
Mechanisms of Lung Cancer Development in Cystic Fibrosis Patients: The Role of Inflammation, Oxidative Stress, and Lung Microbiome Dysbiosis

 , , ,
, , ,  and
and
Abstract
1. Introduction
1.1. Overview of Cystic Fibrosis
1.2. Lung Cancer in CF Patients
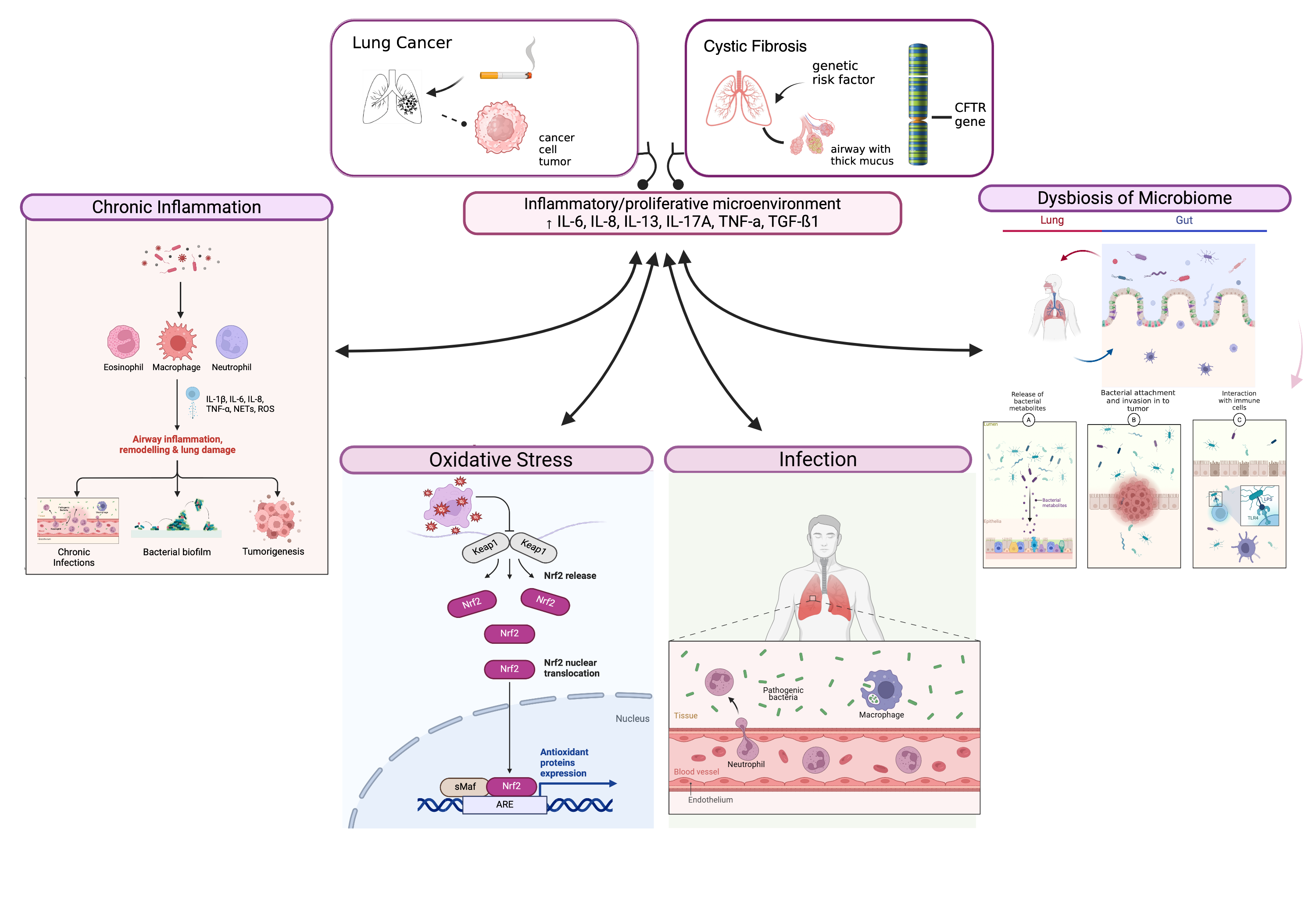
2. Chronic Inflammation in Cystic Fibrosis
2.1. Role of Inflammation in CF Pathogenesis
2.2. Inflammatory Mediators and Their Contribution to Cancer Risk in CF Patients
3. Chronic Lung Infections in Cystic Fibrosis
Infections as Drivers of Inflammation and Tumorigenesis
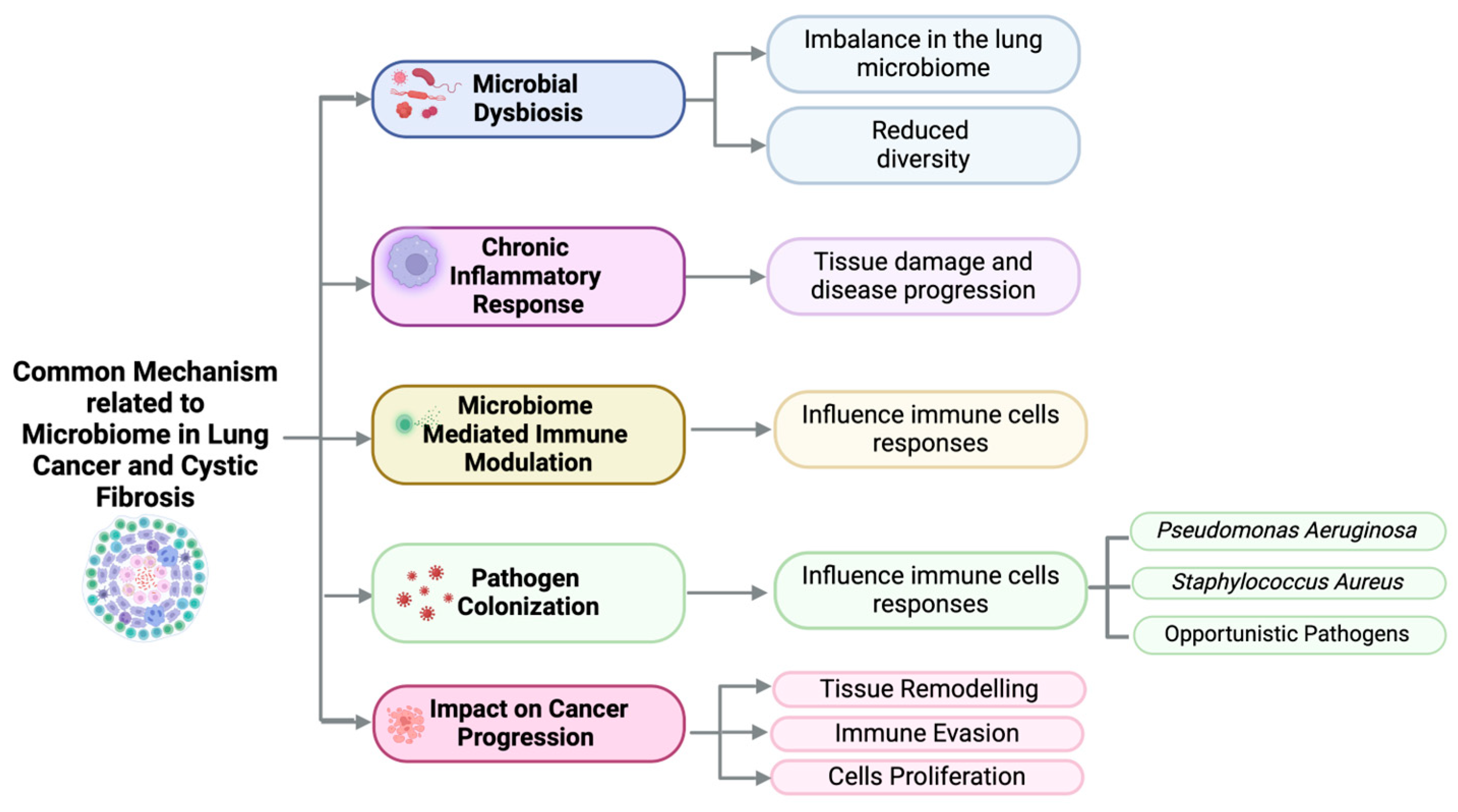
4. Dysbiosis of the Lung Microbiome in Cystic Fibrosis
4.1. Microbiome Composition in Cystic Fibrosis
4.2. Impact of Dysbiosis on Inflammation and Cancer Development
5. The Role of Oxidative Stress in Cystic Fibrosis
6. Interactions Between Inflammation, Oxidative Stress, and Microbiome in Lung Cancer Development
7. Clinical Implications
8. Conclusions and Future Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Shteinberg, M.; Haq, I.J.; Polineni, D.; Davies, J.C. Cystic fibrosis. Lancet 2021, 397, 2195–2211. [Google Scholar] [CrossRef] [PubMed]
- Elborn, J.S. Cystic fibrosis. Lancet 2016, 388, 2519–2531. [Google Scholar] [CrossRef] [PubMed]
- Farinha, C.M.; Callebaut, I. Molecular Mechanisms of Cystic Fibrosis—How Mutations Lead to Misfunction and Guide Therapy; Portland Press Ltd.: London, UK, 2022. [Google Scholar] [CrossRef]
- Almughem, F.A.; Aldossary, A.M.; Tawfik, E.A.; Alomary, M.N.; Alharbi, W.S.; Alshahrani, M.Y.; Alshehri, A.A. Cystic Fibrosis: Overview of the Current Development Trends and Innovative Therapeutic Strategies. Pharmaceutics 2020, 12, 616. [Google Scholar] [CrossRef] [PubMed]
- Boeck, K.D. Cystic fibrosis in the year 2020: A disease with a new face. Acta Paediatr. 2020, 109, 893–899. [Google Scholar] [CrossRef] [PubMed]
- Carnovale, V.; Scialò, F.; Gelzo, M.; Iacotucci, P.; Amato, F.; Zarrilli, F.; Celardo, A.; Castaldo, G.; Corso, G. Cystic Fibrosis Patients with F508del/Minimal Function Genotype: Laboratory and Nutritional Evaluations after One Year of Elexacaftor/Tezacaftor/Ivacaftor Treatment. J. Clin. Med. 2022, 11, 6900. [Google Scholar] [CrossRef]
- Lukacs, G.L.; Verkman, A.S. CFTR: Folding, misfolding and correcting the ΔF508 conformational defect. Trends Mol. Med. 2012, 18, 81–91. [Google Scholar] [CrossRef]
- Lopes-Pacheco, M. CFTR Modulators: The Changing Face of Cystic Fibrosis in the Era of Precision Medicine; Frontiers Media S.A.: Lausanne, Switzerland, 2020. [Google Scholar] [CrossRef]
- Stefania, L.C.; Castelli, G.; Blaconà, G.; Bruno, S.M.; Sette, G.; Pigliucci, R.; Villella, V.R.; Esposito, S.; Zollo, I.; Spadaro, F.; et al. L1077P CFTR pathogenic variant function rescue by Elexacaftor-Tezacaftor-Ivacaftor in cystic fibrosis patient-derived air-liquid interface (ALI) cultures and organoids: In vitro guided personalized therapy of non-F508del patients. Respir Res. 2023, 24, 217. [Google Scholar] [CrossRef]
- Lyczak, J.B.; Cannon, C.L.; Pier, G.B. Lung infections associated with cystic fibrosis. Clin. Microbiol. Rev. 2002, 15, 194–222. [Google Scholar] [CrossRef]
- Morrison, C.B.; Markovetz, M.R.; Ehre, C. Mucus, Mucins, and Cystic Fibrosis; John Wiley and Sons Inc.: Hoboken, NJ, USA, 2019. [Google Scholar] [CrossRef]
- Lisle, R.C.D.; Borowitz, D. The cystic fibrosis intestine. Cold Spring Harb. Perspect. Med. 2013, 3, a009753. [Google Scholar] [CrossRef]
