
Brusatol Inhibits Esophageal Squamous Cell Carcinoma Tumorigenesis Through Bad-Mediated Mitochondrial Apoptosis Induction and Anti-Metastasis by Targeting Akt1

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Reagents
2.2. Cell Culture
2.3. Apoptosis Assays
2.4. Cell Counting Kit-8 (CCK-8) Assay
2.5. Wound-Healing Assay
2.6. Transwell Assay
2.7. Quantitative Real-Time Polymerase Chain Reaction (qRT-PCR)
2.8. Mitochondria Isolation and Mitochondrial Membrane Potential (MMP) Analyses
2.9. Network–Pharmacology Analysis for Bru
2.9.1. Potential Target Screening
2.9.2. Protein-Protein Interaction (PPI) Network and Pathway Enrichment
2.9.3. Western Blotting (WB)
2.9.4. Immunofluorescence (IF) Assay
2.9.5. Drug Affinity Responsive Target Stability (DARTS) Assay
2.9.6. Cellular Thermal Shift Assay (CETSA)
2.9.7. Molecular Docking
2.10. Animal Experiments
2.11. Tissue Staining
2.12. Statistical Analysis
3. Results
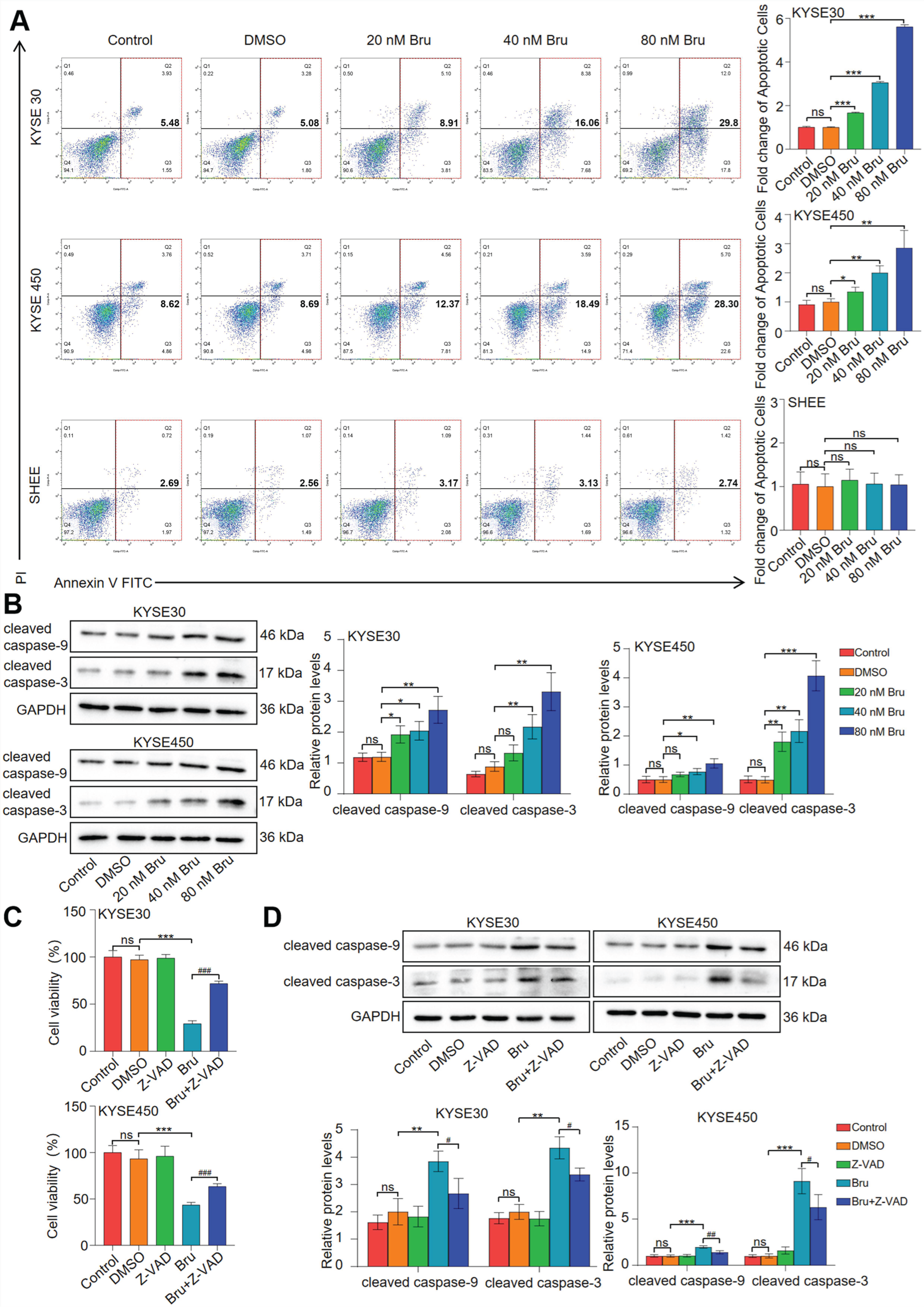
3.1. Bru Induced Caspase-9/3-Dependent Apoptosis in ESCC Cells
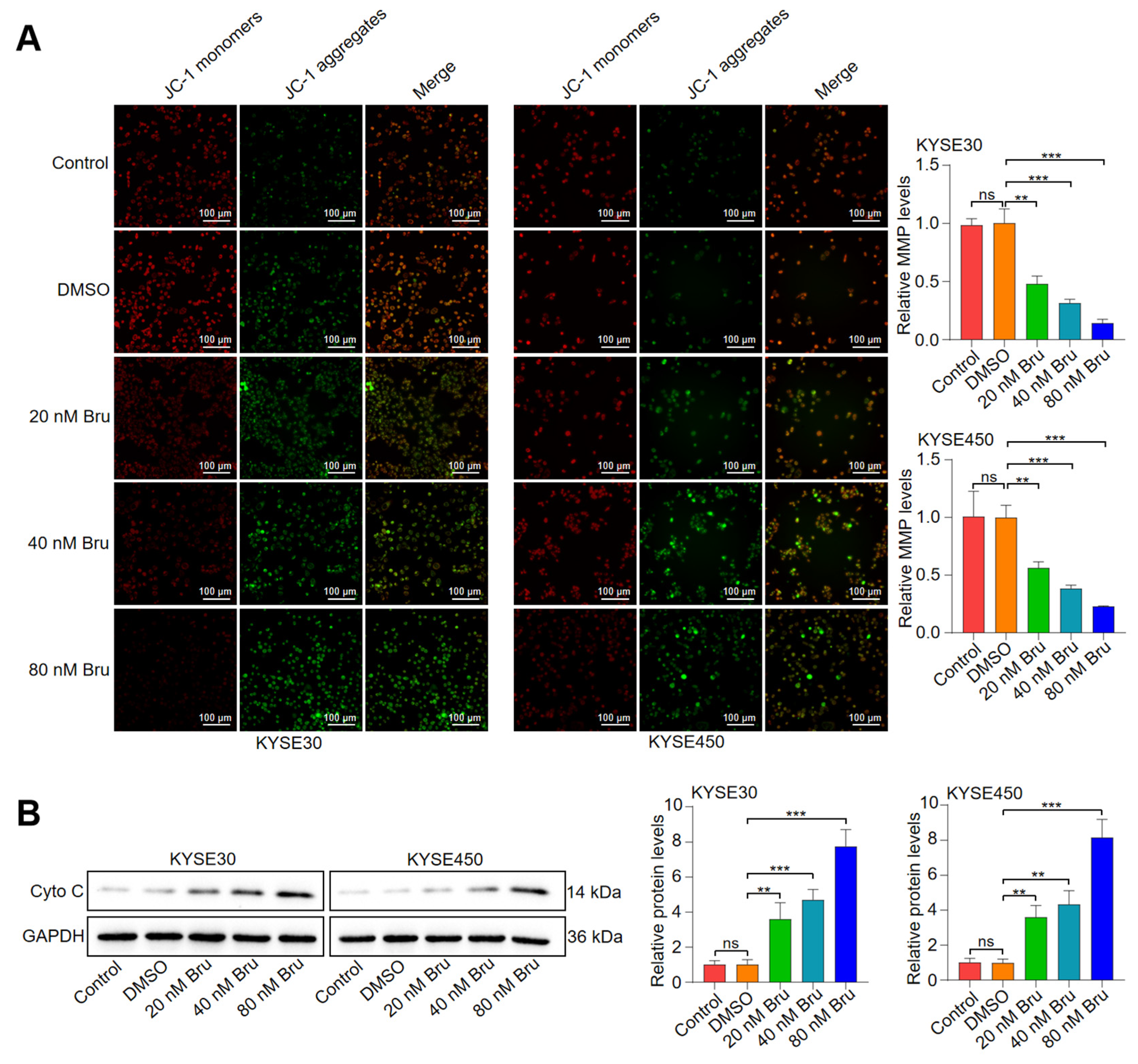
3.2. Bru-Mediated Apoptosis Relied on Mitochondrial Dysfunction in ESCC Cells
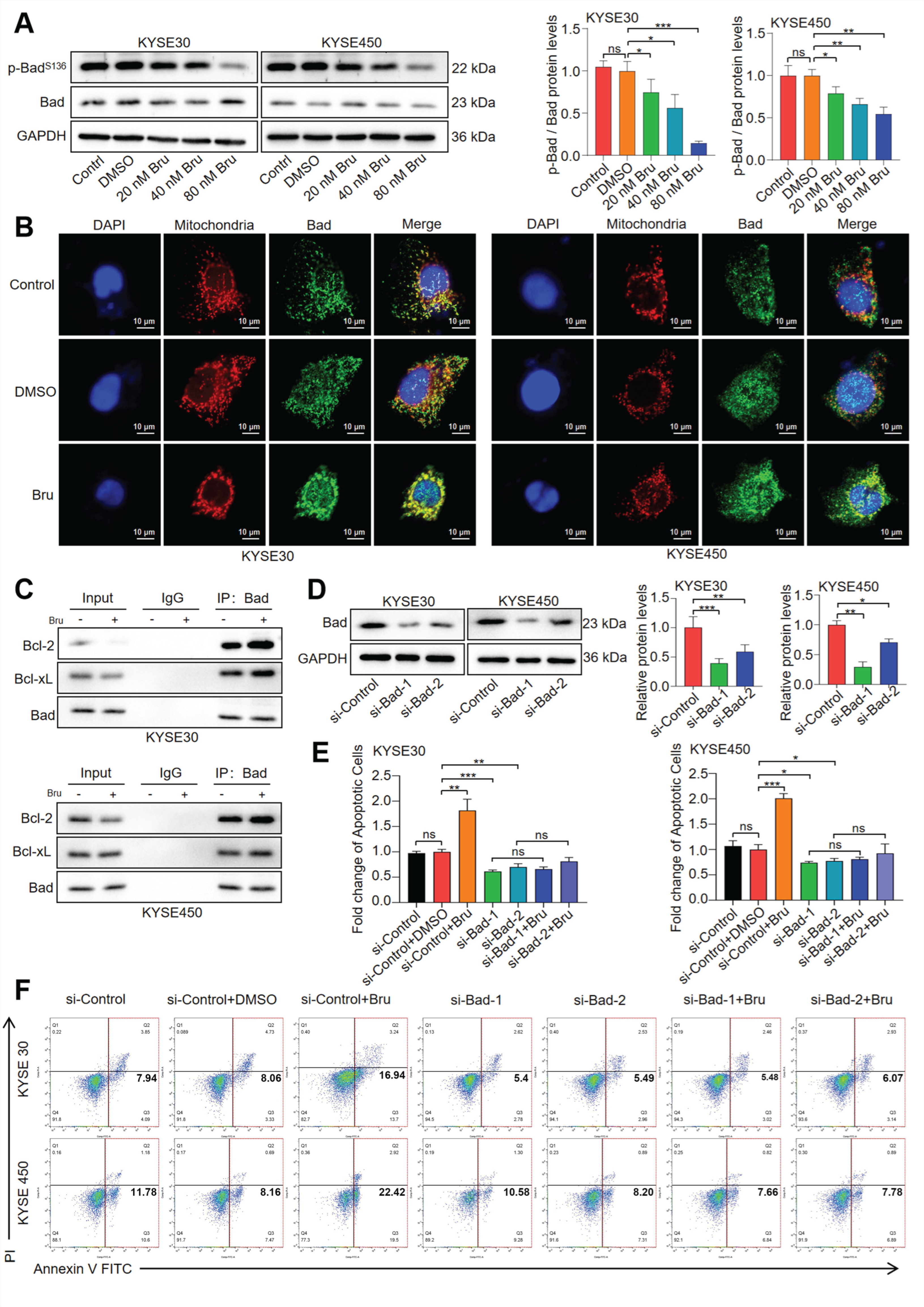
3.3. The Dephosphorylation of Bad Led to Bru-Mediated Apoptosis in ESCC Cells
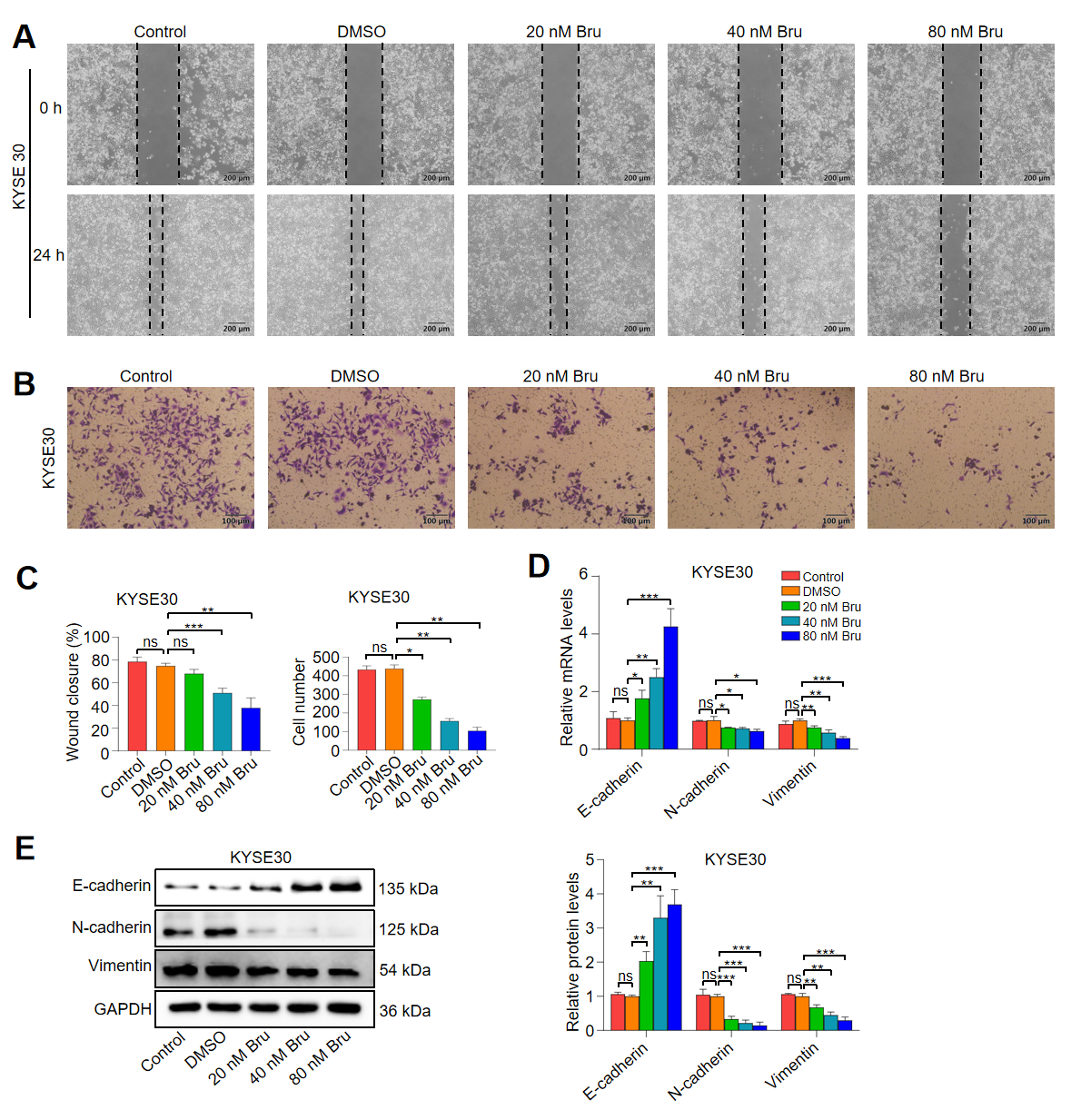
3.4. Bru Exhibited the Anti-Metastasis Activity in ESCC Cells
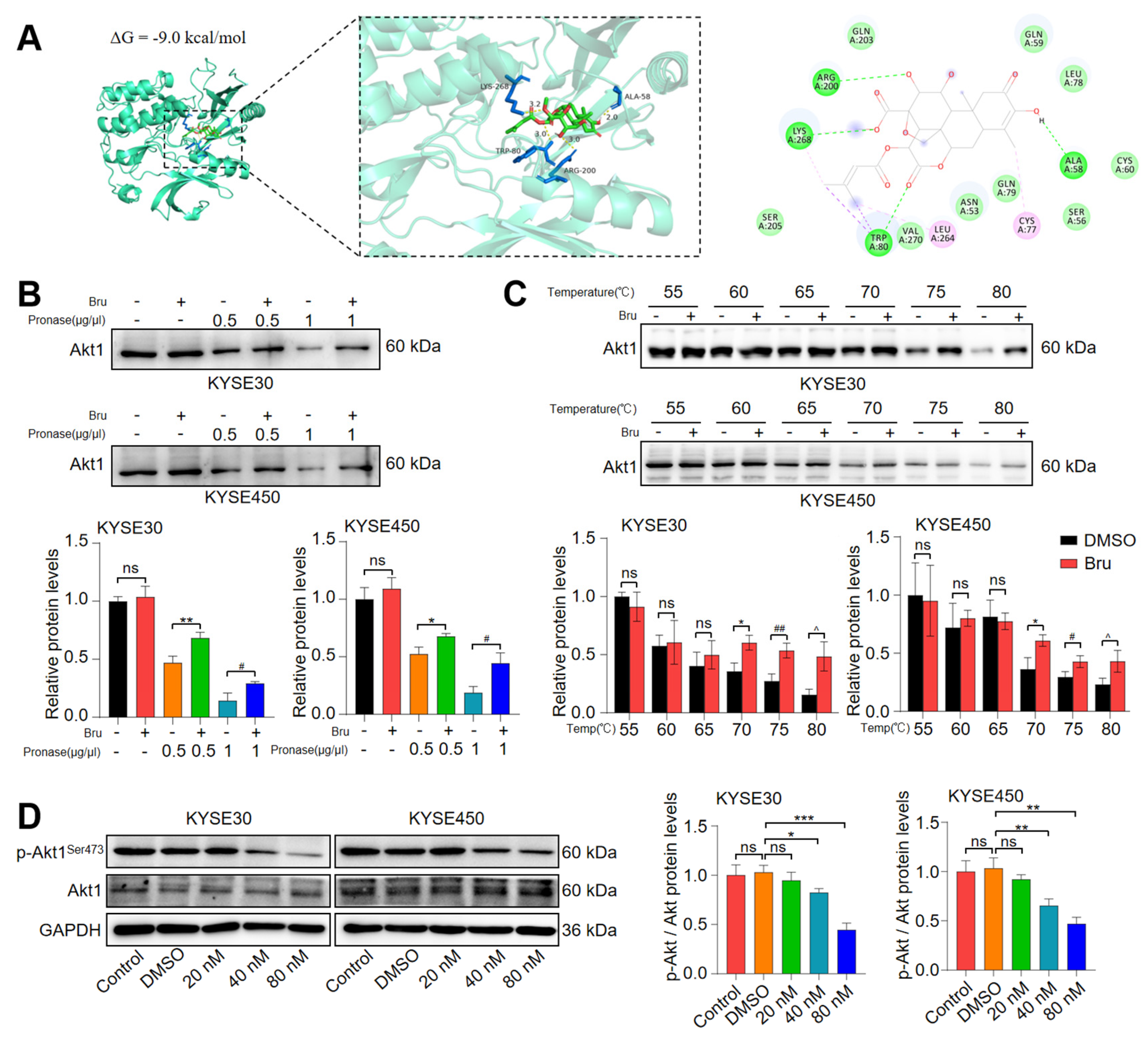
3.5. Akt1 Was a Direct Target of Bru
3.6. Bru Exhibited Mitochondria-Mediated Apoptosis via Inhibiting Akt1-Phospho-Bad Pathway in ESCC Cells
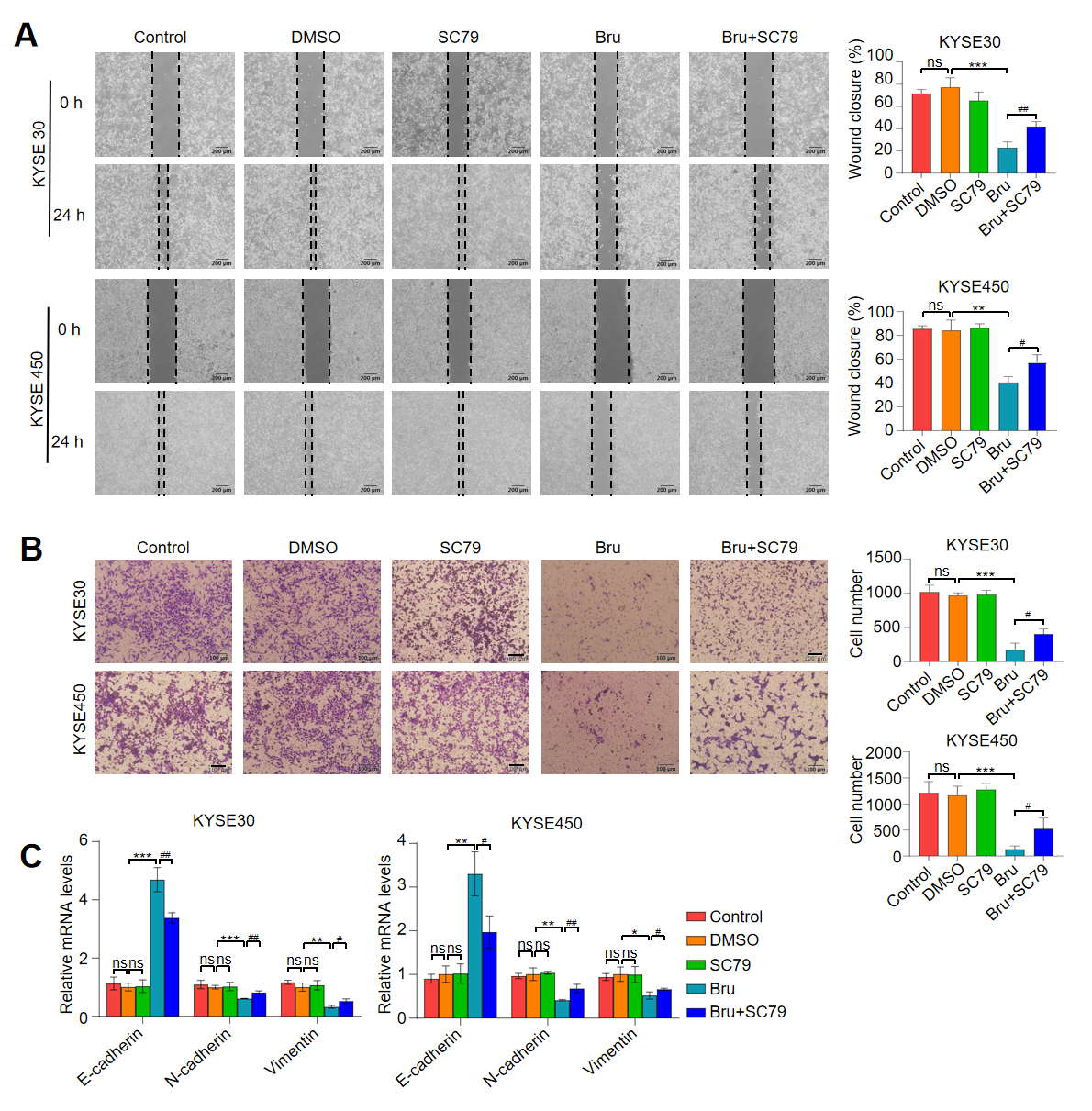
3.7. Activation of Akt1 Pathway Attenuated the Anti-Metastasis Activity of Bru in ESCC Cells In Vitro
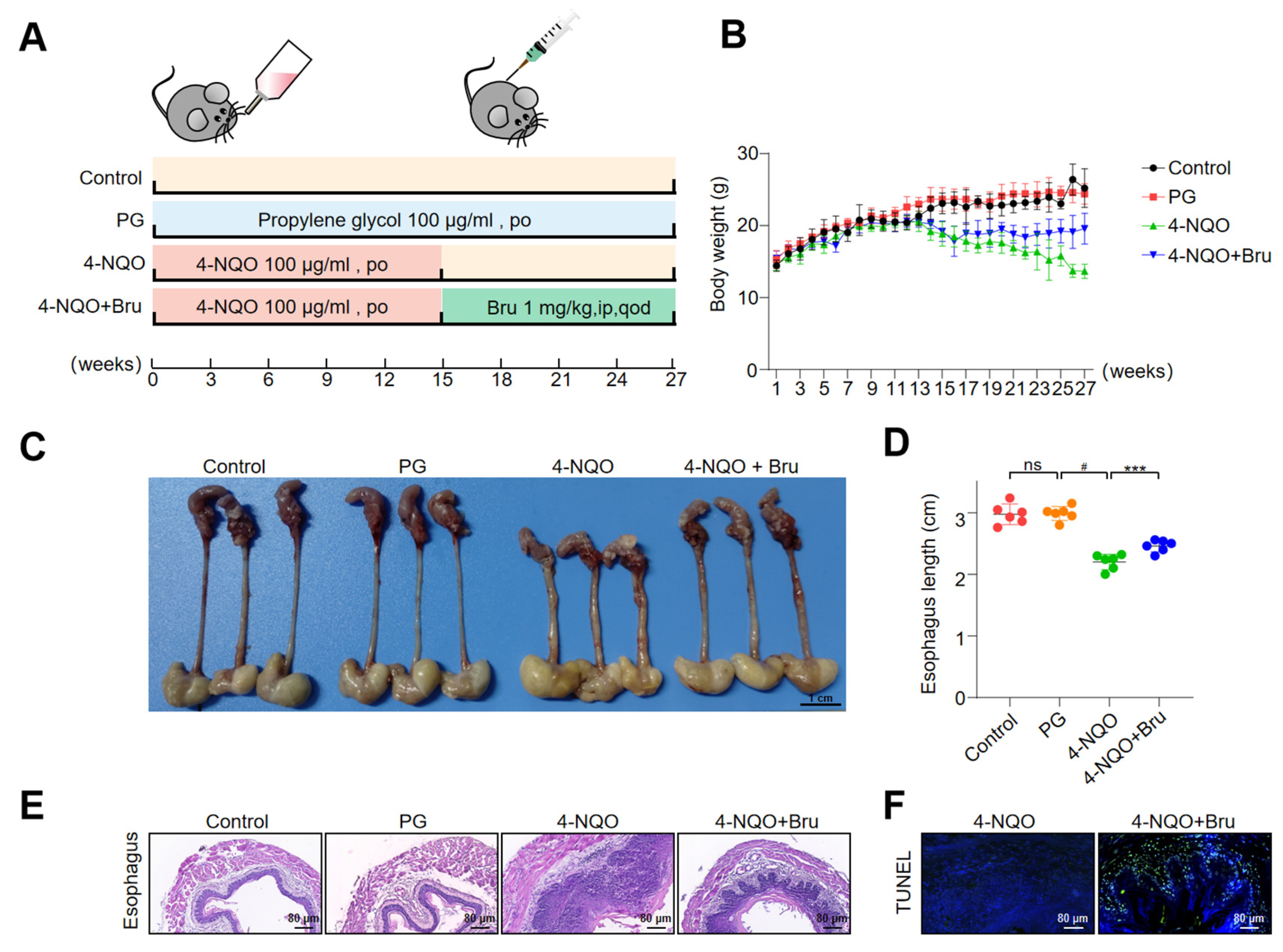
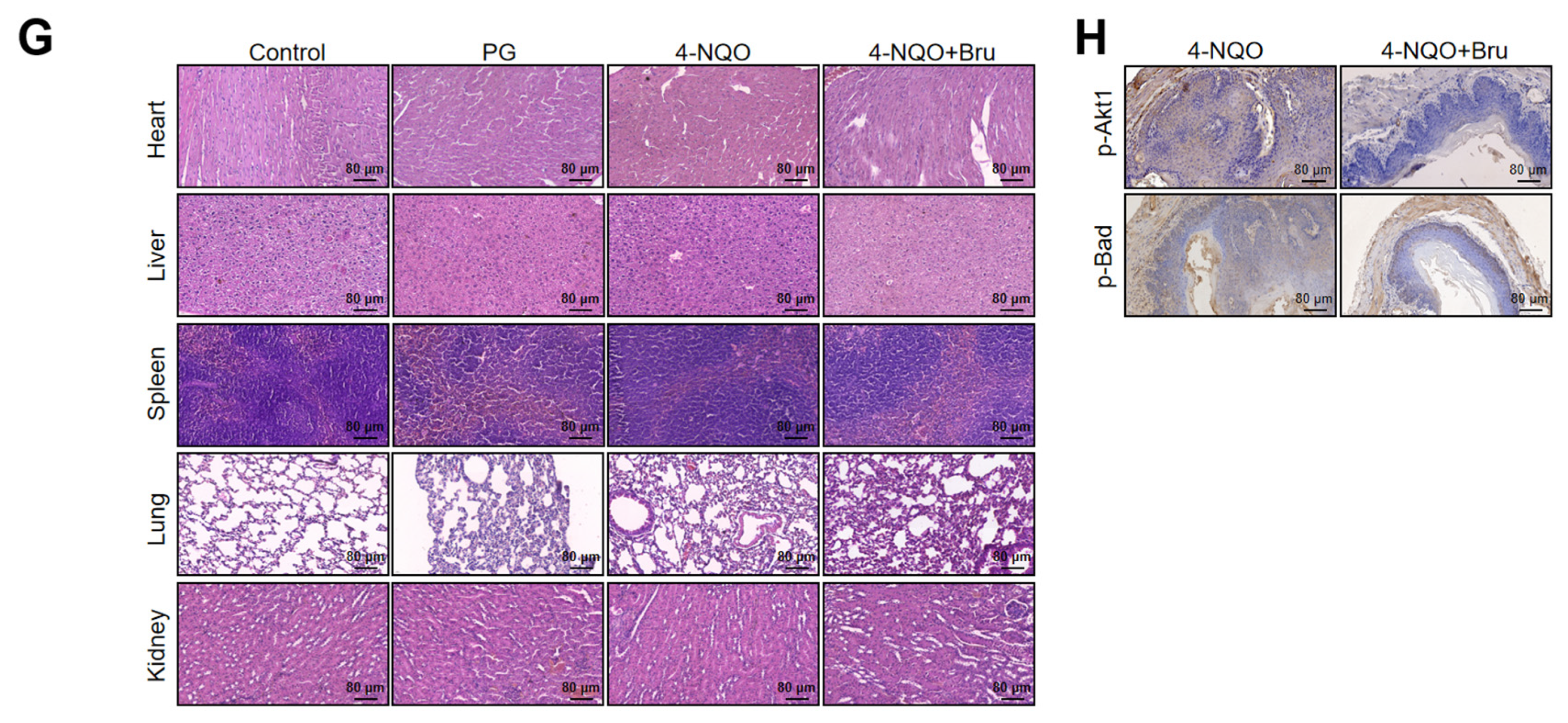
3.8. Bru Retarded ESCC Tumorigenesis
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| Akt1 | Akt serine/threonine kinase 1 gene |
| Bcl-2 | B-cell lymphoma-2 |
| Bru | Brusatol |
| CCK8 | Cell Counting Kit-8 |
| CETSA | Cellular thermal shift assay |
| Cyto C | Cytochrome C |
| DARTS | Drug affinity responsive target stability |
| EC | Esophageal carcinoma |
