
Probing the Active Site of Class 3 L-Asparaginase by Mutagenesis: Mutations of the Ser-Lys Tandems of ReAV

 , , , , and
, , , , and
Abstract
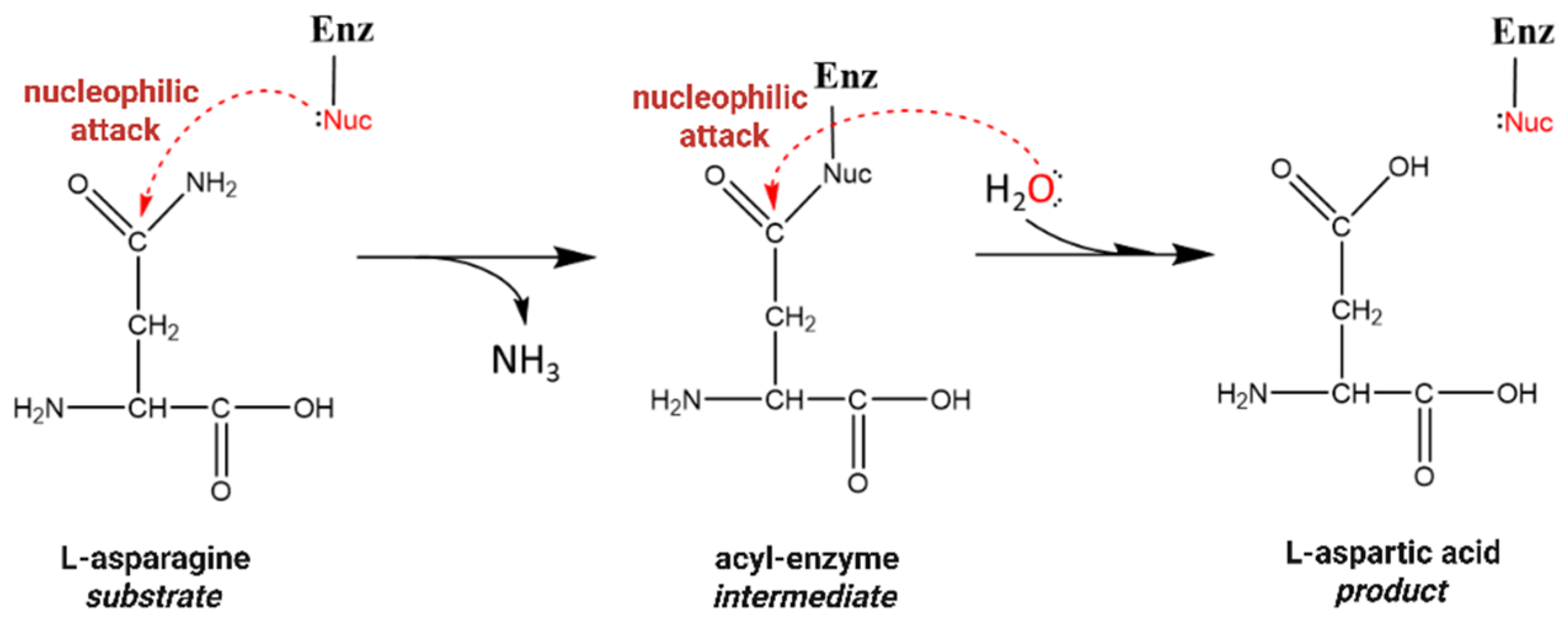
1. Introduction
2. Methods
2.1. Site-Directed Mutagenesis
2.2. Protein Expression and Purification
2.3. Measurements of Enzymatic Activity of ReAV Mutants
2.4. Crystallization
2.5. X-Ray Data Collection, Crystal Structure Solution, and Refinement
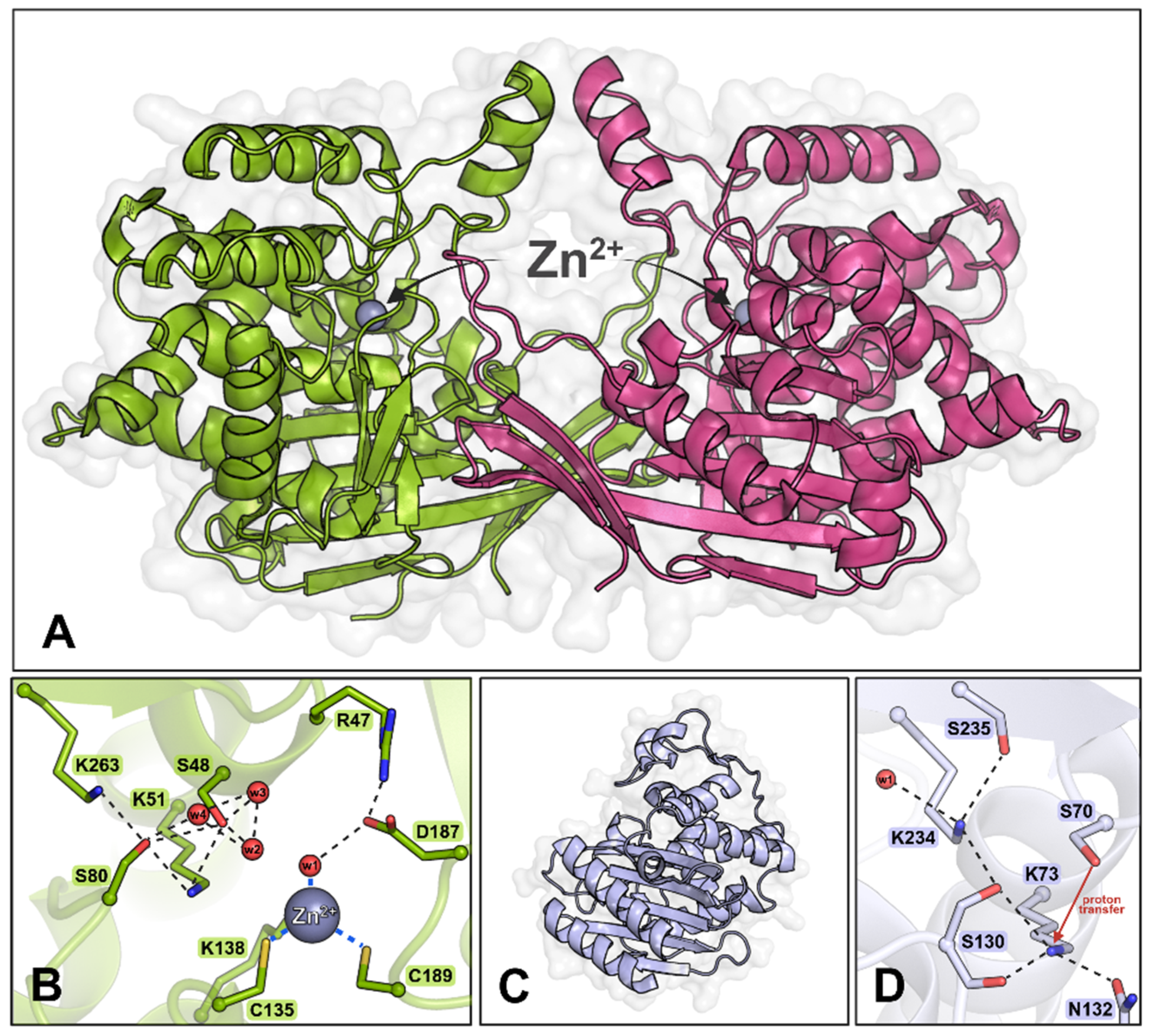
3. Results and Discussion
3.1. Exploring the Importance and Properties of Selected ReAV Residues
3.2. Expression and Purification of the Generated ReAV Mutants
3.3. Enzymatic Activity and Kinetic Measurements
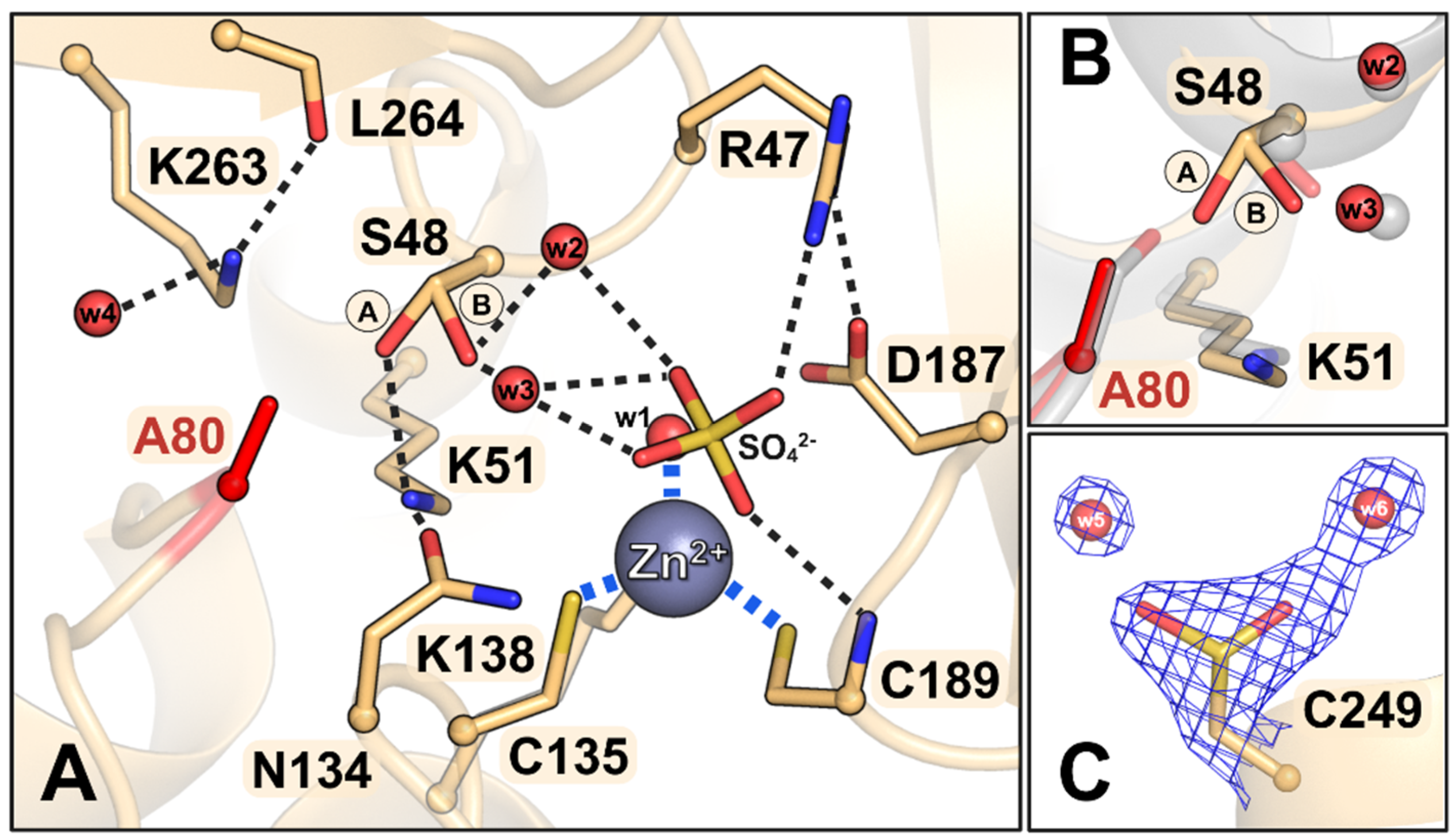
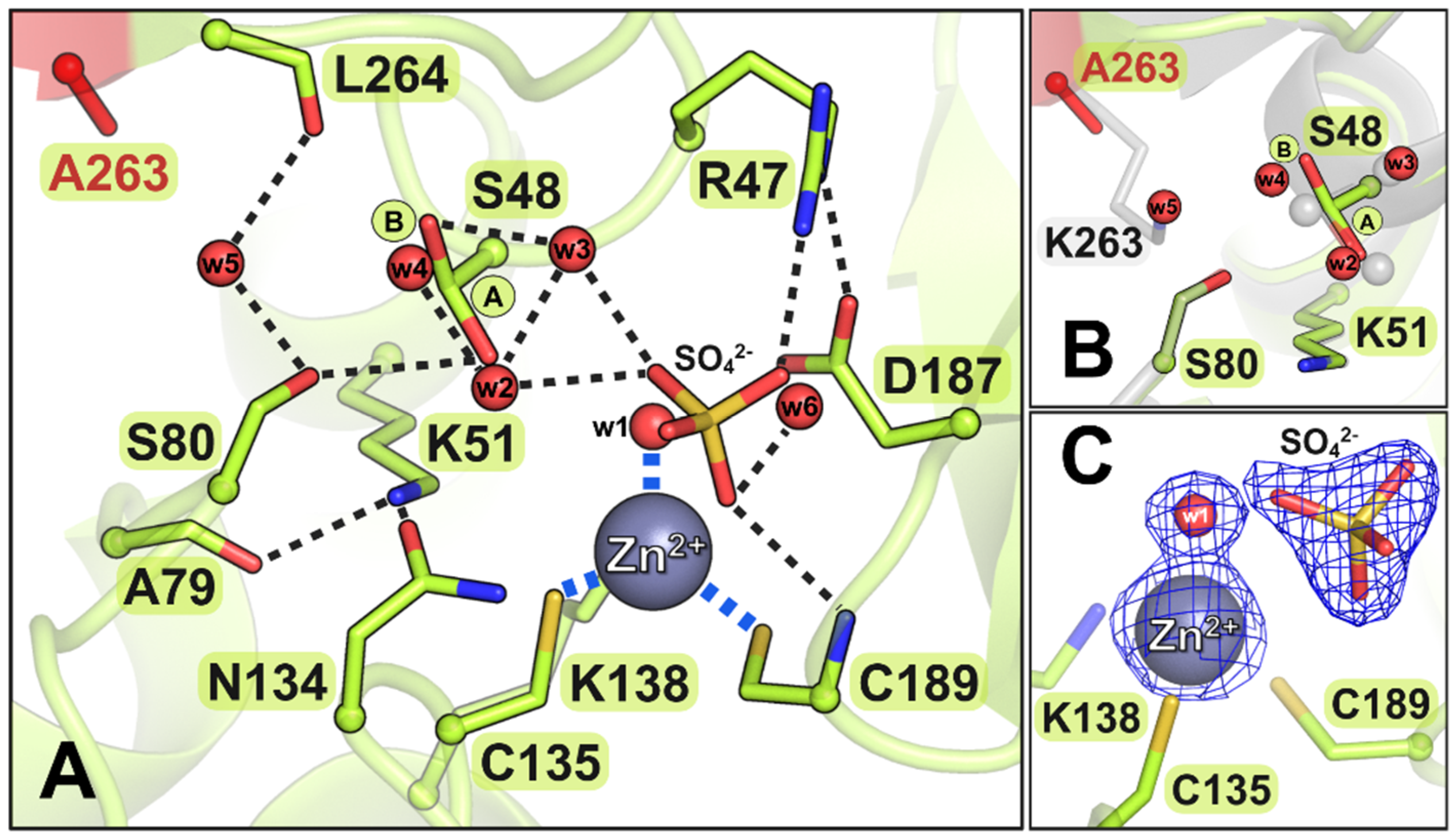
3.4. Description of the Crystal Structures of the Mutant Proteins
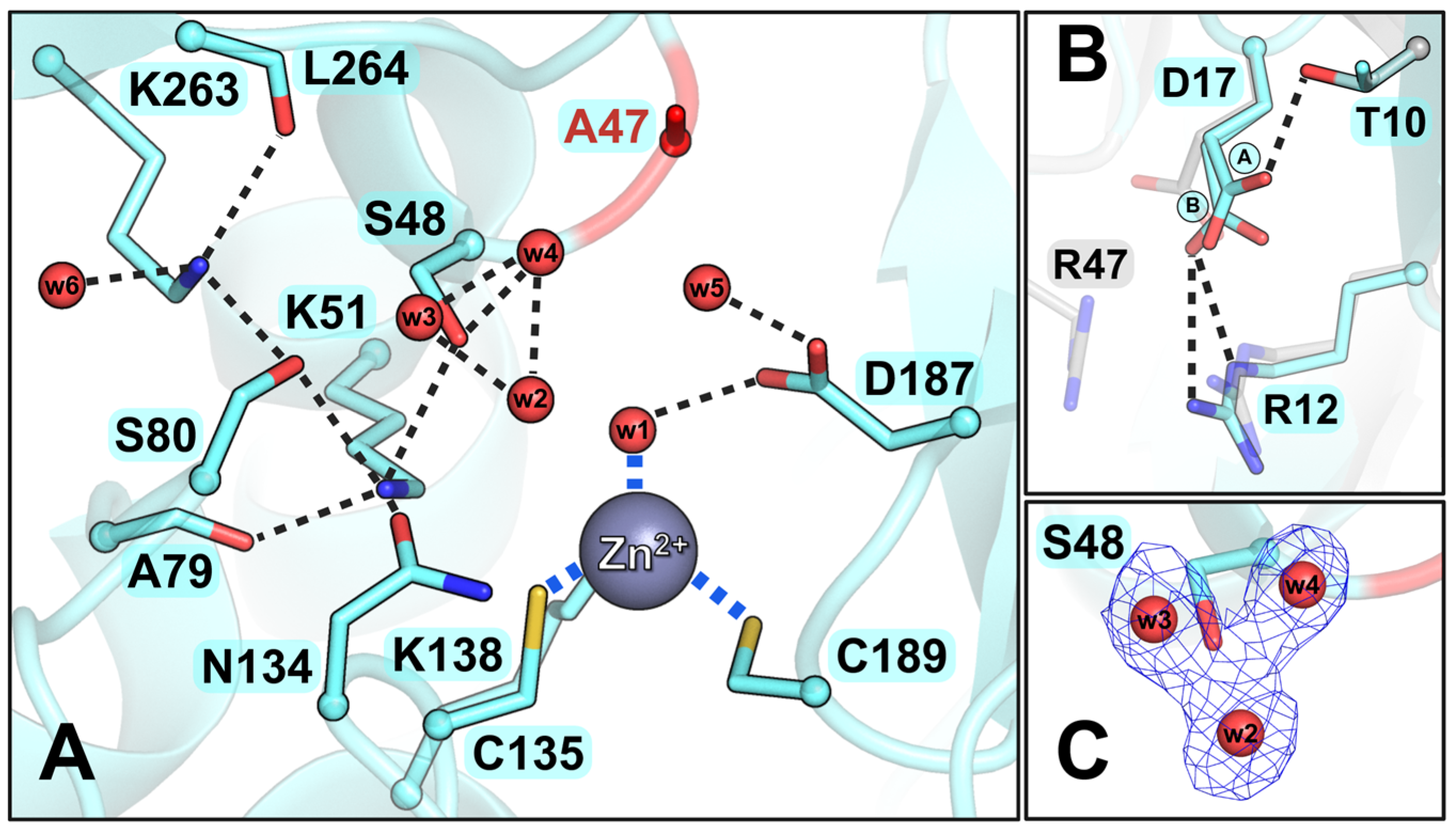
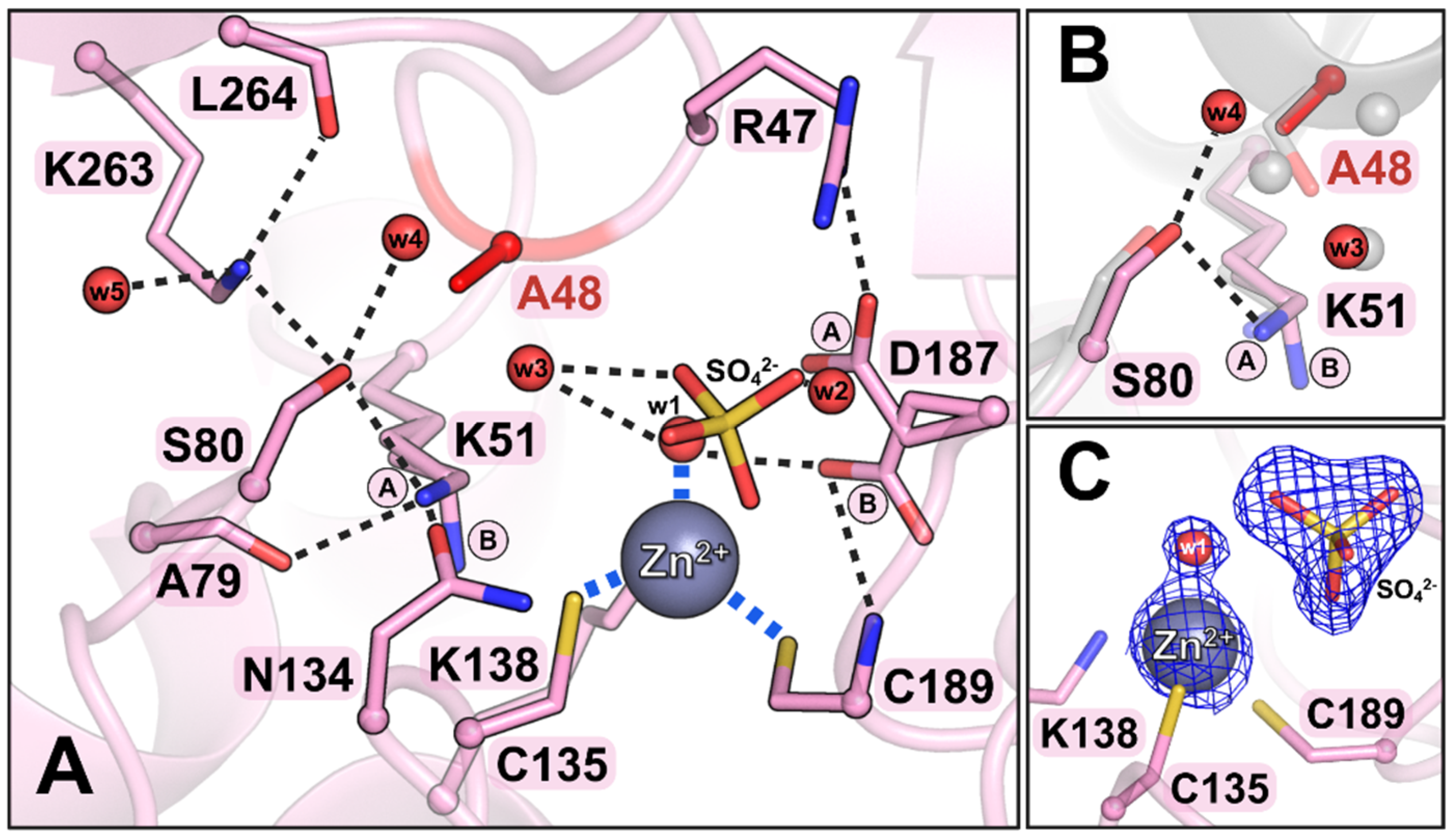
3.4.1. The R47A Mutant
3.4.2. The S48A Mutant
3.4.3. The K51A Mutant
3.4.4. The S80A Mutant
3.4.5. The K263A Mutant
3.5. Insights from the Structural and Functional Analysis of ReAV Mutants
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Loch, J.I.; Jaskolski, M. Structural and biophysical aspects of L-asparaginases: A growing family with amazing diversity. IUCrJ 2021, 8, 514–531. [Google Scholar] [CrossRef]
- da Silva, L.S.; Doonan, L.B.; Pessoa, A., Jr.; de Oliveira, M.A.; Long, P.F. Structural and functional diversity of asparaginases: Overview and recommendations for a revised nomenclature. Biotechnol. Appl. Biochem. 2022, 69, 503–513. [Google Scholar] [CrossRef] [PubMed]
- Srikhanta, Y.N.; Atack, J.M.; Beacham, I.R.; Jennings, M.P. Distinct physiological roles for the two L-asparaginase isozymes of Escherichia coli. Biochem. Biophys. Res. Commun. 2013, 436, 362–365. [Google Scholar] [CrossRef] [PubMed]
- Campbell, H.A.; Mashburn, L.T.; Boyse, E.A.; Old, L.J. Two L-asparaginases from Escherichia coli B. Their separation, purification, and antitumor activity. Biochemistry 1967, 6, 721–730. [Google Scholar] [CrossRef] [PubMed]
