
Unraveling the Multi-Omic Landscape of Extracellular Vesicles in Human Seminal Plasma

 , , , ,
, , , ,  , , ,
, , ,  ,
,  and
and
Abstract

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Human Biological Sample Collection and Analysis
2.2. EV Isolation from Seminal Plasma
2.3. EV Characterization
2.3.1. Nanoparticle Tracking Analysis
2.3.2. Cholesterol Quantification Assay
2.3.3. Enzyme-Linked Immunosorbent Assay
2.3.4. Transmission Electron Microscopy Analysis
2.4. Proteomic Analysis—Gel-Free Analysis
2.4.1. EVs Preparation for Liquid Chromatography–Tandem Mass Spectrometry
2.4.2. LC-MS/MS Analysis
2.5. Proteomic Analysis—Gel-Based Analysis
2.5.1. EV Preparation for Gel-Based Proteomic Analysis
2.5.2. 2D Electrophoresis
2.5.3. MALDI-TOF/TOF MS—Protein Identification
2.5.4. Heatmap and PCA of Proteomic Gel-Based Results
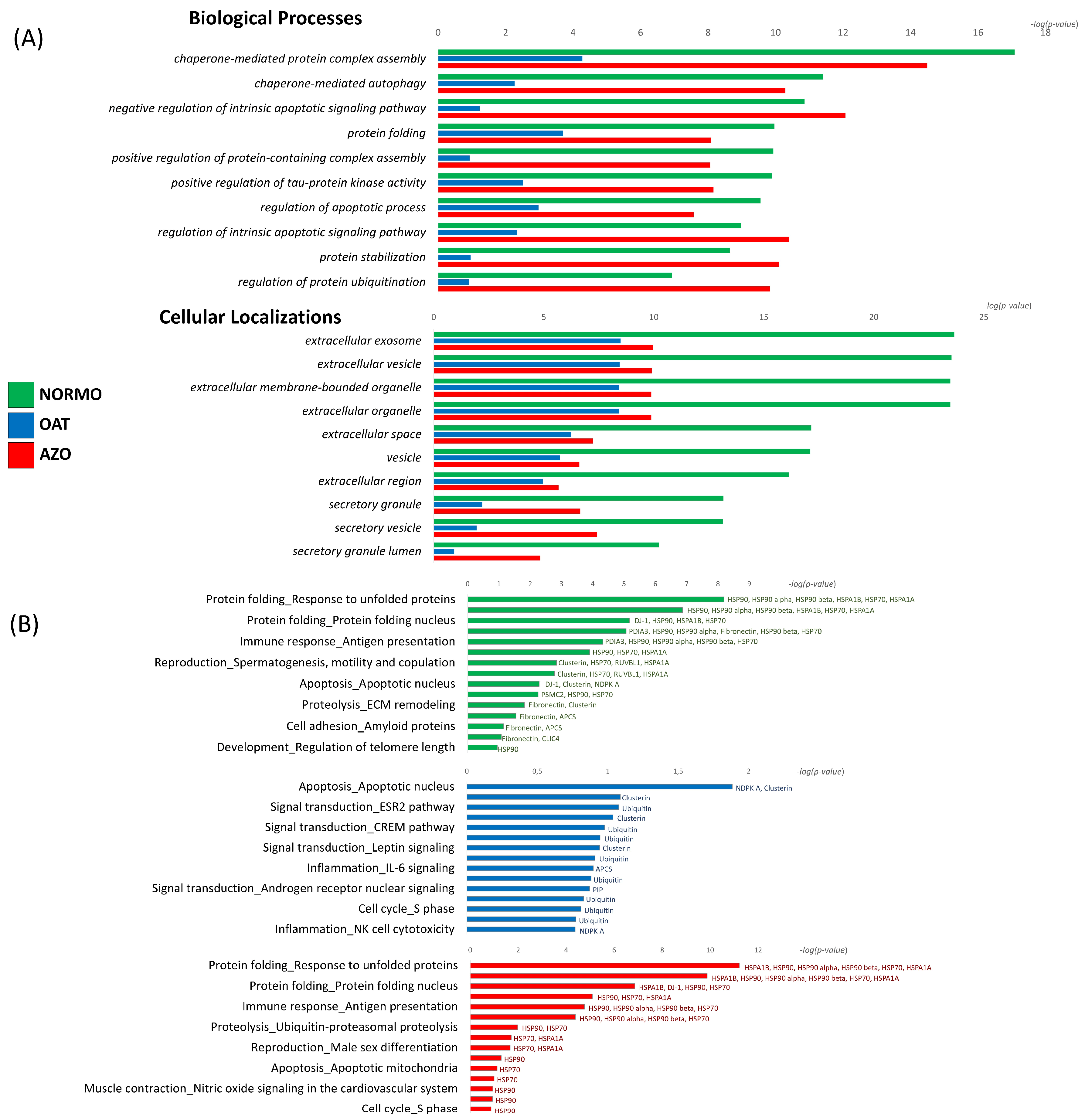
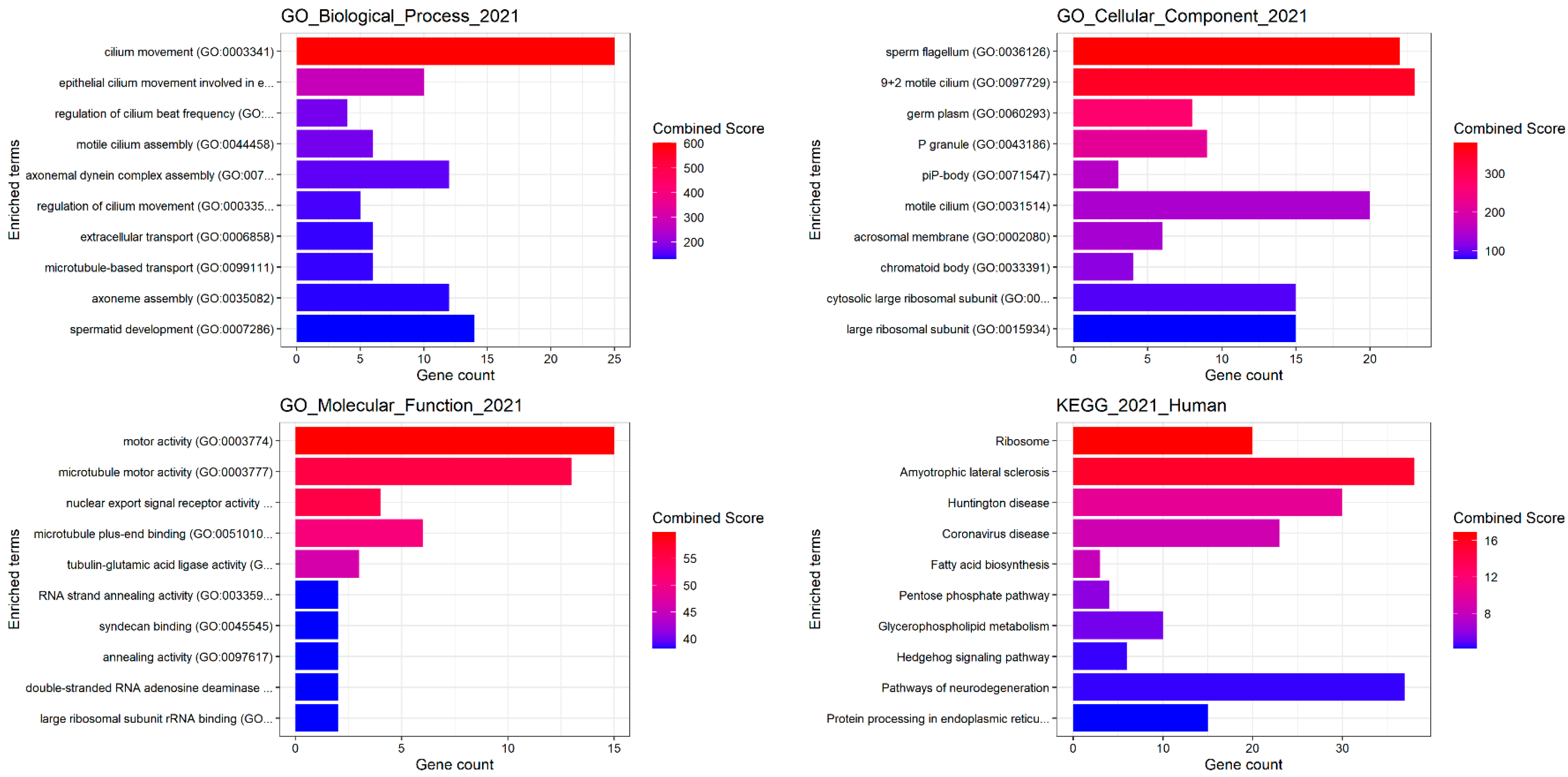
2.5.5. Enrichment Analysis of Gel-Based and Gel-Free Proteomic Results by MetaCore
2.6. Transcriptome Profiling
2.6.1. Total RNA Isolation and Quality Assessment
2.6.2. Library Preparation and RNA-Seq
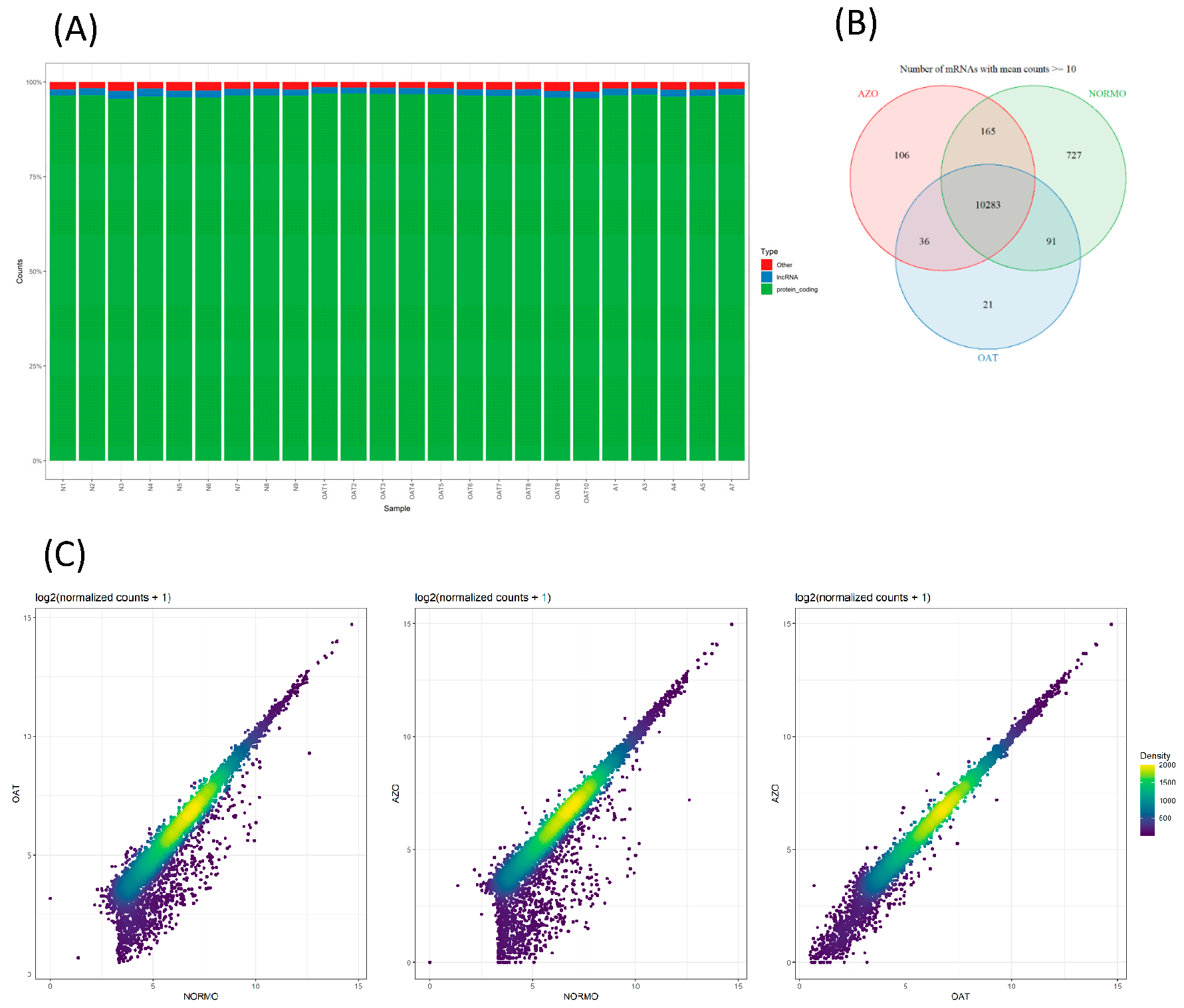
