

Non-Coding RNAs: lncRNA, piRNA, and snoRNA as Robust Plasma Biomarkers of Alzheimer’s Disease

 ,
,
Abstract
1. Introduction
2. Materials and Methods
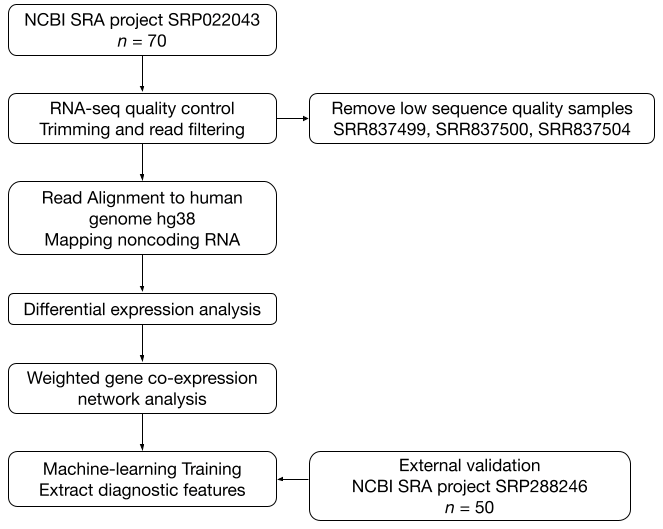
2.1. Data Acquisition
2.2. Quality Control and Preprocessing
2.3. Alignment to Reference Genome and Feature Annotation
2.4. Differential Expression Analysis and WGCNA
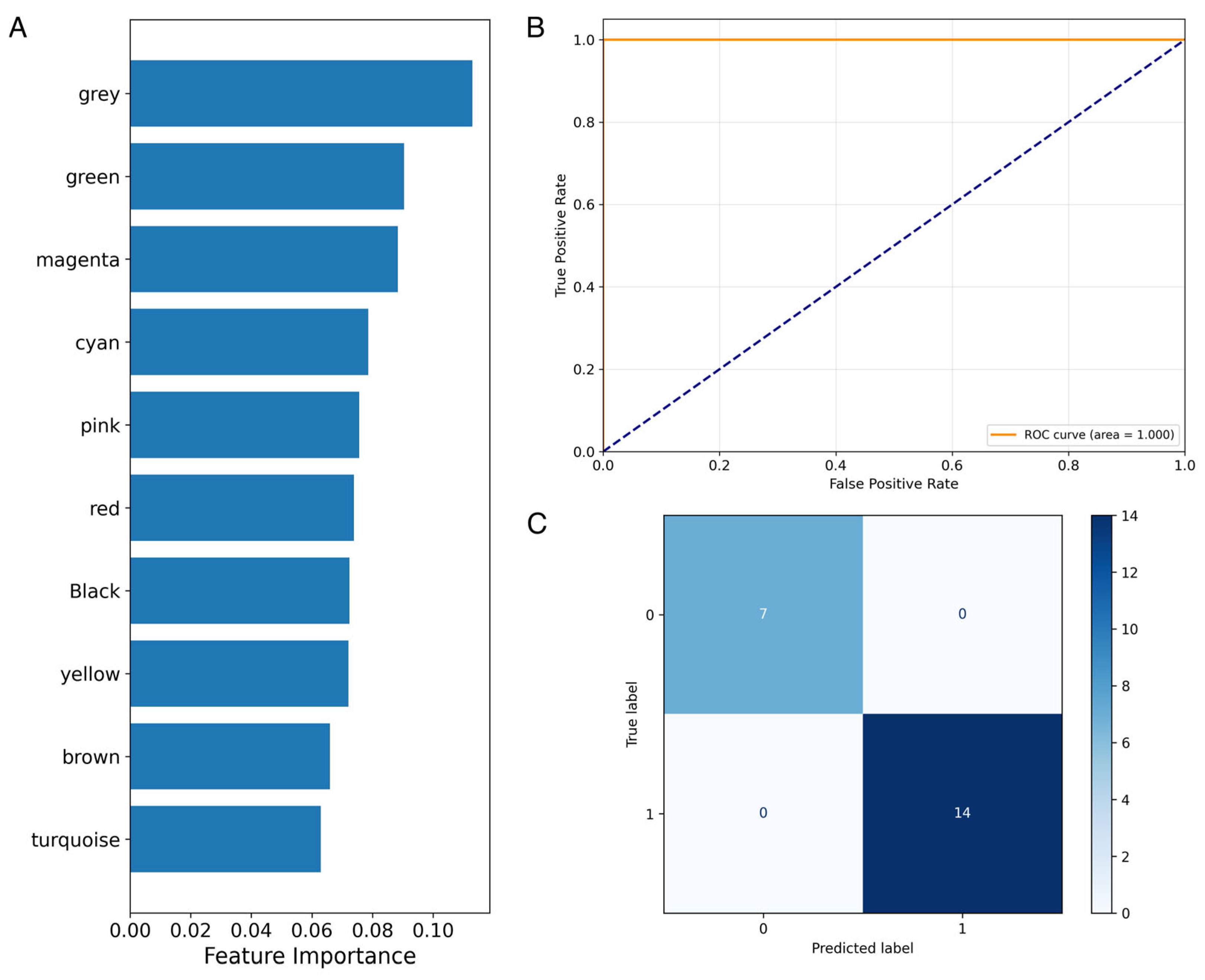
2.5. Machine-Learning Models and Feature Importance
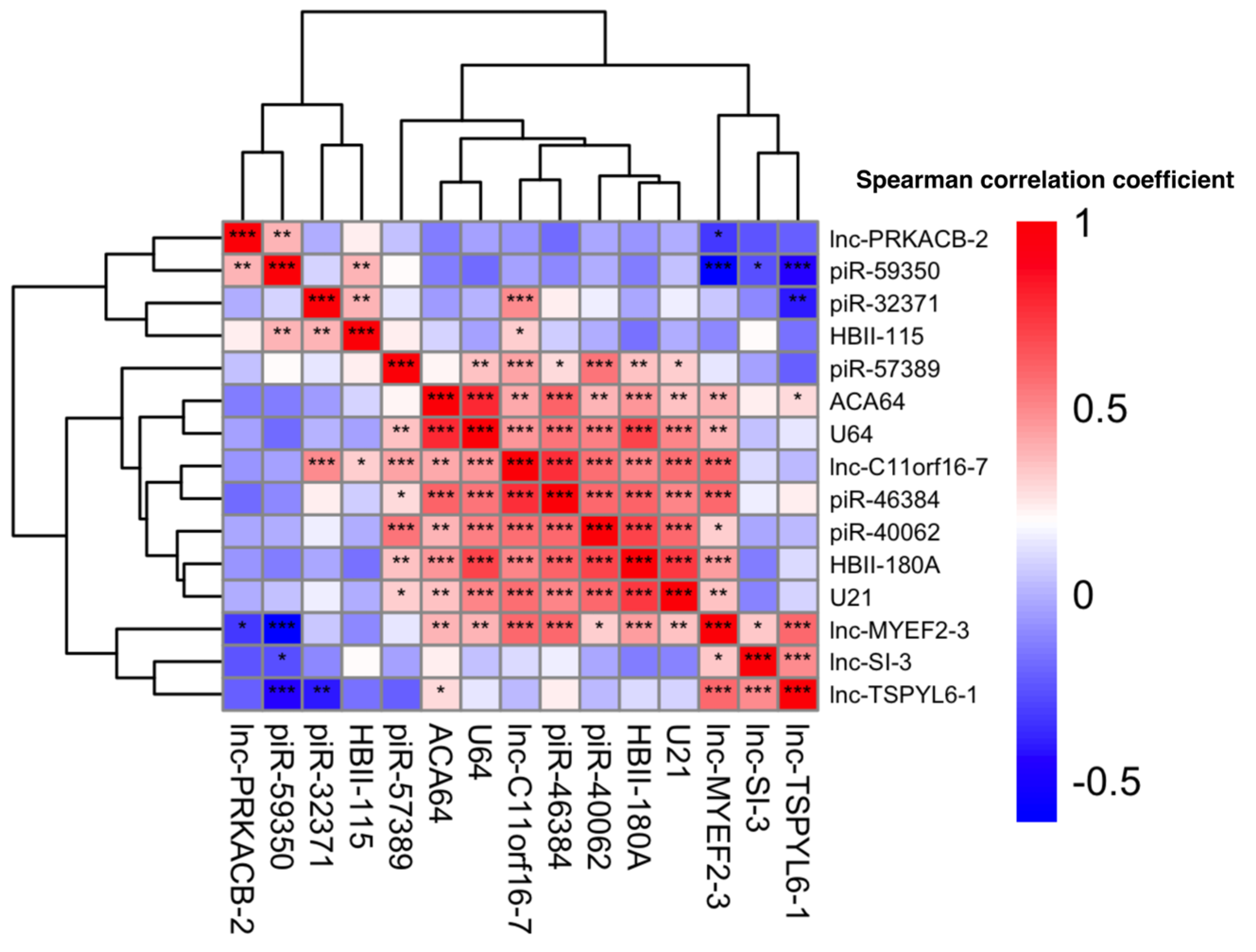
2.6. Pairwise Correlation Analysis and Heatmap Generation
3. Results
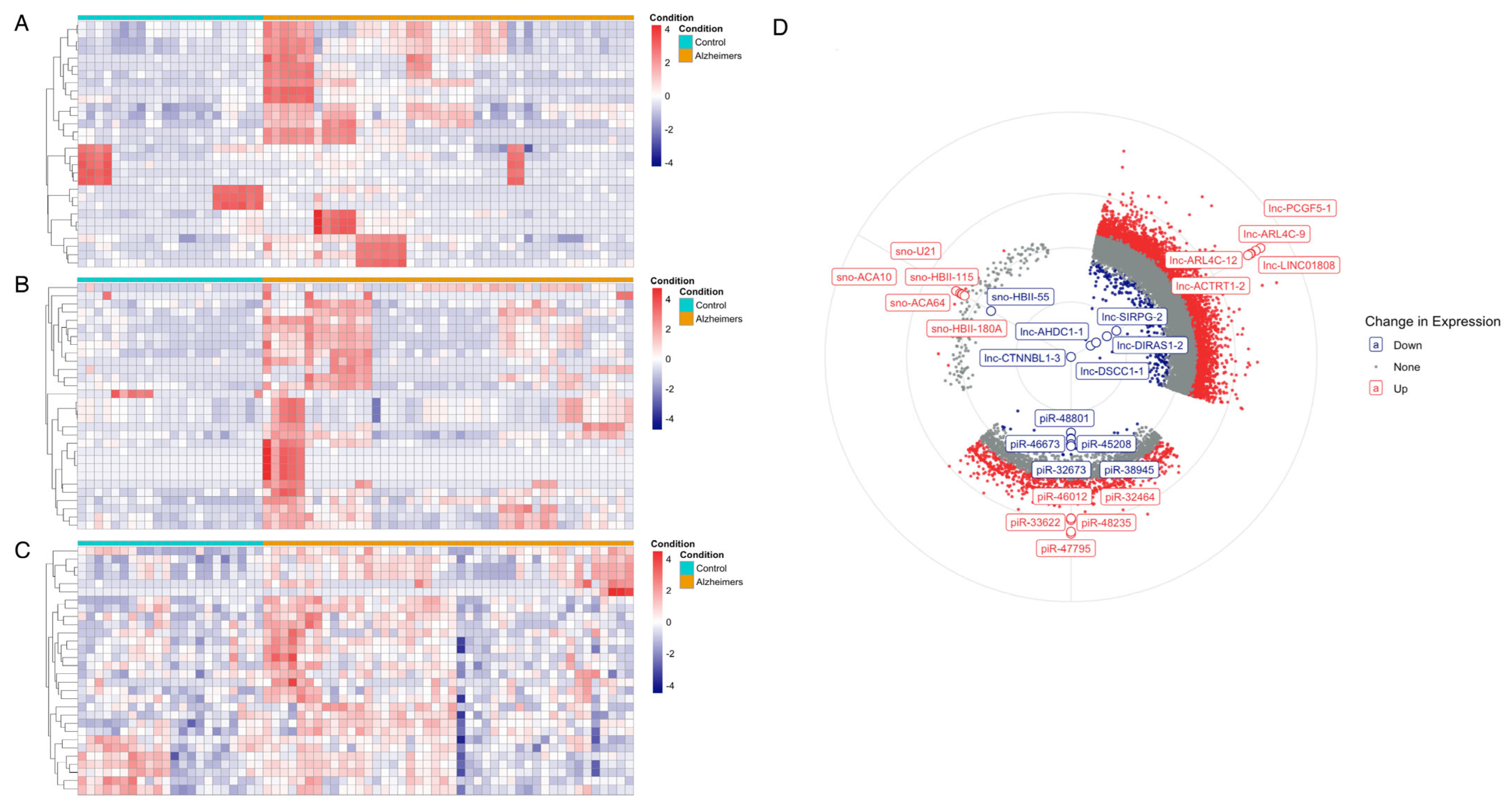
3.1. Differential Expression Analysis of ncRNA
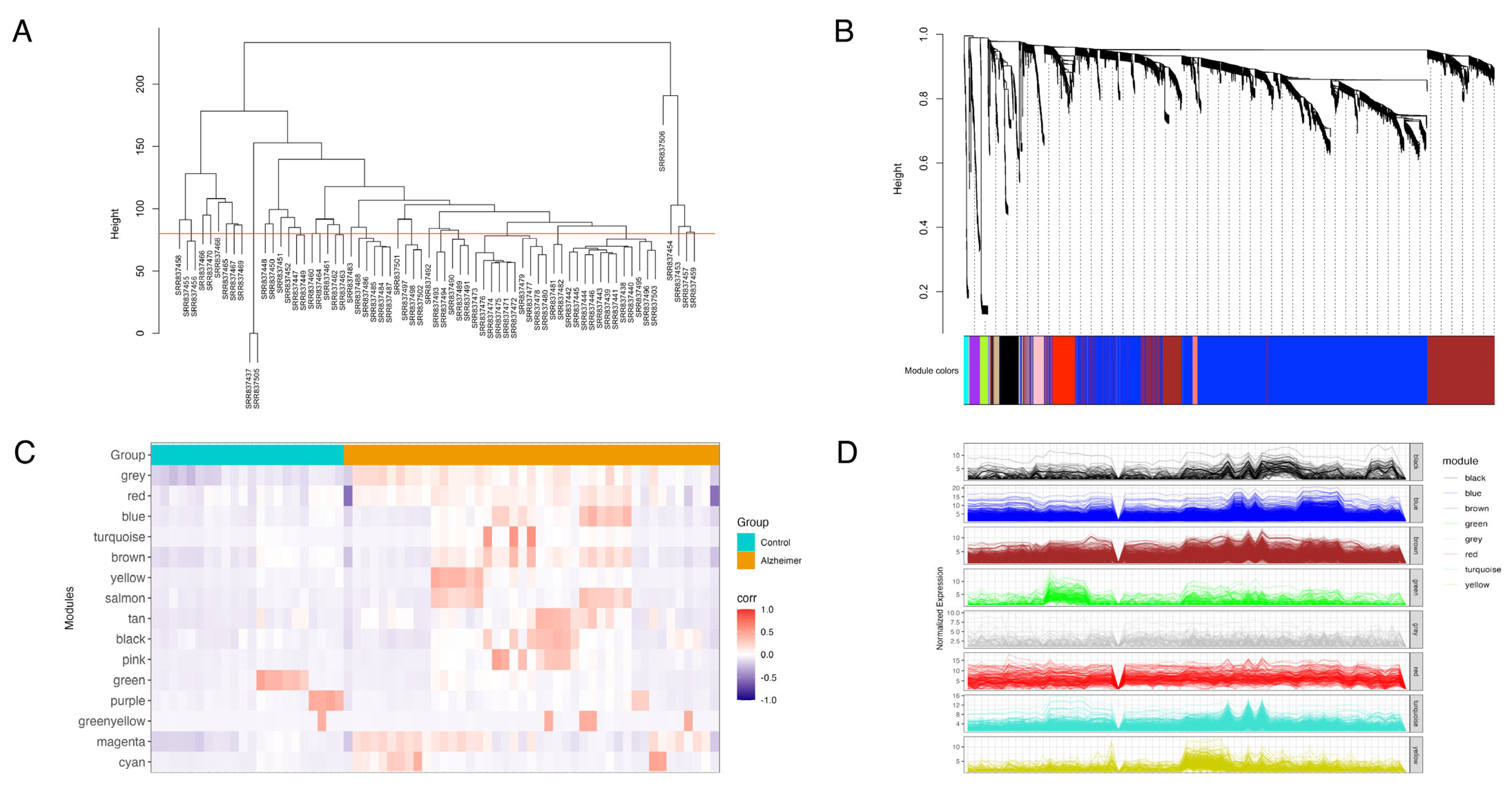
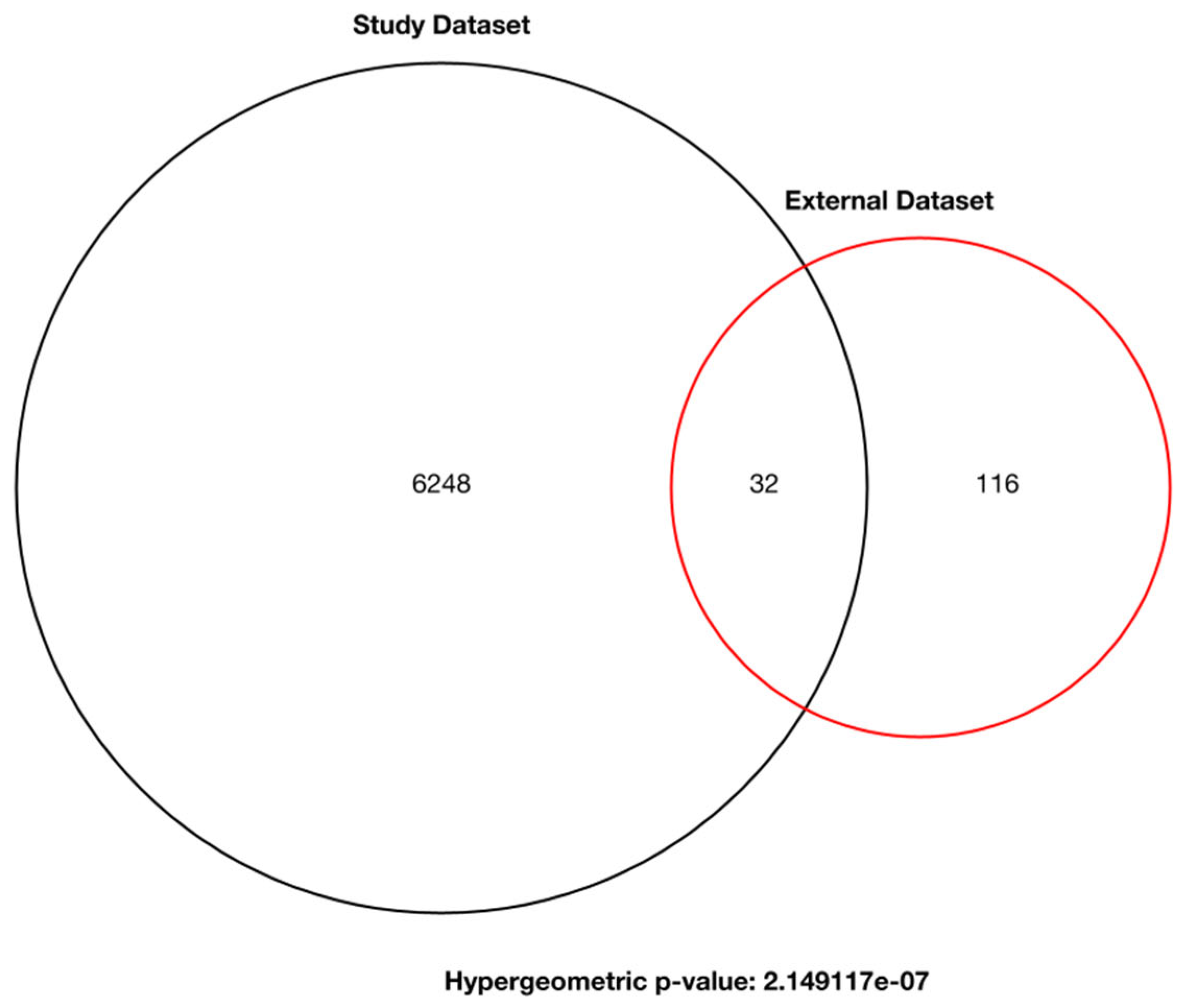
3.2. Weighted Gene Co-Expression Network Analysis
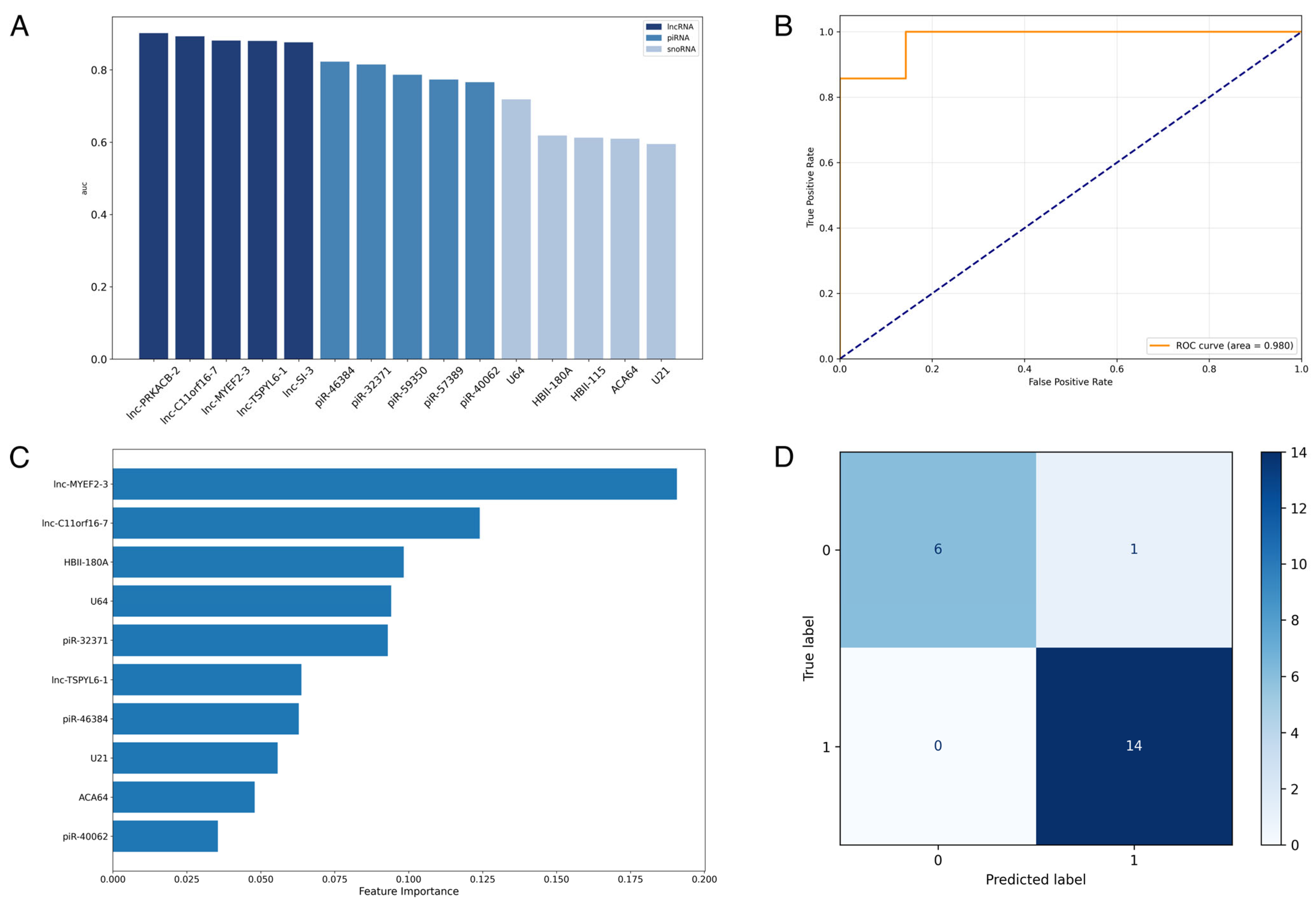
3.3. Supervised Machine-Learning Prediction of Alzheimer’s Disease Using ncRNA Expression Profiles
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| AD | Alzheimer’s disease |
| ncRNA | Non-coding RNA |
| lncRNA | Long non-coding RNA |
| piRNA | PIWI-interacting RNA |
| snoRNA | Small nucleolar RNA |
| WGCNA | Weighted gene co-expression network analysis |