- Parisi, G.F.; Papale, M.; Pecora, G.; Rotolo, N.; Manti, S.; Russo, G.; Leonardi, S. Cystic Fibrosis and Cancer: Unraveling the Complex Role of CFTR Gene in Cancer Susceptibility. Cancers 2023, 15, 4244. [Google Scholar] [CrossRef]
- Zhang, J.; Wang, Y.; Jiang, X.; Chan, H.C. Cystic Fibrosis Transmembrane Conductance Regulator—Emerging Regulator of Cancer; Birkhauser Verlag AG: Basel, Switzerland, 2018. [Google Scholar] [CrossRef]
- Gonzalez, H.; Hagerling, C.; Werb, Z. Roles of the immune system in cancer: From tumor initiation to metastatic progression. Genes Dev. 2018, 32, 1267–1284. [Google Scholar] [CrossRef] [PubMed]
- Purushothaman, A.K.; Nelson, E.J.R. Role of Innate Immunity and Systemic Inflammation in Cystic Fibrosis Disease Progression; Elsevier Ltd.: Amsterdam, The Netherlands, 2023. [Google Scholar] [CrossRef]
- Li, C.; Lei, S.; Ding, L.; Xu, Y.; Wu, X.; Wang, H.; Zhang, Z.; Gao, T.; Zhang, Y.; Li, L. Global burden and trends of lung cancer incidence and mortality. Chin. Med. J. Engl. 2023, 136, 1583–1590. [Google Scholar] [CrossRef] [PubMed]
- Appelt, D.; Fuchs, T.; Steinkamp, G.; Ellemunter, H. Malignancies in patients with cystic fibrosis: A case series. J. Med. Case Rep. 2022, 16, 27. [Google Scholar] [CrossRef]
- Cabrini, G.; Rimessi, A.; Borgatti, M.; Lampronti, I.; Finotti, A.; Pinton, P.; Gambari, R. Role of Cystic Fibrosis Bronchial Epithelium in Neutrophil Chemotaxis; Frontiers Media S.A.: Lausanne, Switzerland, 2020. [Google Scholar] [CrossRef]
- Raju, S.V.; Jackson, P.L.; Courville, C.A.; McNicholas, C.M.; Sloane, P.A.; Sabbatini, G.; Tidwell, S.; Tang, L.P.; Liu, B.; Fortenberry, J.A.; et al. Cigarette smoke induces systemic defects in cystic fibrosis transmembrane conductance regulator function. Am. J. Respir. Crit. Care Med. 2013, 188, 1321–1330. [Google Scholar] [CrossRef] [PubMed]
- Oates, G.R.; Baker, E.; Rowe, S.M.; Gutierrez, H.H.; Schechter, M.S.; Morgan, W.; Harris, W.T. Tobacco smoke exposure and socioeconomic factors are independent predictors of pulmonary decline in pediatric cystic fibrosis. J. Cyst. Fibros. 2020, 19, 783–790. [Google Scholar] [CrossRef]
- Archangelidi, O.; Cullinan, P.; Simmonds, N.J.; Mentzakis, E.; Peckham, D.; Bilton, D.; Carr, S.B. Incidence and risk factors of cancer in individuals with cystic fibrosis in the UK; a case-control study. J. Cyst. Fibros. 2022, 21, 302–308. [Google Scholar] [CrossRef]
- Patel, V.; Majumdar, T.; Samreen, I.; Grewal, H.; Kaleekal, T. Primary lung carcinoma in cystic fibrosis: A case report and literature review. Respir. Med. Case Rep. 2020, 31, 101242. [Google Scholar] [CrossRef]
- Amaral, M.D.; Quaresma, M.C.; Pankonien, I. What Role Does Cftr Play in Development, Differentiation, Regeneration and Cancer? Int. J. Mol. Sci. 2020, 21, 3133. [Google Scholar] [CrossRef]
- Ma, X.; Zang, X.; Yang, L.; Zhou, W.; Li, Y.; Wei, J.; Guo, J.; Han, J.; Liang, J.; Jin, T. Genetic polymorphisms in CYP2B6 may be associated with lung cancer risk in the Chinese Han population. Expert Rev. Respir. Med. 2023, 17, 1297–1305. [Google Scholar] [CrossRef]
- Vekens, K.; Vincken, S.; Hanon, S.; Demuynck, K.; Stylemans, D.; Vanderhelst, E. Lung cancer in a CF patient: Combination of bad luck or is there more to say? Acta Clin. Belg. 2021, 76, 379–380. [Google Scholar] [CrossRef]