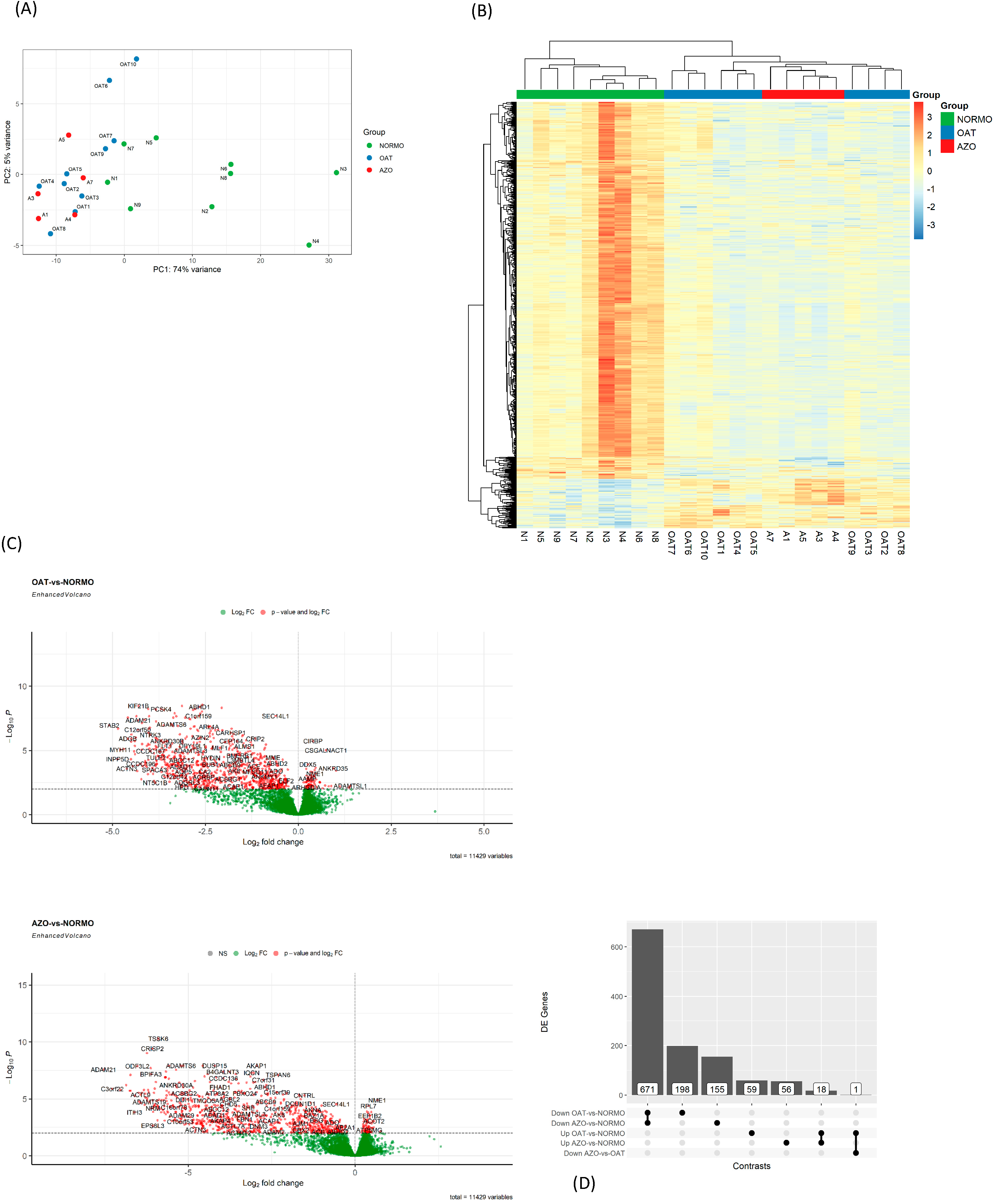
2.6.3. RNA-Seq Data Alignment and Identification of DEGs
2.7. Gene Expression Analysis by Droplets Digital PCR (ddPCR)
3. Results
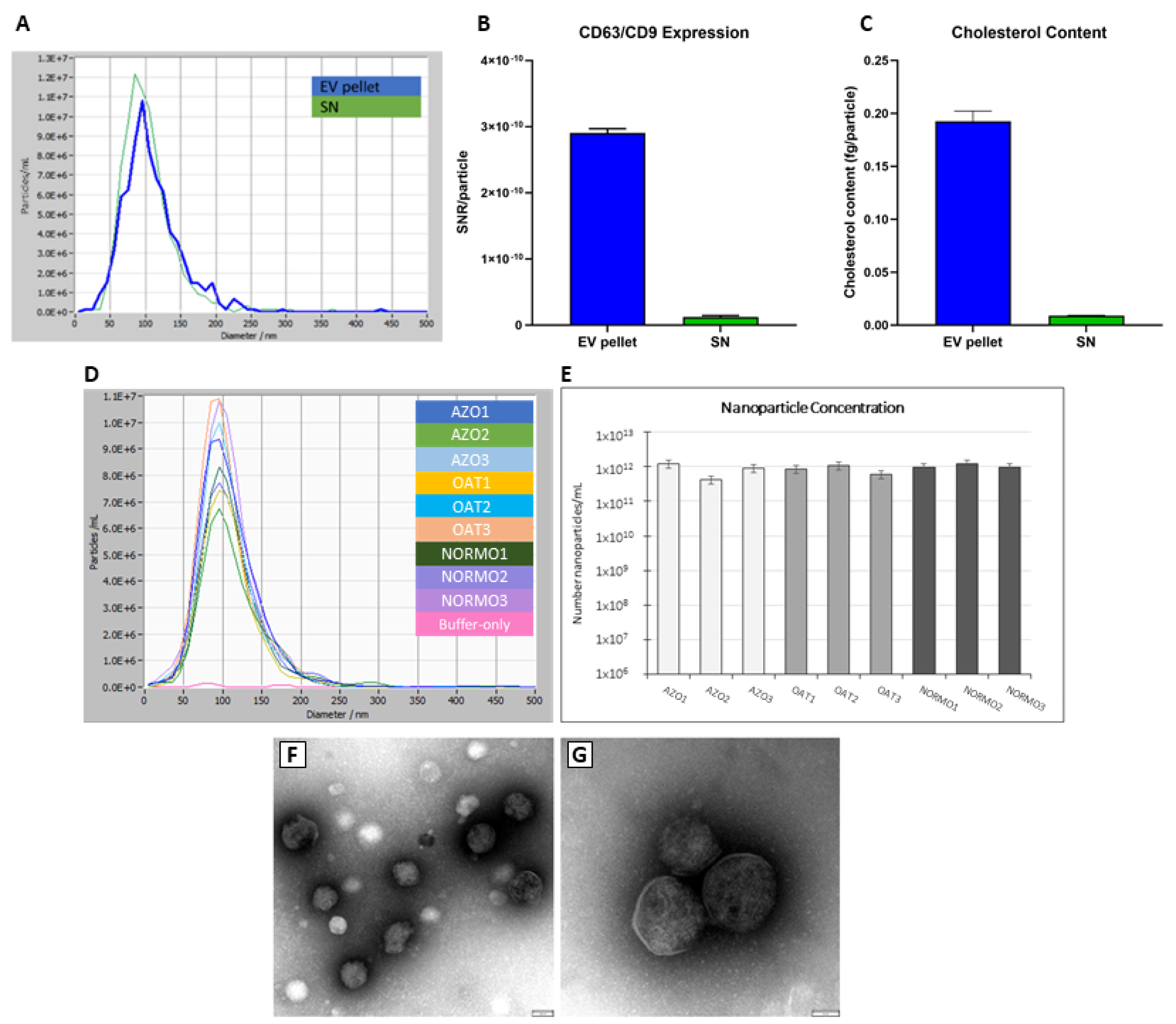
3.1. Assessment of EV Recovery and Enrichment from Seminal Plasma
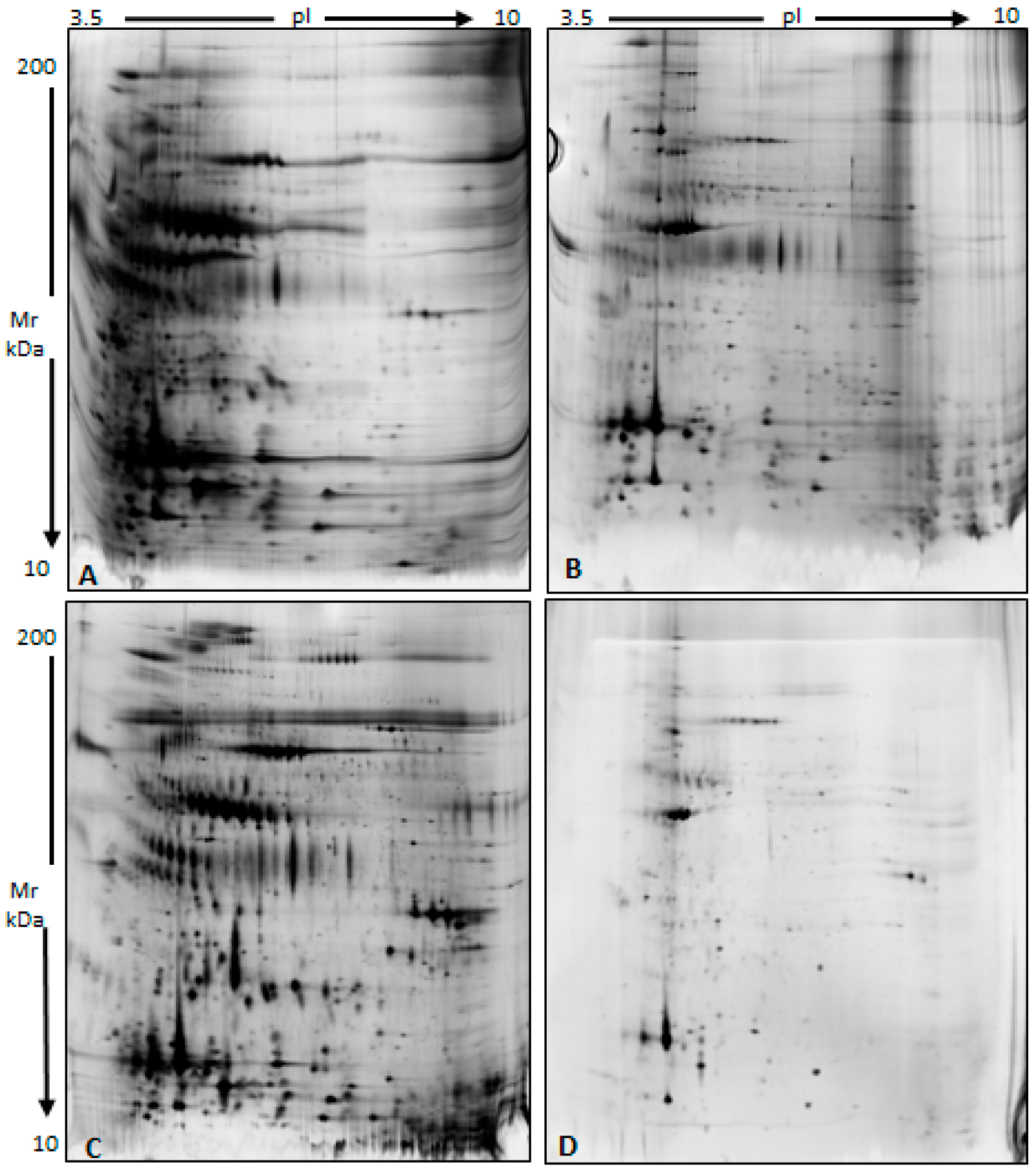
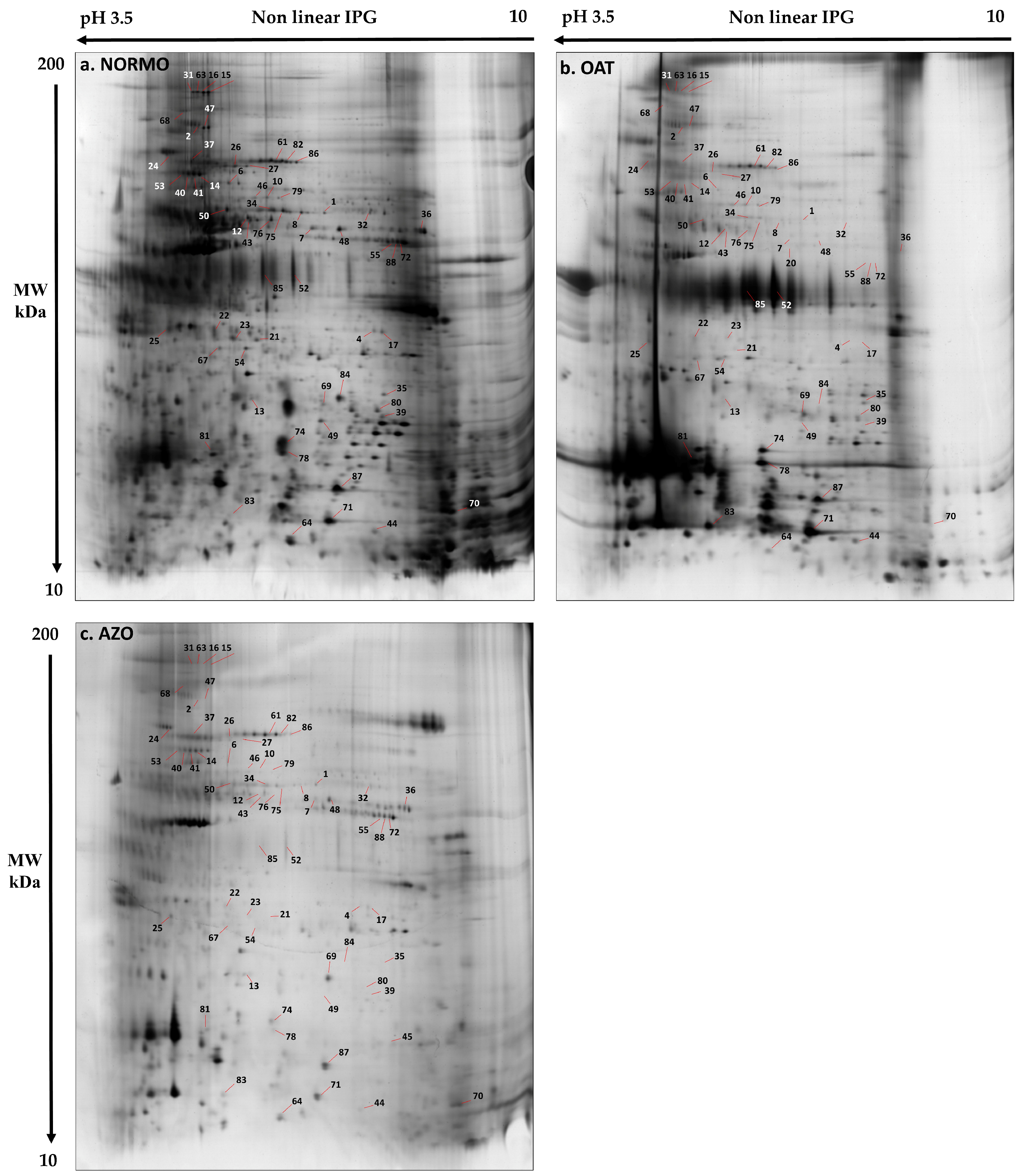
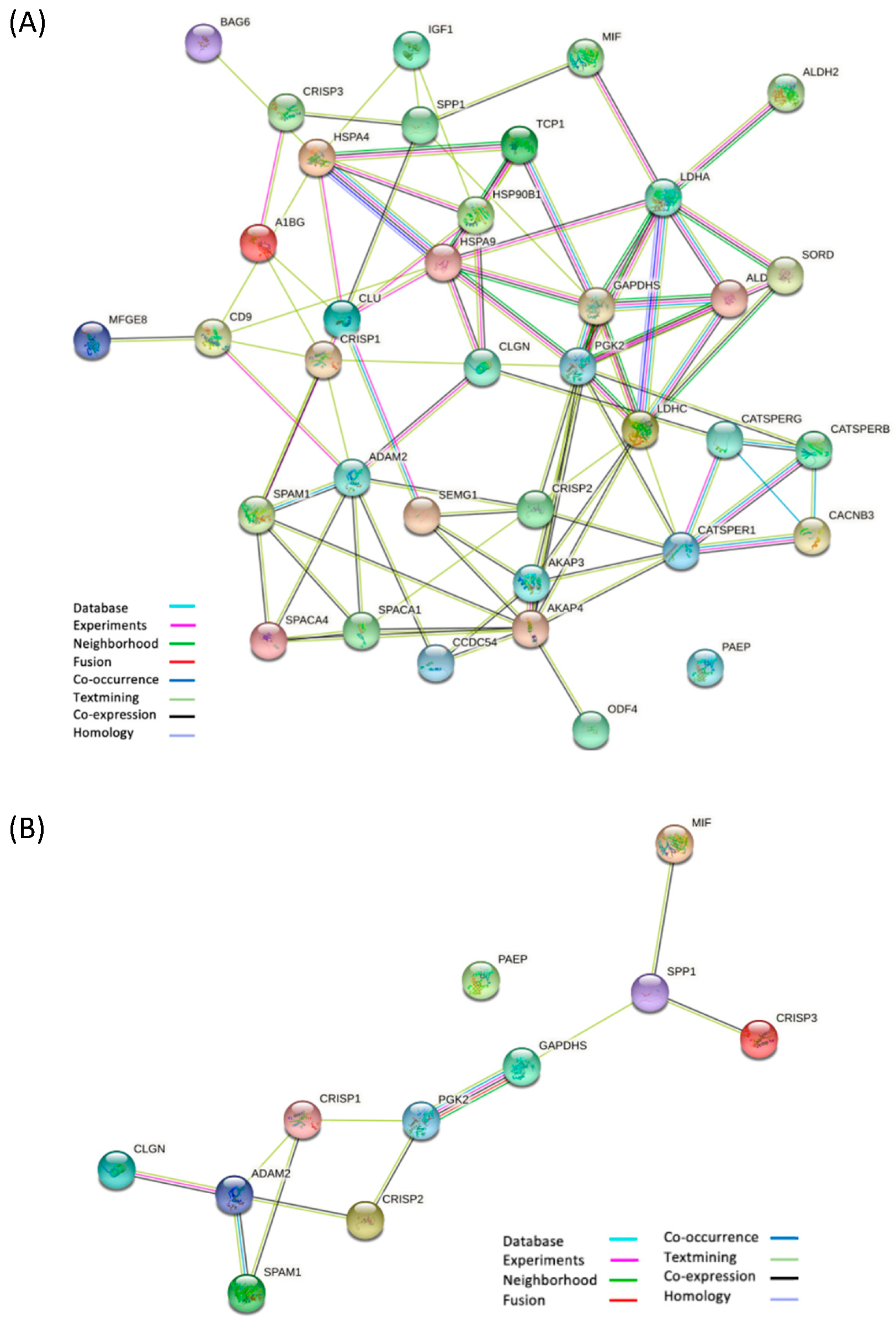
3.2. Characterization of a Normal Protein Cargo in Sperm EVs
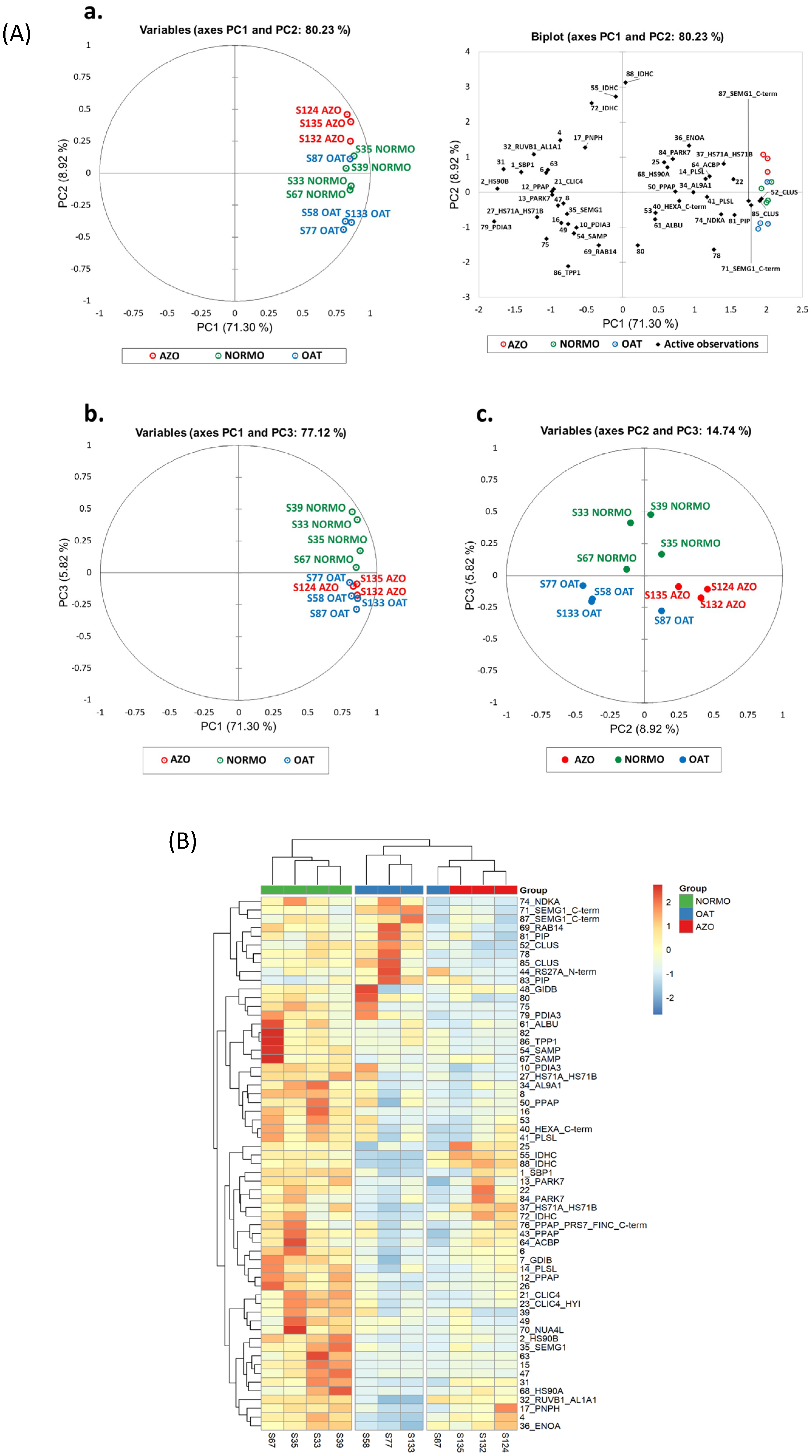
3.3. Differential Proteomic Analysis of EVs from Subjects with Different Sperm Parameters
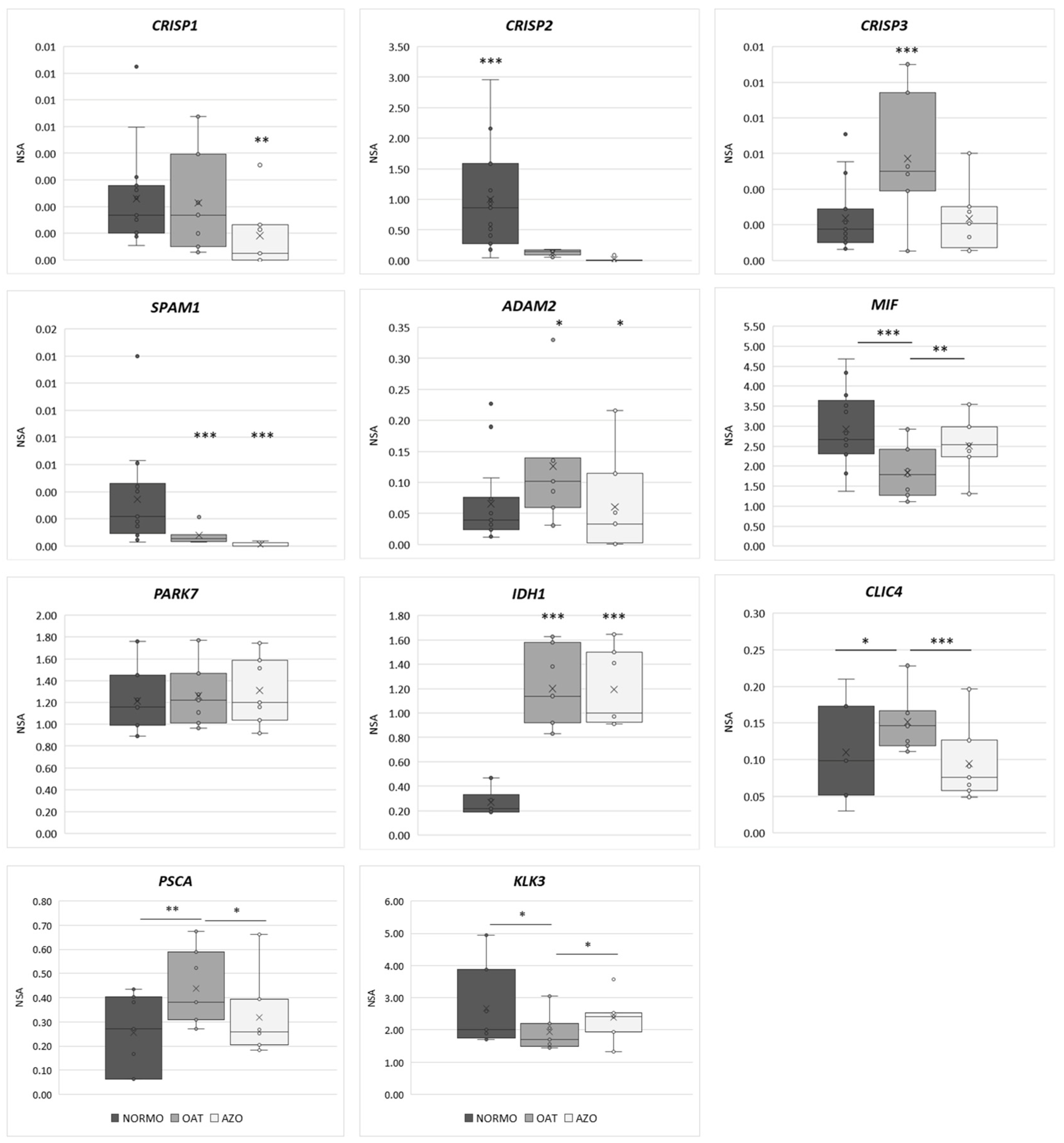
3.4. Differential Gene Expression Analysis of EVs from Subjects with Different Sperm Parameters
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| EVs | Extracellular Vesicles |
| NORMO | Normozoospermia |
| OAT | OligoAsthenoTeratozoospermia |
| AZO | Azoospermia |
| NTA | Nanoparticle Tracking Analysis |
| PCR | Polymerase Chain Reaction |
| GAPDH | Glyceraldehyde-3-Phosphate Dehydrogenase |
| RNY4 | RNA, Ro60-Associated Y4 |
| HSPA70 | Heat shock protein family A (Hsp70) member 4 |
| CRISP1 | Cysteine Rich Secretory Protein 1 |
