
Salutary Effects of Overexpression of Rsm22, an Assembly Factor for the Mitochondrial Ribosome, on Frataxin/Yfh1 Depletion Phenotypes in Saccharomyces cerevisiae


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Chemicals and Reagents
2.2. Yeast Strains and Growth Conditions
2.3. Isolation of Mitochondria and Cytoplasm
2.4. Fe-S Cluster Assembly in Isolated Mitochondria
2.5. Cytoplasmic Fe-S Cluster Assembly
2.6. Miscellaneous
2.7. Data Analysis
3. Results
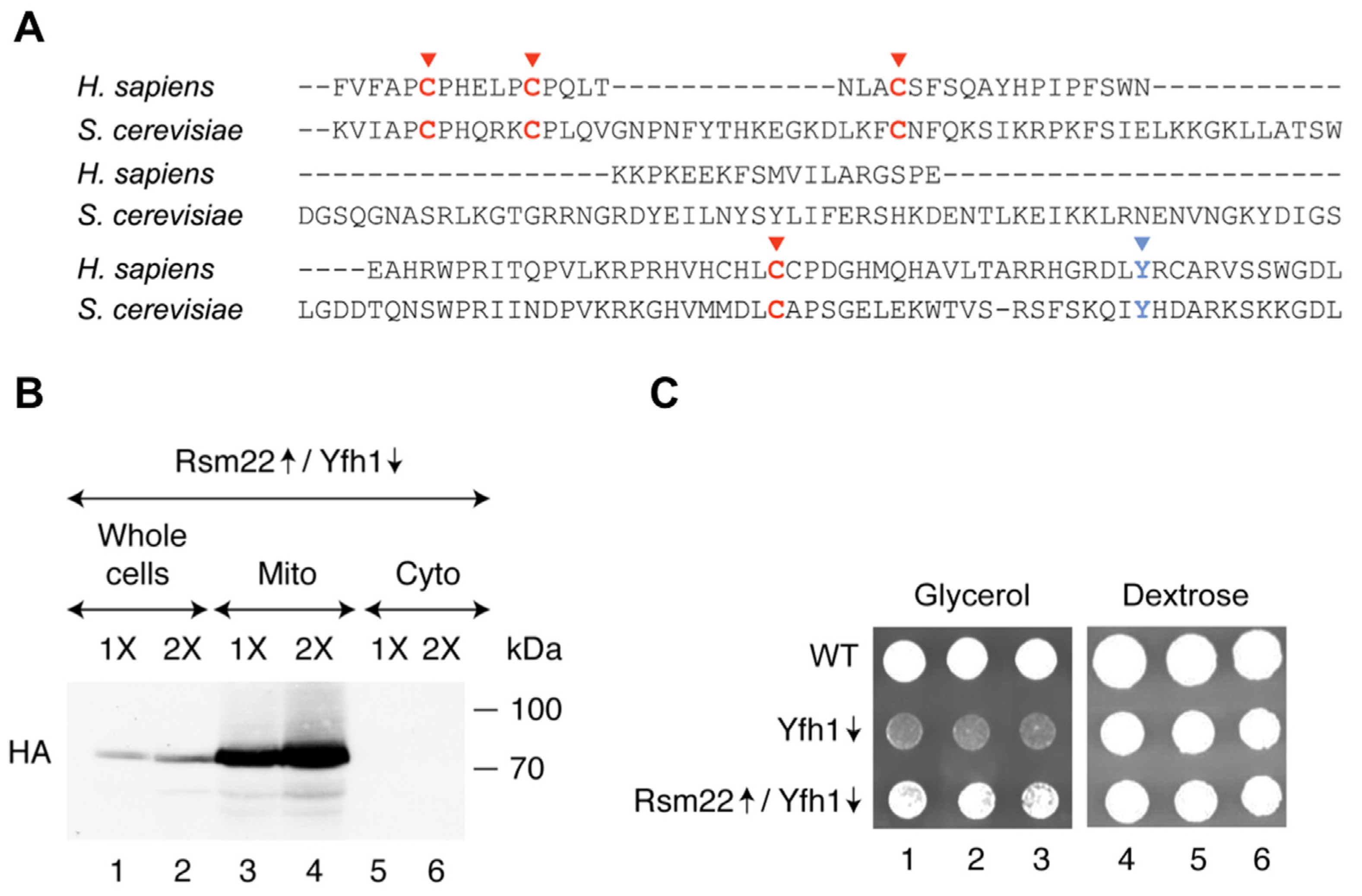
3.1. Rsm22 Overexpression Led to Improved Growth of Yfh1-Depleted Cells on a Non-Fermentable Carbon Source
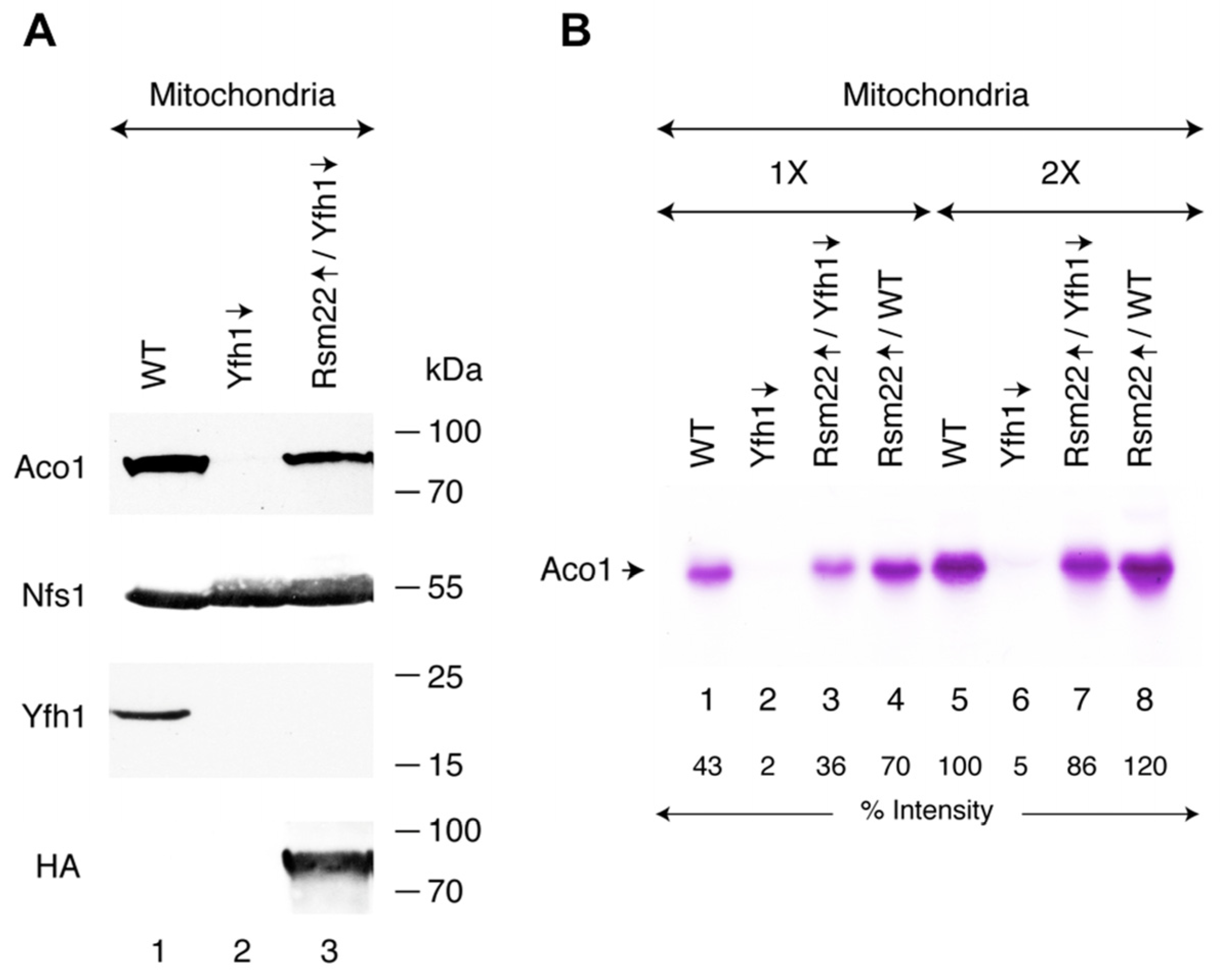
3.2. Rsm22 Overexpression in Yfh1-Depleted Cells Restored Aconitase Protein and Activity
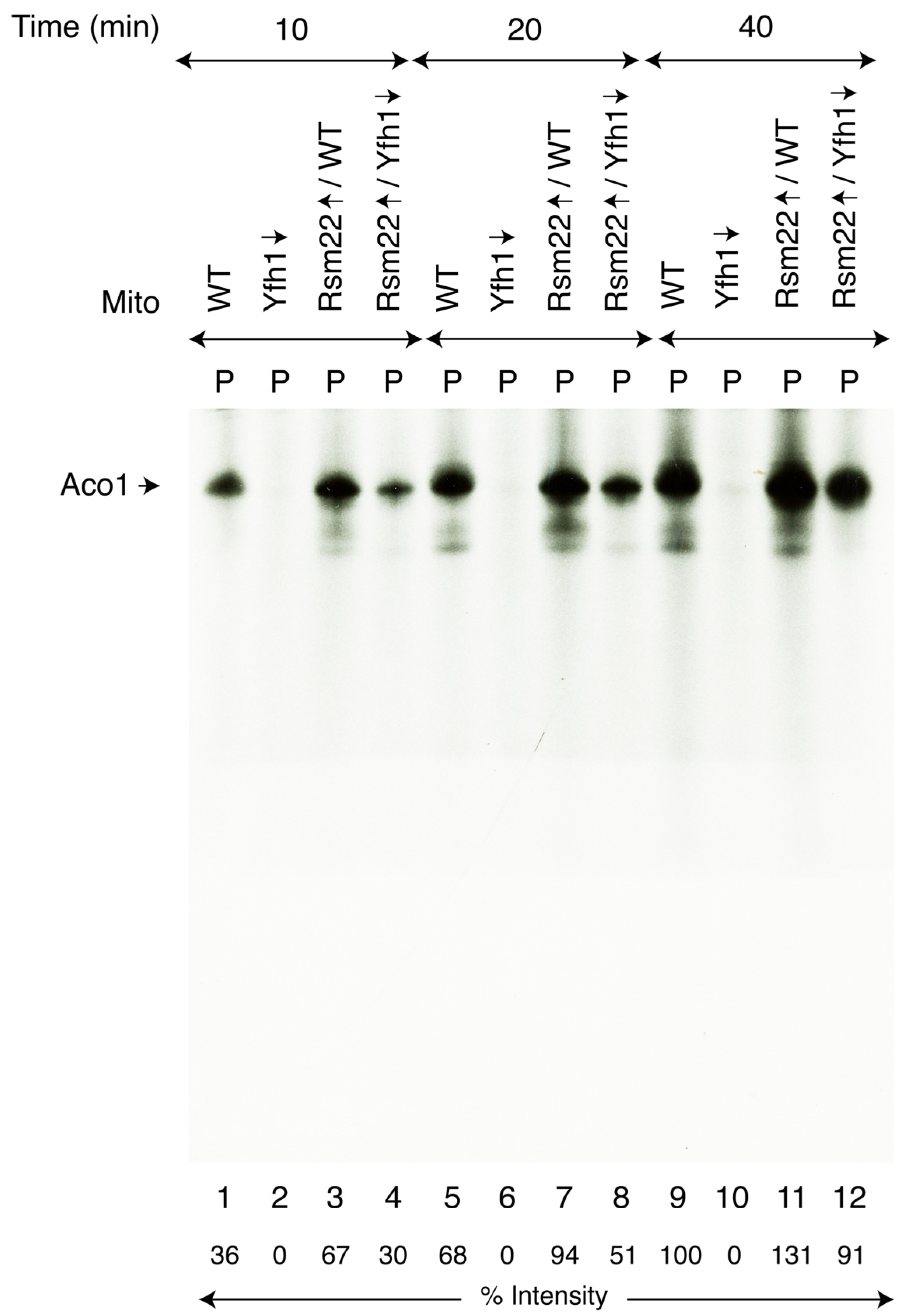
3.3. Rsm22 Overexpression Restored New Fe-S Cluster Synthesis/Assembly in Isolated Mitochondria Lacking Yfh1
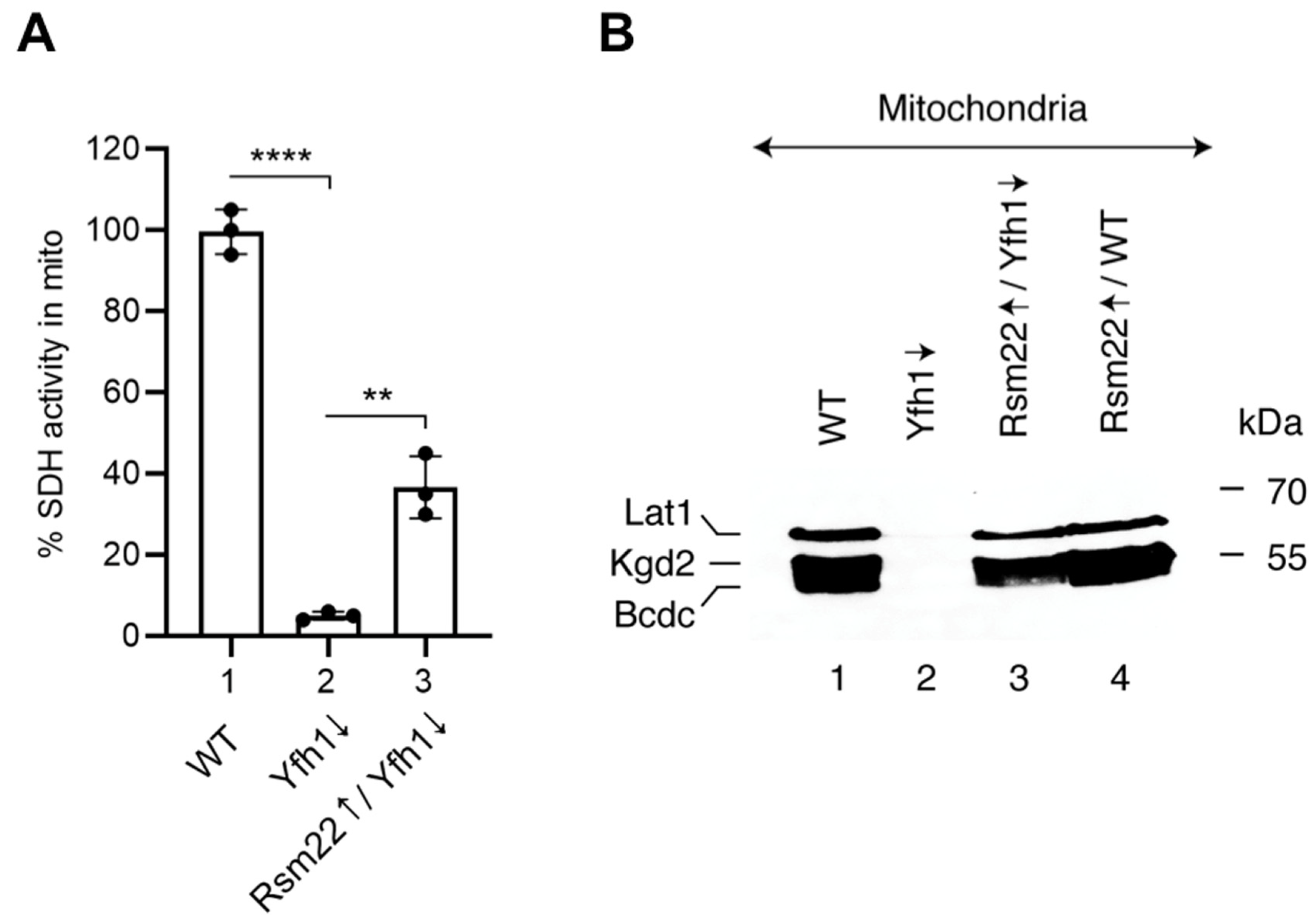
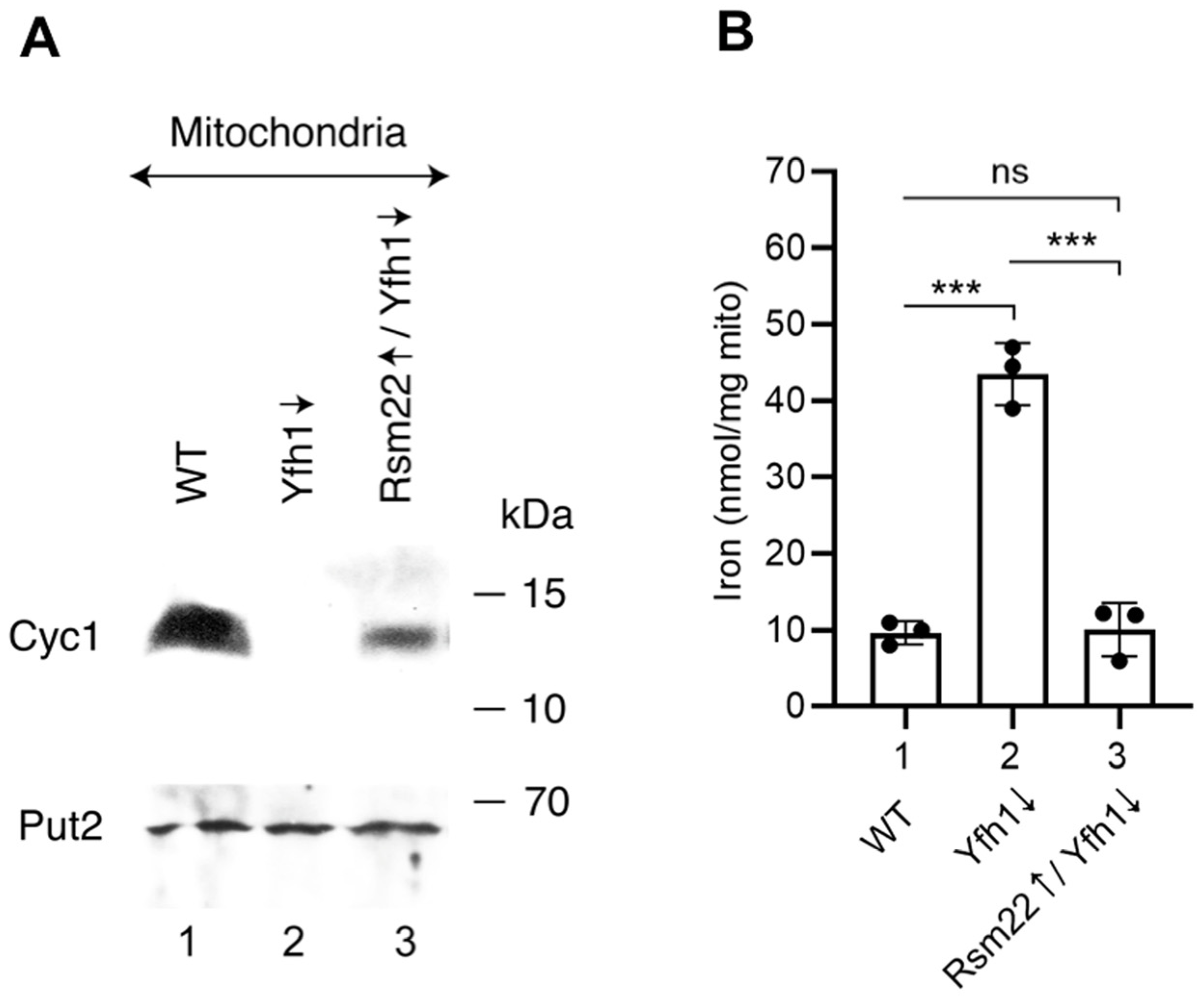
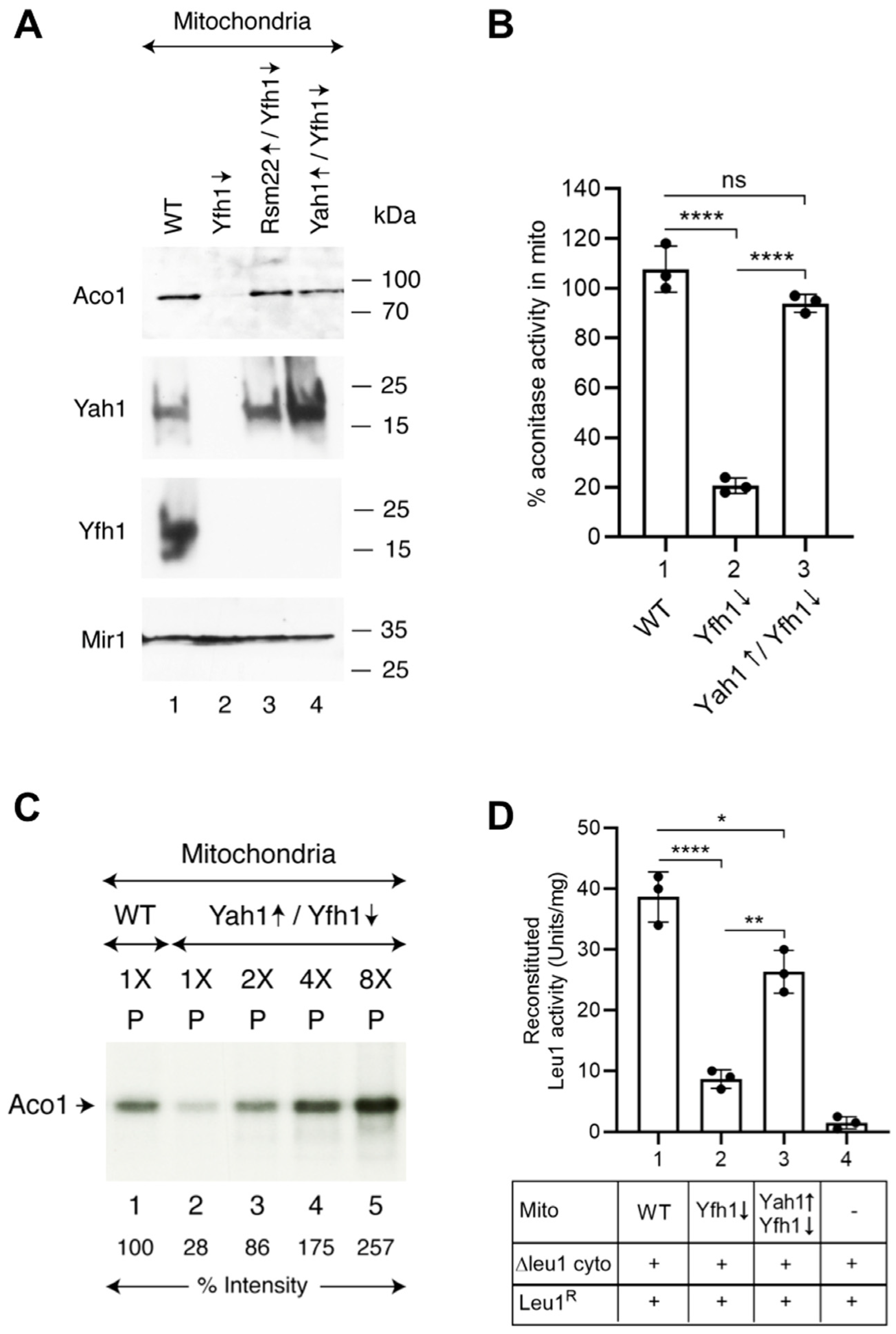
3.4. Overexpression of Rsm22 Rescued Other Iron Proteins, Restored Iron Homeostasis, and Conferred Membrane Potential in Mitochondria Lacking Yfh1
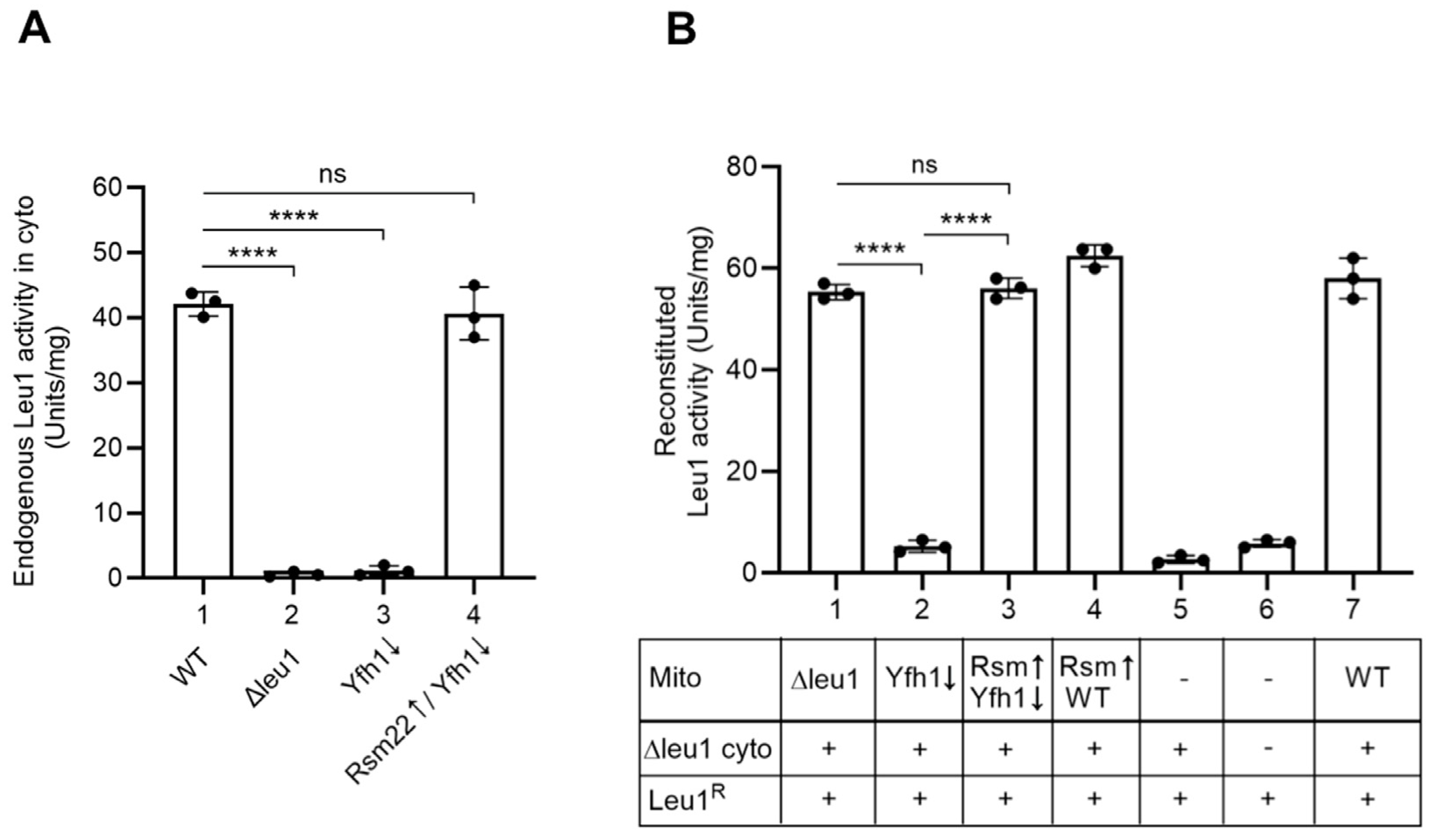
3.5. Yfh1↓ Mitochondria with Overexpressed Rsm22 Regained Capability to Promote Cytoplasmic Fe-S Cluster Assembly
3.6. A Possible Suppression Mechanism
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
Abbreviations
| (Fe-S)int | (Fe-S) intermediate |
| ΔNYah1 | N-terminal 60 amino acids, including the mitochondrial targeting signal, removed from the Yah1 precursor protein (pYah1) |
| Leu1R | Recombinant Leu1 |
| ISC | Mitochondrial iron–sulfur cluster machinery |
| CIA | Cytoplasmic iron–sulfur protein assembly machinery |
References
