
Cell-Type Annotation for scATAC-Seq Data by Integrating Chromatin Accessibility and Genome Sequence


Abstract
1. Introduction
- 1.
- Alleviating batch effects [32,33] between the reference and query datasets, including differences introduced by variations in experimental protocols, sequencing depth, sample handling, or platform-specific biases. These batch-induced discrepancies can obscure true biological signals and negatively impact the accuracy and robustness of downstream cell-type annotation.
- 2.
- The majority of existing methods primarily focus on peak matrices, often overlooking genomic sequence information [34]. Fully leveraging genomic sequence features, such as nucleotide composition, transcription factor binding motifs, and local sequence patterns extracted from accessible chromatin regions, to enhance cell-type annotation accuracy remains an open research question. Addressing this challenge requires the development of more integrative computational approaches that can effectively incorporate such sequence-derived information alongside chromatin accessibility signals.
2. Materials and Methods
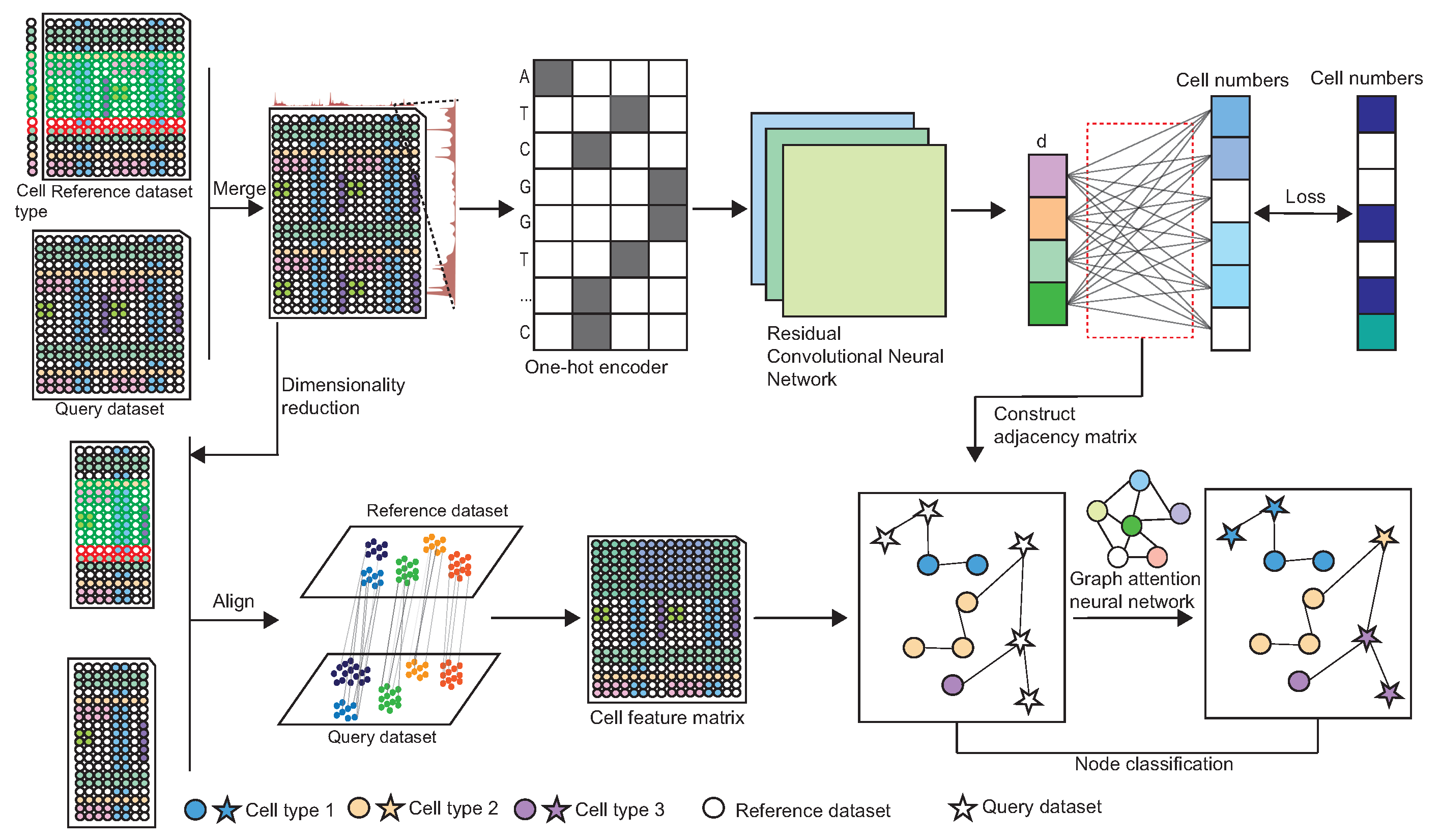
2.1. The Architecture of scAttG
2.2. Construct the Adjacency Matrix Based on the Genome Information
2.2.1. DNA Sequence Information Extraction
2.2.2. Cell Representation Extraction
2.2.3. Training Details of CNN Model
2.2.4. Construct the Adjacency Matrix
2.3. Extract Cell Embedding from Peak-by-Cell Matrix
2.4. Integrate DNA Sequence Information with Chromatin Accessibility Signals to Perform Cell-Type Annotation
Network Architecture and Training Details
2.5. Benchmark Methods
2.6. Benchmark Datasets
- WholeBrainA, WholeBrainB, Cerebellum, and PreFrontalCortex: These datasets were derived from the adult mouse brain atlas, accessible via the GEO accession number GSE111586 or through the project website: http://atlas.gs.washington.edu/mouse-atac/data/, accessed on 14 August 2018. The data were generated using sciATAC-seq technology and aligned to the mm9 reference genome.