| CRISP2 | Cysteine Rich Secretory Protein 2 |
| CRISP3 | Cysteine Rich Secretory Protein 3 |
| SPP1 | Secreted Phosphoprotein 1 |
| MIF | Macrophage Migration Inhibitory Factor |
| SPAM1 | Sperm Adhesion Molecule 1 |
| ADAM2 | ADAM Metallopeptidase Domain 2 |
| PGK2 | Phosphoglycerate Kinase 2 |
| CLGN | Calmegin |
| PAEP | Progestagen-Associated Endometrial Protein |
| CLIC4 | Chloride intracellular channel 4 |
| KLK3 | Kallikrein-related peptidase 3 |
| IDH1 | Ocitrate dehydrogenase (NADP(+)) 1, cytosolic |
| PSCA | Prostate stem cell antigen |
| PARK7 | Parkinsonism-associated deglycase |
References
- Candenas, L.; Chianese, R. Exosome Composition and Seminal Plasma Proteome: A Promising Source of Biomarkers of Male Infertility. Int. J. Mol. Sci. 2020, 21, 7022. [Google Scholar] [CrossRef]
- Moura, A.A.; Memili, E.; Portela, A.M.R.; Viana, A.G.; Velho, A.L.C.; Bezerra, M.J.B.; Vasconselos, F.R. Seminal plasma proteins and metabolites: Effects on sperm function and potential as fertility markers. Anim. Reprod. 2018, 15, 691–702. [Google Scholar] [CrossRef]
- Szczykutowicz, J.; Kałuża, A.; Kaźmierowska-Niemczuk, M.; Ferens-Sieczkowska, M. The Potential Role of Seminal Plasma in the Fertilization Outcomes. BioMed Res. Int. 2019, 2019, 5397804. [Google Scholar] [CrossRef]
