
Dissecting the tRNA Fragment tRF3E–Nucleolin Interaction: Implications in Breast Cancer

 ,
,  , , , ,
, , , , 
 and
and
Abstract
1. Introduction
2. Materials and Methods
2.1. General Procedures
2.2. Electroblotting
2.3. Electrophoretic Mobility Shift Assay (EMSA)
2.4. Cell Cultures and Generation of MCF-7-tRF3E and MCF-7-M19–24 Cell Lines
- TRF3E Forward 5′-CCGGAGGCGGCCCGGGTTCGACTCCCGGTGTGGGAATTTTT-3′
- TRF3E Reverse 5′-AATTAAAAATTCCCACACCGGGAGTCGAACCCGGGCCGCCT-3′
- M19–24 Forward 5′-CCGGAGGCGGCCCGGGTTCGACTAAATGTGTGGGAATTTTT-3′
- M19–24 Reverse 5′-AATTAAAAATTCCCACACATTTAGTCGAACCCGGGCCGCCT-3′
2.5. Cell Viability Assay
2.6. Cell Cycle Analysis
2.7. Growth Curve Analysis
2.8. Colony Assay
2.9. Western Blot
2.10. Immunofluorescence Analysis
2.11. Computational Modeling
2.12. Statistical Analysis
3. Results and Discussion
3.1. Identification of the Molecular Determinants of tRF3E-NCL Interaction

3.2. NCL Cooperatively Binds tRF3E
3.3. Molecular Dynamics of tRF3E and M19–24 NCL Complexes
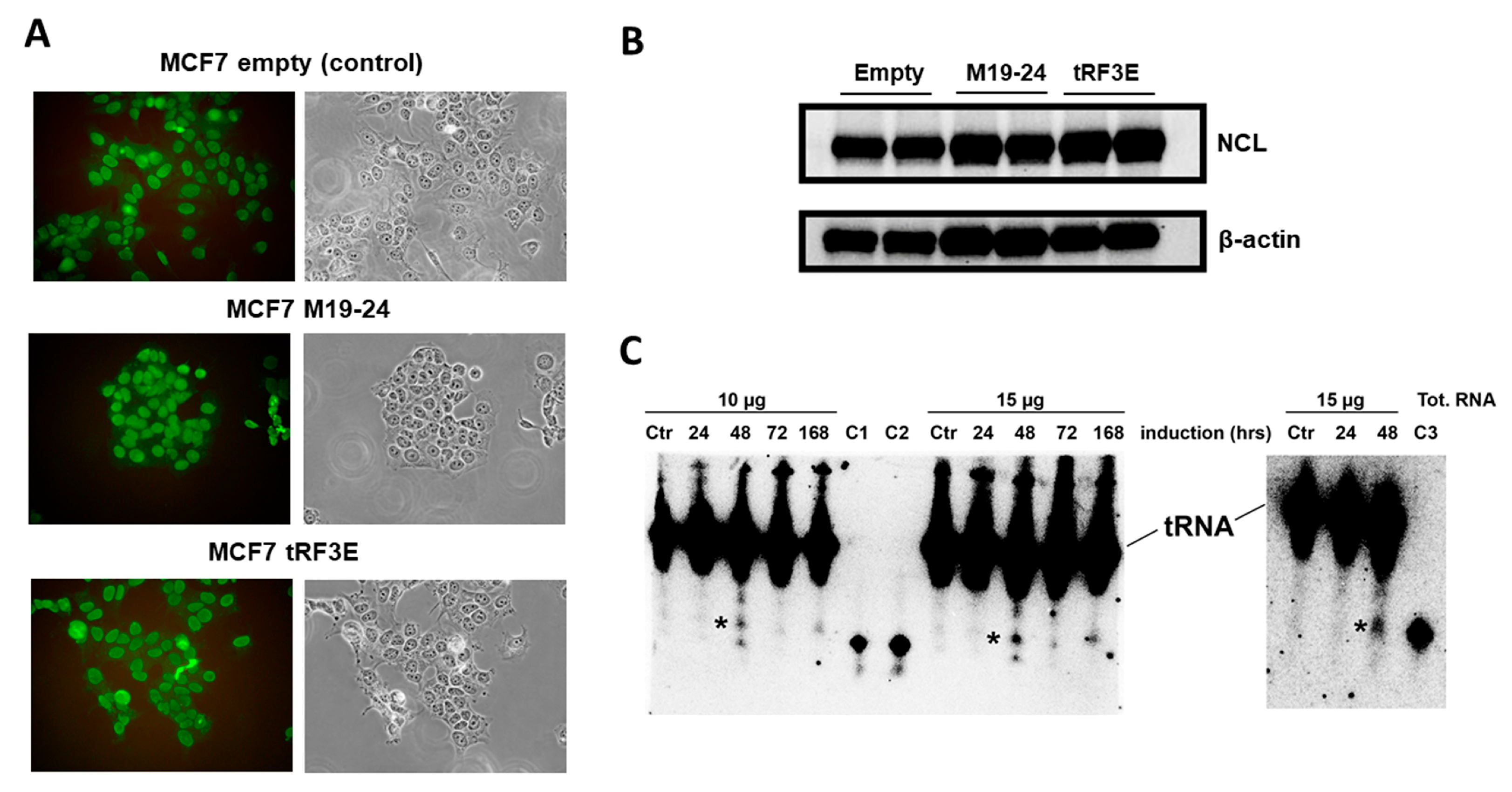
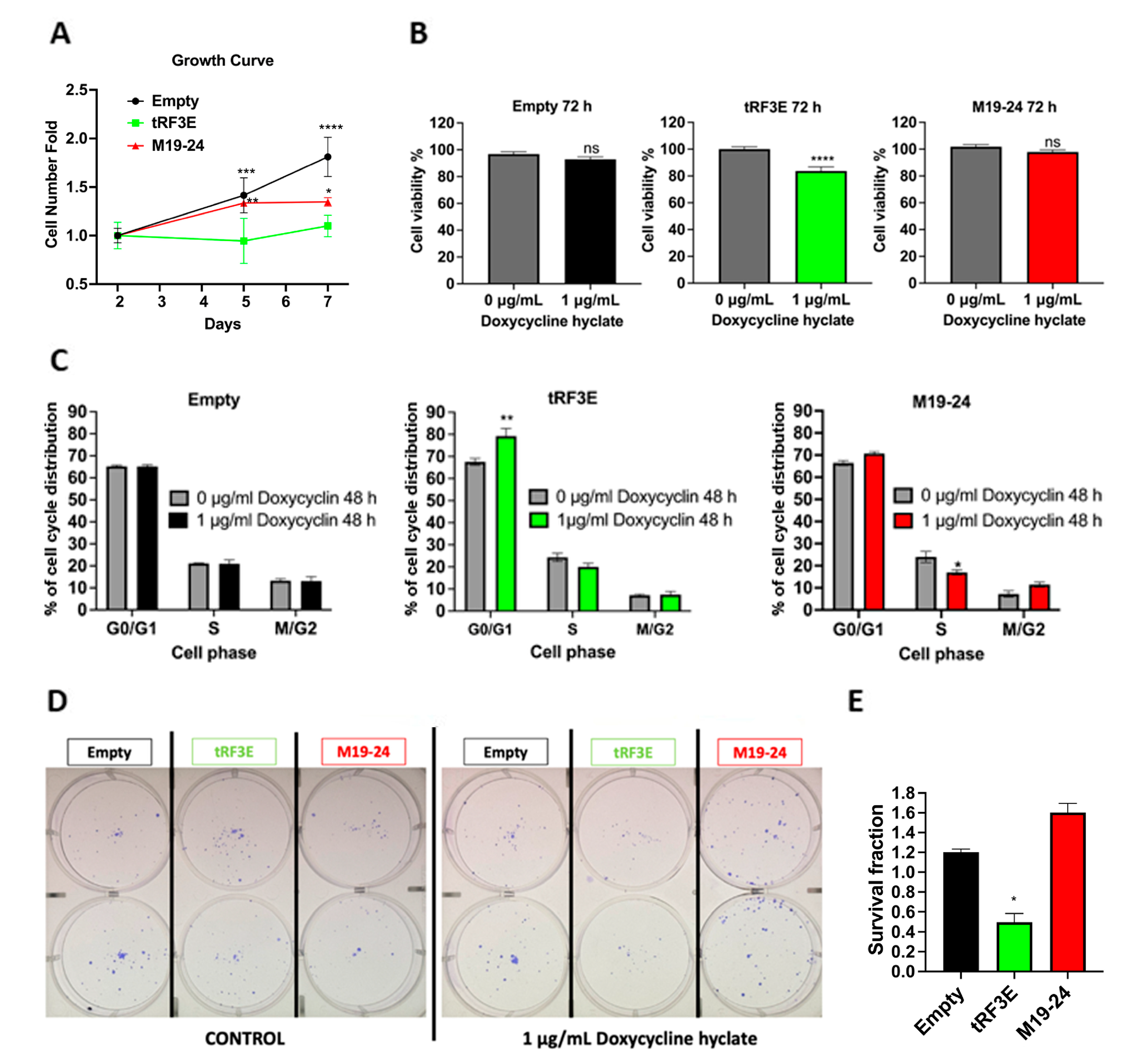
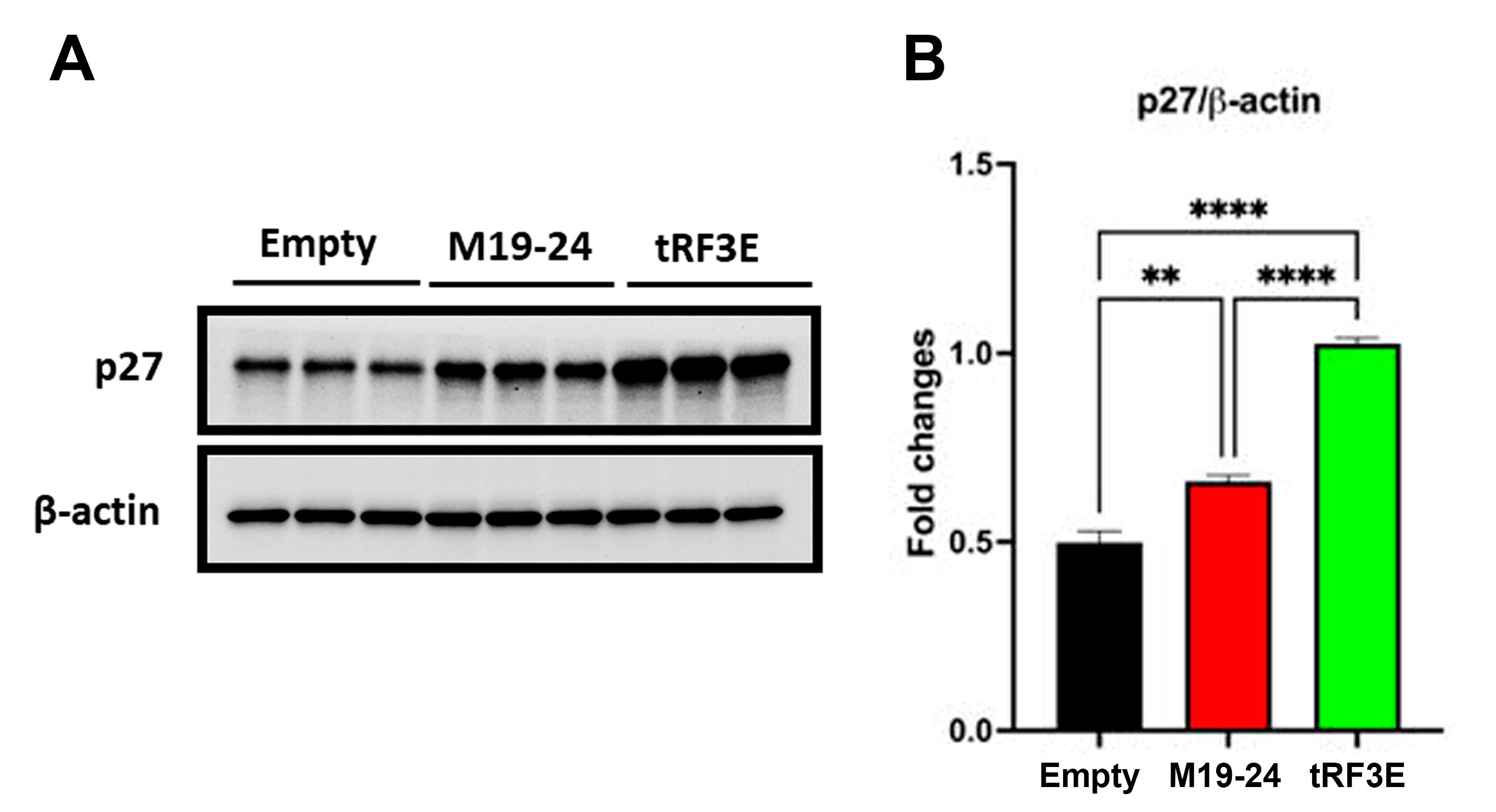
3.4. The Occupancy of Both NCL RBD1–2 and RBD3–4 Is Required for tRF3E Antitumor Function in Breast Cancer Cells
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| NCL | Nucleolin |
| RBD | RNA-Binding Domain |
| EMSA | Electrophoretic Mobility Shift Assay |
| tRF | tRNA-derived Fragment |
References
- Mongelard, F.; Bouvet, P. Nucleolin: A multifaceted protein. Trends Cell Biol. 2007, 17, 80–86. [Google Scholar] [CrossRef] [PubMed]
- Jia, W.; Yao, Z.; Zhao, J.; Guan, Q.; Gao, L. New perspectives of physiological and pathological functions of nucleolin (NCL). Life Sci. 2017, 186, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Abdelmohsen, K.; Tominaga, K.; Lee, E.K.; Srikantan, S.; Kang, M.J.; Kim, M.M.; Selimyan, R.; Martindale, J.L.; Yang, X.; Carrier, F.; et al. Enhanced translation by nucleolin via G-rich elements in coding and non-coding regions of target mRNAs. Nucleic Acids Res. 2011, 39, 8513–8530. [Google Scholar] [CrossRef] [PubMed]
