
Antechodynamics and Antechokinetics: Dynamics and Kinetics of Antibiotic Resistance Biomolecules

 , , , ,
, , , ,
Abstract
1. Introduction: Antibiotic Resistance Dynamics and Kinetics as an Action and Reaction Process
2. Antechodynamics
2.1. Primary Effectors of Antibiotic Resistance: Modifying and Drug-Degrading Biomolecules
2.1.1. Beta-Lactams
2.1.2. Aminoglycosides
2.1.3. Macrolides, Lincosamides, and Streptogramins
2.1.4. Phenicols
2.1.5. Tetracyclines
2.1.6. Fluoroquinolones
2.1.7. Fosfomycin
2.1.8. Rifampicin
2.1.9. Glycopeptides and Lipopeptides
2.1.10. Polymyxins
2.1.11. Sulfonamides
2.1.12. Nitrofurantoin
2.2. Secondary Effector Biomolecules Triggering the Expression of Genes Involved in Antibiotic Resistance
2.2.1. Beta-Lactams
2.2.2. Aminoglycosides
2.2.3. Macrolides, Lincosamides, and Streptogramins
2.2.4. Phenicols
2.2.5. Tetracyclines
2.2.6. Fluoroquinolones
2.2.7. Fosfomycin
2.2.8. Sulfonamides and Trimethoprim
2.2.9. Glycopeptides and Lipopeptides
2.2.10. Polymyxins
2.2.11. Oxazolidinones
2.2.12. Fusidic Acid
2.2.13. Nitrofurantoin
2.2.14. The Combined Effects of Antibiotic Resistance Biomolecules
2.2.15. Metabolic Biomolecules Influencing Antibiotic Detoxification
3. Antechokinetics
3.1. Three Previous Questions on Antechokinetics
3.1.1. The Question of Efflux Pumps
3.1.2. The Question of the Number of Reduced Affinity Genes
3.1.3. The Question of Intracellular Topology in Transcription–Translation Efficiency
3.2. The Antechokinetics of Biomolecules Involved in Resistance to Various Antibiotic Classes
3.2.1. Beta-Lactam Resistance
3.2.2. Aminoglycoside Resistance
3.2.3. Macrolide, Lincosamide, and Streptogramin Resistance
3.2.4. Tetracycline Resistance
3.2.5. Fluoroquinolone Resistance
3.2.6. Trimethoprim Resistance
3.2.7. Glycopeptide and Lipopeptide Resistance
4. The Crossroads Between Antechology (AD/AK) and Pharmacology: Future Directions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Jacobs, M.R. Optimisation of antimicrobial therapy using pharmacokinetic and pharmacodynamic parameters. Clin. Microbiol. Infect. 2001, 7, 589–596. [Google Scholar] [CrossRef] [PubMed]
- Mouton, J.W.; Dudley, M.N.; Cars, O.; Derendorf, H.; Drusano, G.L. Standardization of pharmacokinetic/pharmacodynamic (PK/PD) terminology for anti-infective drugs: An update. J. Antimicrob. Chemother. 2005, 55, 601–607. [Google Scholar] [CrossRef] [PubMed]
- Darby, E.M.; Trampari, E.; Siasat, P.; Gaya, M.S.; Alav, I.; Webber, M.A.; Blair, J.M.A. Molecular mechanisms of antibiotic resistance revisited. Nat. Rev. Microbiol. 2023, 21, 280–295. [Google Scholar] [CrossRef] [PubMed]
- Luz, C.F.; van Niekerk, J.M.; Keizer, J.; Beerlage-de Jong, N.; Braakman-Jansen, L.M.A.; Stein, A.; Sinha, B.; van Gemert-Pijnen, J.E.W.C.; Glasner, C. Mapping twenty years of antimicrobial resistance research trends. Artif. Intell. Med. 2022, 123, 102216. [Google Scholar] [CrossRef] [PubMed]
- Schaenzer, A.J.; Wright, G.D. Antibiotic resistance by enzymatic modification of antibiotic targets. Trends Mol. Med. 2020, 26, 768–782. [Google Scholar] [CrossRef] [PubMed]
- Avery, C.; Baker, L.; Jacobs, D.J. Functional dynamics of substrate recognition in TEM beta-lactamase. Entropy 2022, 24, 729. [Google Scholar] [CrossRef] [PubMed]
- Pantsar, T.; Poso, A. Binding affinity via docking: Fact and fiction. Molecules 2018, 23, 1899. [Google Scholar] [CrossRef] [PubMed]
- Baquero, F.; Martínez, J.L.; Novais, Â.; Rodríguez-Beltrán, J.; Martínez-García, L.; Coque, T.M.; Galán, J.C. Allogenous selection of mutational collateral resistance: Old drugs select for new resistance within antibiotic families. Front. Microbiol. 2021, 12, 757833. [Google Scholar] [CrossRef]
- Albery, W.J.; Knowles, J.R. Evolution of enzyme function and the development of catalytic efficiency. Biochemistry 1976, 15, 5631–5640. [Google Scholar] [CrossRef] [PubMed]
- Bulychev, A.; Mobashery, S. Class C β-lactamases operate at the diffusion limit for turnover of their preferred cephalosporin substrates. Antimicrob. Agents Chemother. 1999, 43, 1743–1746. [Google Scholar] [CrossRef] [PubMed]
- Monterroso, B.; Margolin, W.; Boersma, A.J.; Rivas, G.; Poolman, B.; Zorrilla, S. Macromolecular crowding, phase separation, and homeostasis in the orchestration of bacterial cellular functions. Chem. Rev. 2024, 124, 1899–1949. [Google Scholar] [CrossRef] [PubMed]
- Di Cera, E. Serine proteases. IUBMB Life 2009, 61, 510–515. [Google Scholar] [CrossRef] [PubMed]
- Hedstrom, L. Serine protease mechanism and specificity. Chem. Rev. 2002, 102, 4501–4524. [Google Scholar] [CrossRef] [PubMed]
- Bush, K. Characterization of beta-lactamases. Antimicrob. Agents Chemother. 1989, 33, 259–263. [Google Scholar] [CrossRef] [PubMed]
- Tooke, C.L.; Hinchliffe, P.; Bragginton, E.C.; Colenso, C.K.; Hirvonen, V.H.A.; Takebayashi, Y.; Spencer, J. β-lactamases and β-lactamase Inhibitors in the 21st Century. J. Mol. Biol. 2019, 431, 3472–3500. [Google Scholar] [CrossRef] [PubMed]
- Zdarska, V.; Kolar, M.; Mlynarcik, P. Occurrence of beta-lactamases in bacteria. Infect. Genet. Evol. 2024, 122, 105610. [Google Scholar] [CrossRef] [PubMed]
- Bordeleau, V.; Stogios, P.J.; Evdokimova, E.; Koteva, K.; Savchenko, A.; Wright, G.D. Mechanistic plasticity in ApmA enables aminoglycoside promiscuity for resistance. Nat. Chem. Biol. 2024, 20, 234–242. [Google Scholar] [CrossRef] [PubMed]
- Krause, K.M.; Serio, A.W.; Kane, T.R.; Connolly, L.E. Aminoglycosides: An overview. Cold Spring Harb. Perspect. Med. 2016, 6, a027029. [Google Scholar] [CrossRef] [PubMed]
- Smith, C.A.; Baker, E.N. Aminoglycoside antibiotic resistance by enzymatic deactivation. Curr. Drug Targets Infect. Disord. 2002, 2, 143–160. [Google Scholar] [CrossRef] [PubMed]
- Revillo Imbernon, J.; Weibel, J.M.; Ennifar, E.; Prévost, G.; Kellenberger, E. Structural analysis of neomycin B and kanamycin A binding Aminoglycosides Modifying Enzymes (AME) and bacterial ribosomal RNA. Mol. Inform. 2024, 43, e202300339. [Google Scholar] [CrossRef] [PubMed]
- Wright, G.D.; Thompson, P.R. Aminoglycoside phosphotransferases: Proteins, structure, and mechanism. Front. Biosci. 1999, 4, D9-21. [Google Scholar] [CrossRef] [PubMed]
- McKay, G.A.; Wright, G.D. Kinetic mechanism of aminoglycoside phosphotransferase type IIIa. Evidence for a Theorell-Chance mechanism. J. Biol. Chem. 1995, 270, 24686–24692. [Google Scholar] [CrossRef] [PubMed]
- Golkar, T.; Zieliński, M.; Berghuis, A.M. Look and Outlook on Enzyme-Mediated Macrolide Resistance. Front. Microbiol. 2018, 9, 1942. [Google Scholar] [CrossRef] [PubMed]
- Pawlowski, A.C.; Stogios, P.J.; Koteva, K.; Skarina, T.; Evdokimova, E.; Savchenko, A.; Wright, G.D. The evolution of substrate discrimination in macrolide antibiotic resistance enzymes. Nat. Commun. 2018, 9, 112. [Google Scholar] [CrossRef] [PubMed]
- Barthélémy, P.; Autissier, D.; Gerbaud, G.; Courvalin, P. Enzymic hydrolysis of erythromycin by a strain of Escherichia coli. A new mechanism of resistance. J. Antibiot. 1984, 37, 1692–1696. [Google Scholar] [CrossRef] [PubMed]