- Kleinfelder, K.; Lotti, V.; Eramo, A.; Amato, F.; Cicero, S.L.; Castelli, G.; Spadaro, F.; Farinazzo, A.; Dell, D.; Preato, S.; et al. In silico analysis and theratyping of an ultra-rare CFTR genotype (W57G/A234D) in primary human rectal and nasal epithelial cells. iScience 2023, 26, 108180. [Google Scholar] [CrossRef] [PubMed]
- Neglia, J.P.; FitzSimmons, S.C.; Maisonneuve, P.; Schöni, M.H.; Corey, M.; Lowenfels, A.B.; Schöni-Affolter, F. The risk of cancer among patients with cystic fibrosis. Cystic Fibrosis and Cancer Study Group. N. Engl. J. Med. 1995, 332, 494–499. [Google Scholar] [CrossRef]
- Li, Y.; Sun, Z.; Wu, Y.; Babovic-Vuksanovic, D.; Li, Y.; Cunningham, J.M.; Pankratz, V.S.; Yang, P. Cystic fibrosis transmembrane conductance regulator gene mutation and lung cancer risk. Lung Cancer 2010, 70, 14–21. [Google Scholar] [CrossRef] [PubMed]
- Cantin, A.M.; Hartl, D.; Konstan, M.W.; Chmiel, J.F. Inflammation in cystic fibrosis lung disease: Pathogenesis and therapy. J. Cyst. Fibros. 2015, 14, 419–430. [Google Scholar] [CrossRef]
- Chmiel, J.F.; Berger, M.; Konstan, M.W. The role of inflammation in the pathophysiology of CF lung disease. Clin. Rev. Allergy Immunol. 2002, 23, 5–27. [Google Scholar] [CrossRef]
- Robb, C.T.; Regan, K.H.; Dorward, D.A.; Rossi, A.G. Key Mechanisms Governing Resolution of Lung Inflammation; Springer: Berlin/Heidelberg, Germany, 2016. [Google Scholar] [CrossRef]
- Nourshargh, S.; Alon, R. Leukocyte Migration into Inflamed Tissues. Immunity 2014, 41, 694–707. [Google Scholar] [CrossRef]
- Ghigo, A.; Prono, G.; Riccardi, E.; Rose, V.D. Dysfunctional Inflammation In Cystic Fibrosis Airways: From Mechanisms to Novel Therapeutic Approaches. Int. J. Mol. Sci. 2021, 22, 1952. [Google Scholar] [CrossRef] [PubMed]
- Rose, V.D. Mechanisms and markers of airway inflammation in cystic fibrosis. Eur. Respir. J. 2002, 19, 333–340. [Google Scholar] [CrossRef]
- Wagener, J.S.; Kahn, T.Z.; Copenhaver, S.C.; Accurso, F.J. Early Inflammation and the Development of Pulmonary Disease in Cystic Fibrosis. Pediatr. Pulmonol. Suppl. 1997, 16, 267–268. [Google Scholar] [CrossRef]
- Keir, H.R.; Chalmers, J.D. Neutrophil Extracellular Traps in Chronic Lung Disease: Implications for Pathogenesis and Therapy; European Respiratory Society: Lausanne, Switzerland, 2022. [Google Scholar] [CrossRef]
- Whitsett, J.A.; Alenghat, T. Respiratory epithelial cells orchestrate pulmonary innate immunity. Nat. Immunol. 2015, 16, 27–35. [Google Scholar] [CrossRef]
- Zhang, S.; Meng, Y.; Zhou, L.; Qiu, L.; Wang, H.; Su, D.; Zhang, B.; Chan, K.-M.; Han, J. Targeting epigenetic regulators for inflammation: Mechanisms and intervention therapy. MedComm 2022, 3, e173. [Google Scholar] [CrossRef]
- Villella, V.R.; Castaldo, A.; Scialò, F.; Castaldo, G. How Effectively Can Oxidative Stress and Inflammation Be Reversed When CFTR Function Is Pharmacologically Improved? Antioxidants 2025, 14, 310. [Google Scholar] [CrossRef] [PubMed]
- Bruscia, E.M.; Zhang, P.-X.; Ferreira, E.; Caputo, C.; Emerson, J.W.; Tuck, D.; Krause, D.S.; Egan, M.E. Macrophages directly contribute to the exaggerated inflammatory response in cystic fibrosis transmembrane conductance regulator-/-mice. Am. J. Respir. Cell Mol. Biol. 2009, 40, 295–304. [Google Scholar] [CrossRef]
- Scott, M.; Sario, A.D. DNA methylation changes in cystic fibrosis: Cause or consequence? Clin. Genet. 2020, 98, 3–9. [Google Scholar] [CrossRef]
- Wu, M.; Chen, J.H. CFTR Dysfunction Leads to Defective Bacterial Eradication on Cystic Fibrosis Airways; Frontiers Media S.A.: Lausanne, Switzerland, 2024. [Google Scholar] [CrossRef]