| FDA | Food and Drug Administration |
| CSF | Cerebrospinal fluid |
| AUC | Area under the curve |
| SRA | Sequence Read Archive |
| RF | Random Forest |
| ROC | Receiver operating characteristic |
| DEGs | Differentially expressed genes |
References
- National Institute on Aging. Alzheimer’s Disease Fact Sheet. Available online: https://www.nia.nih.gov/health/alzheimers-and-dementia/alzheimers-disease-fact-sheet (accessed on 31 March 2025).
- Passeri, E.; Elkhoury, K.; Morsink, M.; Broersen, K.; Linder, M.; Tamayol, A.; Malaplate, C.; Yen, F.T.; Arab-Tehrany, E. Alzheimer’s Disease: Treatment Strategies and Their Limitations. Int. J. Mol. Sci. 2022, 23, 13954. [Google Scholar] [CrossRef] [PubMed]
- Bomasang-Layno, E.; Bronsther, R. Diagnosis and Treatment of Alzheimer’s Disease: An Update. Dela. J. Public Health 2021, 7, 74–85. [Google Scholar] [CrossRef] [PubMed]
- Bellio, M.; Oxtoby, N.P.; Walker, Z.; Henley, S.; Ribbens, A.; Blandford, A.; Alexander, D.C.; Yong, K.X.X. Analyzing Large Alzheimer’s Disease Cognitive Datasets: Considerations and Challenges. Alzheimers Dement. (Amst.) 2020, 12, e12135. [Google Scholar] [CrossRef]