- Keita, M.; McIntyre, K.; Rodden, L.N.; Schadt, K.; Lynch, D.R. Friedreich ataxia: Clinical features and new developments. Neurodegener. Dis. Manag. 2022, 12, 267–283. [Google Scholar] [CrossRef] [PubMed]
- Pandolfo, M. Friedreich Ataxia: An (Almost) 30-Year History After Gene Discovery. Neurol. Genet. 2025, 11, e200236. [Google Scholar] [CrossRef]
- Krasilnikova, M.M.; Humphries, C.L.; Shinsky, E.M. Friedreich’s ataxia: New insights. Emerg. Top. Life Sci. 2023, 7, 313–323. [Google Scholar] [PubMed]
- Campuzano, V.; Montermini, L.; Lutz, Y.; Cova, L.; Hindelang, C.; Jiralerspong, S.; Trottier, Y.; Kish, S.J.; Faucheux, B.; Trouillas, P.; et al. Frataxin is reduced in Friedreich ataxia patients and is associated with mitochondrial membranes. Hum. Mol. Genet. 1997, 6, 1771–1780. [Google Scholar] [CrossRef]
- Lill, R.; Freibert, S.A. Mechanisms of Mitochondrial Iron-Sulfur Protein Biogenesis. Annu. Rev. Biochem. 2020, 89, 471–499. [Google Scholar] [CrossRef]
- Dancis, A.; Pandey, A.K.; Pain, D. Mitochondria function in cytoplasmic FeS protein biogenesis. Biochim. Biophys. Acta Mol. Cell Res. 2024, 1871, 119733. [Google Scholar] [CrossRef]
- Uzarska, M.A.; Grochowina, I.; Soldek, J.; Jelen, M.; Schilke, B.; Marszalek, J.; Craig, E.A.; Dutkiewicz, R. During FeS cluster biogenesis, ferredoxin and frataxin use overlapping binding sites on yeast cysteine desulfurase Nfs1. J. Biol. Chem. 2022, 298, 101570. [Google Scholar] [CrossRef]
- Johnson, D.C.; Dean, D.R.; Smith, A.D.; Johnson, M.K. Structure, function, and formation of biological iron-sulfur clusters. Annu. Rev. Biochem. 2005, 74, 247–281. [Google Scholar] [CrossRef] [PubMed]