- Bell, S.C.; Mall, M.A.; Gutierrez, H.; Macek, M.; Madge, S.; Davies, J.C.; Burgel, P.-R.; Tullis, E.; Castaños, C.; Castellani, C.; et al. The Future of Cystic Fibrosis Care: A Global Perspective; Lancet Publishing Group: Amsterdam, The Netherlands, 2020. [Google Scholar] [CrossRef]
- Petrocheilou, A.; Moudaki, A.; Kaditis, A.G. Inflammation and Infection in Cystic Fibrosis: Update for the Clinician. Children 2022, 9, 1898. [Google Scholar] [CrossRef]
- Guerville, F.; Bourdel-Marchasson, I.; Déchanet-Merville, J.; Pellegrin, I.; Soubeyran, P.; Appay, V.; Lemoine, M. Does Inflammation Contribute to Cancer Incidence and Mortality During Aging? A Conceptual Review. Cancers 2022, 14, 1622. [Google Scholar] [CrossRef]
- Xie, Y.; Liu, F.; Wu, Y.; Zhu, Y.; Jiang, Y.; Wu, Q.; Dong, Z.; Liu, K. Inflammation in Cancer: Therapeutic Opportunities from New Insights; BioMed Central Ltd.: London, UK, 2025. [Google Scholar] [CrossRef]
- Wu, B.; Sodji, Q.H.; Oyelere, A.K. Inflammation, Fibrosis and Cancer: Mechanisms, Therapeutic Options and Challenges. Cancers 2022, 14, 552. [Google Scholar] [CrossRef] [PubMed]
- Proctor, M.J.; McMillan, D.C.; Horgan, P.G.; Fletcher, C.D.; Talwar, D.; Morrison, D.S. Systemic inflammation predicts all-cause mortality: A glasgow inflammation outcome study. PLoS ONE 2015, 10, e0116206. [Google Scholar] [CrossRef]
- Oft, M. IL-10: Master switch from tumor-promoting inflammation to antitumor immunity. Cancer Immunol. Res. 2014, 2, 194–199. [Google Scholar] [CrossRef] [PubMed]
- Catellani, C.; Cirillo, F.; Graziano, S.; Montanini, L.; Marmiroli, N.; Gullì, M.; Street, M.E. MicroRNA global profiling in cystic fibrosis cell lines reveals dysregulated pathways related with inflammation, cancer, growth, glucose and lipid metabolism, and fertility: An exploratory study. Acta Biomed. 2022, 93, e2022133. [Google Scholar] [CrossRef]
- Uribe, M.L.; Marrocco, I.; Yarden, Y. EGFR in Cancer: Signaling Mechanisms, Drugs, and Acquired Resistance. Cancers 2021, 13, 2748. [Google Scholar] [CrossRef]
- Stolarczyk, M.; Veit, G.; Schnúr, A.; Veltman, M.; Lukacs, G.L.; Scholte, B.J. Extracellular oxidation in cystic fibrosis airway epithelium causes enhanced EGFR/ADAM17 activity. Am. J. Physiol.-Lung Cell. Mol. Physiol. 2018, 314, 555–568. [Google Scholar] [CrossRef] [PubMed]
- Bartoszewski, R.; Rab, A.; Jurkuvenaite, A.; Mazur, M.; Wakefield, J.; Collawn, J.F.; Bebok, Z. Activation of the unfolded protein response by ΔF508 CFTR. Am. J. Respir. Cell Mol. Biol. 2008, 39, 448–457. [Google Scholar] [CrossRef] [PubMed]
- Cordani, M.; Butera, G.; Pacchiana, R.; Masetto, F.; Mullappilly, N.; Riganti, C.; Donadelli, M. Mutant p53-Associated Molecular Mechanisms of ROS Regulation in Cancer Cells. Biomolecules 2020, 10, 361. [Google Scholar] [CrossRef]
- García-Guede, Á.; Vera, O.; Ibáñez-de-Caceres, I. When Oxidative Stress Meets Epigenetics: Implications in Cancer Development. Antioxidants 2020, 9, 468. [Google Scholar] [CrossRef]
- Roesch, E.A.; Nichols, D.P.; Chmiel, J.F. Inflammation in Cystic Fibrosis: An Update; John Wiley and Sons Inc.: Hoboken, NJ, USA, 2018. [Google Scholar] [CrossRef]
- Maisonneuve, P.; FitzSimmons, S.C.; Neglia, J.P.; Campbell, P.W., III; Lowenfels, A.B. Cancer Risk in Nontransplanted and Transplanted Cystic Fibrosis Patients: A 10-Year Study. JNCI J. Natl. Cancer Inst. 2003, 95, 381–387. [Google Scholar] [CrossRef]
