
Targeted Overexpression of Mitochondrial ALDH2 in Coronary Endothelial Cells Mitigates HFpEF in a Diabetic Mouse Model
 ,
,  , ,
, ,  ,
,  , and
, and {kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Animals: db/db Mouse
2.2. Intracardiac Delivery of ALDH2 Gene Using AAV9 Viral Vector
2.3. Cardiac Ultrasound
2.4. Immunofluorescence Staining
2.5. Western Immunoblotting Using Proteins from Isolated Cardiac Cells and Tissue
2.6. ALDH Activity Assay
2.7. Statistical Analysis
3. Results
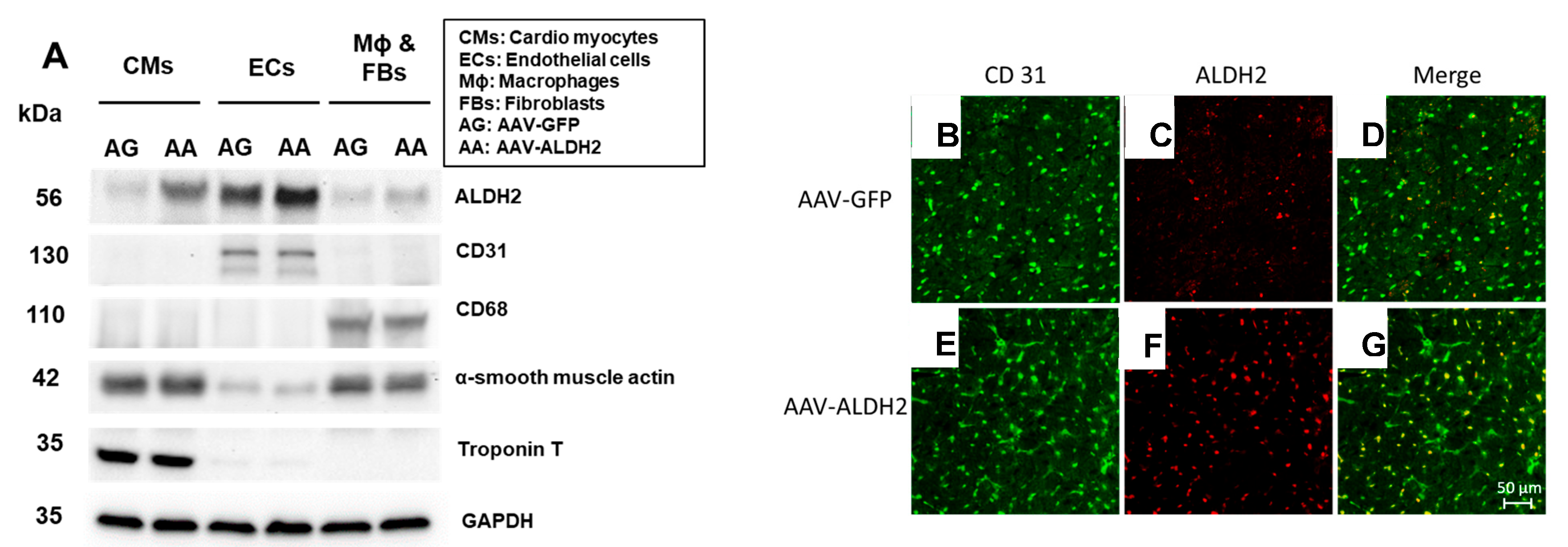
3.1. Effective and Selective Transduction of Injected Genes into CVECs via Intracardiac Transfer
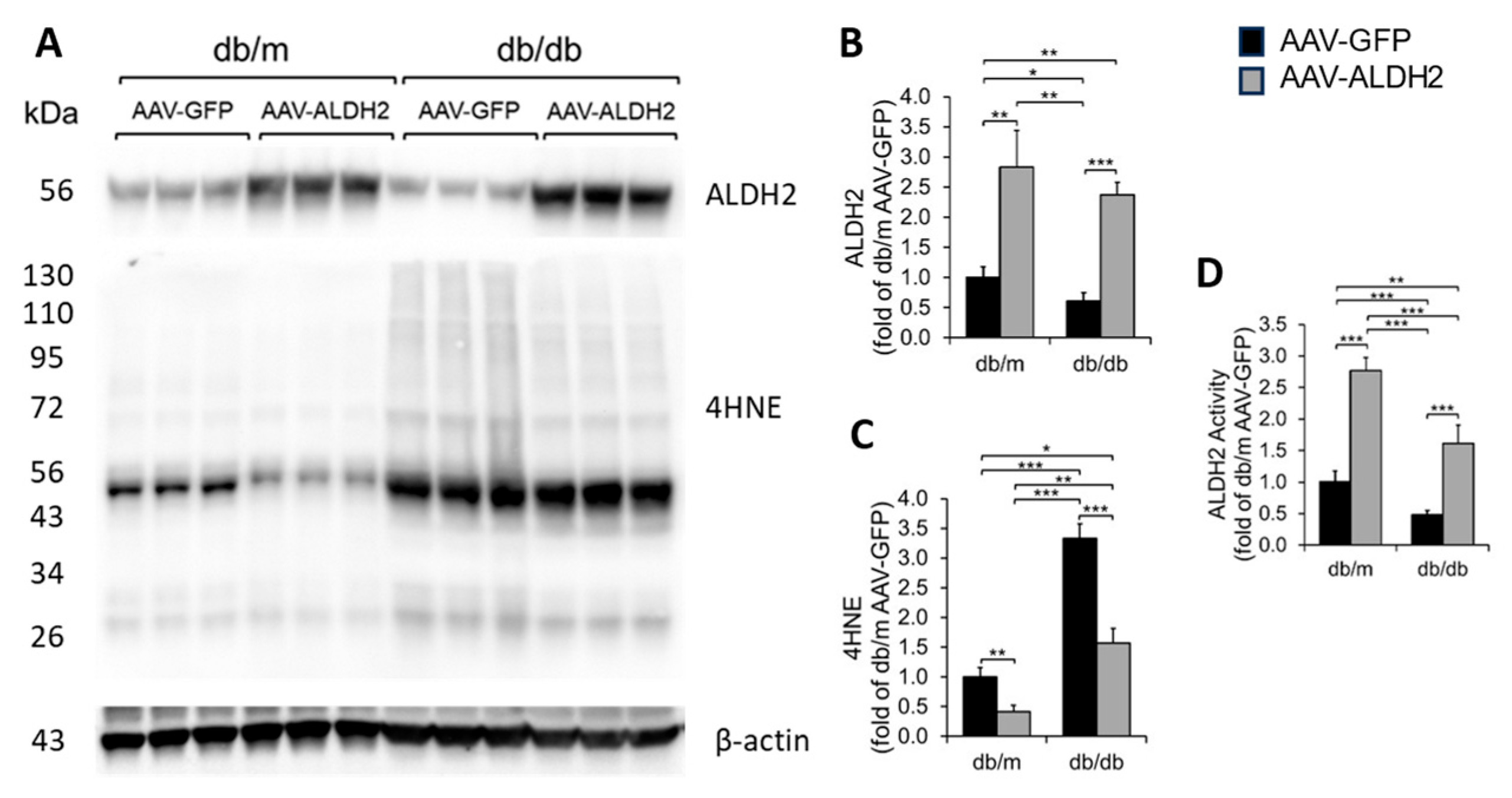
3.2. Intracardiac Injection of ALDH2 Gene with EC-Specific VE Cadherin Promoter Increased the Myocardial ALDH2 Level and ALDH2 Activity and Reduced 4HNE Adducts in db/db and db/m Hearts
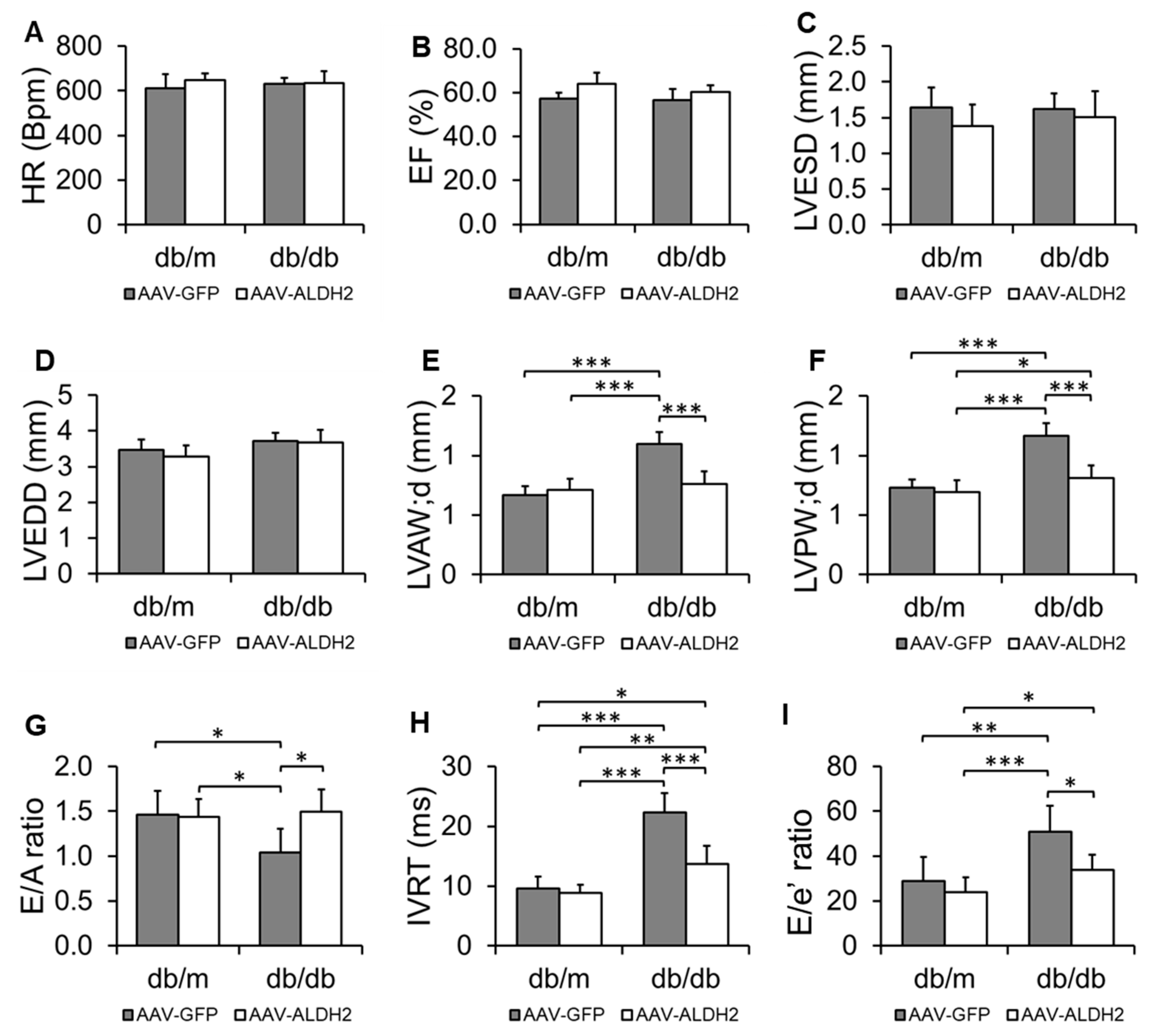
3.3. Specific Overexpression of the ALDH2 Gene in CVECs Improved Diastolic Dysfunction in db/db Mice
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Bozkurt, B.; Ahmad, T.; Alexander, K.M.; Baker, W.L.; Bosak, K.; Breathett, K.; Fonarow, G.C.; Heidenreich, P.; Ho, J.E.; Hsich, E.; et al. Heart Failure Epidemiology and Outcomes Statistics: A Report of the Heart Failure Society of America. J. Card. Fail. 2023, 29, 1412–1451. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Dunlay, S.M.; Roger, V.L.; Redfield, M.M. Epidemiology of heart failure with preserved ejection fraction. Nat. Rev. Cardiol. 2017, 14, 591–602. [Google Scholar] [CrossRef] [PubMed]
- Valero-Munoz, M.; Backman, W.; Sam, F. Murine Models of Heart Failure with Preserved Ejection Fraction: A “Fishing Expedition”. JACC Basic. Transl. Sci. 2017, 2, 770–789. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- van Dalen, B.M.; Chin, J.F.; Motiram, P.A.; Hendrix, A.; Emans, M.E.; Brugts, J.J.; Westenbrink, B.D.; de Boer, R.A. Challenges in the diagnosis of heart failure with preserved ejection fraction in individuals with obesity. Cardiovasc. Diabetol. 2025, 24, 71. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Abudureyimu, M.; Luo, X.; Wang, X.; Sowers, J.R.; Wang, W.; Ge, J.; Ren, J.; Zhang, Y. Heart failure with preserved ejection fraction (HFpEF) in type 2 diabetes mellitus: From pathophysiology to therapeutics. J. Mol. Cell Biol. 2022, 14, mjac028. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Wende, A.R.; Abel, E.D. Lipotoxicity in the heart. Biochim. Biophys. Acta 2010, 1801, 311–319. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Nirengi, S.; Peres Valgas da Silva, C.; Stanford, K.I. Disruption of energy utilization in diabetic cardiomyopathy; a mini review. Curr. Opin. Pharmacol. 2020, 54, 82–90. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Kallikourdis, M.; Cochran, J.D.; Walsh, K.; Condorelli, G. Contributions of Noncardiac Organ-Heart Immune Crosstalk and Somatic Mosaicism to Heart Failure: Current Knowledge and Perspectives. Circ. Res. 2025, 136, 1208–1232. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Franssen, C.; Chen, S.; Unger, A.; Korkmaz, H.I.; De Keulenaer, G.W.; Tschope, C.; Leite-Moreira, A.F.; Musters, R.; Niessen, H.W.; Linke, W.A.; et al. Myocardial Microvascular Inflammatory Endothelial Activation in Heart Failure with Preserved Ejection Fraction. JACC Heart Fail. 2016, 4, 312–324. [Google Scholar] [CrossRef] [PubMed]
- Mali, V.R.; Palaniyandi, S.S. Regulation and therapeutic strategies of 4-hydroxy-2-nonenal metabolism in heart disease. Free Radic. Res. 2014, 48, 251–263. [Google Scholar] [CrossRef] [PubMed]
- Dham, D.; Roy, B.; Gowda, A.; Pan, G.; Sridhar, A.; Zeng, X.; Thandavarayan, R.A.; Palaniyandi, S.S. 4-Hydroxy-2-nonenal, a lipid peroxidation product, as a biomarker in diabetes and its complications: Challenges and opportunities. Free Radic. Res. 2021, 55, 547–561. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Pan, G.; Munukutla, S.; Kar, A.; Gardinier, J.; Thandavarayan, R.A.; Palaniyandi, S.S. Type-2 diabetic aldehyde dehydrogenase 2 mutant mice (ALDH 2*2) exhibiting heart failure with preserved ejection fraction phenotype can be determined by exercise stress echocardiography. PLoS ONE 2018, 13, e0195796. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Pan, G.; Roy, B.; Palaniyandi, S.S. Diabetic Aldehyde Dehydrogenase 2 Mutant (ALDH2*2) Mice Are More Susceptible to Cardiac Ischemic-Reperfusion Injury Due to 4-Hydroxy-2-Nonenal Induced Coronary Endothelial Cell Damage. J. Am. Heart Assoc. 2021, 10, e021140. [Google Scholar] [CrossRef] [PubMed]
- Roy, B.; Pan, G.; Giri, S.; Thandavarayan, R.A.; Palaniyandi, S.S. Aldehyde dehydrogenase 2 augments adiponectin signaling in coronary angiogenesis in HFpEF associated with diabetes. FASEB J. 2022, 36, e22440. [Google Scholar] [CrossRef] [PubMed]