- Macanovic, B.; Vucetic, M.; Jankovic, A.; Stancic, A.; Buzadzic, B.; Garalejic, E.; Korac, A.; Korac, B.; Otasevic, V. Correlation between Sperm Parameters and Protein Expression of Antioxidative Defense Enzymes in Seminal Plasma: A Pilot Study. Dis. Markers 2015, 2015, 436236. [Google Scholar] [CrossRef]
- Ayaz, A.; Houle, E.; Pilsner, J.R. Extracellular vesicle cargo of the male reproductive tract and the paternal preconception environment. Syst. Biol. Reprod. Med. 2021, 67, 103–111. [Google Scholar] [CrossRef]
- Tschuschke, M.; Kocherova, I.; Bryja, A.; Mozdziak, P.; Angelova Volponi, A.; Janowicz, K.; Sibiak, R.; Piotrowska-Kempisty, H.; Iżycki, D.; Bukowska, D.; et al. Inclusion Biogenesis, Methods of Isolation and Clinical Application of Human Cellular Exosomes. J. Clin. Med. 2020, 9, 436. [Google Scholar] [CrossRef]
- Jeppesen, D.K.; Zhang, Q.; Franklin, J.L.; Coffey, R.J. Extracellular vesicles and nanoparticles: Emerging complexities. Trends Cell Biol. 2023, 33, 667–681. [Google Scholar] [CrossRef]