- Costa, I.M.; Schultz, L.; de Araujo Bianchi Pedra, B.; Leite, M.S.; Farsky, S.H.; de Oliveira, M.A.; Pessoa, A.; Monteiro, G. Recombinant L-asparaginase 1 from Saccharomyces cerevisiae: An allosteric enzyme with antineoplastic activity. Sci. Rep. 2016, 6, 36239. [Google Scholar] [CrossRef]
- Anishkin, A.; Vanegas, J.M.; Rogers, D.M.; Lorenzi, P.L.; Chan, W.K.; Purwaha, P.; Weinstein, J.N.; Sukharev, S.; Rempe, S.B. Catalytic Role of the Substrate Defines Specificity of Therapeutic L-Asparaginase. J. Mol. Biol. 2015, 427, 2867–2885. [Google Scholar] [CrossRef]
- Brannigan, J.A.; Dodson, G.; Duggleby, H.J.; Moody, P.C.; Smith, J.L.; Tomchick, D.R.; Murzin, A.G. A protein catalytic framework with an N-terminal nucleophile is capable of self-activation. Nature 1995, 378, 416–419. [Google Scholar] [CrossRef]
- Nomme, J.; Su, Y.; Konrad, M.; Lavie, A. Structures of apo and product-bound human L-asparaginase: Insights into the mechanism of autoproteolysis and substrate hydrolysis. Biochemistry 2012, 51, 6816–6826. [Google Scholar] [CrossRef]
- Schalk, A.M.; Lavie, A. Structural and kinetic characterization of guinea pig L-asparaginase type III. Biochemistry 2014, 53, 2318–2328. [Google Scholar] [CrossRef]
- Michalska, K.; Bujacz, G.; Jaskolski, M. Crystal structure of plant asparaginase. J. Mol. Biol. 2006, 360, 105–116. [Google Scholar] [CrossRef]
- Borek, D.; Michalska, K.; Brzezinski, K.; Kisiel, A.; Podkowinski, J.; Bonthron, D.T.; Krowarsch, D.; Otlewski, J.; Jaskolski, M. Expression, purification and catalytic activity of Lupinus luteus asparagine β-amidohydrolase and its Escherichia coli homolog. Eur. J. Biochem. 2004, 271, 3215–3226. [Google Scholar] [CrossRef] [PubMed]
- Cantor, J.R.; Stone, E.M.; Chantranupong, L.; Georgiou, G. The human asparaginase-like protein 1 hASRGL1 is an Ntn hydrolase with β-aspartyl peptidase activity. Biochemistry 2009, 48, 11026–11031. [Google Scholar] [CrossRef] [PubMed]