- Parent, A.; Elduque, X.; Cornu, D.; Belot, L.; Le Caer, J.P.; Grandas, A.; Toledano, M.B.; D’Autreaux, B. Mammalian frataxin directly enhances sulfur transfer of NFS1 persulfide to both ISCU and free thiols. Nat. Commun. 2015, 6, 5686. [Google Scholar] [CrossRef]
- Schulz, V.; Steinhilper, R.; Oltmanns, J.; Freibert, S.A.; Krapoth, N.; Linne, U.; Welsch, S.; Hoock, M.H.; Schunemann, V.; Murphy, B.J.; et al. Mechanism and structural dynamics of sulfur transfer during de novo [2Fe-2S] cluster assembly on ISCU2. Nat. Commun. 2024, 15, 3269. [Google Scholar] [CrossRef]
- Gervason, S.; Larkem, D.; Mansour, A.B.; Botzanowski, T.; Müller, C.S.; Pecqueur, L.; Le Pavec, G.; Delaunay-Moisan, A.; Brun, O.; Agramunt, J.; et al. Physiologically relevant reconstitution of iron-sulfur cluster biosynthesis uncovers persulfide-processing functions of ferredoxin-2 and frataxin. Nat. Commun. 2019, 10, 3566. [Google Scholar] [CrossRef]
- Steinhilper, R.; Boss, L.; Freibert, S.A.; Schulz, V.; Krapoth, N.; Kaltwasser, S.; Lill, R.; Murphy, B.J. Two-stage binding of mitochondrial ferredoxin-2 to the core iron-sulfur cluster assembly complex. Nat. Commun. 2024, 15, 10559. [Google Scholar] [CrossRef] [PubMed]
- Ast, T.; Itoh, Y.; Sadre, S.; McCoy, J.G.; Namkoong, G.; Wengrod, J.C.; Chicherin, I.; Joshi, P.R.; Kamenski, P.; Suess, D.L.M.; et al. METTL17 is an Fe-S cluster checkpoint for mitochondrial translation. Mol. Cell 2024, 84, 359–374 e358. [Google Scholar] [CrossRef]
- Gerber, J.; Muhlenhoff, U.; Lill, R. An interaction between frataxin and Isu1/Nfs1 that is crucial for Fe/S cluster synthesis on Isu1. EMBO Rep. 2003, 4, 906–911. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.; Craig, E.A. Binding of yeast frataxin to the scaffold for Fe-S cluster biogenesis, Isu. J. Biol. Chem. 2008, 283, 12674–12679. [Google Scholar] [CrossRef]