- Ghisolfi-Nieto, L.; Joseph, G.; Puvion-Dutilleul, F.; Amalric, F.; Bouvet, P. Nucleolin is a sequence-specific RNA-binding protein: Characterization of targets on pre-ribosomal RNA. J. Mol. Biol. 1996, 260, 34–53. [Google Scholar] [CrossRef] [PubMed]
- Johansson, C.; Finger, L.D.; Trantirek, L.; Mueller, T.D.; Kim, S.; Laird-Offringa, I.A.; Feigon, J. Solution structure of the complex formed by the two N-terminal RNA-binding domains of nucleolin and a pre-rRNA target. J. Mol. Biol. 2004, 337, 799–816. [Google Scholar] [CrossRef] [PubMed]
- Tonello, F.; Massimino, M.L.; Peggion, C. Nucleolin: A cell portal for viruses, bacteria, and toxins. Cell. Mol. Life Sci. 2022, 79, 271. [Google Scholar] [CrossRef] [PubMed]
- Qiu, W.; Zhou, F.; Zhang, Q.; Sun, X.; Shi, X.; Liang, Y.; Wang, X.; Yue, L. Overexpression of nucleolin and different expression sites both related to the prognosis of gastric cancer. APMIS 2013, 121, 919–925. [Google Scholar] [CrossRef] [PubMed]
- Pichiorri, F.; Palmieri, D.; De Luca, L.; Consiglio, J.; You, J.; Rocci, A.; Talabere, T.; Piovan, C.; Lagana, A.; Cascione, L.; et al. In vivo NCL targeting affects breast cancer aggressiveness through miRNA regulation. J. Exp. Med. 2013, 210, 951–968, Erratum in J. Exp. Med. 2017, 214, 1557. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.Y.; Lu, S.; Xu, X.Y.; Hu, S.L.; Li, B.; Li, W.X.; Chang, J.Y. Prognostic significance of nuclear or cytoplasmic nucleolin expression in human non-small cell lung cancer and its relationship with DNA-PKcs. Tumour Biol. 2016, 37, 10349–10356. [Google Scholar] [CrossRef] [PubMed]
- Azman, M.S.; Alard, E.L.; Dodel, M.; Capraro, F.; Faraway, R.; Dermit, M.; Fan, W.; Chakraborty, A.; Ule, J.; Mardakheh, F.K. An ERK1/2-driven RNA-binding switch in nucleolin drives ribosome biogenesis and pancreatic tumorigenesis downstream of RAS oncogene. EMBO J. 2023, 42, e110902. [Google Scholar] [CrossRef] [PubMed]