- Svetlov, M.S.; Vázquez-Laslop, N.; Mankin, A.S. Kinetics of drug-ribosome interactions defines the cidality of macrolide antibiotics. Proc. Natl. Acad. Sci. USA 2017, 114, 13673–13678. [Google Scholar] [CrossRef] [PubMed]
- Svetlov, M.S.; Cohen, S.; Alsuhebany, N.; Vázquez-Laslop, N.; Mankin, A.S. A long-distance rRNA base pair impacts the ability of macrolide antibiotics to kill bacteria. Proc. Natl. Acad. Sci. USA 2020, 117, 1971–1975. [Google Scholar] [CrossRef] [PubMed]
- Gil-Gil, T.; Berryhill, B.A.; Manuel, J.A.; Smith, A.P.; McCall, I.C.; Baquero, F.; Levin, B.R. The evolution of heteroresistance via small colony variants in Escherichia coli following long-term exposure to bacteriostatic antibiotics. Nat. Commun. 2024, 15, 7936. [Google Scholar] [CrossRef] [PubMed]
- Douthwaite, S.; Champney, W.S. Structures of ketolides and macrolides determine their mode of interaction with the ribosomal target site. J. Antimicrob. Chemother. 2001, 48 (Suppl. T1), 1–8. [Google Scholar] [CrossRef] [PubMed]
- Stogios, P.J.; Evdokimova, E.; Morar, M.; Koteva, K.; Wright, G.D.; Courvalin, P.; Savchenko, A. Structural and functional plasticity of antibiotic resistance nucleotidyltransferases revealed by molecular characterization of lincosamide nucleotidyltransferases lnu(A) and lnu(D). J. Mol. Biol. 2015, 427, 2229–2243. [Google Scholar] [CrossRef] [PubMed]
- Stogios, P.J.; Kuhn, M.L.; Evdokimova, E.; Courvalin, P.; Anderson, W.F.; Savchenko, A. Potential for reduction of streptogramin A resistance revealed by structural analysis of acetyltransferase VatA. Antimicrob. Agents Chemother. 2014, 58, 7083–7092. [Google Scholar] [CrossRef] [PubMed]
- Roberts, M.C.; Sutcliffe, J.; Courvalin, P.; Jensen, L.B.; Rood, J.; Seppala, H. Nomenclature for macrolide and macrolide-lincosamide-streptogramin B resistance determinants. Antimicrob. Agents Chemother. 1999, 43, 2823–2830. [Google Scholar] [CrossRef] [PubMed]
- Murray, I.A.; Shaw, W.V. O-Acetyltransferases for chloramphenicol and other natural products. Antimicrob. Agents Chemother. 1997, 41, 4004–4010. [Google Scholar] [CrossRef] [PubMed]
- Farrell, D.J.; Castanheira, M.; Chopra, I. Characterization of global patterns and the genetics of fusidic acid resistance. Clin Infect Dis. 2011, 52 (Suppl. 7), S487–S492. [Google Scholar] [CrossRef] [PubMed]
- Gasparrini, A.J.; Markley, J.L.; Kumar, H.; Wang, B.; Fang, L.; Irum, S.; Symister, C.T.; Wallace, M.; Burnham, C.D.; Andleeb, S.; et al. Tetracycline-inactivating enzymes from environmental, human commensal, and pathogenic bacteria cause broad-spectrum tetracycline resistance. Commun. Biol. 2020, 3, 241. [Google Scholar] [CrossRef] [PubMed]
- Forsberg, K.J.; Patel, S.; Wencewicz, T.A.; Dantas, G. The tetracycline Destructases: A novel family of tetracycline-inactivating enzymes. Chem. Biol. 2015, 22, 888–897. [Google Scholar] [CrossRef] [PubMed]
- Cheng, Q.; Cheung, Y.; Liu, C.; Chan, E.W.; Wong, K.Y.; Zhang, R.; Chen, S. Functional and phylogenetic analysis of TetX variants to design a new classification system. Commun. Biol. 2022, 5, 522. [Google Scholar] [CrossRef] [PubMed]
- Park, J.; Gasparrini, A.J.; Reck, M.R.; Symister, C.T.; Elliott, J.L.; Vogel, J.P.; Wencewicz, T.A.; Dantas, G.; Tolia, N.H. Plasticity, dynamics, and inhibition of emerging tetracycline resistance enzymes. Nat. Chem. Biol. 2017, 13, 730–736. [Google Scholar] [CrossRef] [PubMed]
- Robicsek, A.; Strahilevitz, J.; Jacoby, G.A.; Macielag, M.; Abbanat, D.; Park, C.H.; Bush, K.; Hooper, D.C. Fluoroquinolone-modifying enzyme: A new adaptation of a common aminoglycoside acetyltransferase. Nat. Med. 2006, 12, 83–88. [Google Scholar] [CrossRef] [PubMed]
- Maia, A.S.; Tiritan, M.E.; Castro, P.M. Enantioselective degradation of ofloxacin and levofloxacin by the bacterial strains Labrys portucalensis F11 and Rhodococcus sp. FP1. Ecotoxicol. Environ. Saf. 2018, 155, 144–151. [Google Scholar] [CrossRef] [PubMed]
- Bernat, B.A.; Laughlin, L.T.; Armstrong, R.N. Fosfomycin resistance protein (FosA) is a manganese metalloglutathione transferase related to glyoxalase I and the extradiol dioxygenases. Biochemistry 1997, 36, 3050–3055. [Google Scholar] [CrossRef] [PubMed]
- Klontz, E.H.; Tomich, A.D.; Günther, S.; Lemkul, J.A.; Deredge, D.; Silverstein, Z.; Shaw, J.F.; McElheny, C.; Doi, Y.; Wintrode, P.L.; et al. Structure and dynamics of FosA-mediated fosfomycin resistance in Klebsiella pneumoniae and Escherichia coli. Antimicrob. Agents Chemother. 2017, 61, e01572-17. [Google Scholar] [CrossRef] [PubMed]
- Tossounian, M.A.; Zhao, Y.; Yu, B.Y.K.; Markey, S.A.; Malanchuk, O.; Zhu, Y.; Cain, A.; Gout, I. Low-molecular-weight thiol transferases in redox regulation and antioxidant defence. Redox Biol. 2024, 71, 103094. [Google Scholar] [CrossRef] [PubMed]
- Goldstein, B.P. Resistance to rifampicin: A review. J. Antibiot. 2014, 67, 625–630. [Google Scholar] [CrossRef] [PubMed]
- Cao, M.; Feng, Y.; Zhang, Y.; Kang, W.; Lian, K.; Ai, L. Studies on the metabolism and degradation of vancomycin in simulated in vitro and aquatic environment by UHPLC-Triple-TOF-MS/MS. Sci. Rep. 2018, 8, 15471. [Google Scholar] [CrossRef] [PubMed]
- D’Costa, V.M.; Mukhtar, T.A.; Patel, T.; Koteva, K.; Waglechner, N.; Hughes, D.W.; Wright, G.D.; De Pascale, G. Inactivation of the lipopeptide antibiotic daptomycin by hydrolytic mechanisms. Antimicrob. Agents Chemother. 2012, 56, 757–764. [Google Scholar] [CrossRef] [PubMed]
- Paris, L.; Devers-Lamrani, M.; Joly, M.; Viala, D.; De Antonio, M.; Pereira, B.; Rouard, N.; Besse-Hoggan, P.; Hébraud, M.; Topp, E.; et al. Effect of subtherapeutic and therapeutic sulfamethazine concentrations on transcribed genes and translated proteins involved in Microbacterium sp. C448 resistance and degradation. FEMS Microbiol. Ecol. 2023, 99, fiad064. [Google Scholar] [CrossRef] [PubMed]
- Pacholak, A.; Juzwa, W.; Zgoła-Grześkowiak, A.; Kaczorek, E. Multi-faceted analysis of bacterial transformation of nitrofurantoin. Sci. Total Environ. 2023, 874, 162422. [Google Scholar] [CrossRef] [PubMed]
- Dar, D.; Sorek, R. Regulation of antibiotic-resistance by non-coding RNAs in bacteria. Curr. Opin. Microbiol. 2017, 36, 111–117. [Google Scholar] [CrossRef] [PubMed]
- Zeng, X.; Lin, J. Beta-lactamase induction and cell wall metabolism in Gram-negative bacteria. Front. Microbiol. 2013, 4, 128. [Google Scholar] [CrossRef] [PubMed]
- Irazoki, O.; Hernandez, S.B.; Cava, F. Peptidoglycan muropeptides: Release, perception, and functions as signaling molecules. Front. Microbiol. 2019, 10, 500. [Google Scholar] [CrossRef] [PubMed]
- Sverak, H.E.; Yaeger, L.N.; Worrall, L.J.; Vacariu, C.M.; Glenwright, A.J.; Vuckovic, M.; Al Azawi, Z.D.; Lamers, R.P.; Marko, V.A.; Skorupski, C.; et al. Cryo-EM characterization of the anydromuropeptide permease AmpG central to bacterial fitness and β-lactam antibiotic resistance. Nat. Commun. 2024, 15, 9936. [Google Scholar] [CrossRef] [PubMed]
