Biol. 2011, 21, R338–R345. [Google Scholar] [CrossRef]
- Wu, T.Y.; Goh, H.; Azodi, C.B.; Krishnamoorthi, S.; Liu, M.J.; Urano, D. Evolutionarily conserved hierarchical gene regulatory networks for plant salt stress response. Nat. Plants 2021, 7, 787–799. [Google Scholar] [CrossRef]
- Zhang, H.; Liu, X.; Wang, X.; Sun, M.; Song, R.; Mao, P.; Jia, S. Genome-Wide Identification of GRAS Gene Family and Their Responses to Abiotic Stress in Medicago sativa. Int. J. Mol. Sci. 2021, 22, 7729. [Google Scholar] [CrossRef]
- Perez-Perez, J.G.; Syvertsen, J.P.; Botia, P.; Garcia-Sanchez, F. Leaf water relations and net gas exchange responses of salinized Carrizo citrange seedlings during drought stress and recovery. Ann. Bot. 2007, 100, 335–345. [Google Scholar] [CrossRef] [PubMed]
- Chen, K.; Li, H.; Chen, Y.; Zheng, Q.; Li, B.; Li, Z. TaSCL14, a Novel Wheat (Triticum aestivum L.) GRAS Gene, Regulates Plant Growth, Photosynthesis, Tolerance to Photooxidative Stress, and Senescence. J. Genet. Genom. 2015, 42, 21–32. [Google Scholar] [CrossRef] [PubMed]
- Hakoshima, T. Structural basis of the specific interactions of GRAS family proteins. FEBS Lett. 2018, 592, 489–501. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Liu, S.; Liu, D.; Guo, F.; Yang, Y.; Dong, T.; Zhang, Y.; Ma, C.; Tang, Z.; Li, F.; et al. Genome-wide survey and expression analysis of GRAS transcription factor family in sweet potato provides insights into their potential roles in stress response. BMC Plant Biol. 2022, 22, 232. [Google Scholar] [CrossRef]
- Wang, S.; Zhang, N.; Zhu, X.; Yang, J.; Li, S.; Che, Y.; Liu, W.; Si, H. Identification and expression analysis of StGRAS gene family in potato (Solanum tuberosum L.). Comput. Biol. Chem. 2019, 80, 195–205. [Google Scholar] [CrossRef]
- Shabalina, S.A.; Ogurtsov, A.Y.; Spiridonov, A.N.; Novichkov, P.S.; Spiridonov, N.A.; Koonin, E.V. Distinct Patterns of Expression and Evolution of Intronless and Intron-Containing Mammalian Genes. Mol. Biol. Evol. 2010, 27, 1745–1749. [Google Scholar] [CrossRef]
- Goldy, C.; Pedroza-Garcia, J.A.; Breakfield, N.; Cools, T.; Vena, R.; Benfey, P.N.; De Veylder, L.; Palatnik, J.; Rodriguez, R.E. The Arabidopsis GRAS-type SCL28 transcription factor controls the mitotic cell cycle and division plane orientation. Proc. Natl. Acad. Sci. USA 2021, 118, e2005256118. [Google Scholar] [CrossRef]
- Murase, K.; Hirano, Y.; Sun, T.P.; Hakoshima, T. Gibberellin-induced DELLA recognition by the gibberellin receptor GID1. Nature 2008, 456, 459–463. [Google Scholar] [CrossRef]
- Colebrook, E.H.; Thomas, S.G.; Phillips, A.L.; Hedden, P.; Davies, S.A.; Dow, J.A.T.; Lukowiak, K. The role of gibberellin signalling in plant responses to abiotic stress. J. Exp. Biol. 2014, 217, 67–75. [Google Scholar] [CrossRef] [PubMed]
- Achard, P.; Cheng, H.; De Grauwe, L.; Decat, J.; Schoutteten, H.; Moritz, T.; Van Der Straeten, D.; Peng, J.; Harberd, N.P. Integration of Plant Responses to Environmentally Activated Phytohormonal Signals. Science 2006, 311, 91–94. [Google Scholar] [CrossRef] [PubMed]
- Fan, G.F.; Xia, X.H.; Yao, W.J.; Cheng, Z.H.; Zhang, X.M.; Jiang, J.H.; Zhou, B.R.; Jiang, T.B. Genome-Wide Identification and Expression Patterns of the F-box Family in Poplar under Salt Stress. Int. J. Mol. Sci. 2022, 23, 10934. [Google Scholar] [CrossRef] [PubMed]
- Xu, F.; Xiao, W.; Li, J.; Ding, C.; Li, S.; Liu, W.; Guo, H. NtGRAS-R1, a topping responsive transcription regulator in tobacco roots. Acta Physiol. Plant. 2015, 37, 191. [Google Scholar] [CrossRef]
- Stuurman, J.; Jäggi, F.; Kuhlemeier, C. Shoot meristem maintenance is controlled by a GRAS-gene mediated signal from differentiating cells. Genes Dev. 2002, 16, 2213–2218. [Google Scholar] [CrossRef]




Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ren, H.; Xu, R.; Wang, J.; Zhang, Q.; Nie, L.; Zhang, L.; Zhou, X.; Liu, X.; Zhang, H. Genome-Wide Identification of the GRAS Transcription Factor Family in Sweet Orange and the Regulation of Salt Stress-Enhanced Plant Salt Tolerance in Sweet Orange by CsGRAS15 and CsGRAS27. Biomolecules 2025, 15, 946. https://doi.org/10.3390/biom15070946
Ren H, Xu R, Wang J, Zhang Q, Nie L, Zhang L, Zhou X, Liu X, Zhang H. Genome-Wide Identification of the GRAS Transcription Factor Family in Sweet Orange and the Regulation of Salt Stress-Enhanced Plant Salt Tolerance in Sweet Orange by CsGRAS15 and CsGRAS27. Biomolecules. 2025; 15(7):946. https://doi.org/10.3390/biom15070946
Chicago/Turabian StyleRen, Hailin, Rong Xu, Jie Wang, Qian Zhang, Lili Nie, Li Zhang, Xianyan Zhou, Xiaozhen Liu, and Hanyao Zhang. 2025. "Genome-Wide Identification of the GRAS Transcription Factor Family in Sweet Orange and the Regulation of Salt Stress-Enhanced Plant Salt Tolerance in Sweet Orange by CsGRAS15 and CsGRAS27" Biomolecules 15, no. 7: 946. https://doi.org/10.3390/biom15070946
APA StyleRen, H., Xu, R., Wang, J., Zhang, Q., Nie, L., Zhang, L., Zhou, X., Liu, X., & Zhang, H. (2025). Genome-Wide Identification of the GRAS Transcription Factor Family in Sweet Orange and the Regulation of Salt Stress-Enhanced Plant Salt Tolerance in Sweet Orange by CsGRAS15 and CsGRAS27. Biomolecules, 15(7), 946. https://doi.org/10.3390/biom15070946