| EMT | Epithelial–mesenchymal transition |
| ESCC | Esophageal squamous cell carcinoma |
| FBS | Fetal bovine serum |
| GO | Gene ontology |
| H&E | Hematoxylin and eosin |
| IF | Immunofluorescence |
| IHC | Immunohistochemistry |
| IP | Immunoprecipitation |
| KEGG | Kyoto Encyclopedia of Genes and Genomes |
| MMP | Mitochondrial membrane potential |
| NRF2 | Nuclear factor erythroid 2-related factor 2 |
| PPI | Protein-protein interaction |
| qRT-PCR | Quantitative real-time polymerase chain reaction |
| SDS | Sodium dodecyl sulfate |
| WB | Western blot |
| Z-VAD | Z-VAD-FMK |
References
- Tirumani, H.; Rosenthal, M.H.; Tirumani, S.H.; Shinagare, A.B.; Krajewski, K.M.; Ramaiya, N.H. Esophageal Carcinoma: Current Concepts in the Role of Imaging in Staging and Management. Can. Assoc. Radiol. J. 2015, 66, 130–139. [Google Scholar] [CrossRef] [PubMed]
- Li, C.Y.; Zhang, W.W.; Xiang, J.L.; Wang, X.H.; Li, J.; Wang, J.L. Identification of microRNAs as novel biomarkers for esophageal squamous cell carcinoma: A study based on The Cancer Genome Atlas (TCGA) and bioinformatics. Chin. Med. J. 2019, 132, 2213–2222. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.J.; Zhang, J.X.; Wang, Z.Z.; Li, P.; Shang, J.W.; Guo, Y.J. Prognostic evaluation of human papillomavirus and p16 in esophageal squamous cell carcinoma. Chin. Med. J. 2020, 133, 751–752. [Google Scholar] [CrossRef] [PubMed]
- Domper Arnal, M.J.; Ferrandez Arenas, A.; Lanas Arbeloa, A. Esophageal cancer: Risk factors, screening and endoscopic treatment in Western and Eastern countries. World J. Gastroenterol. 2015, 21, 7933–7943. [Google Scholar] [CrossRef]
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer Statistics, 2017. CA Cancer J. Clin. 2017, 67, 7–30. [Google Scholar] [CrossRef]
- Pennathur, A.; Gibson, M.K.; Jobe, B.A.; Luketich, J.D. Oesophageal carcinoma. Lancet 2013, 381, 400–412. [Google Scholar] [CrossRef]
- Jain, S.; Dhingra, S. Pathology of esophageal cancer and Barrett’s esophagus. Ann. Cardiothorac. Surg. 2017, 6, 99–109. [Google Scholar] [CrossRef]