- Barthélemy, N.R.; Salvadó, G.; Schindler, S.E.; He, Y.; Janelidze, S.; Collij, L.E.; Saef, B.; Henson, R.L.; Chen, C.D.; Gordon, B.A.; et al. Highly Accurate Blood Test for Alzheimer’s Disease Is Similar or Superior to Clinical Cerebrospinal Fluid Tests. Nat. Med. 2024, 30, 1085–1095. [Google Scholar] [CrossRef]
- Webster, S.F.; Ghalei, H. Maturation of Small Nucleolar RNAs: From Production to Function. RNA Biol. 2023, 20, 715–736. [Google Scholar] [CrossRef]
- Fitz, N.F.; Wang, J.; Kamboh, M.I.; Koldamova, R.; Lefterov, I. Small Nucleolar RNAs in Plasma Extracellular Vesicles and Their Discriminatory Power as Diagnostic Biomarkers of Alzheimer’s Disease. Neurobiol. Dis. 2021, 159, 105481. [Google Scholar] [CrossRef] [PubMed]
- Chauhan, W.; Sudharshan, S.J.; Kafle, S.; Zennadi, R. SnoRNAs: Exploring Their Implication in Human Diseases. Int. J. Mol. Sci. 2024, 25, 7202. [Google Scholar] [CrossRef]
- Gstir, R.; Schafferer, S.; Scheideler, M.; Misslinger, M.; Griehl, M.; Daschil, N.; Humpel, C.; Obermair, G.J.; Schmuckermair, C.; Striessnig, J.; et al. Generation of a Neuro-Specific Microarray Reveals Novel Differentially Expressed Noncoding RNAs in Mouse Models for Neurodegenerative Diseases. RNA 2014, 20, 1929–1943. [Google Scholar] [CrossRef]