- Vadlamani, G.; Thomas, M.D.; Patel, T.R.; Donald, L.J.; Reeve, T.M.; Stetefeld, J.; Standing, K.G.; Vocadlo, D.J.; Mark, B.L. The β-lactamase gene regulator AmpR is a tetramer that recognizes and binds the D-Ala-D-Ala motif of its repressor UDP-N-acetylmuramic acid (MurNAc)-pentapeptide. J. Biol. Chem. 2015, 290, 2630–2643. [Google Scholar] [CrossRef] [PubMed]
- Mallik, D.; Jain, D.; Bhakta, S.; Ghosh, A.S. Role of AmpC-inducing genes in modulating other serine beta-lactamases in Escherichia coli. Antibiotics 2022, 11, 67. [Google Scholar] [CrossRef] [PubMed]
- Tamma, P.D.; Doi, Y.; Bonomo, R.A.; Johnson, J.K.; Simner, P.J.; Antibacterial Resistance Leadership Group. A Primer on AmpC β-Lactamases: Necessary Knowledge for an Increasingly Multidrug-resistant World. Clin. Infect. Dis. 2019, 69, 1446–1455. [Google Scholar] [CrossRef]
- Aracil-Gisbert, S.; Fernández-De-Bobadilla, M.D.; Guerra-Pinto, N.; Serrano-Calleja, S.; Pérez-Cobas, A.E.; Soriano, C.; de Pablo, R.; Lanza, V.F.; Pérez-Viso, B.; Reuters, S.; et al. The ICU environment contributes to the endemicity of the “Serratia marcescens complex” in the hospital setting. mBio 2024, 15, e0305423. [Google Scholar] [CrossRef] [PubMed]
- Belluzo, B.S.; Abriata, L.A.; Giannini, E.; Mihovilcevic, D.; Dal Peraro, M.; Llarrull, L.I. An experiment-informed signal transduction model for the role of the Staphylococcus aureus MecR1 protein in β-lactam resistance. Sci. Rep. 2019, 9, 19558. [Google Scholar] [CrossRef] [PubMed]
- Clarke, S.R.; Dyke, K.G. The signal transducer (BlaRI) and the repressor (BlaI) of the Staphylococcus aureus beta-lactamase operon are inducible. Microbiology 2001, 147, 803–810. [Google Scholar] [CrossRef] [PubMed]
- Yang, M.J.; Li, M.J.; Huang, L.D.; Zhang, X.W.; Huang, Y.Y.; Gou, X.Y.; Chen, S.N.; Yan, J.; Du, P.; Sun, A.H. Response regulator protein CiaR regulates the transcription of ccn-microRNAs and β-lactam antibiotic resistance conversion of Streptococcus pneumoniae. Int. J. Antimicrob. Agents. 2025, 65, 107387. [Google Scholar] [CrossRef] [PubMed]
- Jia, X.; Zhang, J.; Sun, W.; He, W.; Jiang, H.; Chen, D.; Murchie, A.I. Riboswitch regulation of aminoglycoside resistance acetyl and adenyl transferases. Cell 2013, 153, 1419–1420. [Google Scholar] [CrossRef] [PubMed]
- Hipólito, A.; García-Pastor, L.; Blanco, P.; Trigo da Roza, F.; Kieffer, N.; Vergara, E.; Jové, T.; Álvarez, J.; Escudero, J.A. The expression of aminoglycoside resistance genes in integron cassettes is not controlled by riboswitches. Nucleic Acids Res. 2022, 50, 8566–8579. [Google Scholar] [CrossRef] [PubMed]
- Carvalho, A.; Hipólito, A.; Trigo da Roza, F.; García-Pastor, L.; Vergara, E.; Buendía, A.; García-Seco, T.; Escudero, J.A. The expression of integron arrays is shaped by the translation rate of cassettes. Nat. Commun. 2024, 15, 9232. [Google Scholar] [CrossRef] [PubMed]
- González-Zorn, B.; Teshager, T.; Casas, M.; Porrero, M.C.; Moreno, M.A.; Courvalin, P.; Domínguez, L. armA and aminoglycoside resistance in Escherichia coli. Emerg. Infect. Dis. 2005, 11, 954–956. [Google Scholar] [CrossRef] [PubMed]
- Fritsche, T.R.; Castanheira, M.; Miller, G.H.; Jones, R.N.; Armstrong, E.S. Detection of methyltransferases conferring high-level resistance to aminoglycosides in Enterobacteriaceae from Europe; North America; and Latin America. Antimicrob. Agents Chemother. 2008, 52, 1843–1845. [Google Scholar] [CrossRef] [PubMed]
- Matamoros, B.R.; Serna, C.; Wedel, E.; Montero, N.; Kirpekar, F.; Gonzalez-Zorn, B. NpmC—A novel A1408 16S rRNA methyltransferase in the gut of humans and animals. Int. J. Antimicrob. Agents. 2025, 65, 107382. [Google Scholar] [CrossRef] [PubMed]
- Yuan, M.; Nie, L.; Huang, Z.; Xu, S.; Qiu, X.; Han, L.; Kang, Y.; Li, F.; Yao, J.; Li, Q.; et al. Capture of armA by a novel ISCR element, ISCR28. Int. J. Antimicrob. Agents 2024, 64, 107250. [Google Scholar] [CrossRef] [PubMed]
- Wangkheimayum, J.; Paul, D.; Chanda, D.D.; Melson Singha, K.; Bhattacharjee, A. Elevated expression of rsmI can act as a reporter of aminoglycoside resistance in Escherichia coli using kanamycin as signal molecule. Infect. Genet. Evol. 2022, 98, 105229. [Google Scholar] [CrossRef] [PubMed]
- Rosenberg, E.Y.; Ma, D.; Nikaido, H. AcrD of Escherichia coli is an aminoglycoside efflux pump. J. Bacteriol. 2000, 182, 1754–1756. [Google Scholar] [CrossRef] [PubMed]
- Sidhu, K.; Talbot, M.; Van Mil, K.; Verstraete, M. Treatment with subinhibitory kanamycin induces adaptive resistance to aminoglycoside antibiotics via the AcrD multidrug efflux pump in Escherichia coli K-12 J. Exp. Microbiol. Immunol. 2012, 16, 1611–1616. [Google Scholar]
- Zhang, Z.; Morgan, C.E.; Cui, M.; Yu, E.W. Cryo-EM structures of AcrD illuminate a mechanism for capturing aminoglycosides from its central cavity. mBio 2023, 14, e0338322. [Google Scholar] [CrossRef] [PubMed]
- Leclercq, R. Mechanisms of resistance to macrolides and lincosamides: Nature of the resistance elements and their clinical implications. Clin. Infect. Dis. 2002, 34, 482–492. [Google Scholar] [CrossRef] [PubMed]
- Min, Y.H.; Kwon, A.R.; Yoon, E.J.; Shim, M.J.; Choi, E.C. Translational attenuation and mRNA stabilization as mechanisms of erm(B) induction by erythromycin. Antimicrob. Agents Chemother. 2008, 52, 1782–1789. [Google Scholar] [CrossRef] [PubMed]
- Sothiselvam, S.; Neuner, S.; Rigger, L.; Klepacki, D.; Micura, R.; Vázquez-Laslop, N.; Mankin, A.S. Binding of macrolide antibiotics leads to ribosomal selection against specific substrates based on their charge and size. Cell Rep. 2016, 16, 1789–1799. [Google Scholar] [CrossRef] [PubMed]
- Seefeldt, A.C.; Aguirre Rivera, J.; Johansson, M. Direct measurements of erythromycin’s effect on protein synthesis kinetics in living bacterial cells. J. Mol. Biol. 2021, 433, 166942. [Google Scholar] [CrossRef] [PubMed]
- Vázquez-Laslop, N.; Mankin, A.S. How macrolide antibiotics work. Trends Biochem. Sci. 2018, 43, 668–684. [Google Scholar] [CrossRef] [PubMed]
- Duval, M.; Dar, D.; Carvalho, F.; Rocha, E.P.; Sorek, R.; Cossart, P. HflXr, a homolog of a ribosome-splitting factor, mediates antibiotic resistance. Proc. Natl. Acad. Sci. USA 2018, 115, 13359–13364. [Google Scholar] [CrossRef] [PubMed]
- Lovett, P.S. Translation attenuation regulation of chloramphenicol resistance in bacteria--a review. Gene 1996, 179, 157–162. [Google Scholar] [CrossRef] [PubMed]
- George, A.M.; Hall, R.M. Efflux of chloramphenicol by the CmlA1 protein. FEMS Microbiol. Lett. 2002, 209, 209–213. [Google Scholar] [CrossRef] [PubMed]
- Long, K.S.; Poehlsgaard, J.; Kehrenberg, C.; Schwarz, S.; Vester, B. The Cfr rRNA methyltransferase confers resistance to phenicols, lincosamides, oxazolidinones, pleuromutilins, and streptogramin A antibiotics. Antimicrob Agents Chemother. 2006, 50, 2500–2505. [Google Scholar] [CrossRef] [PubMed]
- Crowe-McAuliffe, C.; Murina, V.; Turnbull, K.J.; Huch, S.; Kasari, M.; Takada, H.; Nersisyan, L.; Sundsfjord, A.; Hegstad, K.; Atkinson, G.C.; et al. Structural basis for PoxtA-mediated resistance to phenicol and oxazolidinone antibiotics. Nat Commun. 2022, 13, 1860. [Google Scholar] [CrossRef] [PubMed]