- MosA1_v1, MosA1_v2, MosA1_v3, MosA1_v4, MosA2, MosM1, MosM2, MosP1, and MosP2: These datasets represent anterior, middle, and posterior regions of the secondary motor cortex in the mouse brain and are available under GEO accession number GSE126724. They were generated using snATAC-seq technology and annotated based on the GRCm38 reference genome.
- PBMC_atlas and PBMC_10K: These human-derived datasets consist of peripheral blood mononuclear cells (PBMCs) profiled using scATAC-seq. Both datasets share the same set of 344,492 peaks and are commonly used for benchmarking cell-type annotation algorithms. PBMC_atlas contains 394,441 cells annotated into 14 cell types and serves as a comprehensive reference, while PBMC_10K includes 10,412 cells and is typically used as a query set for evaluating model generalization performance. It can be downloaded from the work in [9].
- For more detailed information about the benchmark dataset, please refer to Table 1.
Evaluation Metrics
3. Results
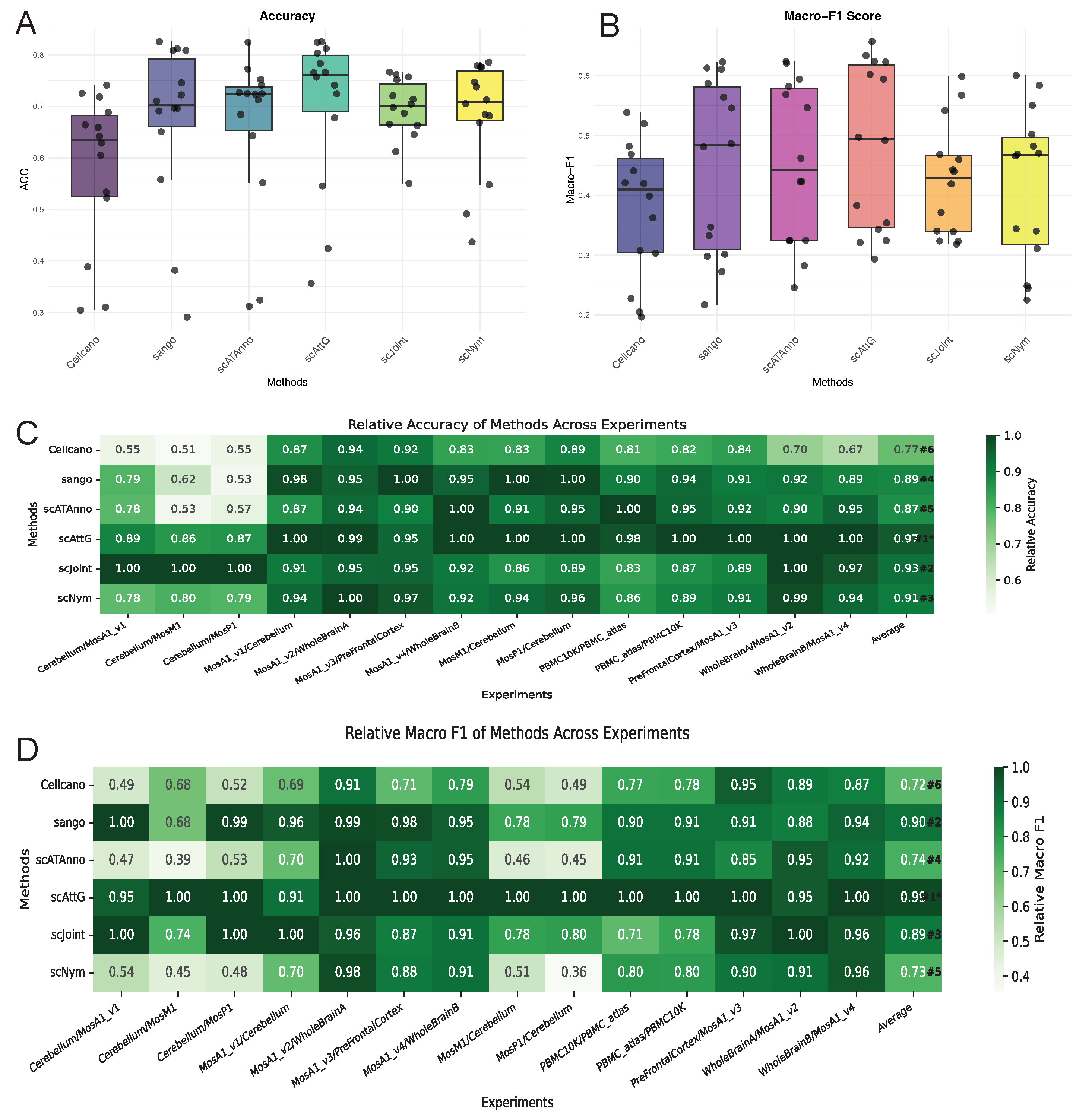
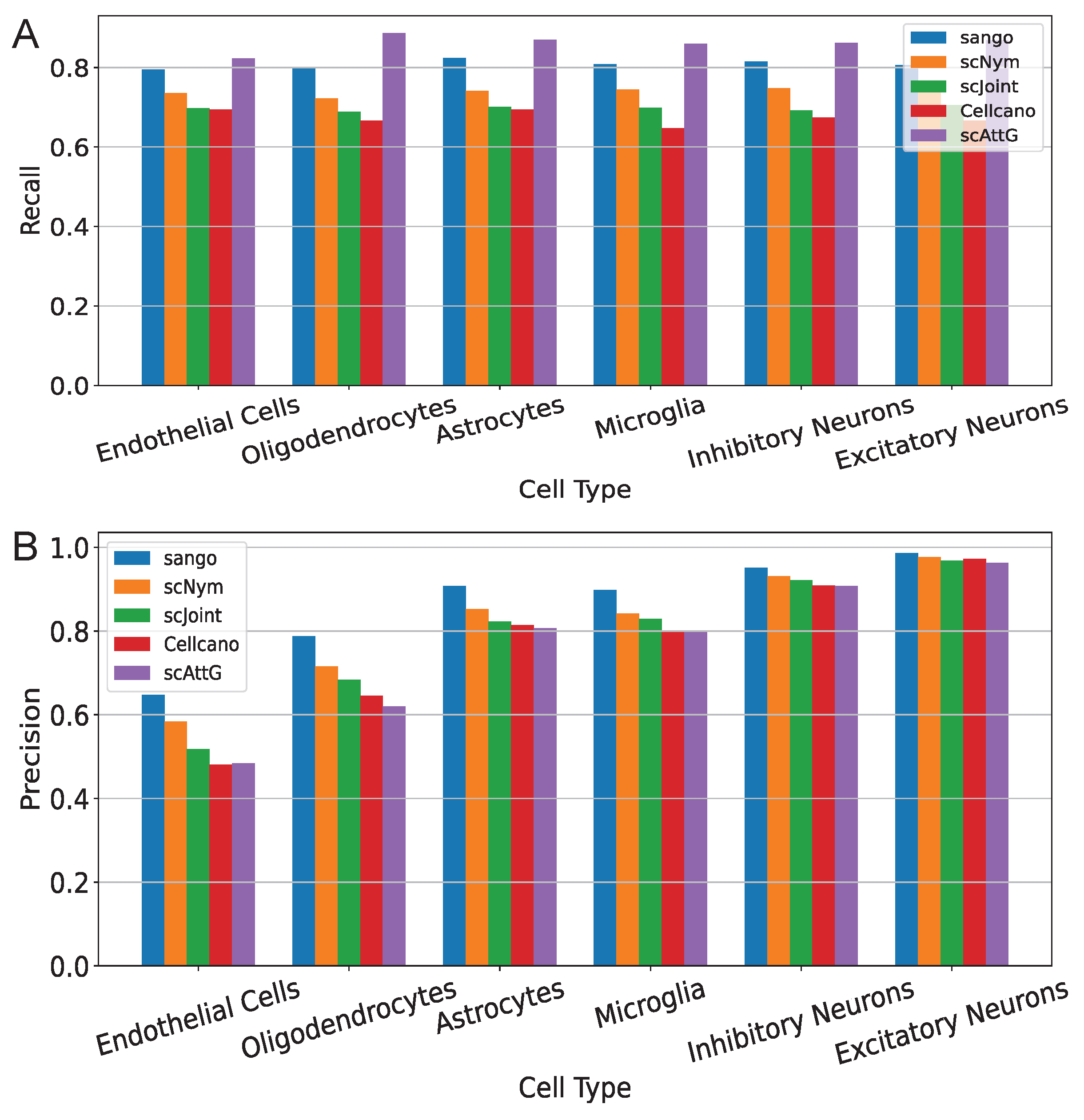
3.1. Performance Evaluation Across Intra-Dataset Experiments
3.2. Performance Evaluation Across Datasets
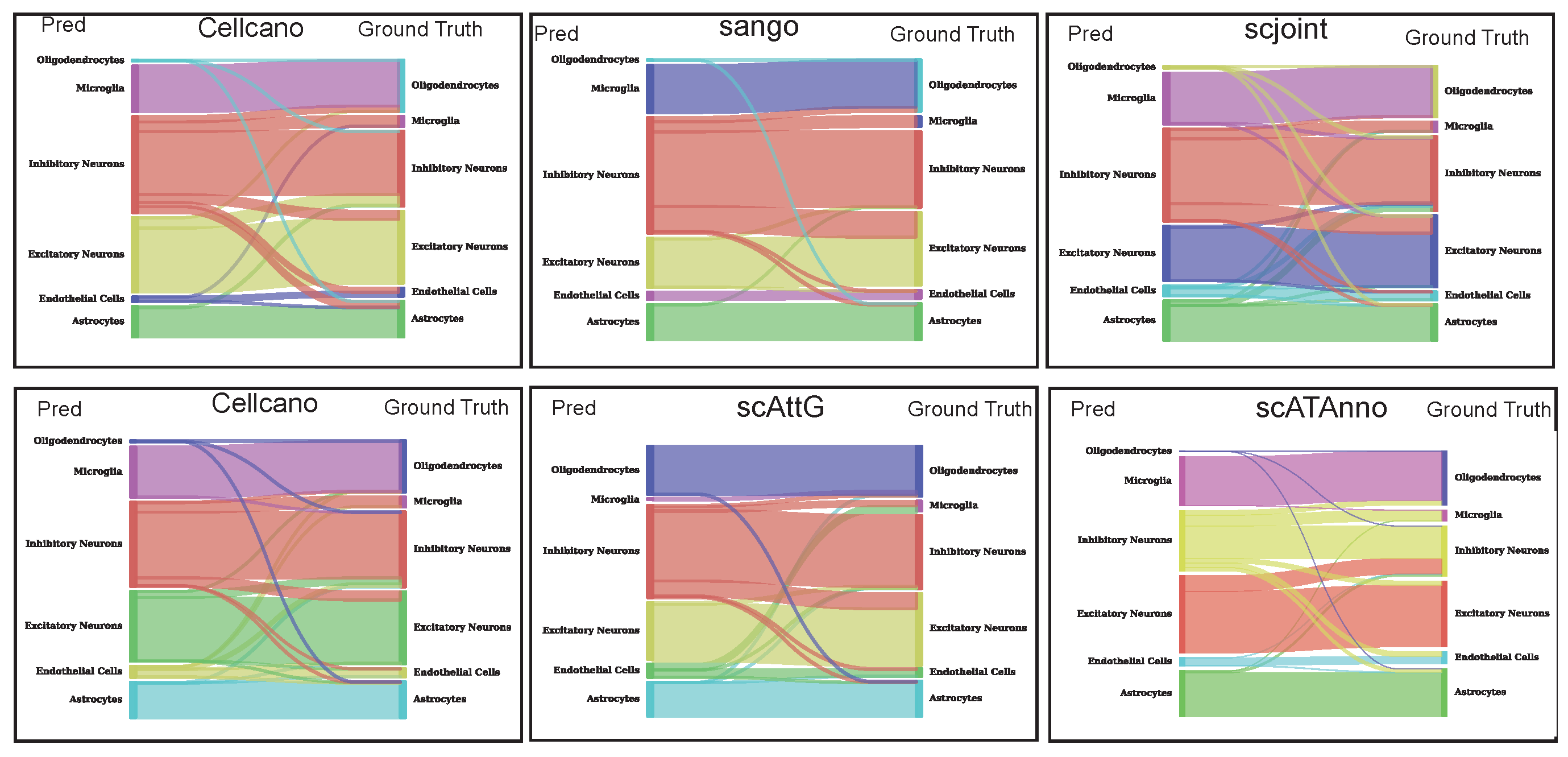
3.3. Sankey Plot Analysis
3.4. Two-Dimensional Embedding Analysis
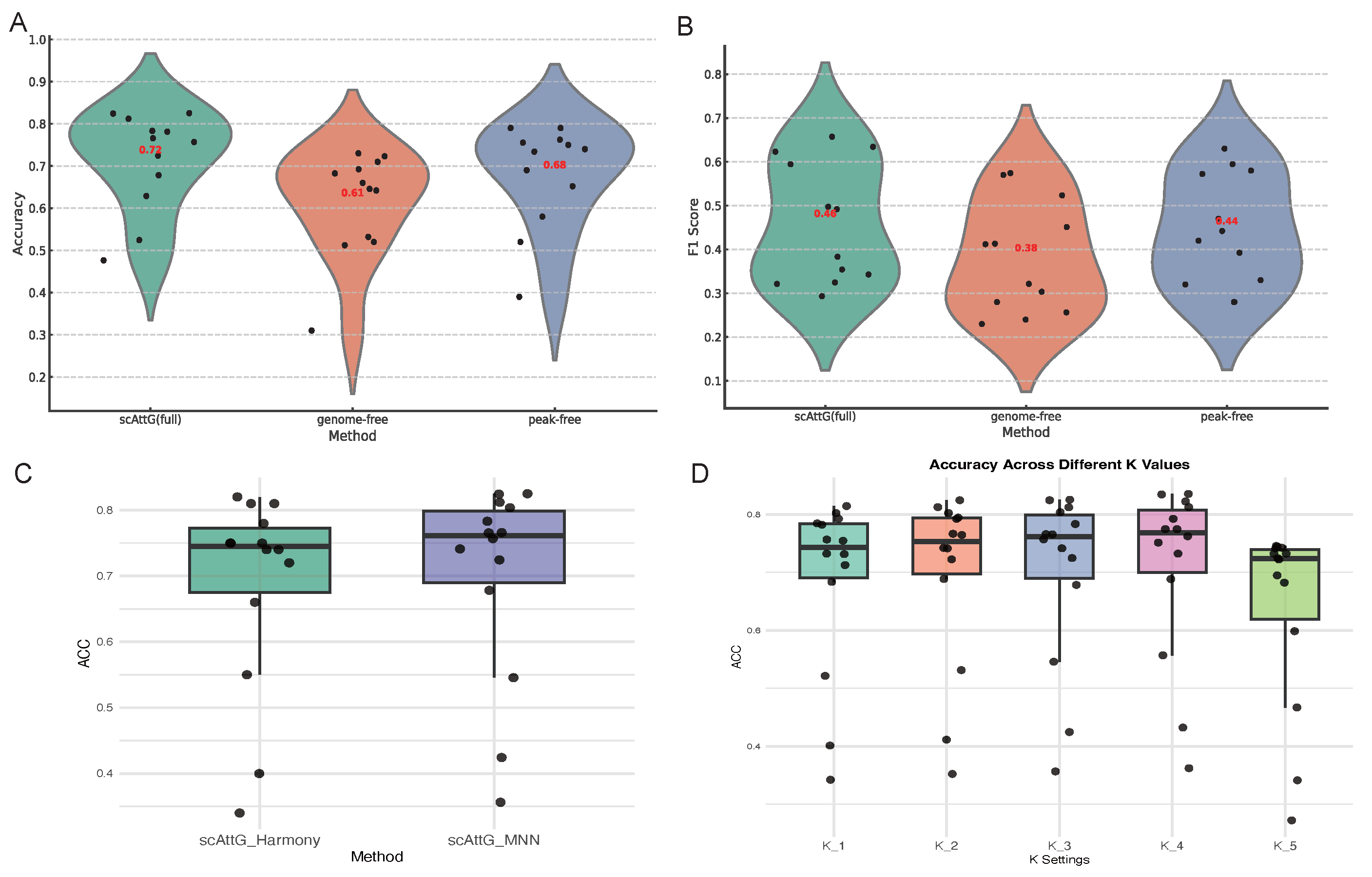
3.5. Ablation Study
3.5.1. Comparative Evaluation of Full and Ablated scAttG Models Accessibility Features
3.5.2. Comparison of Batch Correction Strategies: MNN vs. Harmony in scAttG
3.5.3. Sensitivity Analysis of KNN in scAttG
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Li, H.; Sun, Y.; Hong, H.; Huang, X.; Tao, H.; Huang, Q.; Wang, L.; Xu, K.; Gan, J.; Chen, H.; et al. Inferring transcription factor regulatory networks from single-cell ATAC-seq data based on graph neural networks. Nat. Mach. Intell. 2022, 4, 389–400. [Google Scholar] [CrossRef]