- Maio, N.; Jain, A.; Rouault, T.A. Mammalian iron-sulfur cluster biogenesis: Recent insights into the roles of frataxin, acyl carrier protein and ATPase-mediated transfer to recipient proteins. Curr. Opin. Chem. Biol. 2020, 55, 34–44. [Google Scholar] [CrossRef] [PubMed]
- Yoon, H.; Golla, R.; Lesuisse, E.; Pain, J.; Donald, J.E.; Lyver, E.R.; Pain, D.; Dancis, A. Mutation in the Fe-S scaffold protein Isu bypasses frataxin deletion. Biochem. J. 2012, 441, 473–480. [Google Scholar] [CrossRef]
- Das, D.; Patra, S.; Bridwell-Rabb, J.; Barondeau, D.P. Mechanism of frataxin “bypass” in human iron-sulfur cluster biosynthesis with implications for Friedreich’s ataxia. J. Biol. Chem. 2019, 294, 9276–9284. [Google Scholar] [CrossRef]
- Harper, N.J.; Burnside, C.; Klinge, S. Principles of mitoribosomal small subunit assembly in eukaryotes. Nature 2023, 614, 175–181. [Google Scholar] [CrossRef]
- Boß, L.; Stehling, O.; Elsasser, H.P.; Lill, R. Crucial role and conservation of the three [2Fe-2S] clusters in the human mitochondrial ribosome. J. Biol. Chem. 2025, 301, 108087. [Google Scholar] [CrossRef]
- Zhong, H.; Janer, A.; Khalimonchuk, O.; Antonicka, H.; Shoubridge, E.A.; Barrientos, A. BOLA3 and NFU1 link mitoribosome iron-sulfur cluster assembly to multiple mitochondrial dysfunctions syndrome. Nucleic Acids Res. 2023, 51, 11797–11812. [Google Scholar] [CrossRef] [PubMed]
- Longtine, M.S.; McKenzie, A., 3rd; Demarini, D.J.; Shah, N.G.; Wach, A.; Brachat, A.; Philippsen, P.; Pringle, J.R. Additional modules for versatile and economical PCR-based gene deletion and modification in Saccharomyces cerevisiae. Yeast 1998, 14, 953–961. [Google Scholar] [CrossRef]
- Sherman, F. Getting started with yeast. Methods Enzymol. 2002, 350, 3–41. [Google Scholar]