- Mali, V.R.; Ning, R.; Chen, J.; Yang, X.P.; Xu, J.; Palaniyandi, S.S. Impairment of aldehyde dehydrogenase-2 by 4-hydroxy-2-nonenal adduct formation and cardiomyocyte hypertrophy in mice fed a high-fat diet and injected with low-dose streptozotocin. Exp. Biol. Med. 2014, 239, 610–618. [Google Scholar] [CrossRef] [PubMed]
- Singh, S.; Brocker, C.; Koppaka, V.; Chen, Y.; Jackson, B.C.; Matsumoto, A.; Thompson, D.C.; Vasiliou, V. Aldehyde dehydrogenases in cellular responses to oxidative/electrophilic stress. Free Radic. Biol. Med. 2013, 56, 89–101. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Gao, J.; Hao, Y.; Piao, X.; Gu, X. Aldehyde Dehydrogenase 2 as a Therapeutic Target in Oxidative Stress-Related Diseases: Post-Translational Modifications Deserve More Attention. Int. J. Mol. Sci. 2022, 23, 2682. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Breitzig, M.; Bhimineni, C.; Lockey, R.; Kolliputi, N. 4-Hydroxy-2-nonenal: A critical target in oxidative stress? Am. J. Physiol. Cell Physiol. 2016, 311, C537–C543. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Chen, C.H.; Budas, G.R.; Churchill, E.N.; Disatnik, M.H.; Hurley, T.D.; Mochly-Rosen, D. Activation of aldehyde dehydrogenase-2 reduces ischemic damage to the heart. Science 2008, 321, 1493–1495. [Google Scholar] [CrossRef] [PubMed]
- Ji, W.; Wei, S.; Hao, P.; Xing, J.; Yuan, Q.; Wang, J.; Xu, F.; Chen, Y. Aldehyde Dehydrogenase 2 Has Cardioprotective Effects on Myocardial Ischaemia/Reperfusion Injury via Suppressing Mitophagy. Front. Pharmacol. 2016, 7, 101. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Wu, B.; Yu, L.; Wang, Y.; Wang, H.; Li, C.; Yin, Y.; Yang, J.; Wang, Z.; Zheng, Q.; Ma, H. Aldehyde dehydrogenase 2 activation in aged heart improves the autophagy by reducing the carbonyl modification on SIRT1. Oncotarget 2016, 7, 2175–2188. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Jin, S.; Chen, J.; Chen, L.; Histen, G.; Lin, Z.; Gross, S.; Hixon, J.; Chen, Y.; Kung, C.; Chen, Y.; et al. ALDH2(E487K) mutation increases protein turnover and promotes murine hepatocarcinogenesis. Proc. Natl. Acad. Sci. USA 2015, 112, 9088–9093. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Bilak, J.M.; Alam, U.; Miller, C.A.; McCann, G.P.; Arnold, J.R.; Kanagala, P. Microvascular Dysfunction in Heart Failure with Preserved Ejection Fraction: Pathophysiology, Assessment, Prevalence and Prognosis. Card. Fail. Rev. 2022, 8, e24. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Mohammed, S.F.; Hussain, S.; Mirzoyev, S.A.; Edwards, W.D.; Maleszewski, J.J.; Redfield, M.M. Coronary microvascular rarefaction and myocardial fibrosis in heart failure with preserved ejection fraction. Circulation 2015, 131, 550–559. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Paulus, W.J.; Tschope, C. A novel paradigm for heart failure with preserved ejection fraction: Comorbidities drive myocardial dysfunction and remodeling through coronary microvascular endothelial inflammation. J. Am. Coll. Cardiol. 2013, 62, 263–271. [Google Scholar] [CrossRef] [PubMed]
- Zeng, H.; Chen, J.X. Sirtuin 3, Endothelial Metabolic Reprogramming, and Heart Failure with Preserved Ejection Fraction. J. Cardiovasc. Pharmacol. 2019, 74, 315–323. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Roy, B.; Palaniyandi, S.S. Aldehyde dehydrogenase 2 inhibition potentiates 4-hydroxy-2-nonenal induced decrease in angiogenesis of coronary endothelial cells. Cell Biochem. Funct. 2020, 38, 290–299. [Google Scholar] [CrossRef] [PubMed]
- Roy, B.; Palaniyandi, S.S. A role for aldehyde dehydrogenase (ALDH) 2 in angiotensin II-mediated decrease in angiogenesis of coronary endothelial cells. Microvasc. Res. 2021, 135, 104133. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Roy, B.; Sundar, K.; Palaniyandi, S.S. 4-hydroxy-2-nonenal decreases coronary endothelial cell migration: Potentiation by aldehyde dehydrogenase 2 inhibition. Vasc. Pharmacol. 2020, 131, 106762. [Google Scholar] [CrossRef] [PubMed]
- Cuijpers, I.; Simmonds, S.J.; van Bilsen, M.; Czarnowska, E.; Gonzalez Miqueo, A.; Heymans, S.; Kuhn, A.R.; Mulder, P.; Ratajska, A.; Jones, E.A.V.; et al. Microvascular and lymphatic dysfunction in HFpEF and its associated comorbidities. Basic. Res. Cardiol. 2020, 115, 39. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Schiattarella, G.G.; Altamirano, F.; Tong, D.; French, K.M.; Villalobos, E.; Kim, S.Y.; Luo, X.; Jiang, N.; May, H.I.; Wang, Z.V.; et al. Nitrosative stress drives heart failure with preserved ejection fraction. Nature 2019, 568, 351–356. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Schiattarella, G.G.; Rodolico, D.; Hill, J.A. Metabolic inflammation in heart failure with preserved ejection fraction. Cardiovasc. Res. 2021, 117, 423–434. [Google Scholar] [CrossRef] [PubMed]