- Laisné, M.; Lupien, M.; Vallot, C. Epigenomic heterogeneity as a source of tumour evolution. Nat. Rev. Cancer 2025, 25, 7–26. [Google Scholar] [CrossRef]
- Lu, C.; Wei, Y.; Abbas, M.; Agula, H.; Wang, E.; Meng, Z.; Zhang, R. Application of Single-Cell Assay for Transposase-Accessible Chromatin with High Throughput Sequencing in Plant Science: Advances, Technical Challenges, and Prospects. Int. J. Mol. Sci. 2024, 25, 1479. [Google Scholar] [CrossRef]
- Jin, W.; Ma, J.; Rong, L.; Huang, S.; Li, T.; Jin, G.; Zhou, Z. Semi-automated IT-scATAC-seq profiles cell-specific chromatin accessibility in differentiation and peripheral blood populations. Nat. Commun. 2025, 16, 2635. [Google Scholar] [CrossRef]
- Wang, Y.; Sun, X.; Zhao, H. Benchmarking automated cell type annotation tools for single-cell ATAC-seq data. Front. Genet. 2022, 13, 1063233. [Google Scholar] [CrossRef]
- Chen, H.; Lareau, C.; Andreani, T.; Vinyard, M.E.; Garcia, S.P.; Clement, K.; Andrade-Navarro, M.A.; Buenrostro, J.D.; Pinello, L. Assessment of computational methods for the analysis of single-cell ATAC-seq data. Genome Biol. 2019, 20, 241. [Google Scholar] [CrossRef]
- Ma, W.; Lu, J.; Wu, H. Cellcano: Supervised cell type identification for single cell ATAC-seq data. Nat. Commun. 2023, 14, 1864. [Google Scholar] [CrossRef]
- Popescu, M.C.; Balas, V.E.; Perescu-Popescu, L.; Mastorakis, N. Multilayer perceptron and neural networks. WSEAS Trans. Circuits Syst. 2009, 8, 579–588. [Google Scholar]
- Jiang, Y.; Hu, Z.; Lynch, A.W.; Jiang, J.; Zhu, A.; Zeng, Z.; Zhang, Y.; Wu, G.; Xie, Y.; Li, R.; et al. scATAnno: Automated cell type annotation for single-cell ATAC sequencing data. bioRxiv 2023. [Google Scholar] [CrossRef]
- Zhang, K.; Zemke, N.R.; Armand, E.J.; Ren, B. A fast, scalable and versatile tool for analysis of single-cell omics data. Nat. Methods 2024, 21, 217–227. [Google Scholar] [CrossRef]