- Pandey, A.K.; Pain, J.; Brindha, J.; Dancis, A.; Pain, D. Essential mitochondrial role in iron-sulfur cluster assembly of the cytoplasmic isopropylmalate isomerase Leu1 in Saccharomyces cerevisiae. Mitochondrion 2023, 69, 104–115. [Google Scholar] [CrossRef] [PubMed]
- Pandey, A.K.; Pain, J.; Singh, P.; Dancis, A.; Pain, D. Mitochondrial glutaredoxin Grx5 functions as a central hub for cellular iron-sulfur cluster assembly. J. Biol. Chem. 2025, 301, 108391. [Google Scholar] [CrossRef]
- Pandey, A.K.; Pain, J.; Dancis, A.; Pain, D. Mitochondria export iron-sulfur and sulfur intermediates to the cytoplasm for iron-sulfur cluster assembly and tRNA thiolation in yeast. J. Biol. Chem. 2019, 294, 9489–9502. [Google Scholar] [CrossRef] [PubMed]
- Pierik, A.J.; Netz, D.J.; Lill, R. Analysis of iron-sulfur protein maturation in eukaryotes. Nat. Protoc. 2009, 4, 753–766. [Google Scholar] [CrossRef]
- Sipos, K.; Lange, H.; Fekete, Z.; Ullmann, P.; Lill, R.; Kispal, G. Maturation of cytosolic iron-sulfur proteins requires glutathione. J. Biol. Chem. 2002, 277, 26944–26949. [Google Scholar] [CrossRef]
- Li, J.; Kogan, M.; Knight, S.A.; Pain, D.; Dancis, A. Yeast mitochondrial protein, Nfs1p, coordinately regulates iron-sulfur cluster proteins, cellular iron uptake, and iron distribution. J. Biol. Chem. 1999, 274, 33025–33034. [Google Scholar] [CrossRef]
- Munujos, P.; Coll-Canti, J.; Gonzalez-Sastre, F.; Gella, F.J. Assay of succinate dehydrogenase activity by a colorimetric-continuous method using iodonitrotetrazolium chloride as electron acceptor. Anal. Biochem. 1993, 212, 506–509. [Google Scholar] [CrossRef]