- Iwasaki, Y.W.; Siomi, M.C.; Siomi, H.; Kornberg, R. PIWI-Interacting RNA: Its Biogenesis and Functions. Annu. Rev. Biochem. 2015, 84, 405–433. [Google Scholar] [CrossRef]
- Qiu, W.; Guo, X.; Lin, X.; Yang, Q.; Zhang, W.; Zhang, Y.; Zuo, L.; Zhu, Y.; Li, C.-S.R.; Ma, C.; et al. Transcriptome-Wide piRNA Profiling in Human Brains of Alzheimer’s Disease. Neurobiol. Aging 2017, 57, 170–177. [Google Scholar] [CrossRef]
- Roy, J.; Sarkar, A.; Parida, S.; Ghosh, Z.; Mallick, B. Small RNA Sequencing Revealed Dysregulated piRNAs in Alzheimer’s Disease and Their Probable Role in Pathogenesis. Mol. Biosyst. 2017, 13, 565–576. [Google Scholar] [CrossRef]
- Guo, X.; Qiu, W.; Garcia-Milian, R.; Lin, X.; Zhang, Y.; Cao, Y.; Tan, Y.; Wang, Z.; Shi, J.; Wang, J.; et al. Genome-Wide Significant, Replicated and Functional Risk Variants for Alzheimer’s Disease. J. Neural Transm. 2017, 124, 1455–1471. [Google Scholar] [CrossRef] [PubMed]