- Gilles, M.E.; Maione, F.; Cossutta, M.; Carpentier, G.; Caruana, L.; Di Maria, S.; Houppe, C.; Destouches, D.; Shchors, K.; Prochasson, C.; et al. Nucleolin Targeting Impairs the Progression of Pancreatic Cancer and Promotes the Normalization of Tumor Vasculature. Cancer Res. 2016, 76, 7181–7193. [Google Scholar] [CrossRef] [PubMed]
- Lin, Q.; Ma, X.; Hu, S.; Li, R.; Wei, X.; Han, B.; Ma, Y.; Liu, P.; Pang, Y. Overexpression of nucleolin is a potential prognostic marker in endometrial carcinoma. Cancer Manag. Res. 2021, 13, 1955–1965. [Google Scholar] [CrossRef] [PubMed]
- Takagi, M.; Absalon, M.J.; McLure, K.G.; Kastan, M.B. Regulation of p53 translation and induction after DNA damage by ribosomal protein L26 and nucleolin. Cell 2005, 123, 49–63. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Guo, K.; Kastan, M.B. Interactions of nucleolin and ribosomal proteinL26 (RPL26) in translational control of human p53 mRNA. J. Biol. Chem. 2012, 287, 16467–16476. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Tsaprailis, G.; Bowden, G.T. Nucleolin stabilizes Bcl-X L messenger RNA in response to UVA irradiation. Cancer Res. 2008, 68, 1046–1054. [Google Scholar] [CrossRef] [PubMed]
- Otake, Y.; Soundararajan, S.; Sengupta, T.K.; Kio, E.A.; Smith, J.C.; Pineda-Roman, M.; Stuart, R.K.; Spicer, E.K.; Fernandes, D.J. Overexpression of nucleolin in chronic lymphocytic leukemia cells induces stabilization of bcl2 mRNA. Blood 2007, 109, 3069–3075. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Song, C.; Jiang, C.; Shen, X.; Qiao, Q.; Hu, Y. Nucleolin targeting AS1411 modified protein nanoparticle for antitumor drugs delivery. Mol. Pharm. 2013, 10, 3555–3563. [Google Scholar] [CrossRef]
- Doherty, C.; Wilbanks, B.; Khatua, S.; Maher, L.J. 3rd Aptamers in neuro-oncology: An emerging therapeutic modality. Neuro Oncol. 2024, 26, 38–54. [Google Scholar] [CrossRef] [PubMed]