- Geissler, A.; Krimmer, T.; Bomer, U.; Guiard, B.; Rassow, J.; Pfanner, N. Membrane potential-driven protein import into mitochondria. The sorting sequence of cytochrome b2 modulates the Δψ-dependence of translocation of the matrix-targeting sequence. Mol. Biol. Cell 2000, 11, 3977–3991. [Google Scholar] [CrossRef] [PubMed]
- Alam, J.; Rahman, F.T.; Sah-Teli, S.K.; Venkatesan, R.; Koski, M.K.; Autio, K.J.; Hiltunen, J.K.; Kastaniotis, A.J. Expression and analysis of the SAM-dependent RNA methyltransferase Rsm22 from Saccharomyces cerevisiae. Acta Cryst. 2021, D77, 840–853. [Google Scholar] [CrossRef]
- Maio, N.; Singh, A.; Uhrigshardt, H.; Saxena, N.; Tong, W.H.; Rouault, T.A. Cochaperone binding to LYR motifs confers specificity of iron sulfur cluster delivery. Cell Metab. 2014, 19, 445–457. [Google Scholar] [CrossRef] [PubMed]
- Saveanu, C.; Fromont-Racine, M.; Harington, A.; Ricard, F.; Namane, A.; Jacquier, A. Identification of 12 new yeast mitochondrial ribosomal proteins including 6 that have no prokaryotic homologues. J. Biol. Chem. 2001, 276, 15861–15867. [Google Scholar] [CrossRef]
- Onder, O.; Yoon, H.; Naumann, B.; Hippler, M.; Dancis, A.; Daldal, F. Modifications of the lipoamide-containing mitochondrial subproteome in a yeast mutant defective in cysteine desulfurase. Mol. Cell. Proteomics 2006, 5, 1426–1436. [Google Scholar] [CrossRef] [PubMed]
- Dickinson, J.R. Branched-chain keto acid dehydrogenase of yeast. Methods Enzymol. 2000, 324, 389–398. [Google Scholar]
- Sherman, F. The importance of mutation, then and now: Studies with yeast cytochrome c. Mutat. Res. 2005, 589, 1–16. [Google Scholar] [CrossRef]
- Masud, A.J.; Kastaniotis, A.J.; Rahman, M.T.; Autio, K.J.; Hiltunen, J.K. Mitochondrial acyl carrier protein (ACP) at the interface of metabolic state sensing and mitochondrial function. Biochim. Biophys. Acta Mol. Cell Res. 2019, 1866, 118540. [Google Scholar] [CrossRef]