- Jain, G.; Stuendl, A.; Rao, P.; Berulava, T.; Pena Centeno, T.; Kaurani, L.; Burkhardt, S.; Delalle, I.; Kornhuber, J.; Hüll, M.; et al. A Combined miRNA–piRNA Signature to Detect Alzheimer’s Disease. Transl. Psychiatry 2019, 9, 250. [Google Scholar] [CrossRef] [PubMed]
- Mattick, J.S.; Amaral, P.P.; Carninci, P.; Carpenter, S.; Chang, H.Y.; Chen, L.-L.; Chen, R.; Dean, C.; Dinger, M.E.; Fitzgerald, K.A.; et al. Long Non-Coding RNAs: Definitions, Functions, Challenges and Recommendations. Nat. Rev. Mol. Cell Biol. 2023, 24, 430–447. [Google Scholar] [CrossRef] [PubMed]
- Kazemi, M.; Sadeh, R.N.; Khaniani, M.S.; Rezazadeh, M.; Derakhshan, S.M.; Ghafouri-Fard, S. Identification of RN7SK LncRNA as a Novel Biomarker in Alzheimer’s Disease Using Bioinformatics and Expression Analysis. Sci. Rep. 2024, 14, 31192. [Google Scholar] [CrossRef]
- Feng, L.; Liao, Y.-T.; He, J.-C.; Xie, C.-L.; Chen, S.-Y.; Fan, H.-H.; Su, Z.-P.; Wang, Z. Plasma Long Non-Coding RNA BACE1 as a Novel Biomarker for Diagnosis of Alzheimer Disease. BMC Neurol. 2018, 18, 4. [Google Scholar] [CrossRef]
- National Center for Biotechnology Information. SRP022043-Sequence Read Archive. Available online: https://www.ncbi.nlm.nih.gov/sra?term=SRP022043 (accessed on 23 April 2025).