- Korsunsky, I.; Millard, N.; Fan, J.; Slowikowski, K.; Zhang, F.; Wei, K.; Baglaenko, Y.; Brenner, M.; Loh, P.r.; Raychaudhuri, S. Fast, sensitive and accurate integration of single-cell data with Harmony. Nat. Methods 2019, 16, 1289–1296. [Google Scholar] [CrossRef] [PubMed]
- Taunk, K.; De, S.; Verma, S.; Swetapadma, A. A brief review of nearest neighbor algorithm for learning and classification. In Proceedings of the 2019 IEEE International Conference on Intelligent Computing and Control Systems (ICCS), Chengdu, China, 21–23 June 2019; pp. 1255–1260. [Google Scholar]
- LeRoy, N.J.; Smith, J.P.; Zheng, G.; Rymuza, J.; Gharavi, E.; Brown, D.E.; Zhang, A.; Sheffield, N.C. Fast clustering and cell-type annotation of scATAC data using pre-trained embeddings. NAR Genom. Bioinform. 2024, 6, lqae073. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Chen, S.; Song, S.; Gao, Z.; Hou, L.; Zhang, X.; Lv, H.; Jiang, R. Cell type annotation of single-cell chromatin accessibility data via supervised Bayesian embedding. Nat. Mach. Intell. 2022, 4, 116–126. [Google Scholar] [CrossRef]
- Goan, E.; Fookes, C. Bayesian neural networks: An introduction and survey. In Case Studies in Applied Bayesian Data Science: CIRM Jean-Morlet Chair, Fall 2018; Springer: Berlin/Heidelberg, Germany, 2020; pp. 45–87. [Google Scholar]
- Wang, C.; Krafft, P.; Mahadevan, S.; Ma, Y.; Fu, Y. Manifold alignment. Manifold Learn. Theory Appl. 2011, 510, 95–119. [Google Scholar]
- Granja, J.M.; Corces, M.R.; Pierce, S.E.; Bagdatli, S.T.; Choudhry, H.; Chang, H.Y.; Greenleaf, W.J. ArchR is a scalable software package for integrative single-cell chromatin accessibility analysis. Nat. Genet. 2021, 53, 403–411. [Google Scholar] [CrossRef]
- Welch, J.D.; Hartemink, A.J.; Prins, J.F. MATCHER: Manifold alignment reveals correspondence between single cell transcriptome and epigenome dynamics. Genome Biol. 2017, 18, 138. [Google Scholar] [CrossRef]