- Sohda, M.; Ojima, H.; Sano, A.; Fukai, Y.; Kuwano, H. Primary esophageal adenocarcinoma with distant metastasis to the skeletal muscle. Int. Surg. 2014, 99, 650–655. [Google Scholar] [CrossRef]
- Chen, E.D.; Cheng, P.; Yan, X.Q.; Ye, Y.L.; Chen, C.Z.; Ji, X.H.; Zhang, X.H. Metastasis of distal esophageal carcinoma to the thyroid with presentation simulating primary thyroid carcinoma: A case report and review of the literature. World J. Surg. Oncol. 2014, 12, 106. [Google Scholar] [CrossRef]
- Isono, K.; Sato, H.; Nakayama, K. Results of a nationwide study on the three-field lymph node dissection of esophageal cancer. Oncology 1991, 48, 411–420. [Google Scholar] [CrossRef]
- Xi, K.; Chen, W.; Yu, H. The Prognostic Value of Log Odds of Positive Lymph Nodes in Early-Stage Esophageal Cancer Patients: A Study Based on the SEER Database and a Chinese Cohort. J. Oncol. 2021, 2021, 8834912. [Google Scholar] [CrossRef] [PubMed]
- Harada, K.; Pool Pizzi, M.; Baba, H.; Shanbhag, N.D.; Song, S.; Ajani, J.A. Cancer stem cells in esophageal cancer and response to therapy. Cancer 2018, 124, 3962–3964. [Google Scholar] [CrossRef]
- Ma, L.; Zhang, M.; Zhao, R.; Wang, D.; Ma, Y.; Li, A. Plant Natural Products: Promising Resources for Cancer Chemoprevention. Molecules 2021, 26, 933. [Google Scholar] [CrossRef]
- Nobili, S.; Lippi, D.; Witort, E.; Donnini, M.; Bausi, L.; Mini, E.; Capaccioli, S. Natural compounds for cancer treatment and prevention. Pharmacol. Res. 2009, 59, 365–378. [Google Scholar] [CrossRef]
- Sun, X.; Wang, Q.; Wang, Y.; Du, L.; Xu, C.; Liu, Q. Brusatol Enhances the Radiosensitivity of A549 Cells by Promoting ROS Production and Enhancing DNA Damage. Int. J. Mol. Sci. 2016, 17, 997. [Google Scholar] [CrossRef]
- Zhan, Y.; Tan, T.; Qian, K.; Yang, S.; Feng, Y.; Wen, Q. Quassinoids from seeds of Brucea Javanica and their anticomplement activities. Nat. Prod. Res. 2020, 34, 1186–1191. [Google Scholar] [CrossRef]
- Cai, S.J.; Liu, Y.; Han, S.; Yang, C. Brusatol, an NRF2 inhibitor for future cancer therapeutic. Cell Biosci. 2019, 9, 45. [Google Scholar] [CrossRef]