- National Center for Biotechnology Information. miRNA-Seq Study of Post-Mortem Alzheimer’s Disease Brain Samples. Available online: https://trace.ncbi.nlm.nih.gov/Traces/index.html?view=study&acc=SRP288246 (accessed on 19 May 2025).
- National Center for Biotechnology Information. The NCBI SRA (Sequence Read Archive). Available online: https://github.com/ncbi/sra-tools (accessed on 23 April 2025).
- Andrews, S. FastQC: A Quality Control tool for High Throughput Sequence Data. Available online: http://www.bioinformatics.babraham.ac.uk/projects/fastqc/ (accessed on 24 April 2025).
- Martin, M. Cutadapt Removes Adapter Sequences from High-Throughput Sequencing Reads. EMBnet J. 2011, 17, 10. [Google Scholar] [CrossRef]
- Dobin, A.; Davis, C.A.; Schlesinger, F.; Drenkow, J.; Zaleski, C.; Jha, S.; Batut, P.; Chaisson, M.; Gingeras, T.R. STAR: Ultrafast Universal RNA-Seq Aligner. Bioinformatics 2013, 29, 15–21. [Google Scholar] [CrossRef]
- Leung, Y.Y.; Kuksa, P.P.; Amlie-Wolf, A.; Valladares, O.; Ungar, L.H.; Kannan, S.; Gregory, B.D.; Wang, L.-S. DASHR: Database of Small Human Noncoding RNAs. Nucleic Acids Res. 2016, 44, D216–D222. [Google Scholar] [CrossRef]