- Hinrichs, W.; Kisker, C.; Düvel, M.; Müller, A.; Tovar, K.; Hillen, W.; Saenger, W. Structure of the Tet repressor-tetracycline complex and regulation of antibiotic resistance. Science 1994, 264, 418–420. [Google Scholar] [CrossRef] [PubMed]
- Starosta, A.L.; Lassak, J.; Jung, K.; Wilson, D.N. The bacterial translation stress response. FEMS Microbiol. Rev. 2014, 38, 1172–1201. [Google Scholar] [CrossRef] [PubMed]
- Dönhöfer, A.; Franckenberg, S.; Wickles, S.; Berninghausen, O.; Beckmann, R.; Wilson, D.N. Structural basis for TetM-mediated tetracycline resistance. Proc. Natl. Acad. Sci. USA 2012, 109, 16900–16905. [Google Scholar] [CrossRef] [PubMed]
- Aldred, K.J.; Kerns, R.J.; Osheroff, N. Mechanism of quinolone action and resistance. Biochemistry 2014, 53, 1565–1574. [Google Scholar] [CrossRef] [PubMed]
- Monárrez, R.; Wang, Y.; Fu, Y.; Liao, C.H.; Okumura, R.; Braun, M.R.; Jacoby, G.A.; Hooper, D.C. Genes and proteins involved in qnrS1 induction. Antimicrob. Agents Chemother. 2018, 62, e00806-18. [Google Scholar] [CrossRef] [PubMed]
- Kurabayashi, K.; Hirakawa, Y.; Tanimoto, K.; Tomita, H.; Hirakawa, H. Role of the CpxAR two-component signal transduction system in control of fosfomycin resistance and carbon substrate uptake. J. Bacteriol. 2014, 196, 248–256. [Google Scholar] [CrossRef] [PubMed]
- Venkatesan, M.; Fruci, M.; Verellen, L.A.; Skarina, T.; Mesa, N.; Flick, R.; Pham, C.; Mahadevan, R.; Stogios, P.J.; Savchenko, A. Molecular mechanism of plasmid-borne resistance to sulfonamide antibiotics. Nat. Commun. 2023, 14, 4031. [Google Scholar] [CrossRef] [PubMed]
- Shi, H.; Li, T.; Xu, J.; Yu, J.; Yang, S.; Zhang, X.E.; Tao, S.; Gu, J.; Deng, J.Y. MgrB inactivation confers trimethoprim resistance in Escherichia coli. Front. Microbiol. 2021, 12, 682205. [Google Scholar] [CrossRef] [PubMed]
- Guberman-Pfeffer, M. Resistance: How VanA strains get away with it. Authorea 2023. [Google Scholar] [CrossRef]
- Stogios, P.J.; Savchenko, A. Molecular mechanisms of vancomycin resistance. Protein Sci. 2020, 29, 654–669. [Google Scholar] [CrossRef] [PubMed]
- Grissom-Arnold, J.; Alborn, W.E.; Nicas, T.I.; Jaskunas, S.R. Induction of VanA vancomycin resistance genes in Enterococcus faecalis: Use of a promoter fusion to evaluate glycopeptide and nonglycopeptide induction signals. Microb. Drug Resist. 1997, 3, 53–64. [Google Scholar] [CrossRef] [PubMed]
- Roch, M.; Clair, P.; Renzoni, A.; Reverdy, M.E.; Dauwalder, O.; Bes, M.; Martra, A.; Freydière, A.M.; Laurent, F.; Reix, P.; et al. Exposure of Staphylococcus aureus to subinhibitory concentrations of β-lactam antibiotics induces heterogeneous vancomycin-intermediate Staphylococcus aureus. Antimicrob. Agents Chemother. 2014, 58, 5306–5314. [Google Scholar] [CrossRef] [PubMed]
- Axell-House, D.B.; Simar, S.R.; Panesso, D.; Rincon, S.; Miller, W.R.; Khan, A.; Pemberton, O.A.; Valdez, L.; Nguyen, A.H.; Hood, K.S.; et al. LiaX is a surrogate marker for cell envelope stress and daptomycin non-susceptibility in Enterococcus faecium. Antimicrob. Agents Chemother. 2024, 68, e0106923. [Google Scholar] [CrossRef] [PubMed]
- Olaitan, A.O.; Morand, S.; Rolain, J.M. Mechanisms of polymyxin resistance: Acquired and intrinsic resistance in bacteria. Front Microbiol. 2014, 5, 643. [Google Scholar] [CrossRef] [PubMed]
- Gallardo, A.; Iglesias, M.R.; Ugarte-Ruiz, M.; Hernández, M.; Miguela-Villoldo, P.; Gutiérrez, G.; Rodríguez-Lázaro, D.; Domínguez, L.; Quesada, A. Plasmid-mediated Kluyvera-like arnBCADTEF operon confers colistin (hetero)resistance to Escherichia coli. Antimicrob. Agents Chemother. 2023, 65, e00091-21. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Srinivas, S.; Xu, Y.; Wei, W.; Feng, Y. Genetic and biochemical mechanisms for bacterial lipid A modifiers associated with polymyxin resistance. Trends Biochem. Sci. 2019, 44, 973–988. [Google Scholar] [CrossRef] [PubMed]
- Kieffer, N.; Royer, G.; Decousser, J.W.; Bourrel, A.S.; Palmieri, M.; Ortiz De La Rosa, J.M.; Jacquier, H.; Denamur, E.; Nordmann, P.; Poirel, L. mcr-9, an Inducible Gene Encoding an Acquired phosphoethanolamine transferase in Escherichia coli, and Its Origin. Antimicrob. Agents Chemother. 2019, 63, e00965-19. [Google Scholar] [CrossRef] [PubMed]
- Locke, J.B.; Finn, J.; Hilgers, M.; Morales, G.; Rahawi, S.; Kedar, G.C.; Picazo, J.J.; Im, W.; Shaw, K.J.; Stein, J.L. Structure-activity relationships of diverse oxazolidinones for linezolid-resistant Staphylococcus aureus strains possessing the cfr methyltransferase gene or ribosomal mutations. Antimicrob. Agents Chemother. 2010, 54, 5337–5343. [Google Scholar] [CrossRef] [PubMed]
- Tomlinson, J.H.; Thompson, G.S.; Kalverda, A.P.; Zhuravleva, A.; O’Neill, A.J. A target-protection mechanism of antibiotic resistance at atomic resolution: Insights into FusB-type fusidic acid resistance. Sci. Rep. 2016, 6, 19524. [Google Scholar] [CrossRef] [PubMed]
- O’Neill, A.J.; McLaws, F.; Kahlmeter, G.; Henriksen, A.S.; Chopra, I. Genetic basis of resistance to fusidic acid in staphylococci. Antimicrob. Agents Chemother. 2007, 51, 1737–1740. [Google Scholar] [CrossRef] [PubMed]
- O’Neill, A.J.; Chopra, I. Molecular basis of fusB-mediated resistance to fusidic acid in Staphylococcus aureus. Mol. Microbiol. 2006, 59, 664–676. [Google Scholar] [CrossRef] [PubMed]
- Roemhild, R.; Linkevicius, M.; Andersson, D.I. Molecular mechanisms of collateral sensitivity to the antibiotic nitrofurantoin. PLoS Biol. 2020, 18, e3000612. [Google Scholar] [CrossRef] [PubMed]
- Hausladen, A.; Privalle, C.T.; Keng, T.; DeAngelo, J.; Stamler, J.S. Nitrosative stress: Activation of the transcription factor OxyR. Cell 1996, 86, 719–729. [Google Scholar] [CrossRef] [PubMed]
- Niu, J.; Straubinger, R.M.; Mager, D.E. Pharmacodynamic drug-drug interactions. Clin. Pharmacol. Ther. 2019, 105, 1395–1406. [Google Scholar] [CrossRef] [PubMed]
- Cabello, M.; Hernández-García, M.; Maruri-Aransolo, A.; Michelena, M.; Pérez-Viso, B.; Ponce-Alonso, M.; Cantón, R.; Ruiz-Garbajosa, P. Occurrence of multi-carbapenemase-producing Enterobacterales in a tertiary hospital in Madrid (Spain): A new epidemiologic scenario. J. Glob. Antimicrob. Resist. 2024, 38, 281–291. [Google Scholar] [CrossRef] [PubMed]
- Li, X.Z.; Nikaido, H. Efflux-mediated drug resistance in bacteria: An update. Drugs 2009, 69, 1555–1623. [Google Scholar] [CrossRef] [PubMed]
- Berryhill, B.A.; Gil-Gil, T.; Manuel, J.A.; Smith, A.P.; Margollis, E.; Baquero, F.; Levin, B.R. What’s the matter with MICs: Bacterial nutrition, limiting resources; and antibiotic pharmacodynamics. Microbiol. Spectr. 2023, 11, e0409122. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Ran, M.; Wang, J.; Ouyang, Q.; Luo, C. Studies of antibiotic resistance of beta-lactamase bacteria under different nutrition limitations at the single-cell level. PLoS ONE 2015, 10, e0127115. [Google Scholar] [CrossRef] [PubMed]
- Peng, B.; Su, Y.B.; Li, H.; Han, Y.; Guo, C.; Tian, Y.M.; Peng, X.X. Exogenous alanine and/or glucose plus kanamycin kills antibiotic-resistant bacteria. Cell Metab. 2015, 21, 249–262. [Google Scholar] [CrossRef] [PubMed]