- Ren, D.; Villeneuve, N.F.; Jiang, T.; Wu, T.; Lau, A.; Toppin, H.A.; Zhang, D.D. Brusatol enhances the efficacy of chemotherapy by inhibiting the Nrf2-mediated defense mechanism. Proc. Natl. Acad. Sci. USA 2011, 108, 1433–1438. [Google Scholar] [CrossRef]
- Olayanju, A.; Copple, I.M.; Bryan, H.K.; Edge, G.T.; Sison, R.L.; Wong, M.W.; Lai, Z.Q.; Lin, Z.X.; Dunn, K.; Sanderson, C.M.; et al. Brusatol provokes a rapid and transient inhibition of Nrf2 signaling and sensitizes mammalian cells to chemical toxicity-implications for therapeutic targeting of Nrf2. Free Radic. Biol. Med. 2015, 78, 202–212. [Google Scholar] [CrossRef]
- Zhang, X.; Wang, Y.; Velkov, T.; Tang, S.; Dai, C. T-2 toxin-induced toxicity in neuroblastoma-2a cells involves the generation of reactive oxygen, mitochondrial dysfunction and inhibition of Nrf2/HO-1 pathway. Food Chem. Toxicol. 2018, 114, 88–97. [Google Scholar] [CrossRef]
- Zhu, X.; Huang, N.; Ji, Y.; Sheng, X.; Huo, J.; Zhu, Y.; Huang, M.; He, W.; Ma, J. Brusatol induces ferroptosis in oesophageal squamous cell carcinoma by repressing GSH synthesis and increasing the labile iron pool via inhibition of the NRF2 pathway. Biomed. Pharmacother. 2023, 167, 115567. [Google Scholar] [CrossRef] [PubMed]
- Carneiro, B.A.; El-Deiry, W.S. Targeting apoptosis in cancer therapy. Nat. Rev. Clin. Oncol. 2020, 17, 395–417. [Google Scholar] [CrossRef] [PubMed]
- Yang, E.; Zha, J.; Jockel, J.; Boise, L.H.; Thompson, C.B.; Korsmeyer, S.J. Bad, a heterodimeric partner for Bcl-XL and Bcl-2, displaces Bax and promotes cell death. Cell 1995, 80, 285–291. [Google Scholar] [CrossRef] [PubMed]
- Craik, A.C.; Veldhoen, R.A.; Czernick, M.; Buckland, T.W.; Kyselytzia, K.; Ghosh, S.; Lai, R.; Damaraju, S.; Underhill, D.A.; Mackey, J.R.; et al. The BH3-only protein Bad confers breast cancer taxane sensitivity through a nonapoptotic mechanism. Oncogene 2010, 29, 5381–5391. [Google Scholar] [CrossRef]
- Wang, T.; Chen, Z.; Chen, H.; Yu, X.; Wang, L.; Liu, X. Brusatol inhibits the growth of renal cell carcinoma by regulating the PTEN/PI3K/AKT pathway. J. Ethnopharmacol. 2022, 288, 115020. [Google Scholar] [CrossRef]