- Stark, S.G.; Ficek, J.; Locatello, F.; Bonilla, X.; Chevrier, S.; Singer, F.; Rätsch, G.; Lehmann, K.V. SCIM: Universal single-cell matching with unpaired feature sets. Bioinformatics 2020, 36, i919–i927. [Google Scholar] [CrossRef]
- Liu, J.; Huang, Y.; Singh, R.; Vert, J.P.; Noble, W.S. Jointly embedding multiple single-cell omics measurements. In Proceedings of the Algorithms in Bioinformatics: The 19th International Workshop, WABI 2019 Proceedings, WABI (Workshop), Dagstuhl, Germany, 8–10 September 2019; Volume 143, p. 10. [Google Scholar]
- Cao, Z.J.; Gao, G. Multi-omics single-cell data integration and regulatory inference with graph-linked embedding. Nat. Biotechnol. 2022, 40, 1458–1466. [Google Scholar] [CrossRef]
- Lowd, D.; Meek, C. Adversarial learning. In Proceedings of the Eleventh ACM SIGKDD International Conference on Knowledge Discovery in Data Mining, Chicago, IL, USA, 21–24 August 2005; pp. 641–647. [Google Scholar]
- Dziugaite, G.K.; Roy, D.M.; Ghahramani, Z. Training generative neural networks via maximum mean discrepancy optimization. arXiv 2015, arXiv:1505.03906. [Google Scholar]
- Scarselli, F.; Gori, M.; Tsoi, A.C.; Hagenbuchner, M.; Monfardini, G. The graph neural network model. IEEE Trans. Neural Netw. 2008, 20, 61–80. [Google Scholar] [CrossRef]
- Lin, Y.; Wu, T.Y.; Wan, S.; Yang, J.Y.; Wong, W.H.; Wang, Y.R. scJoint integrates atlas-scale single-cell RNA-seq and ATAC-seq data with transfer learning. Nat. Biotechnol. 2022, 40, 703–710. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Yang, C.; Zhang, X. scDART: Integrating unmatched scRNA-seq and scATAC-seq data and learning cross-modality relationship simultaneously. Genome Biol. 2022, 23, 139. [Google Scholar] [CrossRef] [PubMed]
- Tian, L.; Xie, Y.; Xie, Z.; Tian, J.; Tian, W. AtacAnnoR: A reference-based annotation tool for single cell ATAC-seq data. Briefings Bioinform. 2023, 24, bbad268. [Google Scholar] [CrossRef] [PubMed]