- Briottet, M.; Shum, M.; Urbach, V. The Role of Specialized Pro-Resolving Mediators in Cystic Fibrosis Airways Disease; Frontiers Media S.A.: Lausanne, Switzerland, 2020. [Google Scholar] [CrossRef]
- Strizova, Z.; Benesova, I.; Bartolini, R.; Novysedlak, R.; Cecrdlova, E.; Foley, L.K.; Striz, I. M1/M2 Macrophages and Their Overlaps—Myth or Reality? Portland Press Ltd.: London, UK, 2023. [Google Scholar] [CrossRef]
- Lévêque, M.; Le Trionnaire, S.; Del Porto, P.; Martin-Chouly, C. The Impact of Impaired Macrophage Functions in Cystic Fibrosis Disease Progression; Elsevier B.V.: Amsterdam, The Netherlands, 2017. [Google Scholar] [CrossRef]
- Camus, L.; Briaud, P.; Vandenesch, F.; Moreau, K. How Bacterial Adaptation to Cystic Fibrosis Environment Shapes Interactions Between Pseudomonas Aeruginosa and Staphylococcus Aureus; Frontiers Media S.A.: Lausanne, Switzerland, 2021. [Google Scholar] [CrossRef]
- Bhagirath, A.Y.; Li, Y.; Somayajula, D.; Dadashi, M.; Badr, S.; Duan, K. Cystic fibrosis lung environment and Pseudomonas aeruginosa infection. BMC Pulm. Med. 2016, 16, 174. [Google Scholar] [CrossRef]
- Döring, G.; Hoiby, N.; Consensus Study Group. Early intervention and prevention of lung disease in cystic fibrosis: A European consensus. J. Cyst. Fibros. 2004, 3, 67–91. [Google Scholar] [CrossRef] [PubMed]
- Larsen, J.M. The Immune Response to Prevotella Bacteria in Chronic Inflammatory Disease; Blackwell Publishing Ltd.: Oxford, UK, 2017. [Google Scholar] [CrossRef]
- Singh, J.; Hunt, S.; Simonds, S.; Boyton, C.; Middleton, A.; Elias, M.; Towns, S.; Pandit, C.; Robinson, P.; Fitzgerald, D.A.; et al. The changing epidemiology of pulmonary infection in children and adolescents with cystic fibrosis: An 18-year experience. Sci. Rep. 2024, 14, 9056. [Google Scholar] [CrossRef]
- Martin, I.; Waters, V.; Grasemann, H. Approaches to Targeting Bacterial Biofilms in Cystic Fibrosis Airways. Int. J. Mol. Sci. 2021, 22, 2155. [Google Scholar] [CrossRef]
- Majka, G.; Mazurek, H.; Strus, M.; Ciszek-Lenda, M.; Szatanek, R.; Pac, A.; Golińska, E.; Marcinkiewicz, J. Chronic bacterial pulmonary infections in advanced cystic fibrosis differently affect the level of sputum neutrophil elastase, IL-8 and IL-6. Clin. Exp. Immunol. 2021, 205, 391–405. [Google Scholar] [CrossRef] [PubMed]
- Blanchard, A.C.; Waters, V.J. Opportunistic Pathogens in Cystic Fibrosis: Epidemiology and Pathogenesis of Lung Infection. J. Pediatr. Infect. Dis. Soc. 2022, 11, S3–S12. [Google Scholar] [CrossRef] [PubMed]
- Hibino, S.; Kawazoe, T.; Kasahara, H.; Itoh, S.; Ishimoto, T.; Sakata-Yanagimoto, M.; Taniguchi, K. Inflammation-Induced Tumorigenesis and Metastasis. Int. J. Mol. Sci. 2021, 22, 5421. [Google Scholar] [CrossRef] [PubMed]
- Guilbaud, E.; Gautier, E.L.; Yvan-Charvet, L. Macrophage Origin, Metabolic Reprogramming and IL-1β Signaling: Promises and Pitfalls in Lung Cancer. Cancers 2019, 11, 298. [Google Scholar] [CrossRef]
- Conway, E.M.; Pikor, L.A.; Kung, S.H.Y.; Hamilton, M.J.; Lam, S.; Lam, W.L.; Bennewith, K.L. Macrophages, Inflammation, and Lung Cancer; American Thoracic Society: New York, NY, USA, 2016. [Google Scholar] [CrossRef]
- Prevaes, S.M.P.J.; de Steenhuijsen Piters, W.A.A.; de Winter-de Groot, K.M.; Janssens, H.M.; Tramper-Stranders, G.A.; Chu, M.L.J.N.; Tiddens, H.A.; van Westreenen, M.; van der Ent, C.K.; Sanders, E.A.M.; et al. Concordance between upper and lower airway microbiota in infants with cystic fibrosis. Eur. Respir. J. 2017, 49, 1602235. [Google Scholar] [CrossRef]