- Mashouri, L.; Yousefi, H.; Aref, A.R.; Ahadi, A.M.; Molaei, F.; Alahari, S.K. Exosomes: Composition, biogenesis, and mechanisms in cancer metastasis and drug resistance. Mol. Cancer 2019, 18, 75. [Google Scholar] [CrossRef]
- Tamessar, C.T.; Trigg, N.A.; Nixon, B.; Skerrett-Byrne, D.A.; Sharkey, D.J.; Robertson, S.A.; Bromfield, E.G.; Schjenken, J.E. Roles of male reproductive tract extracellular vesicles in reproduction. Am. J. Reprod. Immunol. 2021, 85, e13338. [Google Scholar] [CrossRef]
- Dai, J.; Su, Y.; Zhong, S.; Cong, L.; Liu, B.; Yang, J.; Tao, Y.; He, Z.; Chen, C.; Jiang, Y. Exosomes: Key players in cancer and potential therapeutic strategy. Signal Transduct. Target. Ther. 2020, 5, 145. [Google Scholar] [CrossRef]
- Baskaran, S.; Panner Selvam, M.K.; Agarwal, A. Exosomes of male reproduction. In Advances in Clinical Chemistry; Elsevier: Amsterdam, The Netherlands, 2020; Volume 95, pp. 149–163. ISBN 978-0-12-821165-6. [Google Scholar]
- Vickram, A.S.; Srikumar, P.S.; Srinivasan, S.; Jeyanthi, P.; Anbarasu, K.; Thanigaivel, S.; Nibedita, D.; Jenila Rani, D.; Rohini, K. Seminal exosomes—An important biological marker for various disorders and syndrome in human reproduction. Saudi J. Biol. Sci. 2021, 28, 3607–3615. [Google Scholar] [CrossRef]