- Yan, X.; Zheng, R.; Chen, J.; Li, M. scNCL: Transferring labels from scRNA-seq to scATAC-seq data with neighborhood contrastive regularization. Bioinformatics 2023, 39, btad505. [Google Scholar] [CrossRef]
- Khosla, P.; Teterwak, P.; Wang, C.; Sarna, A.; Tian, Y.; Isola, P.; Maschinot, A.; Liu, C.; Krishnan, D. Supervised contrastive learning. Adv. Neural Inf. Process. Syst. 2020, 33, 18661–18673. [Google Scholar]
- Wang, X.; Qi, G.J. Contrastive learning with stronger augmentations. IEEE Trans. Pattern Anal. Mach. Intell. 2022, 45, 5549–5560. [Google Scholar] [CrossRef]
- Yigit, H. A weighting approach for KNN classifier. In Proceedings of the 2013 IEEE International Conference on Electronics, Computer and Computation (ICECCO), Ankara, Turkey, 7–9 November 2013; pp. 228–231. [Google Scholar]
- De Rop, F.V.; Hulselmans, G.; Flerin, C.; Soler-Vila, P.; Rafels, A.; Christiaens, V.; Gonzalez-Blas, C.B.; Marchese, D.; Caratu, G.; Poovathingal, S.; et al. Systematic benchmarking of single-cell ATAC-sequencing protocols. Nat. Biotechnol. 2024, 42, 916–926. [Google Scholar] [CrossRef]
- Scherer, A. Batch Effects and Noise in Microarray Experiments: Sources and Solutions; Wiley Online Library: Hoboken, NJ, USA, 2009; Volume 868. [Google Scholar]
- Yuan, H.; Kelley, D.R. scBasset: Sequence-based modeling of single-cell ATAC-seq using convolutional neural networks. Nat. Methods 2022, 19, 1088–1096. [Google Scholar] [CrossRef]
- Veličković, P.; Cucurull, G.; Casanova, A.; Romero, A.; Lio, P.; Bengio, Y. Graph attention networks. arXiv 2017, arXiv:1710.10903. [Google Scholar]
- Li, Z.; Liu, F.; Yang, W.; Peng, S.; Zhou, J. A survey of convolutional neural networks: Analysis, applications, and prospects. IEEE Trans. Neural Netw. Learn. Syst. 2021, 33, 6999–7019. [Google Scholar] [CrossRef]
- Xiao, S.; Wang, S.; Dai, Y.; Guo, W. Graph neural networks in node classification: Survey and evaluation. Mach. Vis. Appl. 2022, 33, 4. [Google Scholar] [CrossRef]