- Tong, X.; Ga, L.; Ai, J.; Wang, Y. Progress in cancer drug delivery based on AS1411 oriented nanomaterials. J. Nanobiotechnol. 2022, 20, 57. [Google Scholar] [CrossRef] [PubMed]
- Gong, M.; Deng, Y.; Xiang, Y.; Ye, D. The role and mechanism of action of tRNA-derived fragments in the diagnosis and treatment of malignant tumors. Cell Commun. Signal 2023, 21, 62. [Google Scholar] [CrossRef] [PubMed]
- Kuhle, B.; Chen, Q.; Schimmel, P. tRNA renovatio: Rebirth through fragmentation. Mol. Cell 2023, 83, 3953–3971. [Google Scholar] [CrossRef] [PubMed]
- Ivanov, P.; Emara, M.M.; Villen, J.; Gygi, S.P.; Anderson, P. Angiogenin-induced tRNA fragments inhibit translation initiation. Mol. Cell 2011, 43, 613–623. [Google Scholar] [CrossRef] [PubMed]
- Gebetsberger, J.; Wyss, L.; Mleczko, A.M.; Reuther, J.; Polacek, N. A tRNA-derived fragment competes with mRNA for ribosome binding and regulates translation during stress. RNA Biol. 2017, 14, 1364–1373. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Mei, W.; Padmanaban, V.; Alwaseem, H.; Molina, H.; Passarelli, M.C.; Tavora, B.; Tavazoie, S.F. A pro-metastatic tRNA fragment drives Nucleolin oligomerization and stabilization of its bound metabolic mRNAs. Mol. Cell 2022, 82, 2604–2617.e8. [Google Scholar] [CrossRef] [PubMed]
- Goodarzi, H.; Liu, X.; Nguyen, H.C.; Zhang, S.; Fish, L.; Tavazoie, S.F. Endogenous tRNA-derived fragments suppress breast cancer progression via YBX1 displacement. Cell 2015, 161, 790–802. [Google Scholar] [CrossRef] [PubMed]
- Falconi, M.; Giangrossi, M.; Zabaleta, M.E.; Wang, J.; Gambini, V.; Tilio, M.; Bencardino, D.; Occhipinti, S.; Belletti, B.; Laudadio, E.; et al. A novel 3′-tRNAGlu-derived fragment acts as a tumor suppressor in breast cancer by targeting nucleolin. FASEB J. 2019, 33, 13228–13240. [Google Scholar] [CrossRef] [PubMed]
- Sambrook, J.; Russell, D.W. Molecular Cloning. A Laboratory Manual, 3rd ed.; CSHL Press, Cold Spring Harbor: Long Island, NY, USA, 2001. [Google Scholar]