- Sullivan, R. Epididymosomes: Role of extracellular microvesicles in sperm maturation. Front Biosci (Sch. Ed) 2016, 8, 106–114. [Google Scholar] [CrossRef]
- Kalluri, R.; LeBleu, V.S. The biology, function, and biomedical applications of exosomes. Science 2020, 367, eaau6977. [Google Scholar] [CrossRef]
- Machtinger, R.; Laurent, L.C.; Baccarelli, A.A. Extracellular vesicles: Roles in gamete maturation, fertilization and embryo implantation. Hum. Reprod. Update 2016, 22, 182–193. [Google Scholar] [CrossRef]
- Méar, L.O.; Tsai, P.-S.; Tamessar, C.T.; Schjenken, J.E.; Nixon, B. Epididymosomes: Composition and Functions for Sperm Maturation. In Advances in Anatomy, Embryology and Cell Biology; Springer: Berlin/Heidelberg, Germany, 2024. [Google Scholar] [CrossRef]
- Saez, F.; Sullivan, R. Prostasomes, post-testicular sperm maturation and fertility. Front Biosci (Landmark Ed) 2016, 21, 1464–1473. [Google Scholar] [CrossRef]
- WHO Laboratory Manual for the Examination and Processing of Human Semen. Available online: https://www.who.int/publications-detail-redirect/9789240030787 (accessed on 2 August 2023).