- Mali, V.R.; Pan, G.; Deshpande, M.; Thandavarayan, R.A.; Xu, J.; Yang, X.P.; Palaniyandi, S.S. Cardiac Mitochondrial Respiratory Dysfunction and Tissue Damage in Chronic Hyperglycemia Correlate with Reduced Aldehyde Dehydrogenase-2 Activity. PLoS ONE 2016, 11, e0163158. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Incalza, M.A.; D’Oria, R.; Natalicchio, A.; Perrini, S.; Laviola, L.; Giorgino, F. Oxidative stress and reactive oxygen species in endothelial dysfunction associated with cardiovascular and metabolic diseases. Vasc. Pharmacol. 2018, 100, 1–19. [Google Scholar] [CrossRef] [PubMed]
- Haybar, H.; Shahrabi, S.; Rezaeeyan, H.; Shirzad, R.; Saki, N. Endothelial Cells: From Dysfunction Mechanism to Pharmacological Effect in Cardiovascular Disease. Cardiovasc. Toxicol. 2019, 19, 13–22. [Google Scholar] [CrossRef] [PubMed]
- Jaganjac, M.; Milkovic, L.; Gegotek, A.; Cindric, M.; Zarkovic, K.; Skrzydlewska, E.; Zarkovic, N. The relevance of pathophysiological alterations in redox signaling of 4-hydroxynonenal for pharmacological therapies of major stress-associated diseases. Free Radic. Biol. Med. 2020, 157, 128–153. [Google Scholar] [CrossRef] [PubMed]
- Shah, S.A.; Reagan, C.E.; Bresticker, J.E.; Wolpe, A.G.; Good, M.E.; Macal, E.H.; Billcheck, H.O.; Bradley, L.A.; French, B.A.; Isakson, B.E.; et al. Obesity-Induced Coronary Microvascular Disease Is Prevented by iNOS Deletion and Reversed by iNOS Inhibition. JACC Basic. Transl. Sci. 2023, 8, 501–514. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Borlaug, B.A.; Jensen, M.D.; Kitzman, D.W.; Lam, C.S.P.; Obokata, M.; Rider, O.J. Obesity and heart failure with preserved ejection fraction: New insights and pathophysiological targets. Cardiovasc. Res. 2023, 118, 3434–3450. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- van der Heijden, D.J.; van Leeuwen, M.A.H.; Janssens, G.N.; Lenzen, M.J.; van de Ven, P.M.; Eringa, E.C.; van Royen, N. Body Mass Index Is Associated with Microvascular Endothelial Dysfunction in Patients With Treated Metabolic Risk Factors and Suspected Coronary Artery Disease. J. Am. Heart Assoc. 