- Chen, Z.; He, B.; Zhao, J.; Li, J.; Zhu, Y.; Li, L.; Bao, W.; Zheng, J.; Yu, H.; Chen, G. Brusatol suppresses the growth of intrahepatic cholangiocarcinoma by PI3K/Akt pathway. Phytomedicine 2022, 104, 154323. [Google Scholar] [CrossRef]
- Yin, H.; Chao, L.; Chao, J. Kallikrein/kinin protects against myocardial apoptosis after ischemia/reperfusion via Akt-glycogen synthase kinase-3 and Akt-Bad.14-3-3 signaling pathways. J. Biol. Chem. 2005, 280, 8022–8030. [Google Scholar] [CrossRef]
- Szanto, A.; Bognar, Z.; Szigeti, A.; Szabo, A.; Farkas, L.; Gallyas, F., Jr. Critical role of bad phosphorylation by Akt in cytostatic resistance of human bladder cancer cells. Anticancer Res. 2009, 29, 159–164. [Google Scholar]
- Burnette, W.N. “Western blotting”: Electrophoretic transfer of proteins from sodium dodecyl sulfate-polyacrylamide gels to unmodified nitrocellulose and radiographic detection with antibody and radioiodinated protein A. Anal. Biochem. 1981, 112, 195–203. [Google Scholar] [CrossRef]
- Chaffer, C.L.; Weinberg, R.A. A perspective on cancer cell metastasis. Science 2011, 331, 1559–1564. [Google Scholar] [CrossRef]
- Shibue, T.; Weinberg, R.A. EMT, CSCs, and drug resistance: The mechanistic link and clinical implications. Nat. Rev. Clin. Oncol. 2017, 14, 611–629. [Google Scholar] [CrossRef] [PubMed]
- Pastushenko, I.; Blanpain, C. EMT Transition States during Tumor Progression and Metastasis. Trends Cell Biol. 2019, 29, 212–226. [Google Scholar] [CrossRef]
- Ackler, S.; Ahmad, S.; Tobias, C.; Johnson, M.D.; Glazer, R.I. Delayed mammary gland involution in MMTV-AKT1 transgenic mice. Oncogene 2002, 21, 198–206. [Google Scholar] [CrossRef]
- Wang, J.; Ni, J.; Song, D.; Ding, M.; Huang, J.; Li, W.; He, G. MAT1 facilitates the lung metastasis of osteosarcoma through upregulation of AKT1 expression. Life Sci. 2019, 234, 116771. [Google Scholar] [CrossRef]
- Huang, L.; Zhang, X.O.; Rozen, E.J.; Sun, X.; Sallis, B.; Verdejo-Torres, O.; Wigglesworth, K.; Moon, D.; Huang, T.; Cavaretta, J.P.; et al. PRMT5 activates AKT via methylation to promote tumor metastasis. Nat. Commun. 2022, 13, 3955. [Google Scholar] [CrossRef]