- Hogan, D.A.; Willger, S.D.; Dolben, E.L.; Hampton, T.H.; Stanton, B.A.; Morrison, H.G.; Sogin, M.L.; Czum, J.; Ashare, A. Analysis of lung microbiota in bronchoalveolar lavage, protected brush and sputum samples from subjects with Mild-To- Moderate cystic fibrosis lung disease. PLoS ONE 2016, 11, e0149998. [Google Scholar] [CrossRef]
- Coburn, B.; Wang, P.W.; Caballero, J.D.; Clark, S.T.; Brahma, V.; Donaldson, S.; Zhang, Y.; Surendra, A.; Gong, Y.; Tullis, D.E.; et al. Lung microbiota across age and disease stage in cystic fibrosis. Sci. Rep. 2015, 5, 10241. [Google Scholar] [CrossRef]
- Raghuvanshi, R.; Vasco, K.; Vázquez-Baeza, Y.; Jiang, L.; Morton, J.T.; Li, D.; Gonzalez, A.; Goldasich, L.D.; Humphrey, G.; Ackermann, G.; et al. High-Resolution Longitudinal Dynamics of the Cystic Fibrosis Sputum Microbiome and Metabolome through Antibiotic Therapy. mSystems 2020, 5. [Google Scholar] [CrossRef]
- Cuthbertson, L.; Walker, A.W.; Oliver, A.E.; Rogers, G.B.; Rivett, D.W.; Hampton, T.H.; Ashare, A.; Elborn, J.S.; De Soyza, A.; Carroll, M.P.; et al. Lung function and microbiota diversity in cystic fibrosis. Microbiome 2020, 8, 45. [Google Scholar] [CrossRef]
- Dickson, R.P.; Martinez, F.J.; Huffnagle, G.B. The Role of the Microbiome in Exacerbations of Chronic Lung Diseases; Elsevier B.V.: Amsterdam, The Netherlands, 2014. [Google Scholar] [CrossRef]
- Scialo, F.; Amato, F.; Cernera, G.; Gelzo, M.; Zarrilli, F.; Comegna, M.; Pastore, L.; Bianco, A.; Castaldo, G. Lung Microbiome in Cystic Fibrosis. Life 2021, 11, 94. [Google Scholar] [CrossRef]
- Ramírez-Labrada, A.G.; Isla, D.; Artal, A.; Arias, M.; Rezusta, A.; Pardo, J.; Gálvez, E.M. The Influence of Lung Microbiota on Lung Carcinogenesis, Immunity, and Immunotherapy; Cell Press: Cambridge, MS, USA, 2020. [Google Scholar] [CrossRef]
- Wang, D.; Cheng, J.; Zhang, J.; Zhou, F.; He, X.; Shi, Y.; Tao, Y. The Role of Respiratory Microbiota in Lung Cancer; Ivyspring International Publisher: Sydney, Australia, 2021. [Google Scholar] [CrossRef]
- Samareh-Fekri, M.; Bajgani, S.M.H.; Shafahi, A.; Asadi-Zarandi, M.; Mollaie, H.; Paghalhe, A.J. Detection of Helicobacter pylori in the bronchoalveolar lavage of patients with lung cancer using real-time PCR. Jundishapur J. Microbiol. 2016, 9, e32144. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Cheng, Y.; Zang, D.; Zhang, M.; Li, X.; Liu, D.; Gao, B.; Zhou, H.; Sun, J.; Han, X.; et al. The Role of Gut Microbiota in Lung Cancer: From Carcinogenesis to Immunotherapy; Frontiers Media S.A.: Lausanne, Switzerland, 2021. [Google Scholar] [CrossRef]
- Jenkins, S.V.; Robeson, M.S.; Griffin, R.J.; Quick, C.M.; Siegel, E.R.; Cannon, M.J.; Vang, K.B.; Dings, R.P.M. Gastrointestinal tract dysbiosis enhances distal tumor progression through suppression of leukocyte trafficking. Cancer Res. 2019, 79, 5999–6009. [Google Scholar] [CrossRef] [PubMed]
- Lurienne, L.; Cervesi, J.; Duhalde, L.; de Gunzburg, J.; Andremont, A.; Zalcman, G.; Buffet, R.; Bandinelli, P.-A. NSCLC Immunotherapy Efficacy and Antibiotic Use: A Systematic Review and Meta-Analysis; Elsevier Inc.: Amsterdam, The Netherlands, 2020. [Google Scholar] [CrossRef]
- Oxidative Stress and Regulation of Glutathione in Lung Inflammation. Available online: https://publications.ersnet.org (accessed on 21 April 2025).