- Huerta-Zepeda, A.; Ortuno, L.; Du Pont, G.; Duran, S.; Lloret, A.; Merchant-Larios, H.; Calderon, J. Isolation and characterization of Rhizobium etli mutants altered in degradation of asparagine. J. Bacteriol. 1997, 179, 2068–2072. [Google Scholar] [CrossRef] [PubMed]
- Moreno-Enriquez, A.; Evangelista-Martinez, Z.; Gonzalez-Mondragon, E.G.; Calderon-Flores, A.; Arreguin, R.; Perez-Rueda, E.; Huerta-Saquero, A. Biochemical characterization of recombinant L-asparaginase (AnsA) from Rhizobium etli, a member of an increasing rhizobial-type family of L-asparaginases. J. Microbiol. Biotechnol. 2012, 22, 292–300. [Google Scholar] [CrossRef]
- Loch, J.I.; Imiolczyk, B.; Sliwiak, J.; Wantuch, A.; Bejger, M.; Gilski, M.; Jaskolski, M. Crystal structures of the elusive Rhizobium etli L-asparaginase reveal a peculiar active site. Nat. Commun. 2021, 12, 6717. [Google Scholar] [CrossRef]
- Loch, J.I.; Worsztynowicz, P.; Sliwiak, J.; Grzechowiak, M.; Imiolczyk, B.; Pokrywka, K.; Chwastyk, M.; Gilski, M.; Jaskolski, M. Rhizobium etli has two L-asparaginases with low sequence identity but similar structure and catalytic center. Acta Cryst. D 2023, 79, 775–791. [Google Scholar] [CrossRef]
- Sliwiak, J.; Worsztynowicz, P.; Pokrywka, K.; Loch, J.I.; Grzechowiak, M.; Jaskolski, M. Biochemical characterization of L-asparaginase isoforms from Rhizobium etli-the boosting effect of zinc. Front. Chem. 2024, 12, 1373312. [Google Scholar] [CrossRef]
- Broome, J.D. Evidence that the L-Asparaginase Activity of Guinea Pig Serum is responsible for its Antilymphoma Effects. Nature 1961, 191, 1114–1115. [Google Scholar] [CrossRef]
- Abaji, R.; Krajinovic, M. Pharmacogenetics of asparaginase in acute lymphoblastic leukemia. Cancer Drug Resist. 2019, 2, 242–255. [Google Scholar] [CrossRef]
- Radadiya, A.; Zhu, W.; Coricello, A.; Alcaro, S.; Richards, N.G.J. Improving the Treatment of Acute Lymphoblastic Leukemia. Biochemistry 2020, 59, 3193–3200. [Google Scholar] [CrossRef]
- Prager, M.D.; Bachynsky, N. Asparagine synthetase in asparaginase resistant and susceptible mouse lymphomas. Biochem. Biophys. Res. Commun. 1968, 31, 43–47. [Google Scholar] [CrossRef] [PubMed]