- Gabbouj, S.; Ryhanen, S.; Marttinen, M.; Wittrahm, R.; Takalo, M.; Kemppainen, S.; Martiskainen, H.; Tanila, H.; Haapasalo, A.; Hiltunen, M.; et al. Altered Insulin Signaling in Alzheimer’s Disease Brain—Special Emphasis on PI3K-Akt Pathway. Front. Neurosci. 2019, 13, 629. [Google Scholar] [CrossRef]
- Na, D.; Lim, D.H.; Hong, J.S.; Lee, H.M.; Cho, D.; Yu, M.S.; Shaker, B.; Ren, J.; Lee, B.; Song, J.G.; et al. A multi-layered network model identifies Akt1 as a common modulator of neurodegeneration. Mol. Syst. Biol. 2023, 19, e11801. [Google Scholar] [CrossRef]
- Ferlay, J.; Soerjomataram, I.; Dikshit, R.; Eser, S.; Mathers, C.; Rebelo, M.; Parkin, D.M.; Forman, D.; Bray, F. Cancer incidence and mortality worldwide: Sources, methods and major patterns in GLOBOCAN 2012. Int. J. Cancer 2015, 136, E359–E386. [Google Scholar] [CrossRef]
- Knapp, K.; Verchio, V.; Coburn-Flynn, O.; Li, Y.; Xiong, Z.; Morrison, J.C.; Shersher, D.D.; Spitz, F.; Chen, X. Exploring cell competition for the prevention and therapy of esophageal squamous cell carcinoma. Biochem. Pharmacol. 2023, 214, 115639. [Google Scholar] [CrossRef]
- Liu, Y.; Chen, H.; Sun, Z.; Chen, X. Molecular mechanisms of ethanol-associated oro-esophageal squamous cell carcinoma. Cancer Lett. 2015, 361, 164–173. [Google Scholar] [CrossRef]
- Ilson, D.H.; van Hillegersberg, R. Management of Patients with Adenocarcinoma or Squamous Cancer of the Esophagus. Gastroenterology 2018, 154, 437–451. [Google Scholar] [CrossRef] [PubMed]
- Xing, S.; Nong, F.; Wang, Y.; Huang, D.; Qin, J.; Chen, Y.F.; He, D.H.; Wu, P.E.; Huang, H.; Zhan, R.; et al. Brusatol has therapeutic efficacy in non-small cell lung cancer by targeting Skp1 to inhibit cancer growth and metastasis. Pharmacol. Res. 2022, 176, 106059. [Google Scholar] [CrossRef] [PubMed]
- Yu, X.; He, Z.; Wang, Z.; Ke, S.; Wang, H.; Wang, Q.; Li, S. Brusatol hinders the progression of bladder cancer by Chac1/Nrf2/SLC7A11 pathway. Exp. Cell Res. 2024, 438, 114053. [Google Scholar] [CrossRef]
- Gong, Z.; Xue, L.; Vlantis, A.C.; van Hasselt, C.A.; Chan, J.Y.K.; Fang, J.; Wang, R.; Yang, Y.; Li, D.; Zeng, X.; et al. Brusatol attenuated proliferation and invasion induced by KRAS in differentiated thyroid cancer through inhibiting Nrf2. J. Endocrinol. Investig. 2024, 47, 1271–1280. [Google Scholar] [CrossRef]