- Hoffman, M.; Gora, M.; Rytka, J. Identification of rate-limiting steps in yeast heme biosynthesis. Biochem. Biophys. Res. Commun. 2003, 310, 1247–1253. [Google Scholar] [CrossRef]
- Outten, C.E.; Albetel, A.N. Iron sensing and regulation in Saccharomyces cerevisiae: Ironing out the mechanistic details. Curr. Opin. Microbiol. 2013, 16, 662–668. [Google Scholar] [CrossRef]
- Miao, R.; Kim, H.; Koppolu, U.M.; Ellis, E.A.; Scott, R.A.; Lindahl, P.A. Biophysical characterization of the iron in mitochondria from Atm1p-depleted Saccharomyces cerevisiae. Biochemistry 2009, 48, 9556–9568. [Google Scholar] [CrossRef] [PubMed]
- Kispal, G.; Csere, P.; Prohl, C.; Lill, R. The mitochondrial proteins Atm1p and Nfs1p are essential for biogenesis of cytosolic Fe/S proteins. EMBO J. 1999, 18, 3981–3989. [Google Scholar] [CrossRef] [PubMed]
- Wilson, R.B.; Roof, D.M. Respiratory deficiency due to loss of mitochondrial DNA in yeast lacking the frataxin homologue. Nat. Genet. 1997, 16, 352–357. [Google Scholar] [CrossRef] [PubMed]
- Babcock, M.; de Silva, D.; Oaks, R.; Davis-Kaplan, S.; Jiralerspong, S.; Montermini, L.; Pandolfo, M.; Kaplan, J. Regulation of mitochondrial iron accumulation by Yfh1p, a putative homolog of frataxin. Science 1997, 276, 1709–1712. [Google Scholar] [CrossRef]
- Yan, R.; Konarev, P.V.; Iannuzzi, C.; Adinolfi, S.; Roche, B.; Kelly, G.; Simon, L.; Martin, S.R.; Py, B.; Barras, F.; et al. Ferredoxin competes with bacterial frataxin in binding to the desulfurase IscS. J. Biol. Chem. 2013, 288, 24777–24787. [Google Scholar] [CrossRef]