- Fonseca, M.H.G.; Fiuza, T.D.S.; Morais, S.B.; Souza, T.; Trevizani, R. Circumventing the side effects of L-asparaginase. Biomed. Pharmacother. 2021, 139, 111616. [Google Scholar] [CrossRef]
- Schmiegelow, K.; Rank, C.U.; Stock, W.; Dworkin, E.; van der Sluis, I. SOHO State of the Art Updates and Next Questions: Management of Asparaginase Toxicity in Adolescents and Young Adults with Acute Lymphoblastic Leukemia. Clin. Lymphoma Myeloma Leuk. 2021, 21, 725–733. [Google Scholar] [CrossRef]
- Asselin, B.L.; Lorenson, M.Y.; Whitin, J.C.; Coppola, D.J.; Kende, A.S.; Blakley, R.L.; Cohen, H.J. Measurement of serum L-asparagine in the presence of L-asparaginase requires the presence of an L-asparaginase inhibitor. Cancer Res. 1991, 51, 6568–6573. [Google Scholar]
- Stegink, L.D.; Filer, L.J., Jr.; Brummel, M.C.; Baker, G.L.; Krause, W.L.; Bell, E.F.; Ziegler, E.E. Plasma amino acid concentrations and amino acid ratios in normal adults and adults heterozygous for phenylketonuria ingesting a hamburger and milk shake meal. Am. J. Clin. Nutr. 1991, 53, 670–675. [Google Scholar] [CrossRef]
- Xu, F.; Oruna-Concha, M.J.; Elmore, J.S. The use of asparaginase to reduce acrylamide levels in cooked food. Food Chem. 2016, 210, 163–171. [Google Scholar] [CrossRef]
- Jana, A.; Biswas, S.; Ghosh, R.; Modak, R. Recent advances in L-Asparaginase enzyme production and formulation development for acrylamide reduction during food processing. Food Chem. X 2025, 25, 102055. [Google Scholar] [CrossRef] [PubMed]
- Pokrywka, K.; Grzechowiak, M.; Sliwiak, J.; Worsztynowicz, P.; Loch, J.I.; Ruszkowski, M.; Gilski, M.; Jaskolski, M. Probing the active site of Class 3 L-asparaginase by mutagenesis. I. Tinkering with the zinc coordination site of ReAV. Front. Chem. 2024, 12, 1381032. [Google Scholar] [CrossRef] [PubMed]