- Wiederschain, D.; Wee, S.; Chen, L.; Loo, A.; Yang, G.; Huang, A.; Chen, Y.; Caponigro, G.; Yao, Y.M.; Lengauer, C.; et al. Single-vector inducible lentiviral RNAi system for oncology target validation. Cell Cycle 2009, 8, 498–504. [Google Scholar] [CrossRef] [PubMed]
- Bisaro, B.; Montani, M.; Konstantinidou, G.; Marchini, C.; Pietrella, L.; Iezzi, M.; Galié, M.; Orso, F.; Camporeale, A.; Colombo, S.M.; et al. p130Cas/Cyclooxygenase-2 axis in the control of mesenchymal plasticity of breast cancer cells. Breast Cancer Res. 2012, 14, R137. [Google Scholar] [CrossRef] [PubMed]
- Segatto, I.; Zompit, M.M.; Citron, F.; D’Andrea, S.; Vinciguerra, G.L.R.; Perin, T.; Berton, S.; Mungo, G.; Schiappacassi, M.; Marchini, C.; et al. Stathmin Is Required for Normal Mouse Mammary Gland Development and Δ16HER2-Driven Tumorigenesis. Cancer Res. 2019, 79, 397–409. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Huang, Y.; Xiao, Y. 3dRNA v2.0: An Updated Web Server for RNA 3D Structure Prediction. Int. J. Mol. Sci. 2019, 20, 4116. [Google Scholar] [CrossRef] [PubMed]
- Dominguez, C.; Boelens, R.; Bonvin, A.M.J.J. HADDOCK: A protein-protein docking approach based on biochemical or biophysical information. J. Am. Chem. Soc. 2003, 125, 1731–1737. [Google Scholar] [CrossRef] [PubMed]
- Tuszynska, I.; Matelska, D.; Magnus, M.; Chojnowski, G.; Kasprzak, J.M.; Kozlowski, L.P.; Dunin-Horkawicz, S.; Bujnicki, J.M. Computational modeling of protein–RNA complex structures. Methods 2014, 65, 310–319. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Duan, Y.; Hong, X.; Xie, J.; Liu, S. Challenges in structural modeling of RNA-protein interactions. Curr. Opin. Struct. Biol. 2023, 81, 102623. [Google Scholar] [CrossRef] [PubMed]
- van Zundert, G.C.P.; Rodrigues, J.P.G.L.M.; Trellet, M.; Schmitz, C.; Kastritis, P.L.; Karaca, E.; Melquiond, A.S.J.; van Dijk, M.; de Vries, S.J.; Bonvin, A.M.J.J. The HADDOCK2.2 webserver: User-friendly integrative modeling of biomolecular complexes. J. Mol. Biol. 2016, 428, 720–725. [Google Scholar] [CrossRef] [PubMed]