- Kim, J.H.; Frederick, R.O.; Reinen, N.M.; Troupis, A.T.; Markley, J.L. [2Fe-2S]-ferredoxin binds directly to cysteine desulfurase and supplies an electron for iron-sulfur cluster assembly but is displaced by the scaffold protein or bacterial frataxin. J. Am. Chem. Soc. 2013, 135, 8117–8120. [Google Scholar] [CrossRef]
- Weiler, B.D.; Brück, M.C.; Kothe, I.; Bill, E.; Lill, R.; Mühlenhoff, U. Mitochondrial [4Fe-4S] protein assembly involves reductive [2Fe-2S] cluster fusion on ISCA1-ISCA2 by electron flow from ferredoxin FDX2. Proc. Natl. Acad. Sci. USA 2020, 117, 20555–20565. [Google Scholar] [CrossRef]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pandey, A.K.; Singh, P.; Pain, J.; Dancis, A.; Pain, D. Salutary Effects of Overexpression of Rsm22, an Assembly Factor for the Mitochondrial Ribosome, on Frataxin/Yfh1 Depletion Phenotypes in Saccharomyces cerevisiae. Biomolecules 2025, 15, 785. https://doi.org/10.3390/biom15060785
Pandey AK, Singh P, Pain J, Dancis A, Pain D. Salutary Effects of Overexpression of Rsm22, an Assembly Factor for the Mitochondrial Ribosome, on Frataxin/Yfh1 Depletion Phenotypes in Saccharomyces cerevisiae. Biomolecules. 2025; 15(6):785. https://doi.org/10.3390/biom15060785
Chicago/Turabian StylePandey, Ashutosh K., Pratibha Singh, Jayashree Pain, Andrew Dancis, and Debkumar Pain. 2025. "Salutary Effects of Overexpression of Rsm22, an Assembly Factor for the Mitochondrial Ribosome, on Frataxin/Yfh1 Depletion Phenotypes in Saccharomyces cerevisiae" Biomolecules 15, no. 6: 785. https://doi.org/10.3390/biom15060785
APA StylePandey, A. K., Singh, P., Pain, J., Dancis, A., & Pain, D. (2025). Salutary Effects of Overexpression of Rsm22, an Assembly Factor for the Mitochondrial Ribosome, on Frataxin/Yfh1 Depletion Phenotypes in Saccharomyces cerevisiae. Biomolecules, 15(6), 785. https://doi.org/10.3390/biom15060785