- Tian, C.; Zheng, M.; Jiao, T.; Zuo, W.; Zhang, Y.; Lin, C.W. A self-supervised CNN for image watermark removal. IEEE Trans. Circuits Syst. Video Technol. 2024, 34, 7566–7576. [Google Scholar] [CrossRef]
- Yin, W.; Kann, K.; Yu, M.; Schütze, H. Comparative study of CNN and RNN for natural language processing. arXiv 2017, arXiv:1702.01923. [Google Scholar]
- Sasse, A.; Ng, B.; Spiro, A.E.; Tasaki, S.; Bennett, D.A.; Gaiteri, C.; De Jager, P.L.; Chikina, M.; Mostafavi, S. Benchmarking of deep neural networks for predicting personal gene expression from DNA sequence highlights shortcomings. Nat. Genet. 2023, 55, 2060–2064. [Google Scholar] [CrossRef]
- Pedregosa, F.; Varoquaux, G.; Gramfort, A.; Michel, V.; Thirion, B.; Grisel, O.; Blondel, M.; Prettenhofer, P.; Weiss, R.; Dubourg, V.; et al. Scikit-learn: Machine learning in Python. J. Mach. Learn. Res. 2011, 12, 2825–2830. [Google Scholar]
- Haghverdi, L.; Lun, A.T.; Morgan, M.D.; Marioni, J.C. Batch effects in single-cell RNA-sequencing data are corrected by matching mutual nearest neighbors. Nat. Biotechnol. 2018, 36, 421–427. [Google Scholar] [CrossRef]
- Clevert, D.A.; Unterthiner, T.; Hochreiter, S. Fast and accurate deep network learning by exponential linear units (elus). arXiv 2015, arXiv:1511.07289. [Google Scholar]
- Zhang, Z. Improved adam optimizer for deep neural networks. In Proceedings of the 2018 IEEE/ACM 26th International Symposium on Quality of Service (IWQoS), Banff, AB, Canada, 4–6 June 2018; pp. 1–2. [Google Scholar]
- Zeng, Y.; Luo, M.; Shangguan, N.; Shi, P.; Feng, J.; Xu, J.; Chen, K.; Lu, Y.; Yu, W.; Yang, Y. Deciphering cell types by integrating scATAC-seq data with genome sequences. Nat. Comput. Sci. 2024, 4, 285–298. [Google Scholar] [CrossRef]
- Kimmel, J.C.; Kelley, D.R. Semisupervised adversarial neural networks for single-cell classification. Genome Res. 2021, 31, 1781–1793. [Google Scholar] [CrossRef]