- Peng, B.; Li, H.; Peng, X.X. Functional metabolomics: From biomarker discovery to metabolome reprogramming. Protein Cell 2015, 6, 628–637. [Google Scholar] [CrossRef] [PubMed]
- Lopatkin, A.J.; Bening, S.C.; Manson, A.L.; Stokes, J.M.; Kohanski, M.A.; Badran, A.H.; Earl, A.M.; Cheney, N.J.; Yang, J.H.; Collins, J.J. Clinically relevant mutations in core metabolic genes confer antibiotic resistance. Science 2021, 371, eaba0862. [Google Scholar] [CrossRef] [PubMed]
- Jiang, M.; Su, Y.B.; Ye, J.Z.; Li, H.; Kuang, S.F.; Wu, J.H.; Li, S.H.; Peng, X.X.; Peng, B. Ampicillin-controlled glucose metabolism manipulates the transition from tolerance to resistance in bacteria. Sci. Adv. 2023, 9, eade8582. [Google Scholar] [CrossRef] [PubMed]
- Yi, X.; Feng, M.; He, F.; Xiao, Z.; Wang, Y.; Wang, S.; Yao, H. Multi-omics analysis explores the impact of ofloxacin pressure on the metabolic state in Escherichia coli. J. Glob. Antimicrob. Resist. 2024, 39, 59–68. [Google Scholar] [CrossRef] [PubMed]
- Vestergaard, M.; Nøhr-Meldgaard, K.; Bojer, M.S.; Krogsgård Nielsen, C.; Meyer, R.L.; Slavetinsky, C.; Peschel, A.; Ingmer, H. Inhibition of the ATP synthase eliminates the intrinsic resistance of Staphylococcus aureus towards polymyxins. mBio 2017, 8, e01114-17. [Google Scholar] [CrossRef] [PubMed]
- Baquero, F.; Martínez, J.L. Interventions on metabolism: Making antibiotic-susceptible bacteria. mBio 2017, 8, e01950-17. [Google Scholar] [CrossRef] [PubMed]
- Baquero, F.; Martínez, J.L.; Sánchez, A.; Fernández-de-Bobadilla, M.D.; San-Millán, A.; Rodríguez-Beltrán, J. Bacterial subcellular architecture, structural epistasis; and antibiotic resistance. Biology 2023, 12, 640. [Google Scholar] [CrossRef] [PubMed]
- Stevanovic, M.; Teuber Carvalho, J.P.; Bittihn, P.; Schultz, D. A dynamical model of antibiotic responses linking the expression of resistance genes to metabolism explains the emergence of heterogeneity during drug exposure. Phys. Biol. 2024, 21, 036002. [Google Scholar] [CrossRef] [PubMed]
- Landy, M.; Larkum, N.W.; Oswald, E.J.; Streightoff, F. Increased synthesis of p-aminobenzoic acid associated with the development of sulfonamide resistance in Staphylococcus aureus. Science 1943, 97, 265–267. [Google Scholar] [CrossRef] [PubMed]
- Baquero, F.; Coque, T.M.; Martínez, J.L.; Aracil-Gisbert, S.; Lanza, V.F. Gene transmission in the one health microbiosphere and the channels of antimicrobial resistance. Front. Microbiol. 2019, 10, 2892. [Google Scholar] [CrossRef] [PubMed]
- MacNair, C.R.; Tan, M.W. The role of bacterial membrane vesicles in antibiotic resistance. Ann. N. Y. Acad. Sci. 2023, 1519, 63–73. [Google Scholar] [CrossRef] [PubMed]
- Novelli, M.; Bolla, J.M. RND efflux pump induction: A crucial network unveiling adaptive antibiotic resistance mechanisms of Gram-negative bacteria. Antibiotics 2024, 13, 501. [Google Scholar] [CrossRef] [PubMed]
- Du, D.; Wang-Kan, X.; Neuberger, A.; van Veen, H.W.; Pos, K.M.; Piddock, L.J.V.; Luisi, B.F. Multidrug efflux pumps: Structure, function and regulation. Nat. Rev. Microbiol. 2018, 16, 523–539. [Google Scholar] [CrossRef] [PubMed]
- Dar, D.; Shamir, M.; Mellin, J.R.; Koutero, M.; Stern-Ginossar, N.; Cossart, P.; Sorek, R. Term-seq reveals abundant ribo-regulation of antibiotics resistance in bacteria. Science 2016, 352, aad9822. [Google Scholar] [CrossRef] [PubMed]
- Møller, T.S.; Overgaard, M.; Nielsen, S.S.; Bortolaia, V.; Sommer, M.O.; Guardabassi, L.; Olsen, J.E. Relation between tetR and tetA expression in tetracycline resistant Escherichia coli. BMC Microbiol. 2016, 16, 39. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Wilhelm, M.J.; Wu, T.; Hu, X.H.; Ruiz, O.N.; Dai, H.L. Quantifying bacterial efflux within subcellular domains of Pseudomonas aeruginosa. Appl. Environ. Microbiol. 2024, 90, e0144724. [Google Scholar] [CrossRef] [PubMed]
- Costafrolaz, J.; Panis, G.; Casu, B.; Ardissone, S.; Degeorges, L.; Pilhofer, M.; Viollier, P.H. Adaptive β-lactam resistance from an inducible efflux pump that is post-translationally regulated by the DjlA co-chaperone. PLoS Biol. 2023, 21, e3002040. [Google Scholar] [CrossRef] [PubMed]
- Gallagher, L.A.; Coughlan, S.; Black, N.S.; Lalor, P.; Waters, E.M.; Wee, B.; Watson, M.; Downing, T.; Fitzgerald, J.R.; Fleming, G.T.A.; et al. Tandem amplification of the Staphylococcal Cassette Chromosome mec Element Can Drive High-Level methicillin Resistance in Methicillin-Resistant Staphylococcus aureus. Antimicrob. Agents Chemother. 2017, 61, e00869-17. [Google Scholar] [CrossRef] [PubMed]
- Peters, K.; Schweizer, I.; Hakenbeck, R.; Denapaite, D. New insights into beta-lactam resistance of Streptococcus pneumoniae: Serine protease HtrA degrades altered penicillin-binding Protein 2x. Microorganisms 2021, 9, 1685. [Google Scholar] [CrossRef] [PubMed]
- Martínez, J.L.; Alonso, A.; Gómez-Gómez, J.M.; Baquero, F. Quinolone resistance by mutations in chromosomal gyrase genes. Just the tip of the iceberg? J. Antimicrob. Chemother. 1998, 42, 683–688. [Google Scholar] [CrossRef] [PubMed]
- Hernandez-Beltran, J.C.; Rodríguez-Beltrán, J.; Aguilar-Luviano, O.B.; Velez-Santiago, J.; Mondragón-Palomino, O.; MacLean, R.C.; Fuentes-Hernández, A.; San Millán, A.; Peña-Miller, R. Plasmid-mediated phenotypic noise leads to transient antibiotic resistance in bacteria. Nat. Commun. 2024, 15, 2610. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Penkul, P.; Milstein, J.N. Quantitative localization microscopy reveals a novel organization of a high-copy number plasmid. Biophys. J. 2016, 111, 467–479. [Google Scholar] [CrossRef] [PubMed]
- Niki, H.; Hiraga, S. Subcellular distribution of actively partitioning F plasmid during the cell division cycle in E. coli. Cell 1997, 90, 951–957. [Google Scholar] [CrossRef] [PubMed]
- Kannaiah, S.; Amster-Choder, O. Protein targeting via mRNA in bacteria. Biochim. Biophys. Acta 2014, 1843, 1457–1465. [Google Scholar] [CrossRef] [PubMed]
- Castellana, M.; Hsin-Jung Li, S.; Wingreen, N.S. Spatial organization of bacterial transcription and translation. Proc. Natl. Acad. Sci. USA 2016, 113, 9286–9291. [Google Scholar] [CrossRef] [PubMed]
- Rudner, D.Z.; Losick, R. Protein subcellular localization in bacteria. Cold Spring Harb. Perspect. Biol. 2010, 2, a000307. [Google Scholar] [CrossRef] [PubMed]
- Manting, E.H.; Driessen, A.J. Escherichia coli translocase: The unravelling of a molecular machine. Mol. Microbiol. 2000, 37, 226–238. [Google Scholar] [CrossRef] [PubMed]
- Kaderabkova, N.; Bharathwaj, M.; Furniss, R.C.; Gonzalez, D.; Palmer, T.; Mavridou, D.A. The biogenesis of β-lactamase enzymes. Microbiology 2022, 168, 001217. [Google Scholar] [CrossRef] [PubMed]
- Plückthun, A.; Pfitzinger, I. Membrane-bound beta-lactamase forms in Escherichia coli. J. Biol. Chem. 1988, 263, 14315–14322. [Google Scholar] [CrossRef] [PubMed]