- Xiang, Y.; Ye, W.; Huang, C.; Lou, B.; Zhang, J.; Yu, D.; Huang, X.; Chen, B.; Zhou, M. Brusatol inhibits growth and induces apoptosis in pancreatic cancer cells via JNK/p38 MAPK/NF-kappab/Stat3/Bcl-2 signaling pathway. Biochem. Biophys. Res. Commun. 2017, 487, 820–826. [Google Scholar] [CrossRef]
- Salomoni, P.; Condorelli, F.; Sweeney, S.M.; Calabretta, B. Versatility of BCR/ABL-expressing leukemic cells in circumventing proapoptotic BAD effects. Blood 2000, 96, 676–684. [Google Scholar] [CrossRef]
- Shi, Y.; Yuan, Q.; Chen, Y.; Li, X.; Zhou, Y.; Zhou, H.; Peng, F.; Jiang, Y.; Qiao, Y.; Zhao, J.; et al. Corynoline inhibits esophageal squamous cell carcinoma growth via targeting Pim-3. Phytomedicine 2024, 123, 155235. [Google Scholar] [CrossRef]
- Zhao, J.; Ma, X.; Xu, H. miR-29b-3p inhibits 22Rv1 prostate cancer cell proliferation through the YWHAE/BCL-2 regulatory axis. Oncol. Lett. 2022, 24, 289. [Google Scholar] [CrossRef]
- Ke, J.; Bian, X.; Liu, H.; Li, B.; Huo, R. Edaravone reduces oxidative stress and intestinal cell apoptosis after burn through up-regulating miR-320 expression. Mol. Med. 2019, 25, 54. [Google Scholar] [CrossRef]
- Zha, J.; Harada, H.; Yang, E.; Jockel, J.; Korsmeyer, S.J. Serine phosphorylation of death agonist BAD in response to survival factor results in binding to 14-3-3 not BCL-X(L). Cell 1996, 87, 619–628. [Google Scholar] [CrossRef]
- Yan, J.; Xiang, J.; Lin, Y.; Ma, J.; Zhang, J.; Zhang, H.; Sun, J.; Danial, N.N.; Liu, J.; Lin, A. Inactivation of BAD by IKK inhibits TNFalpha-induced apoptosis independently of NF-kappaB activation. Cell 2013, 152, 304–315. [Google Scholar] [CrossRef] [PubMed]
- Cheng, C.; Yuan, F.; Chen, X.P.; Zhang, W.; Zhao, X.L.; Jiang, Z.P.; Zhou, H.H.; Zhou, G.; Cao, S. Inhibition of Nrf2-mediated glucose metabolism by brusatol synergistically sensitizes acute myeloid leukemia to Ara-C. Biomed. Pharmacother. 2021, 142, 111652. [Google Scholar] [CrossRef]
- Lamouille, S.; Xu, J.; Derynck, R. Molecular mechanisms of epithelial-mesenchymal transition. Nat. Rev. Mol. Cell Biol. 2014, 15, 178–196. [Google Scholar] [CrossRef] [PubMed]