- Barceló, M.; Mata, A.; Bassas, L.; Larriba, S. Exosomal microRNAs in seminal plasma are markers of the origin of azoospermia and can predict the presence of sperm in testicular tissue. Hum. Reprod. 2018, 33, 1087–1098. [Google Scholar] [CrossRef]
- Mercadal, M.; Herrero, C.; López-Rodrigo, O.; Castells, M.; de la Fuente, A.; Vigués, F.; Bassas, L.; Larriba, S. Impact of Extracellular Vesicle Isolation Methods on Downstream miRNA Analysis in Semen: A Comparative Study. Int. J. Mol. Sci. 2020, 21, 5949. [Google Scholar] [CrossRef]
- Fortunato, D.; Mladenović, D.; Criscuoli, M.; Loria, F.; Veiman, K.-L.; Zocco, D.; Koort, K.; Zarovni, N. Opportunities and Pitfalls of Fluorescent Labeling Methodologies for Extracellular Vesicle Profiling on High-Resolution Single-Particle Platforms. Int. J. Mol. Sci. 2021, 22, 10510. [Google Scholar] [CrossRef]
- Luddi, A.; Zarovni, N.; Maltinti, E.; Governini, L.; De Leo, V.; Cappelli, V.; Quintero, L.; Paccagnini, E.; Loria, F.; Piomboni, P. Clues to Non-Invasive Implantation Window Monitoring: Isolation and Characterisation of Endometrial Exosomes. Cells 2019, 8, 811. [Google Scholar] [CrossRef]
- Sinha, P.; Poland, J.; Schnölzer, M.; Rabilloud, T. A new silver staining apparatus and procedure for matrix-assisted laser desorption/ionization-time of flight analysis of proteins after two-dimensional electrophoresis. Proteomics 2001, 1, 835–840. [Google Scholar] [CrossRef]
- Deutsch, E.W.; Bandeira, N.; Perez-Riverol, Y.; Sharma, V.; Carver, J.J.; Mendoza, L.; Kundu, D.J.; Wang, S.; Bandla, C.; Kamatchinathan, S.; et al. The ProteomeXchange consortium at 10 years: 2023 update. Nucleic Acids Res. 2023, 51, D1539–D1548. [Google Scholar] [CrossRef] [PubMed]
- Perez-Riverol, Y.; Bai, J.; Bandla, C.; García-Seisdedos, D.; Hewapathirana, S.; Kamatchinathan, S.; Kundu, D.J.; Prakash, A.; Frericks-Zipper, A.; Eisenacher, M.; et al. The PRIDE database resources in 2022: A hub for mass spectrometry-based proteomics evidences. Nucleic Acids Res. 2022, 50, D543–D552. [Google Scholar] [CrossRef]
- Ponchia, R.; Bruno, A.; Renzi, A.; Landi, C.; Shaba, E.; Luongo, F.P.; Haxhiu, A.; Artini, P.G.; Luddi, A.; Governini, L.; et al. Oxidative Stress Measurement in Frozen/Thawed Human Sperm: The Protective Role of an In Vitro Treatment with Myo-Inositol. Antioxidants 2021, 11, 10. [Google Scholar] [CrossRef]
- Wingett, S.W.; Andrews, S. FastQ Screen: A tool for multi-genome mapping and quality control. F1000Research 2018, 7, 1338. [Google Scholar] [CrossRef]
- Ewels, P.; Magnusson, M.; Lundin, S.; Käller, M. MultiQC: Summarize analysis results for multiple tools and samples in a single report. Bioinformatics 2016, 32, 3047–3048. [Google Scholar] [CrossRef]
- Chen, S.; Zhou, Y.; Chen, Y.; Gu, J. fastp: An ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 2018, 34, i884–i890. [Google Scholar] [CrossRef]
- Frankish, A.; Diekhans, M.; Ferreira, A.-M.; Johnson, R.; Jungreis, I.; Loveland, J.; Mudge, J.M.; Sisu, C.; Wright, J.; Armstrong, J.; et al. GENCODE reference annotation for the human and mouse genomes. Nucleic Acids Res. 2019, 47, D766–D773. [Google Scholar] [CrossRef]
- Dobin, A.; Davis, C.A.; Schlesinger, F.; Drenkow, J.; Zaleski, C.; Jha, S.; Batut, P.; Chaisson, M.; Gingeras, T.R. STAR: Ultrafast universal RNA-seq aligner. Bioinformatics 2013, 29, 15–21. [Google Scholar] [CrossRef]
- Danecek, P.; Bonfield, J.K.; Liddle, J.; Marshall, J.; Ohan, V.; Pollard, M.O.; Whitwham, A.; Keane, T.; McCarthy, S.A.; Davies, R.M.; et al. Twelve years of SAMtools and BCFtools. Gigascience 2021, 10, giab008. [Google Scholar] [CrossRef]
- Smith, T.; Heger, A.; Sudbery, I. UMI-tools: Modeling sequencing errors in Unique Molecular Identifiers to improve quantification accuracy. Genome Res. 2017, 27, 491–499. [Google Scholar] [CrossRef]
- Okonechnikov, K.; Conesa, A.; García-Alcalde, F. Qualimap 2: Advanced multi-sample quality control for high-throughput sequencing data. Bioinformatics 2016, 32, 292–294. [Google Scholar] [CrossRef] [PubMed]
- Liao, Y.; Smyth, G.K.; Shi, W. featureCounts: An efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 2014, 30, 923–930. [Google Scholar] [CrossRef] [PubMed]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef]
- Wickham, H.; Averick, M.; Bryan, J.; Chang, W.; McGowan, L.D.; François, R.; Grolemund, G.; Hayes, A.; Henry, L.; Hester, J.; et al. Welcome to the Tidyverse. J. Open Source Softw. 2019, 4, 1686. [Google Scholar] [CrossRef]
- Allaire, J.J. RStudio: Integrated Development Environment for R. UseR! 2011 Conference. 2011. Available online: https://www.r-project.org/conferences/useR-2011/abstracts/180111-allairejj.pdf (accessed on 9 April 2024).
- Chen VennDiagram: Generate High-Resolution Venn and Euler Plots. Software: VennDiagram (Generate High-Resolution Venn and Euler Plots) (R-Packages). Available online: https://cran.r-project.org/web/packages/VennDiagram/index.html (accessed on 9 April 2024).
- Schloerke, B.; Cook, D.; Larmarange, J.; Briatte, F.; Marbach, M.; Thoen, E.; Elberg, A.; Toomet, O.; Crowley, J.; Hofmann, H.; et al. GGally: Extension to “ggplot2”. Software: GGally (Extension to ‘Ggplot2’) (R-Packages). 2024. Available online: https://cran.r-project.org/web/packages/GGally/index.html (accessed on 9 April 2024).
- Kremer, L.P.M.; Anders, S. Ggpointdensity: A Cross Between a 2D Density Plot and a Scatter Plot. Software: Ggpointdensity (A Cross Between a 2D Density Plot and a Scatter Plot) (R-Packages). Available online: https://cran.r-project.org/web/packages/ggpointdensity/index.html (accessed on 9 April 2024).