2017, 6, e006082. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Aroor, A.R.; Habibi, J.; Kandikattu, H.K.; Garro-Kacher, M.; Barron, B.; Chen, D.; Hayden, M.R.; Whaley-Connell, A.; Bender, S.B.; Klein, T.; et al. Dipeptidyl peptidase-4 (DPP-4) inhibition with linagliptin reduces western diet-induced myocardial TRAF3IP2 expression, inflammation and fibrosis in female mice. Cardiovasc. Diabetol. 2017, 16, 61. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Matsiukevich, D.; Kovacs, A.; Li, T.; Kokkonen-Simon, K.; Matkovich, S.J.; Oladipupo, S.S.; Ornitz, D.M. Characterization of a robust mouse model of heart failure with preserved ejection fraction. Am. J. Physiol. Heart Circ. Physiol. 2023, 325, H203–H231. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Alex, L.; Russo, I.; Holoborodko, V.; Frangogiannis, N.G. Characterization of a mouse model of obesity-related fibrotic cardiomyopathy that recapitulates features of human heart failure with preserved ejection fraction. Am. J. Physiol. Heart Circ. Physiol. 2018, 315, H934–H949. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Cross, M.J.; Claesson-Welsh, L. FGF and VEGF function in angiogenesis: Signalling pathways, biological responses and therapeutic inhibition. Trends Pharmacol. Sci. 2001, 22, 201–207. [Google Scholar] [CrossRef] [PubMed]
- Avogaro, A.; de Kreutzenberg, S.; Fadini, G. Dipeptidyl-peptidase 4 inhibition: Linking metabolic control to cardiovascular protection. Curr. Pharm. Des. 2014, 20, 2387–2394. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Chapple, S.J.; Cheng, X.; Mann, G.E. Effects of 4-hydroxynonenal on vascular endothelial and smooth muscle cell redox signaling and function in health and disease. Redox Biol. 2013, 1, 319–331. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- McHugh, K.; DeVore, A.D.; Wu, J.; Matsouaka, R.A.; Fonarow, G.C.; Heidenreich, P.A.; Yancy, C.W.; Green, J.B.; Altman, N.; Hernandez, A.F. Heart Failure with Preserved Ejection Fraction and Diabetes: JACC State-of-the-Art Review. J. Am. Coll. Cardiol. 2019, 73, 602–611. [Google Scholar] [CrossRef] [PubMed]
- Jasinska-Stroschein, M. The current state of preclinical modeling of human diabetic cardiomyopathy using rodents. Biomed. Pharmacother. 2023, 168, 115843. [Google Scholar] [CrossRef] [PubMed]