- Quinlan, A.R.; Hall, I.M. BEDTools: A Flexible Suite of Utilities for Comparing Genomic Features. Bioinformatics 2010, 26, 841–842. [Google Scholar] [CrossRef]
- Love, M.I.; Huber, W.; Anders, S. Moderated Estimation of Fold Change and Dispersion for RNA-Seq Data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [PubMed]
- Langfelder, P.; Horvath, S. WGCNA: An R Package for Weighted Correlation Network Analysis. BMC Bioinform. 2008, 9, 559. [Google Scholar] [CrossRef] [PubMed]
- Pedregosa, F.; Varoquaux, G.; Gramfort, A.; Michel, V.; Thirion, B.; Grisel, O.; Blondel, M.; Prettenhofer, P.; Weiss, R.; Dubourg, V.; et al. Scikit-Learn: Machine Learning in Python. J. Mach. Learn. 2011, 12, 2825–2830. [Google Scholar]
- Wickham, H.; Averick, M.; Bryan, J.; Chang, W.; McGowan, L.D.; François, R.; Grolemund, G.; Hayes, A.; Henry, L.; Hester, J.; et al. Welcome to the Tidyverse. J. Open Source Softw. 2019, 4, 1686. [Google Scholar] [CrossRef]
- Kolde, R.; Metsalu, T.; Lizee, A. pheatmap: Pretty Heatmaps, R package version 1.0.12; GitHub, Inc.: San Francisco, CA, USA. Available online: https://github.com/raivokolde/pheatmap (accessed on 26 April 2025).