- McInnes, L.; Healy, J.; Melville, J. Umap: Uniform manifold approximation and projection for dimension reduction. arXiv 2018, arXiv:1802.03426. [Google Scholar]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Dataset | Tissue | #Peaks | #Cell Types | #Cells | GEO Accession |
|---|---|---|---|---|---|
| MosP1 | nervous | 68,555 | 6 | 10,889 | GSE126724 |
| Cerebellum | Cerebellum | 68,555 | 16 | 2,278 | GSE111586 |
| MosA1_v1 | nervous | 67,300 | 6 | 11,034 | GSE126724 |
| MosM1 | nervous | 67,637 | 6 | 10,294 | GSE126724 |
| MosA1_v2 | nervous | 68,576 | 6 | 11,034 | GSE126724 |
| WholeBrainA | WholeBrain | 68,576 | 17 | 5,494 | GSE111586 |
| MosA1_v3 | nervous | 68,578 | 6 | 11,034 | GSE126724 |
| PreFrontalCortex | PreFrontalCortex | 68,578 | 18 | 5,959 | GSE111586 |
| MosA1_v4 | nervous | 68,567 | 6 | 11,034 | GSE126724 |
| WholeBrainB | WholeBrain | 68,567 | 16 | 3,272 | GSE111586 |
| PBMC_atlas | PBMC | 344492 | 14 | 394441 | - |
| PBMC_10K | PBMC | 344492 | 14 | 10412 | - |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wei, G.; Wang, L.; Liu, Y.; Zhang, X. Cell-Type Annotation for scATAC-Seq Data by Integrating Chromatin Accessibility and Genome Sequence. Biomolecules 2025, 15, 938. https://doi.org/10.3390/biom15070938
Wei G, Wang L, Liu Y, Zhang X. Cell-Type Annotation for scATAC-Seq Data by Integrating Chromatin Accessibility and Genome Sequence. Biomolecules. 2025; 15(7):938. https://doi.org/10.3390/biom15070938
Chicago/Turabian StyleWei, Guo, Long Wang, Yan Liu, and Xiaohui Zhang. 2025. "Cell-Type Annotation for scATAC-Seq Data by Integrating Chromatin Accessibility and Genome Sequence" Biomolecules 15, no. 7: 938. https://doi.org/10.3390/biom15070938
APA StyleWei, G., Wang, L., Liu, Y., & Zhang, X. (2025). Cell-Type Annotation for scATAC-Seq Data by Integrating Chromatin Accessibility and Genome Sequence. Biomolecules, 15(7), 938. https://doi.org/10.3390/biom15070938