- Rahman, I.; Macnee, W. Role of transcription factors in inflammatory lung diseases. Thorax 1998, 53, 601–612. [Google Scholar] [CrossRef]
- Brown, L.A. Glutathione protects signal transduction in type II cells under oxidant stress. Am. J. Physiol. 1994, 266, L172-7. [Google Scholar] [CrossRef]
- Van Klaveren, R.J.; Demedts, M.; Nemery, B. Cellular glutathione turnover in vitro, with emphasis on type II pneumocytes. Eur. Respir. J. 1997, 10, 1392–1400. [Google Scholar] [CrossRef]
- Hector, A.; Griese, M.; Hartl, D. Oxidative Stress in Cystic Fibrosis Lung Disease: An Early Event, but Worth Targeting? European Respiratory Society: Lausanne, Switzerland, 2014. [Google Scholar] [CrossRef]
- Sosinski, L.M.; Martin, H.C.; Neugebauer, K.A.; Ghuneim, L.-A.J.; Guzior, D.V.; Castillo-Bahena, A.; Mielke, J.; Thomas, R.; McClelland, M.; Conrad, D.; et al. A restructuring of microbiome niche space is associated with Elexacaftor-Tezacaftor-Ivacaftor therapy in the cystic fibrosis lung. J. Cyst. Fibros. 2022, 21, 996–1005. [Google Scholar] [CrossRef]
- Hisert, K.B.; Heltshe, S.L.; Pope, C.; Jorth, P.; Wu, X.; Edwards, R.M.; Radey, M.; Accurso, F.J.; Wolter, D.J.; Cooke, G.; et al. Restoring cystic fibrosis transmembrane conductance regulator function reduces airway bacteria and inflammation in people with cystic fibrosis and chronic lung infections. Am. J. Respir. Crit. Care Med. 2017, 195, 1617–1628. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Kinter, M.; Shank, S.; Cotton, C.; Kelley, T.J.; Ziady, A.G. Dysfunction of Nrf-2 in CF epithelia leads to excess intracellular H2O2 and inflammatory cytokine production. PLoS ONE 2008, 3, e3367. [Google Scholar] [CrossRef]
- Di Pietro, C.; Öz, H.H.; Murray, T.S.; Bruscia, E.M. Targeting the Heme Oxygenase 1/Carbon Monoxide Pathway to Resolve Lung Hyper-Inflammation and Restore a Regulated Immune Response in Cystic Fibrosis; Frontiers Media S.A.: Lausanne, Switzerland, 2020. [Google Scholar] [CrossRef]
- Signorelli, P.; Pivari, F.; Barcella, M.; Merelli, I.; Zulueta, A.; Cas, M.D.; Rosso, L.; Ghidoni, R.; Caretti, A.; Paroni, R.; et al. Myriocin modulates the altered lipid metabolism and storage in cystic fibrosis. Cell Signal. 2021, 81, 109928. [Google Scholar] [CrossRef]
- Bezerra, F.S.; Lanzetti, M.; Nesi, R.T.; Nagato, A.C.; Silva, C.P.E.; Kennedy-Feitosa, E.; Melo, A.C.; Cattani-Cavalieri, I.; Porto, L.C.; Valenca, S.S. Oxidative Stress and Inflammation in Acute and Chronic Lung Injuries. Antioxidants 2023, 12, 548. [Google Scholar] [CrossRef] [PubMed]
- Pizzino, G.; Irrera, N.; Cucinotta, M.; Pallio, G.; Mannino, F.; Arcoraci, V.; Squadrito, F.; Altavilla, D.; Bitto, A. Oxidative Stress: Harms and Benefits for Human Health; Hindawi Limited: Bognor Regis, UK, 2017. [Google Scholar] [CrossRef]
- Fabbrizzi, A.; Amedei, A.; Lavorini, F.; Renda, T.; Fontana, G. The Lung Microbiome: Clinical and Therapeutic Implications; Springer-Verlag Italia s.r.l.: Berlin/Heidelberg, Germany, 2019. [Google Scholar] [CrossRef]
- Perrone, F.; Belluomini, L.; Mazzotta, M.; Bianconi, M.; Di Noia, V.; Meacci, F.; Montrone, M.; Pignataro, D.; Prelaj, A.; Rinaldi, S. Exploring the Role of Respiratory Microbiome in Lung Cancer: A Systematic Review; Elsevier Ireland Ltd.: Amsterdam, The Netherlands, 2021. [Google Scholar] [CrossRef]
- Garratt, L.W.; Breuer, O.; Schofield, C.J.; McLean, S.A.; Laucirica, D.R.; Tirouvanziam, R.; Clements, B.S.; Kicic, A.; Ranganathan, S.; Stick, S.M. Changes in airway inflammation with pseudomonas eradication in early cystic fibrosis. J. Cyst. Fibros. 2021, 20, 941–948. [Google Scholar] [CrossRef] [PubMed]
- Mariniello, D.F.; D’Agnano, V.; Cennamo, D.; Conte, S.; Quarcio, G.; Notizia, L.; Pagliaro, R.; Schiattarella, A.; Salvi, R.; Bianco, A.; et al. Comorbidities in COPD: Current and Future Treatment Challenges. J. Clin. Med. 2024, 13, 743. [Google Scholar] [CrossRef]
- Parisi, G.F.; Terlizzi, V.; Manti, S.; Papale, M.; Pecora, G.; Presti, S.; Tosto, M.; Leonardi, S. Cutting-Edge Advances in Cystic Fibrosis: From Gene Therapy to Personalized Medicine and Holistic Management. Genes 2025, 16, 402. [Google Scholar] [CrossRef]
- Middleton, P.G.; Mall, M.A.; Dřevínek, P.; Lands, L.C.; McKone, E.F.; Polineni, D.; Ramsey, B.W.; Taylor-Cousar, J.L.; Tullis, E.; Vermeulen, F.; et al. Elexacaftor–Tezacaftor–Ivacaftor for Cystic Fibrosis with a Single Phe508del Allele. N. Engl. J. Med. 2019, 381, 1809–1819. [Google Scholar] [CrossRef]
- Taylor-Cousar, J.L.; Robinson, P.D.; Shteinberg, M.; Downey, D.G. CFTR modulator therapy: Transforming the landscape of clinical care in cystic fibrosis. Lancet 2023, 402, 1171–1184. [Google Scholar] [CrossRef]
- Ridley, K.; Condren, M. Elexacaftor-tezacaftor-ivacaftor: The first triple-combination cystic fibrosis transmembrane conductance regulator modulating therapy. J. Pediatr. Pharmacol. Ther. 2022, 25, 192–197. [Google Scholar] [CrossRef]