- Gupta, G.P.; Massague, J. Cancer metastasis: Building a framework. Cell 2006, 127, 679–695. [Google Scholar] [CrossRef]
- Steeg, P.S. Tumor metastasis: Mechanistic insights and clinical challenges. Nat. Med. 2006, 12, 895–904. [Google Scholar] [CrossRef]
- Li, W.; Zhang, X.; Wu, F.; Zhou, Y.; Bao, Z.; Li, H.; Zheng, P.; Zhao, S. Gastric cancer-derived mesenchymal stromal cells trigger M2 macrophage polarization that promotes metastasis and EMT in gastric cancer. Cell Death Dis. 2019, 10, 918. [Google Scholar] [CrossRef]
- Gonzalez, D.M.; Medici, D. Signaling mechanisms of the epithelial-mesenchymal transition. Sci. Signal. 2014, 7, re8. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ji, Y.; Zhu, X.; Shi, Y.; Fang, R.; Sun, Y.; Ruan, Y.; Zhou, L.; Ge, Y.; Luo, Q.; Zhang, J.; et al. Brusatol Inhibits Esophageal Squamous Cell Carcinoma Tumorigenesis Through Bad-Mediated Mitochondrial Apoptosis Induction and Anti-Metastasis by Targeting Akt1. Biomolecules 2025, 15, 812. https://doi.org/10.3390/biom15060812
Ji Y, Zhu X, Shi Y, Fang R, Sun Y, Ruan Y, Zhou L, Ge Y, Luo Q, Zhang J, et al. Brusatol Inhibits Esophageal Squamous Cell Carcinoma Tumorigenesis Through Bad-Mediated Mitochondrial Apoptosis Induction and Anti-Metastasis by Targeting Akt1. Biomolecules. 2025; 15(6):812. https://doi.org/10.3390/biom15060812
Chicago/Turabian StyleJi, Yao, Xinxin Zhu, Yi Shi, Rui Fang, Yimeng Sun, Yurong Ruan, Liying Zhou, Yuanyuan Ge, Qichao Luo, Junyan Zhang, and et al. 2025. "Brusatol Inhibits Esophageal Squamous Cell Carcinoma Tumorigenesis Through Bad-Mediated Mitochondrial Apoptosis Induction and Anti-Metastasis by Targeting Akt1" Biomolecules 15, no. 6: 812. https://doi.org/10.3390/biom15060812
APA StyleJi, Y., Zhu, X., Shi, Y., Fang, R., Sun, Y., Ruan, Y., Zhou, L., Ge, Y., Luo, Q., Zhang, J., & Ma, J. (2025). Brusatol Inhibits Esophageal Squamous Cell Carcinoma Tumorigenesis Through Bad-Mediated Mitochondrial Apoptosis Induction and Anti-Metastasis by Targeting Akt1. Biomolecules, 15(6), 812. https://doi.org/10.3390/biom15060812