- Bradford, P.A.; Bonomo, R.A.; Bush, K.; Carattoli, A.; Feldgarden, M.; Haft, D.H.; Ishii, Y.; Jacoby, G.A.; Klimke, W.; Palzkill, T.; et al. Consensus on β-lactamase nomenclature. Antimicrob. Agents Chemother. 2022, 66, e0033322. [Google Scholar] [CrossRef] [PubMed]
- Francisco, J.A.; Earhart, C.F.; Georgiou, G. Transport and anchoring of beta-lactamase to the external surface of Escherichia coli. Proc. Natl. Acad. Sci. USA 1992, 89, 2713–2717. [Google Scholar] [CrossRef] [PubMed]
- Livermore, D.M. Beta-lactamases: Quantity and resistance. Clin. Microbiol. Infect. 1997, 3 (Suppl. 4), S10–S19. [Google Scholar] [CrossRef] [PubMed]
- Kadonaga, J.T.; Gautier, A.E.; Straus, D.R.; Charles, A.D.; Edge, M.D.; Knowles, J.R. The role of the beta-lactamase signal sequence in the secretion of proteins by Escherichia coli. J. Biol. Chem. 1984, 259, 2149–2154. [Google Scholar] [CrossRef] [PubMed]
- Balchin, D.; Hayer-Hartl, M.; Hartl, F.U. Recent advances in understanding catalysis of protein folding by molecular chaperones. FEBS Lett. 2020, 594, 2770–2781. [Google Scholar] [CrossRef] [PubMed]
- Bowden, G.A.; Paredes, A.M.; Georgiou, G. Structure and morphology of protein inclusion bodies in Escherichia coli. Biotechnology 1991, 9, 725–730. [Google Scholar] [CrossRef]
- Khodaparast, L.; Khodaparast, L.; Wu, G.; Michiels, E.; Gallardo, R.; Houben, B.; Garcia, T.; De Vleeschouwer, M.; Ramakers, M.; Wilkinson, H.; et al. Exploiting the aggregation propensity of beta-lactamases to design inhibitors that induce enzyme misfolding. Nat. Commun. 2023, 14, 5571. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.W.; Seo, J.S.; Park, S.B.; Lee, A.R.; Lee, J.S.; Jung, J.W.; Chun, J.H.; Lazarte, J.M.S.; Kim, J.; Kim, J.H.; et al. Significant increase in the secretion of extracellular vesicles and antibiotic resistance from methicillin-resistant Staphylococcus aureus induced by ampicillin stress. Sci. Rep. 2020, 10, 21066. [Google Scholar] [CrossRef] [PubMed]
- Jiang, B.; Lai, Y.; Xiao, W.; Zhong, T.; Liu, F.; Gong, J.; Huang, J. Microbial extracellular vesicles contribute to antimicrobial resistance. PLoS Pathog. 2024, 20, e1012143. [Google Scholar] [CrossRef] [PubMed]
- Herencias, C.; Álvaro-Llorente, L.; Ramiro-Martínez, P.; Fernández-Calvet, A.; Muñoz-Cazalla, A.; DelaFuente, J.; Graf, F.E.; Jaraba-Soto, L.; Castillo-Polo, J.A.; Cantón, R.; et al. β-lactamase expression induces collateral sensitivity in Escherichia coli. Nat. Commun. 2024, 15, 4731. [Google Scholar] [CrossRef] [PubMed]
- Reguera, J.A.; Baquero, F.; Perez-Diaz, J.C.; Martinez, J.L. Synergistic effect of dosage and bacterial inoculum in TEM-1 mediated antibiotic resistance. Eur. J. Clin. Microbiol. Infect. Dis. 1988, 7, 778–779. [Google Scholar] [CrossRef] [PubMed]
- San Millan, A.; Escudero, J.A.; Gifford, D.R.; Mazel, D.; MacLean, R.C. Multicopy plasmids potentiate the evolution of antibiotic resistance in bacteria. Nat. Ecol. Evol. 2016, 1, 10. [Google Scholar] [CrossRef] [PubMed]
- Canabal, R.; González-Bello, C. Chemical sensors for the early diagnosis of bacterial resistance to β-lactam antibiotics. Bioorg. Chem. 2024, 150, 107528. [Google Scholar] [CrossRef] [PubMed]
- Gross, R.; Yelin, I.; Lázár, V.; Datta, M.S.; Kishony, R. Beta-lactamase dependent and independent evolutionary paths to high-level ampicillin resistance. Nat. Commun. 2024, 15, 5383. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Wang, Q.; Zhang, H.; Yang, M.; Khan, M.I.; Zhou, X. Sensor histidine kinase is a β-lactam receptor and induces resistance to β-lactam antibiotics. Proc. Natl. Acad. Sci. USA 2016, 113, 1648–1653. [Google Scholar] [CrossRef] [PubMed]
- Balasubramanian, D.; Schneper, L.; Merighi, M.; Smith, R.; Narasimhan, G.; Lory, S.; Mathee, K. The regulatory repertoire of Pseudomonas aeruginosa AmpC ß-lactamase regulator AmpR includes virulence genes. PLoS ONE 2012, 7, e34067. [Google Scholar] [CrossRef] [PubMed]
- Morè, N.; Martorana, A.M.; Biboy, J.; Otten, C.; Winkle, M.; Serrano, C.K.G.; Montón Silva, A.; Atkinson, L.; Yau, H.; Breukink, E.; et al. Peptidoglycan remodeling enables Escherichia coli to survive severe outer membrane assembly defect. mBio 2019, 10, e02729-18. [Google Scholar] [CrossRef] [PubMed]
- Martinez, J.L.; Vicente, M.F.; Delgado-Iribarren, A.; Perez-Diaz, J.C.; Baquero, F. Small plasmids are involved in amoxicillin-clavulanate resistance in Escherichia coli. Antimicrob. Agents Chemother. 1989, 33, 595. [Google Scholar] [CrossRef] [PubMed]
- Martinez, J.L.; Cercenado, E.; Rodriguez-Creixems, M.; Vincente-Perez, M.F.; Delgado-Iribarren, A.; Baquero, F. Resistance to beta-lactam/clavulanate. Lancet 1987, 2, 1473. [Google Scholar] [CrossRef] [PubMed]
- San Millan, A.; Santos-Lopez, A.; Ortega-Huedo, R.; Bernabe-Balas, C.; Kennedy, S.P.; Gonzalez-Zorn, B. Small-plasmid-mediated antibiotic resistance is enhanced by increases in plasmid copy number and bacterial fitness. Antimicrob. Agents Chemother. 2015, 59, 3335–3341. [Google Scholar] [CrossRef] [PubMed]
- Sandegren, L.; Andersson, D.I. Bacterial gene amplification: Implications for the evolution of antibiotic resistance. Nat. Rev. Microbiol. 2009, 7, 7578–7588. [Google Scholar] [CrossRef] [PubMed]
- Culp, E.; Wright, G.D. Bacterial proteases, untapped antimicrobial drug targets. J. Antibiot. 2017, 70, 366–377. [Google Scholar] [CrossRef] [PubMed]
- Schultz, S.C.; Dalbadie-McFarland, G.; Neitzel, J.J.; Richards, J.H. Stability of wild-type and mutant RTEM-1 beta-lactamases: Effect of the disulfide bond. Proteins 1987, 2, 290–297. [Google Scholar] [CrossRef] [PubMed]
- Shimizu-Ibuka, A.; Matsuzawa, H.; Sakai, H. Effect of disulfide-bond introduction on the activity and stability of the extended-spectrum class A beta-lactamase Toho-1. Biochim. Biophys. Acta 2006, 1764, 1349–1355. [Google Scholar] [CrossRef] [PubMed]
- Beadle, B.M.; McGovern, S.L.; Patera, A.; Shoichet, B.K. Functional analyses of AmpC beta-lactamase through differential stability. Protein Sci. 1999, 8, 1816–1824. [Google Scholar] [CrossRef] [PubMed]
- Baquero, F.; Levin, B.R. Proximate and ultimate causes of the bactericidal action of antibiotics. Nat. Rev. Microbiol. 2021, 19, 123–312. [Google Scholar] [CrossRef] [PubMed]
- Hancock, R.E.; Farmer, S.W.; Li, Z.S.; Poole, K. Interaction of aminoglycosides with the outer membranes and purified lipopolysaccharide and OmpF porin of Escherichia coli. Antimicrob. Agents Chemother. 1991, 35, 1309–1314. [Google Scholar] [CrossRef] [PubMed]
- Shaw, K.J.; Rather, P.N.; Hare, R.S.; Miller, G.H. Molecular genetics of aminoglycoside resistance genes and familial relationships of the aminoglycoside-modifying enzymes. Microbiol. Rev. 1993, 57, 138–163. [Google Scholar] [CrossRef] [PubMed]
- Dery, K.J.; Søballe, B.; Witherspoon, M.S.; Bui, D.; Koch, R.; Sherratt, D.J.; Tolmasky, M.E. The aminoglycoside 6′-N-acetyltransferase type Ib encoded by Tn1331 is evenly distributed within the cell’s cytoplasm. Antimicrob. Agents Chemother. 2003, 47, 2897–2902. [Google Scholar] [CrossRef] [PubMed]