- Comella, P.; Frasci, G.; Cataldis, G.D.; Panza, N.; Cioffi, R.; Curcio, C.; Belli, M.; Bianco, A.; Ianniello, G.; Maiorino, L.; et al. Cisplatin/carboplatin + etoposide + vinorelbine in advanced non-small-cell lung cancer: A multicentre randomised trial. Br. J. Cancer 1996, 74, 1805–1811. [Google Scholar] [CrossRef]
- Medusa, P.M.; Gilli, M.; Notizia, L.; Pagliaro, R.; Carro, N.; Moriello, A.; D’Agnano, V.; Bianco, A.; Perrotta, F.; Vitiello, F. Complete response to pembrolizumab as a single agent in a patient with stage III NSCLC with high PD-L1 expression: A case report. Monaldi Arch. Chest Dis. 2022, 93. [Google Scholar] [CrossRef] [PubMed]
- Pagliaro, R.; Medusa, P.M.; Vitiello, F.; Aronne, L.; Campbell, S.F.M.; Perrotta, F.; Bianco, A. Case report: Selpercatinib in the treatment of RET fusion-positive advanced lung adenocarcinoma: A challenging clinical case. Front. Oncol. 2024, 14, 1500449. [Google Scholar] [CrossRef] [PubMed]
- Stella, G.M.; Scialò, F.; Bortolotto, C.; Agustoni, F.; Sanci, V.; Saddi, J.; Casali, L.; Corsico, A.G.; Bianco, A. Pragmatic Expectancy on Microbiota and Non-Small Cell Lung Cancer: A Narrative Review. Cancers 2022, 14, 3131. [Google Scholar] [CrossRef] [PubMed]
- Cauwenberghs, E.; De Boeck, I.; Spacova, I.; Van Tente, I.; Bastiaenssen, J.; Lammertyn, E.; Verhulst, S.; Van Hoorenbeeck, K.; Lebeer, S. Positioning the Preventive Potential of Microbiome Treatments for Cystic Fibrosis in the Context of Current Therapies; Cell Press: Cambridge, MS, USA, 2024. [Google Scholar] [CrossRef]
- Hadhud, M.; Arnon, J.; Hershko-Moshe, A.; Hollander, A.; Hurvitz-Lehmann, N.; Potruch, A.; Azmanov, H.; Kuint, R.; Hiller, N.; Picard, E.; et al. Non-classical pulmonary exacerbation in cystic fibrosis revealing ALK-Translocated lung cancer: A case report. Respir. Med. Case Rep. 2025, 53, 102171. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
| Pathway/Mediator | Role in Cystic Fibrosis | Contribution to Lung Cancer |
|---|---|---|
| Chronic Inflammation | Persistent neutrophilic inflammation due to CFTR dysfunction and infections | Promotes epithelial-mesenchymal transition, DNA damage, angiogenesis, and tumor progression |
| Pro-inflammatory Cytokines | Elevated IL-6, IL-8, TNF-α, IL-1β in CF airways | Activate STAT3 and NF-κB pathways, enhancing cell proliferation and survival |
| Epigenetic Modifications | DNA methylation and histone changes in immune cells | Promote pro-tumorigenic immune phenotypes and chronic inflammation |
| Oxidative Stress (ROS) | Excess ROS from activated neutrophils and impaired antioxidant response (e.g., reduced glutathione) | Causes DNA damage, supports tumorigenesis, and disrupts epithelial integrity |
| Microbiome Dysbiosis | Loss of diversity, dominance of P. aeruginosa and Burkholderia, antibiotic overuse | Alters immune responses, sustains chronic inflammation, and may directly promote carcinogenesis |
| UPR (Unfolded Protein Response) | Activated by F508del-CFTR mutation and ER stress | Enhances macrophage inflammation and may promote survival of transformed cells |
| EGFR Pathway Activation | Upregulated in CF airway epithelium | Contributes to cell proliferation and tumor development |
| IL-10 / Treg Imbalance | Dysregulation contributes to unbalanced immune responses | Supports tumor immune evasion and progression |
| CFTR Modulator Effects | May modulate inflammation and microbiome composition | Potential indirect role in reducing LC risk (needs further research) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pagliaro, R.; Scialò, F.; Schiattarella, A.; Cianci, R.; Campbell, S.F.M.; Perrotta, F.; Bianco, A.; Castaldo, G. Mechanisms of Lung Cancer Development in Cystic Fibrosis Patients: The Role of Inflammation, Oxidative Stress, and Lung Microbiome Dysbiosis. Biomolecules 2025, 15, 828. https://doi.org/10.3390/biom15060828
Pagliaro R, Scialò F, Schiattarella A, Cianci R, Campbell SFM, Perrotta F, Bianco A, Castaldo G. Mechanisms of Lung Cancer Development in Cystic Fibrosis Patients: The Role of Inflammation, Oxidative Stress, and Lung Microbiome Dysbiosis. Biomolecules. 2025; 15(6):828. https://doi.org/10.3390/biom15060828
Chicago/Turabian StylePagliaro, Raffaella, Filippo Scialò, Angela Schiattarella, Roberta Cianci, Susan F. M. Campbell, Fabio Perrotta, Andrea Bianco, and Giuseppe Castaldo. 2025. "Mechanisms of Lung Cancer Development in Cystic Fibrosis Patients: The Role of Inflammation, Oxidative Stress, and Lung Microbiome Dysbiosis" Biomolecules 15, no. 6: 828. https://doi.org/10.3390/biom15060828
APA StylePagliaro, R., Scialò, F., Schiattarella, A., Cianci, R., Campbell, S. F. M., Perrotta, F., Bianco, A., & Castaldo, G. (2025). Mechanisms of Lung Cancer Development in Cystic Fibrosis Patients: The Role of Inflammation, Oxidative Stress, and Lung Microbiome Dysbiosis. Biomolecules, 15(6), 828. https://doi.org/10.3390/biom15060828