- Zielezinski, A.; Loch, J.I.; Karlowski, W.M.; Jaskolski, M. Massive annotation of bacterial L-asparaginases reveals their puzzling distribution and frequent gene transfer events. Sci. Rep. 2022, 12, 15797. [Google Scholar] [CrossRef]
- Strynadka, N.C.; Adachi, H.; Jensen, S.E.; Johns, K.; Sielecki, A.; Betzel, C.; Sutoh, K.; James, M.N. Molecular structure of the acyl-enzyme intermediate in beta-lactam hydrolysis at 1.7 Å resolution. Nature 1992, 359, 700–705. [Google Scholar] [CrossRef]
- Gibson, R.M.; Christensen, H.; Waley, S.G. Site-directed mutagenesis of beta-lactamase I. Single and double mutants of Glu-166 and Lys-73. Biochem. J. 1990, 272, 613–619. [Google Scholar] [CrossRef] [PubMed]
- Madgwick, P.J.; Waley, S.G. β-lactamase I from Bacillus cereus. Structure and site-directed mutagenesis. Biochem. J. 1987, 248, 657–662. [Google Scholar] [CrossRef]
- Brannigan, J.; Matagne, A.; Jacob, F.; Damblon, C.; Joris, B.; Klein, D.; Spratt, B.G.; Frere, J.M. The mutation Lys234His yields a class A β-lactamase with a novel pH-dependence. Biochem. J. 1991, 278, 673–678. [Google Scholar] [CrossRef] [PubMed]
- Pokrywka, K.; Grzechowiak, M.; Sliwiak, J.; Worsztynowicz, P.; Loch, J.I.; Ruszkowski, M.; Gilski, M.; Jaskolski, M. Controlling enzyme activity by mutagenesis and metal exchange to obtain crystal structures of stable substrate complexes of Class 3 L-asparaginase. FEBS J. 2025, 292, 1159–1173. [Google Scholar] [CrossRef]
- Klock, H.E.; Lesley, S.A. The Polymerase Incomplete Primer Extension (PIPE) method applied to high-throughput cloning and site-directed mutagenesis. Methods Mol. Biol. 2009, 498, 91–103. [Google Scholar]
- Simas, R.G.; Krebs Kleingesinds, E.; Pessoa Junior, A.; Long, P.F. An improved method for simple and accurate colorimetric determination of l-asparaginase enzyme activity using Nessler’s reagent. J. Chem. Technol. Biotechnol. 2021, 96, 1326–1332. [Google Scholar] [CrossRef]
- Mazzei, L.; Ciurli, S.; Zambelli, B. Isothermal Titration Calorimetry to Characterize Enzymatic Reactions. Methods Enzymol. 2016, 567, 215–236. [Google Scholar]
- Kabsch, W. Xds. Acta Cryst. D 2010, 66, 125–132. [Google Scholar] [CrossRef]