- d’Udekem d’Acoz, O.; Hue, F.; Ye, T.; Wang, L.; Leroux, M.; Rajngewerc, L.; Tran, T.; Phan, K.; Ramirez, M.S.; Reisner, W.; et al. Dynamics and quantitative contribution of the aminoglycoside 6′-N-acetyltransferase type Ib to amikacin resistance. mSphere 2024, 9, e0078923. [Google Scholar] [CrossRef]
- McGann, P.; Courvalin, P.; Snesrud, E.; Clifford, R.J.; Yoon, E.J.; Onmus-Leone, F.; Ong, A.C.; Kwak, Y.I.; Grillot-Courvalin, C.; Lesho, E.; et al. Amplification of the aminoglycoside resistance gene aphA1 in Acinetobacter baumannii results in tobramycin therapy failure. mBio 2014, 5, e00915. [Google Scholar] [CrossRef] [PubMed]
- Martinez, J.L.; Blazquez, J.; Vicente, M.F.; Martinez-Ferrer, M.; Reguera, J.A.; Culebras, E.; Baquero, F. Influence of gene dosing on antibiotic resistance mediated by inactivating enzymes. J. Chemother. 1989, 1 (Suppl. S4), 265–266. [Google Scholar] [PubMed]
- Blazquez, J.; Martinez, J.L.; Baquero, F. Bleomycin increases amikacin and streptomycin resistance in Escherichia coli harboring transposon Tn5. Antimicrob. Agents Chemother. 1993, 37, 1982–1985. [Google Scholar] [CrossRef] [PubMed]
- Vázquez-Laslop, N.; Mankin, A.S. Context-specific action of ribosomal antibiotics. Annu. Rev. Microbiol. 2018, 72, 185–207. [Google Scholar] [CrossRef] [PubMed]
- Dzyubak, E.; Yap, M.N. The expression of antibiotic resistance methyltransferase correlates with mRNA stability independently of ribosome stalling. Antimicrob. Agents Chemother. 2016, 60, 7178–7188. [Google Scholar] [CrossRef] [PubMed]
- Chopra, I.; Roberts, M. Tetracycline antibiotics: Mode of action, applications, molecular biology, and epidemiology of bacterial resistance. Microbiol. Mol. Biol. Rev. 2001, 65, 232–260. [Google Scholar] [CrossRef] [PubMed]
- Cramariuc, O.; Rog, T.; Javanainen, M.; Monticelli, L.; Polishchuk, A.V.; Vattulainen, I. Mechanism for translocation of fluoroquinolones across lipid membranes. Biochim. Biophys. Acta 2012, 1818, 2563–2571. [Google Scholar] [CrossRef] [PubMed]
- Dumont, E.; Vergalli, J.; Conraux, L.; Taillier, C.; Vassort, A.; Pajovic, J.; Réfrégiers, M.; Mourez, M.; Pagès, J.M. Antibiotics and efflux: Combined spectrofluorimetry and mass spectrometry to evaluate the involvement of concentration and efflux activity in antibiotic intracellular accumulation. J. Antimicrob. Chemother. 2019, 74, 58–65. [Google Scholar] [CrossRef] [PubMed]
- García-López, M.; Megias, D.; Ferrándiz, M.J.; de la Campa, A.G. The balance between gyrase and topoisomerase I activities determines levels of supercoiling, nucleoid compaction, and viability in bacteria. Front. Microbiol. 2022, 13, 1094692. [Google Scholar] [CrossRef] [PubMed]
- Mustaev, A.; Malik, M.; Zhao, X.; Kurepina, N.; Luan, G.; Oppegard, L.M.; Hiasa, H.; Marks, K.R.; Kerns, R.J.; Berger, J.M.; et al. Fluoroquinolone-gyrase-DNA complexes: Two modes of drug binding. J. Biol. Chem. 2014, 289, 12300–12312. [Google Scholar] [CrossRef] [PubMed]
- Murawski, A.M.; Brynildsen, M.P. Ploidy is an important determinant of fluoroquinolone persister survival. Curr. Biol. 2021, 31, 2039–2050.e7. [Google Scholar] [CrossRef] [PubMed]
- Gaona, M.; Corral, J.; Campoy, S.; Barbé, J.; Pérez Varela, M.; Aranda, J. The novel MFS efflux pump SxtP, regulated by the LysR-type transcriptional activator SxtR, is involved in the susceptibility to sulfamethoxazole/trimethoprim (SXT) and the pathogenesis of Acinetobacter baumannii. Antimicrob. Agents Chemother. 2024, 68, e0071224. [Google Scholar] [CrossRef] [PubMed]
- Nicolaou, K.C.; Boddy, C.N.; Bräse, S.; Winssinger, N. Chemistry, biology, and medicine of the glycopeptide antibiotics. Angew. Chem. Int. Ed. Engl. 1999, 38, 2096–2152. [Google Scholar] [CrossRef] [PubMed]
- Masi, M.; Réfregiers, M.; Pos, K.M.; Pagès, J.M. Mechanisms of envelope permeability and antibiotic influx and efflux in Gram-negative bacteria. Nat. Microbiol. 2017, 2, 17001. [Google Scholar] [CrossRef] [PubMed]
- Nicoloff, H.; Hjort, K.; Andersson, D.I.; Wang, H. Three concurrent mechanisms generate gene copy number variation and transient antibiotic heteroresistance. Nat. Commun. 2024, 15, 3981. [Google Scholar] [CrossRef] [PubMed]
- Stracy, M.; Wollman, A.J.M.; Kaja, E.; Gapinski, J.; Lee, J.E.; Leek, V.A.; McKie, S.J.; Mitchenall, L.A.; Maxwell, A.; Sherratt, D.J.; et al. Single-molecule imaging of DNA gyrase activity in living Escherichia coli. Nucleic Acids Res. 2019, 47, 210–220. [Google Scholar] [CrossRef] [PubMed]
- Thornton, M.; Armitage, M.; Maxwell, A.; Dosanjh, B.; Howells, A.J.; Norris, V.; Sigee, D.C. Immunogold localization of GyrA and GyrB proteins in Escherichia coli. Microbiology 1994, 140, 2371–2382. [Google Scholar] [CrossRef] [PubMed]
- Garoff, L.; Yadav, K.; Hughes, D. Increased expression of Qnr is sufficient to confer clinical resistance to ciprofloxacin in Escherichia coli. J. Antimicrob. Chemother. 2018, 73, 348–352. [Google Scholar] [CrossRef] [PubMed]
- Xiong, X.; Bromley, E.H.; Oelschlaeger, P.; Woolfson, D.N.; Spencer, J. Structural insights into quinolone antibiotic resistance mediated by pentapeptide repeat proteins: Conserved surface loops direct the activity of a Qnr protein from a gram-negative bacterium. Nucleic Acids Res. 2011, 39, 3917–3927. [Google Scholar] [CrossRef] [PubMed]
- Stock, J.B.; Rauch, B.; Roseman, S. Periplasmic space in Salmonella typhimurium and Escherichia coli. J. Biol. Chem. 1977, 252, 7850–7861. [Google Scholar] [CrossRef] [PubMed]
- Garde, S.; Chodisetti, P.K.; Reddy, M. Peptidoglycan: Structure, synthesis, and regulation. EcoSal Plus. 2021, 9, eESP-0010-2020. [Google Scholar] [CrossRef] [PubMed]
- Scheffers, D.J.; Pinho, M.G. Bacterial cell wall synthesis: New insights from localization studies. Microbiol. Mol. Biol. Rev. 2005, 69, 585–607. [Google Scholar] [CrossRef] [PubMed]
- Bailey, B.W.; Stansfeld, P.J. The bacterial cytoskeleton alters the cell membrane in molecular dynamics simulations. Biophys. J. 2024, 123, 374a. [Google Scholar] [CrossRef]
- Pucci, M.J.; Dougherty, T.J. Direct quantitation of the numbers of individual penicillin-binding proteins per cell in Staphylococcus aureus. J. Bacteriol. 2002, 184, 588–591. [Google Scholar] [CrossRef] [PubMed]
- Dougherty, T.J.; Kennedy, K.; Kessler, R.E.; Pucci, M.J. Direct quantitation of the number of individual penicillin-binding proteins per cell in Escherichia coli. J. Bacteriol. 1996, 178, 6110–6115. [Google Scholar] [CrossRef] [PubMed]
- Allam, A.; Maigre, L.; Vergalli, J.; Dumont, E.; Cinquin, B.; Alves de Sousa, R.; Pajovic, J.; Pinet, E.; Smith, N.; Herbeuval, J.P.; et al. Microspectrofluorimetry to dissect the permeation of ceftazidime in Gram-negative bacteria. Sci. Rep. 2017, 7, 986. [Google Scholar] [CrossRef] [PubMed]
- Geyer, C.N.; Fowler, R.C.; Johnson, J.R.; Johnston, B.; Weissman, S.J.; Hawkey, P.; Hanson, N.D. Evaluation of CTX-M steady-state mRNA, mRNA half-life, and protein production in various STs of Escherichia coli. J. Antimicrob. Chemother. 2016, 71, 607–616. [Google Scholar] [CrossRef] [PubMed]