- Guterres, H.; Wonpil, I. CHARMM-GUI-Based Induced Fit Docking Workflow to Generate Reliable Protein–Ligand Binding Modes. J. Chem. Inf. Model 2023, 63, 4772–4779. [Google Scholar] [CrossRef] [PubMed]
- Valdés-Tresanco, M.S.; Valdés-Tresanco, M.E.; Valiente, P.A.; Moreno, E. gmx_MMPBSA: A New Tool to Perform End-State Free Energy Calculations with GROMACS. J. Chem. Theory Comput. 2021, 17, 6281–6291. [Google Scholar] [CrossRef] [PubMed]
- Abraham, M.J.; Murtola, T.; Schulz, R.; Páll, S.; Smith, J.C.; Hess, B.; Lindahl, E. GROMACS: High performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX 2015, 1–2, 19–25. [Google Scholar] [CrossRef]
- Stitzinger, S.H.; Sohrabi-Jahromi, S.; Söding, J. Cooperativity boosts affinity and specificity of proteins with multiple RNA-binding domains. NAR Genom. Bioinform. 2023, 5, lqad057. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Dickerhoff, J.; Zheng, K.W.; Erramilli, S.; Feng, H.; Wu, G.; Onel, B.; Chen, Y.; Wang, K.B.; Carver, M.; et al. Structural basis for nucleolin recognition of MYC promoter G-quadruplex. Science 2025, 388, 6744. [Google Scholar] [CrossRef] [PubMed]
- Varshney, D.; Spiegel, J.; Zyner, K.; Tannahill, D.; Balasubramanian, S. The regulation and functions of DNA and RNA G-quadruplexes. Nat. Rev. Mol. Cell Biol. 2020, 21, 459–474. [Google Scholar] [CrossRef] [PubMed]
- Lago, S.; Tosoni, E.; Nadai, M.; Palumbo, M.; Richter, S.N. The cellular protein nucleolin preferentially binds long-looped G-quadruplex nucleic acids. Biochim. Biophys. Acta Gen. Subj. 2017, 1861, 1371–1381. [Google Scholar] [CrossRef] [PubMed]
- Razavipour, S.F.; Harikumar, K.B.; Slingerland, J.M. p27 as a Transcriptional Regulator: New Roles in Development and Cancer. Cancer Res. 2020, 80, 3451–3458. [Google Scholar] [CrossRef] [PubMed]
- Reynisdottir, I.; Polyak, K.; Iavarone, A.; Massague, J. Kip/Cip and Ink4 Cdk inhibitors cooperate to induce cell cycle arrest in response to TGF-beta. Genes Dev. 1995, 9, 1831–1845. [Google Scholar] [CrossRef] [PubMed]