- McCoy, A.J.; Grosse-Kunstleve, R.W.; Adams, P.D.; Winn, M.D.; Storoni, L.C.; Read, R.J. Phaser crystallographic software. J. Appl. Crystallogr. 2007, 40, 658–674. [Google Scholar] [CrossRef]
- Afonine, P.V.; Grosse-Kunstleve, R.W.; Echols, N.; Headd, J.J.; Moriarty, N.W.; Mustyakimov, M.; Terwilliger, T.C.; Urzhumtsev, A.; Zwart, P.H.; Adams, P.D. Towards automated crystallographic structure refinement with phenix.refine. Acta Cryst. D 2012, 68, 352–367. [Google Scholar] [CrossRef]
- Emsley, P.; Lohkamp, B.; Scott, W.G.; Cowtan, K. Features and development of Coot. Acta Cryst. D 2010, 66, 486–501. [Google Scholar] [CrossRef] [PubMed]
- Kowiel, M.; Jaskolski, M.; Dauter, Z. ACHESYM: An algorithm and server for standardized placement of macromolecular models in the unit cell. Acta Cryst. D 2014, 70, 3290–3298. [Google Scholar] [CrossRef] [PubMed]
- DeLano, W. Pymol: An open-source molecular graphics tool. CCP4 Newsl. Protein Crystallogr. 2002, 40, 82–92. [Google Scholar]
- Jayaram, H.N.; Cooney, D.A.; Jayaram, S.; Rosenblum, L. A simple and rapid method for the estimation of L-asparaginase in chromatographic and electrophoretic effluents: Comparison with other methods. Anal. Biochem. 1974, 59, 327–346. [Google Scholar] [CrossRef]
- Freyer, M.W.; Lewis, E.A. Isothermal titration calorimetry: Experimental design, data analysis, and probing macromolecule/ligand binding and kinetic interactions. Methods Cell Biol. 2008, 84, 79–113. [Google Scholar]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Mutant | Forward Primer | Reverse Primer |
|---|---|---|
| R47A | ACGCTCGCCGCGTCTGCGGCGAAGCCGG | GCCGCAGACGCGGCGAGCGTCATACGCGTC |
| S48A | GCTCGCCCGGGCTGCGGCGAAGCCGGC | TTCGCCGCAGCCCGGGCGAGCGTCATACG |
| K51A | GTCTGCGGCGGCGCCGGCGCAG | CGGGCGAGCGTCATACGC |
| S80A | GATGTGCGCGGCCCACAGCAG | AGTGCAATATCCGCATCATCGAAGC |
| K263A | CCTCGTCGGCGCGCTCGGGGCCG | GCGCCGTCGAATGCGCGC |
| Mutant | Crystal Form | Protein Concentration | Precipitant |
|---|---|---|---|