- Vergalli, J.; Dumont, E.; Pajović, J.; Cinquin, B.; Maigre, L.; Masi, M.; Réfrégiers, M.; Pagés, J.M. Spectrofluorimetric quantification of antibiotic drug concentration in bacterial cells for the characterization of translocation across bacterial membranes. Nat. Protoc. 2018, 13, 1348–1361. [Google Scholar] [CrossRef] [PubMed]
- Ukleja, M.; Kricks, L.; Torrens, G.; Peschiera, I.; Rodrigues-Lopes, I.; Krupka, M.; García-Fernández, J.; Melero, R.; Del Campo, R.; Eulalio, A.; et al. Flotillin-mediated stabilization of unfolded proteins in bacterial membrane microdomains. Nat. Commun. 2024, 15, 5583. [Google Scholar] [CrossRef] [PubMed]
- Welling, P.G. Differences between pharmacokinetics and toxicokinetics. Toxicol. Pathol. 1995, 23, 143–147. [Google Scholar] [CrossRef] [PubMed]
- Andersson, D.I.; Hughes, D. Antibiotic resistance and its cost: Is it possible to reverse resistance? Nat. Rev. Microbiol. 2010, 8, 260–271. [Google Scholar] [CrossRef] [PubMed]
- San Millan, A.; MacLean, R.C. Fitness costs of plasmids: A limit to plasmid transmission. Microbiol. Spectr. 2017, 5, e10-1128. [Google Scholar] [CrossRef] [PubMed]
- Baquero, F.; Rodríguez-Beltrán, J.; Coque, T.M.; Del Campo, R. Boosting fitness costs associated with antibiotic resistance in the gut: On the way to biorestoration of susceptible populations. Biomolecules 2024, 14, 76. [Google Scholar] [CrossRef] [PubMed]
- Munita, J.M.; Arias, C.A. Mechanisms of antibiotic resistance. Microbiol. Spectr. 2016, 4, e481–e511. [Google Scholar] [CrossRef] [PubMed]

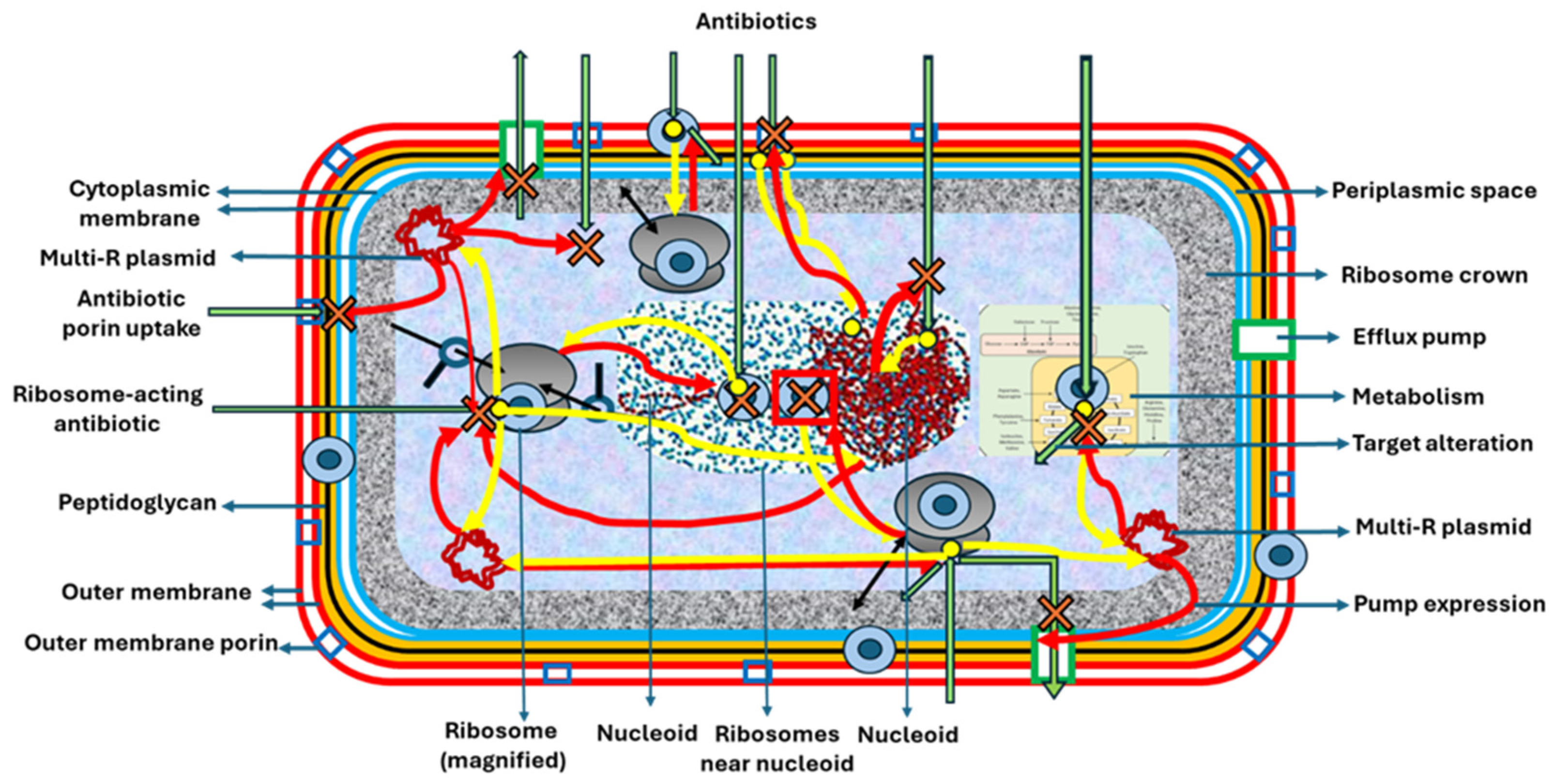

{kind=link}
{kind=link}
| Antibiotics | Primary Detoxifying Effector Molecules |
|---|---|
| Beta-lactams | Beta-lactamases (proteases-hydrolases) |
| Aminoglycosides | Acetyl-transferases, Phospho-transferases, Nucleotydyl-transferases |
| Macrolides, Lincosamides, Streptogramins | Phospho-transferases, Esterases, Nucleotydyl-transferases, Acetyl-transferases, Hydrolases. |
| Phenicols | Acetyltransferases |
| Tetracyclines | Monooxygenases |
| Fluoroquinolones | Acetyl-transferases, Monooxygenases |
| Fosfomycin | Metallo-glutathione-transferases |
| Rifampicin | Glycosyl-transferases, Nucleotydyl-transferases, Phospho-transferases, Monooxygenases |
| Glyco-Lipopeptides | Monooxygenases (?), Deacylases, Serin-protease-hydrolases |
| Sulphonamides | Flavin-Monooxygenases, Flavin-Reductases |
| Antibiotics | Secondary Effector Biomolecules Triggering Antibiotic Detoxification | Detoxification Mechanism |
|---|---|---|
| Beta-lactams | Muropeptides (murein fragments), Transmembrane sensor transducers MicroRNA transcriptases | Induction beta-lactamases Induction beta-lactamases PBP degradation |
| Aminoglycosides | AttC-site integron recombinases 16SrRNA methyl-transferases AcrD efflux pump synthases | Increased acetyl-transferases Increased nucleotidyl-transferases Reduced ribosome binding Efflux pump AcrD |
| Macrolides Lincosamides, Streptogramins | 23S-rRNA methyl-transferase | Reduced ribosome binding |
| Phenicols | 23S-rRNA methyltransferase ATP binding cassette proteins | Reduced ribosome binding |
| Tetracyclines | tetR repressor-tetracycline complex TetM and TetO proteins | Expression efflux pump TetA Tetracycline target displacement |
| Fluoroquinolones | Qnr pentapeptide repeat protein, requiring integration host factors | DNA target protection |
| Fosfomycin | Two-component signal transduction | Decreased uptake |
| Sulfonamides | Two-component signal transduction activated by reduced thymidine levels | Increase in thymidine levels |
| Glyco-Lipopeptides | Two-component signal transduction | d-Ala-d-lac ligase, modifying the target in the cell wall |
| Polymyxins | Two-component signal transduction | Induction of lipid A acetylase, phosphoethanolamine, or 4-amino-4-deoxy-L-arabinose transferases: target modification |
| Oxazolidinones | 23S-rRNA methyltransferase ATP-binding cassette | Reduced ribosome binding Target modification |
| Fusidic acid | Elongation Factor-G-binding protein | Target protection |
| Nitrofurantoin | Two-component signal transduction | Lower transcription of nitroreductases with reduced nitrofurantoin effect. |
| Pharmacokinetics (antimicrobial drugs) | Antechokinetics (resistance molecules) |
| Antibiotic absorption | Expression of the resistance genes |
| Maximal antibiotic concentration (Cmax) | Maximal resistance-effector concentration |
| Drug concentration over time | Effector concentration over time |
| Elimination constant (Ke) | Elimination or degradation of resistance |
| Half-life (t1/2) | Half-life of the resistance mechanism |
| Area under the time curve (AUC) | Area under the time curve of the inhibitor |
| Antibiotic time of exposure over the MIC | Inhibitor time of exposure over the MPC |
| Distribution volume in the body (Vd) | Resistance molecules/bacterial cell volume |
| Antibiotic molecules in the infected site | Resistance molecules in bacterial compartment (i.e., periplasm) |
| Number of microbial molecular targets | Resistance molecules/number of targets |
| Clearance (CL) | Resistance cleared per unit of time |
| Diffusion constraints | Intracellular diffusion constraints |
| Protein binding, non-specific binding | Non-specific binding, self-aggregation |
| Pharmacodynamics (antimicrobial drugs) | Antechodynamics (resistance molecules) |
| Minimal inhibitory concentration (MIC) | Minimal protective concentration (MPC) |
| Cellular target substrate affinity (Km) | Antibiotic substrate affinity |
| Maximum rate of action on target (Vmax) | Maximum rate of antibiotic inactivation |
| Antibiotic bioavailability | Resistance molecule bioavailability |
| Hill function (dose–response curve) | Resistance expression and cell protection |
| Reversibility of the effect (bacteriostasis) | Reversibility of the resistance mechanism |
| Synergy, antagonism between antibiotics | Synergy, antagonism between resistances |
| Minimal antibiotic toxic concentration | Minimal concentration reducing fitness |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Baquero, F.; Cantón, R.; Pérez-Cobas, A.E.; Coque, T.M.; Levin, B.; Rodríguez-Beltrán, J. Antechodynamics and Antechokinetics: Dynamics and Kinetics of Antibiotic Resistance Biomolecules. Biomolecules 2025, 15, 823. https://doi.org/10.3390/biom15060823
Baquero F, Cantón R, Pérez-Cobas AE, Coque TM, Levin B, Rodríguez-Beltrán J. Antechodynamics and Antechokinetics: Dynamics and Kinetics of Antibiotic Resistance Biomolecules. Biomolecules. 2025; 15(6):823. https://doi.org/10.3390/biom15060823
Chicago/Turabian StyleBaquero, F., R. Cantón, A. E. Pérez-Cobas, T. M. Coque, B. Levin, and J. Rodríguez-Beltrán. 2025. "Antechodynamics and Antechokinetics: Dynamics and Kinetics of Antibiotic Resistance Biomolecules" Biomolecules 15, no. 6: 823. https://doi.org/10.3390/biom15060823
APA StyleBaquero, F., Cantón, R., Pérez-Cobas, A. E., Coque, T. M., Levin, B., & Rodríguez-Beltrán, J. (2025). Antechodynamics and Antechokinetics: Dynamics and Kinetics of Antibiotic Resistance Biomolecules. Biomolecules, 15(6), 823. https://doi.org/10.3390/biom15060823