- Thongchot, S.; Aksonnam, K.; Thuwajit, P.; Yenchitsomanus, P.T.; Thuwajit, C. Nucleolin-based targeting strategies in cancer treatment: Focus on cancer immunotherapy. Int. J. Mol. Med. 2023, 52, 81. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| tRF3E-NCL-tRF3E | tRF3E-NCL-M19–24 | |
|---|---|---|
| ΔGcomplex (complex stability, 100 ns) | −22,165 ± 135 kcal/mol | −20,373 ± 128 kcal/mol |
| ΔGcomplex (complex stability, last 50 ns) | −22,224 ± 105 kcal/mol | −20,397 ± 130 kcal/mol |
| ΔGbinding (100 ns) | −32.88 ± 12 kcal/mol | 371 ± 50 kcal/mol |
| ΔGbinding (last 50 ns) | −41.21 ± 14 kcal/mol | 361 ± 66 kcal/mol |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Falconi, M.; Wang, J.; Costamagna, A.; Giangrossi, M.; Alimi, S.S.; Turco, E.; Bramucci, M.; Quassinti, L.; Petrilli, R.; Buccioni, M.; et al. Dissecting the tRNA Fragment tRF3E–Nucleolin Interaction: Implications in Breast Cancer. Biomolecules 2025, 15, 1054. https://doi.org/10.3390/biom15071054
Falconi M, Wang J, Costamagna A, Giangrossi M, Alimi SS, Turco E, Bramucci M, Quassinti L, Petrilli R, Buccioni M, et al. Dissecting the tRNA Fragment tRF3E–Nucleolin Interaction: Implications in Breast Cancer. Biomolecules. 2025; 15(7):1054. https://doi.org/10.3390/biom15071054
Chicago/Turabian StyleFalconi, Maurizio, Junbiao Wang, Andrea Costamagna, Mara Giangrossi, Sunday Segun Alimi, Emilia Turco, Massimo Bramucci, Luana Quassinti, Rossana Petrilli, Michela Buccioni, and et al. 2025. "Dissecting the tRNA Fragment tRF3E–Nucleolin Interaction: Implications in Breast Cancer" Biomolecules 15, no. 7: 1054. https://doi.org/10.3390/biom15071054
APA StyleFalconi, M., Wang, J., Costamagna, A., Giangrossi, M., Alimi, S. S., Turco, E., Bramucci, M., Quassinti, L., Petrilli, R., Buccioni, M., Marucci, G., Amici, A., Defilippi, P., Galeazzi, R., & Marchini, C. (2025). Dissecting the tRNA Fragment tRF3E–Nucleolin Interaction: Implications in Breast Cancer. Biomolecules, 15(7), 1054. https://doi.org/10.3390/biom15071054