| R47A | monoclinic | 16 mg·mL−1 | 22% PEG 4000, 0.2 M MgCl2, 0.1 M Bicine pH 9.0, 1% v/v isopropanol |
| S48A | orthorhombic | 21 mg·mL−1 | 20% PEG 8000, 0.2 M Li2SO4, 0.1 M Bicine pH 9.0 + 0.2 M Glycine |
| K51A | orthorhombic | 16 mg·mL−1 | 30% PEG 4000, 0.2 M Li2SO4, 0.1 M Tris pH 8.5 |
| S80A | monoclinic | 15 mg·mL−1 | 22% PEG 4000, 0.2 M Li2SO4, 0.1 M Bicine pH 9.0 |
| K263A | monoclinic | 15 mg·mL−1 | 22% PEG 3350, 0.2 M Li2SO4, 0.1 M Tris pH 8.5 |
| Structure | ReAV R47A | ReAV S48A | ReAV K51A | ReAV S80A | ReAV K263A |
|---|---|---|---|---|---|
| DATA COLLECTION | |||||
| Beamline | EMBL P13/Petra III, DESY Hamburg | ||||
| Wavelength (Å) | 0.9184 | 0.9150 | 0.9763 | 0.9763 | 0.9770 |
| Temperature (K) | 100 | 100 | 100 | 100 | 100 |
| Space group | P21 | P212121 | P212121 | P21 | P21 |
| Unit cell parameters (Å,°) | a = 78.06, b = 91.55, c = 114.46, β = 96.90 | a = 78.04, b = 91.34, c = 105.87 | a = 78.00, b = 91.27, c = 106.11 | a = 78.06, b = 91.48, c = 104.31, β = 105.61 | a = 77.89, b = 91.51, c = 114.34, β = 97.08 |
| Oscillation range (°) | 0.10 | 0.20 | 0.10 | 0.10 | 0.10 |
| Number of images | 1800 | 1800 | 1800 | 1800 | 1800 |
| Resolution range (Å) | 71.29–1.75 (1.86–1.75) * | 78.04–1.40 (1.49–1.40) | 91.27–1.95 (2.07–1.95) | 75.18–1.70 (1.80–1.70) | 91.51–1.60 (1.70–1.60) |
| Reflections collected/unique | 559,271/160,568 | 1,982,046/148,671 | 36,9270/55,856 | 519,510/154,207 | 698,497/207,205 |
| Completeness (%) | 99.6 (99.1) | 100.0 (99.8) | 99.7 (99.2) | 99.5 (98.5) | 99.3 (96.7) |
| Multiplicity | 3.5 (3.5) | 13.3 (12.9) | 6.6 (6.8) | 3.4 (3.1) | 3.4 (3.1) |
| Wilson B (Å2) | 18.9 | 17.1 | 24.6 | 21.3 | 17.8 |
| Rmerge (%) | 11.5 (89.2) | 8.4 (178.3) | 14.3 (104.6) | 9.8 (100.7) | 9.6 (78.7) |
| Rmeas (%) | 13.5 (105.7) | 8.7 (185.7) | 15.5 (113.2) | 11.6 (121.9) | 11.4 (95.2) |
| <I/σ> | 8.38 (1.38) | 17.97 (1.48) | 10.75 (2.06) | 7.94 (1.14) | 7.98 (1.44) |
| CC1/2 (%) | 99.7 (57.0) | 100.0 (67.1) | 99.8 (81.9) | 99.7 (52.2) | 99.7 (62.9) |
| REFINEMENT | |||||