- Hinkel, R.; Howe, A.; Renner, S.; Ng, J.; Lee, S.; Klett, K.; Kaczmarek, V.; Moretti, A.; Laugwitz, K.L.; Skroblin, P.; et al. Diabetes Mellitus-Induced Microvascular Destabilization in the Myocardium. J. Am. Coll. Cardiol. 2017, 69, 131–143. [Google Scholar] [CrossRef] [PubMed]
- Katz, M.G.; Fargnoli, A.S.; Weber, T.; Hajjar, R.J.; Bridges, C.R. Use of Adeno-Associated Virus Vector for Cardiac Gene Delivery in Large-Animal Surgical Models of Heart Failure. Hum. Gene Ther. Clin. Dev. 2017, 28, 157–164. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Penaud-Budloo, M.; Le Guiner, C.; Nowrouzi, A.; Toromanoff, A.; Cherel, Y.; Chenuaud, P.; Schmidt, M.; von Kalle, C.; Rolling, F.; Moullier, P.; et al. Adeno-associated virus vector genomes persist as episomal chromatin in primate muscle. J. Virol. 2008, 82, 7875–7885. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Mingozzi, F.; High, K.A. Immune responses to AAV vectors: Overcoming barriers to successful gene therapy. Blood 2013, 122, 23–36. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Vrtovec, B.; Frljak, S.; Poglajen, G.; Zemljic, G.; Cerar, A.; Sever, M.; Haddad, F.; Wu, J.C. A pilot clinical trial of cell therapy in heart failure with preserved ejection fraction. Eur. J. Heart Fail. 2022, 24, 1441–1449. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Vrtovec, B.; Poglajen, G.; Lezaic, L.; Sever, M.; Domanovic, D.; Cernelc, P.; Socan, A.; Schrepfer, S.; Torre-Amione, G.; Haddad, F.; et al. Effects of intracoronary CD34+ stem cell transplantation in nonischemic dilated cardiomyopathy patients: 5-year follow-up. Circ. Res. 2013, 112, 165–173. [Google Scholar] [CrossRef] [PubMed]
- Pan, G.; Roy, B.; Harding, P.; Lanigan, T.; Hilgarth, R.; Thandavarayan, R.A.; Palaniyandi, S.S. Effects of intracardiac delivery of aldehyde dehydrogenase 2 gene in myocardial salvage. Gene Ther. 2023, 30, 115–121. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Hahn, V.S.; Knutsdottir, H.; Luo, X.; Bedi, K.; Margulies, K.B.; Haldar, S.M.; Stolina, M.; Yin, J.; Khakoo, A.Y.; Vaishnav, J.; et al. Myocardial Gene Expression Signatures in Human Heart Failure with Preserved Ejection Fraction. Circulation 2021, 143, 120–134. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Louis Jeune, V.; Joergensen, J.A.; Hajjar, R.J.; Weber, T. Pre-existing anti-adeno-associated virus antibodies as a challenge in AAV gene therapy. Hum. Gene Ther. Methods 2013, 24, 59–67. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Mingozzi, F.; Anguela, X.M.; Pavani, G.; Chen, Y.; Davidson, R.J.; Hui, D.J.; Yazicioglu, M.; Elkouby, L.; Hinderer, C.J.; Faella, A.; et al. Overcoming preexisting humoral immunity to AAV using capsid decoys. Sci. Transl. Med. 2013, 5, 194ra192. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Hordeaux, J.; Lamontagne, R.J.; Song, C.; Buchlis, G.; Dyer, C.; Buza, E.L.; Ramezani, A.; Wielechowski, E.; Greig, J.A.; Chichester, J.A.; et al. High-dose systemic adeno-associated virus vector administration causes liver and sinusoidal endothelial cell injury. Mol. Ther. 2024, 32, 952–968. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Stone, D.; Aubert, M.; Jerome, K.R. Adeno-associated virus vectors and neurotoxicity-lessons from preclinical and human studies. Gene Ther. 2025, 32, 60–73. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pan, G.; Roy, B.; Yeboah, E.O.; Lanigan, T.; Hilgarth, R.; Thandavarayan, R.A.; Petriello, M.C.; Giri, S.; Palaniyandi, S.S. Targeted Overexpression of Mitochondrial ALDH2 in Coronary Endothelial Cells Mitigates HFpEF in a Diabetic Mouse Model. Biomolecules 2025, 15, 1029. https://doi.org/10.3390/biom15071029
Pan G, Roy B, Yeboah EO, Lanigan T, Hilgarth R, Thandavarayan RA, Petriello MC, Giri S, Palaniyandi SS. Targeted Overexpression of Mitochondrial ALDH2 in Coronary Endothelial Cells Mitigates HFpEF in a Diabetic Mouse Model. Biomolecules. 2025; 15(7):1029. https://doi.org/10.3390/biom15071029
Chicago/Turabian StylePan, Guodong, Bipradas Roy, Emmanuel Oppong Yeboah, Thomas Lanigan, Roland Hilgarth, Rajarajan A. Thandavarayan, Michael C. Petriello, Shailendra Giri, and Suresh Selvaraj Palaniyandi. 2025. "Targeted Overexpression of Mitochondrial ALDH2 in Coronary Endothelial Cells Mitigates HFpEF in a Diabetic Mouse Model" Biomolecules 15, no. 7: 1029. https://doi.org/10.3390/biom15071029
APA StylePan, G., Roy, B., Yeboah, E. O., Lanigan, T., Hilgarth, R., Thandavarayan, R. A., Petriello, M. C., Giri, S., & Palaniyandi, S. S. (2025). Targeted Overexpression of Mitochondrial ALDH2 in Coronary Endothelial Cells Mitigates HFpEF in a Diabetic Mouse Model. Biomolecules, 15(7), 1029. https://doi.org/10.3390/biom15071029