- Slowikowski, K.; Schep, A.; Hughes, S.; Dang, T.K.; Lukauskas, S.; Irisson, J.-O.; Kamvar, Z.N.; Ryan, T.; Christophe, D.; Hiroaki, Y.; et al. Ggrepel: Automatically Position Non-Overlapping Text Labels with “Ggplot2”. Software: Ggrepel (Automatically Position Non-Overlapping Text Labels with ‘Ggplot2’) (R-Packages). Available online: https://cran.r-project.org/web/packages/ggrepel/index.html (accessed on 9 April 2024).
- Kolde, R. Pheatmap: Pretty Heatmaps. Software: Pheatmap (Pretty Heatmaps) (R-Rackages). Available online: https://cran.r-project.org/web/packages/pheatmap/index.html (accessed on 9 April 2024).
- Blighe, K.; Rana, S.; Lewis, M. EnhancedVolcano: Publication-Ready Volcano Plots with Enhanced Colouring and Labeling. Available online: https://bioconductor.org/packages/devel/bioc/vignettes/EnhancedVolcano/inst/doc/EnhancedVolcano.html (accessed on 9 April 2024).
- Ahlmann-Eltze, C. Ggupset: Combination Matrix Axis for “Ggplot2” to Create “UpSet” Plots 2020. Software: Ggupset (Combination Matrix Axis for ‘Ggplot2’ to Create ‘UpSet’ Plots) (R-Packages). Available online: https://cran.r-project.org/web/packages/ggupset/index.html (accessed on 9 April 2024).
- Jawaid, W. EnrichR: Provides an R Interface to “Enrichr”. Software: EnrichR (Provides an R Interface to ‘Enrichr’) (R-Packages). 2023. Available online: https://cran.r-project.org/web/packages/enrichR/index.html (accessed on 9 April 2024).
- Gene Ontology Consortium; Aleksander, S.A.; Balhoff, J.; Carbon, S.; Cherry, J.M.; Drabkin, H.J.; Ebert, D.; Feuermann, M.; Gaudet, P.; Harris, N.L.; et al. The Gene Ontology knowledgebase in 2023. Genetics 2023, 224, iyad031. [Google Scholar] [CrossRef]
- Kanehisa, M.; Furumichi, M.; Sato, Y.; Kawashima, M.; Ishiguro-Watanabe, M. KEGG for taxonomy-based analysis of pathways and genomes. Nucleic Acids Res. 2023, 51, D587–D592. [Google Scholar] [CrossRef]
- Shaba, E.; Landi, C.; Carleo, A.; Vantaggiato, L.; Paccagnini, E.; Gentile, M.; Bianchi, L.; Lupetti, P.; Bargagli, E.; Prasse, A.; et al. Proteome Characterization of BALF Extracellular Vesicles in Idiopathic Pulmonary Fibrosis: Unveiling Undercover Molecular Pathways. Int. J. Mol. Sci. 2021, 22, 5696. [Google Scholar] [CrossRef] [PubMed]
- Combe, M.; Isaac, K.S.; Plews, J.R.; Sokolenko, S. Quantifying extracellular vesicle heterogeneity: The effect of process conditions on protein cargo for skin therapy. Stem Cell Res. Ther. 2025, 16, 224. [Google Scholar] [CrossRef]
- Cannarella, R.; Barbagallo, F.; Crafa, A.; La Vignera, S.; Condorelli, R.A.; Calogero, A.E. Seminal Plasma Transcriptome and Proteome: Towards a Molecular Approach in the Diagnosis of Idiopathic Male Infertility. Int. J. Mol. Sci. 2020, 21, 7308. [Google Scholar] [CrossRef]
- Koppers, A.J.; Reddy, T.; O’Bryan, M.K. The role of cysteine-rich secretory proteins in male fertility. Asian J. Androl. 2011, 13, 111–117. [Google Scholar] [CrossRef]
- Da Ros, V.G.; Muñoz, M.W.; Battistone, M.A.; Brukman, N.G.; Carvajal, G.; Curci, L.; Gómez-ElIas, M.D.; Cohen, D.B.J.; Cuasnicu, P.S. From the epididymis to the egg: Participation of CRISP proteins in mammalian fertilization. Asian J. Androl. 2015, 17, 711–715. [Google Scholar] [CrossRef] [PubMed]
- Tamessar, C.T.; Anderson, A.L.; Bromfield, E.G.; Trigg, N.A.; Parameswaran, S.; Stanger, S.J.; Weidenhofer, J.; Zhang, H.-M.; Robertson, S.A.; Sharkey, D.J.; et al. The efficacy and functional consequences of interactions between human spermatozoa and seminal fluid extracellular vesicles. Reprod. Fertil. 2024, 5, e230088. [Google Scholar] [CrossRef] [PubMed]
- Mateo-Otero, Y.; Yeste, M.; Roca, J.; Llavanera, M.; Bucci, D.; Galeati, G.; Spinaci, M.; Barranco, I. Seminal extracellular vesicles subsets modulate gene expression in cumulus cells of porcine in vitro matured oocytes. Sci. Rep. 2022, 12, 19096. [Google Scholar] [CrossRef] [PubMed]
- An, C.-N.; Jiang, H.; Wang, Q.; Yuan, R.-P.; Liu, J.-M.; Shi, W.-L.; Zhang, Z.-Y.; Pu, X.-P. Down-regulation of DJ-1 protein in the ejaculated spermatozoa from Chinese asthenozoospermia patients. Fertil. Steril. 2011, 96, 19–23.e2. [Google Scholar] [CrossRef]
- Recuero, S.; Delgado-Bermúdez, A.; Mateo-Otero, Y.; Garcia-Bonavila, E.; Llavanera, M.; Yeste, M. Parkinson Disease Protein 7 (PARK7) Is Related to the Ability of Mammalian Sperm to Undergo In Vitro Capacitation. Int. J. Mol. Sci. 2021, 22, 10804. [Google Scholar] [CrossRef]
- Myers, K.; Somanath, P.R.; Berryman, M.; Vijayaraghavan, S. Identification of chloride intracellular channel proteins in spermatozoa. FEBS Lett. 2004, 566, 136–140. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Governini, L.; Haxhiu, A.; Shaba, E.; Vantaggiato, L.; Mori, A.; Bruttini, M.; Loria, F.; Zarovni, N.; Piomboni, P.; Landi, C.; et al. Unraveling the Multi-Omic Landscape of Extracellular Vesicles in Human Seminal Plasma. Biomolecules 2025, 15, 836. https://doi.org/10.3390/biom15060836
Governini L, Haxhiu A, Shaba E, Vantaggiato L, Mori A, Bruttini M, Loria F, Zarovni N, Piomboni P, Landi C, et al. Unraveling the Multi-Omic Landscape of Extracellular Vesicles in Human Seminal Plasma. Biomolecules. 2025; 15(6):836. https://doi.org/10.3390/biom15060836
Chicago/Turabian StyleGovernini, Laura, Alesandro Haxhiu, Enxhi Shaba, Lorenza Vantaggiato, Alessia Mori, Marco Bruttini, Francesca Loria, Natasa Zarovni, Paola Piomboni, Claudia Landi, and et al. 2025. "Unraveling the Multi-Omic Landscape of Extracellular Vesicles in Human Seminal Plasma" Biomolecules 15, no. 6: 836. https://doi.org/10.3390/biom15060836
APA StyleGovernini, L., Haxhiu, A., Shaba, E., Vantaggiato, L., Mori, A., Bruttini, M., Loria, F., Zarovni, N., Piomboni, P., Landi, C., & Luddi, A. (2025). Unraveling the Multi-Omic Landscape of Extracellular Vesicles in Human Seminal Plasma. Biomolecules, 15(6), 836. https://doi.org/10.3390/biom15060836