| Unique/test reflections | 159,564/1001 | 147,184/1487 | 54,853/1000 | 152,192/2000 | 206,165/1037 |
| Rwork/Rfree (%) | 17.1/21.1 | 14.9/17.9 | 18.1/22.8 | 20.5/24.8 | 16.0/19.2 |
| Protein chains in ASU | 4 | 2 | 2 | 4 | 4 |
| Matthews coeff. (Å3/Da)/solvent (%) | 2.42/49.2 | 2.25/45.3 | 2.25/45.4 | 2.14/42.5 | 2.41/49.0 |
| ADP model | TLS | Aniso | TLS | TLS | TLS |
| <B> (Å2) | 22.1 | 24.4 | 30.5 | 26.9 | 23.8 |
| Protein/solvent/ Zn atoms | 10,561/1485/4 | 5554/771/2 | 5383/466/2 | 10,524/911/4 | 10,664/1549/4 |
| Rmsd bonds (Å)/angles (°) | 0.012/1.11 | 0.010/1.18 | 0.011/1.15 | 0.011/1.12 | 0.012/1.05 |
| Ramachandran plot (%) favored/allowed/outliers | 98/2/0 | 98/2/0 | 97/3/0 | 97/3/0 | 97/3/0 |
| PDB code | 9qct | 9qcu | 9qcw | 9qcy | 9qcz |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pokrywka, K.; Grzechowiak, M.; Sliwiak, J.; Worsztynowicz, P.; Loch, J.I.; Ruszkowski, M.; Gilski, M.; Jaskolski, M. Probing the Active Site of Class 3 L-Asparaginase by Mutagenesis: Mutations of the Ser-Lys Tandems of ReAV. Biomolecules 2025, 15, 944. https://doi.org/10.3390/biom15070944
Pokrywka K, Grzechowiak M, Sliwiak J, Worsztynowicz P, Loch JI, Ruszkowski M, Gilski M, Jaskolski M. Probing the Active Site of Class 3 L-Asparaginase by Mutagenesis: Mutations of the Ser-Lys Tandems of ReAV. Biomolecules. 2025; 15(7):944. https://doi.org/10.3390/biom15070944
Chicago/Turabian StylePokrywka, Kinga, Marta Grzechowiak, Joanna Sliwiak, Paulina Worsztynowicz, Joanna I. Loch, Milosz Ruszkowski, Miroslaw Gilski, and Mariusz Jaskolski. 2025. "Probing the Active Site of Class 3 L-Asparaginase by Mutagenesis: Mutations of the Ser-Lys Tandems of ReAV" Biomolecules 15, no. 7: 944. https://doi.org/10.3390/biom15070944
APA StylePokrywka, K., Grzechowiak, M., Sliwiak, J., Worsztynowicz, P., Loch, J. I., Ruszkowski, M., Gilski, M., & Jaskolski, M. (2025). Probing the Active Site of Class 3 L-Asparaginase by Mutagenesis: Mutations of the Ser-Lys Tandems of ReAV. Biomolecules, 15(7), 944. https://doi.org/10.3390/biom15070944