- Shobeiri, P.; Alilou, S.; Jaberinezhad, M.; Zare, F.; Karimi, N.; Maleki, S.; Teixeira, A.L.; Perry, G.; Rezaei, N. Circulating long non-coding RNAs as novel diagnostic biomarkers for Alzheimer’s disease (AD): A systematic review and meta-analysis. PLoS ONE 2023, 18, e0281784. [Google Scholar] [CrossRef]
- Wang, L.; Liu, J.; Wang, Q.; Jiang, H.; Zeng, L.; Li, Z.; Liu, R. MicroRNA-200a-3p Mediates Neuroprotection in Alzheimer- Related Deficits and Attenuates Amyloid-Beta Overproduction and Tau Hyperphosphorylation via Coregulating BACE1 and PRKACB. Front. Pharmacol. 2019, 10, 806. [Google Scholar] [CrossRef]
- Ren, J.; Zhang, S.; Wang, X.; Deng, Y.; Zhao, Y.; Xiao, Y.; Liu, J.; Chu, L.; Qi, X. MEF2C Ameliorates Learning, Memory, and Molecular Pathological Changes in Alzheimer’s Disease in Vivo and in Vitro. Acta Biochim. Biophys. Sin. 2022, 54, 77–90. [Google Scholar] [CrossRef]
- Galardi, S.; Fatica, A.; Bachi, A.; Scaloni, A.; Presutti, C.; Bozzoni, I. Purified Box C/D snoRNPs Are Able To Reproduce Site- Specific 2′-O-Methylation of Target RNA In Vitro. Mol. Cell. Biol. 2002, 22, 6663–6668. [Google Scholar] [CrossRef]
- Idda, M.L.; Munk, R.; Abdelmohsen, K.; Gorospe, M. Noncoding RNAs in Alzheimer’s Disease. Wiley Interdiscip. Rev. RNA 2018, 9, e1463. [Google Scholar] [CrossRef]
- Salta, E.; De Strooper, B. Noncoding RNAs in Neurodegeneration. Nat. Rev. Neurosci. 2017, 18, 627–640. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Co-Expression Modules (Color) | Module Size (Gene Count) |
|---|---|
| Turquoise | 2261 |
| Blue | 2046 |
| Brown | 783 |
| Yellow | 425 |
| Green | 155 |
| Red | 149 |
| Grey | 139 |
| Black | 136 |
| Pink | 71 |
| Magenta | 70 |
| Purple | 67 |
| Green-yellow | 48 |
| Tan | 40 |
| Salmon | 39 |
| Cyan | 30 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Xin, R.; Kim, E.; Li, W.T.; Wang-Rodriguez, J.; Ongkeko, W.M. Non-Coding RNAs: lncRNA, piRNA, and snoRNA as Robust Plasma Biomarkers of Alzheimer’s Disease. Biomolecules 2025, 15, 806. https://doi.org/10.3390/biom15060806
Xin R, Kim E, Li WT, Wang-Rodriguez J, Ongkeko WM. Non-Coding RNAs: lncRNA, piRNA, and snoRNA as Robust Plasma Biomarkers of Alzheimer’s Disease. Biomolecules. 2025; 15(6):806. https://doi.org/10.3390/biom15060806
Chicago/Turabian StyleXin, Ruomin, Elizabeth Kim, Wei Tse Li, Jessica Wang-Rodriguez, and Weg M. Ongkeko. 2025. "Non-Coding RNAs: lncRNA, piRNA, and snoRNA as Robust Plasma Biomarkers of Alzheimer’s Disease" Biomolecules 15, no. 6: 806. https://doi.org/10.3390/biom15060806
APA StyleXin, R., Kim, E., Li, W. T., Wang-Rodriguez, J., & Ongkeko, W. M. (2025). Non-Coding RNAs: lncRNA, piRNA, and snoRNA as Robust Plasma Biomarkers of Alzheimer’s Disease. Biomolecules, 15(6), 806. https://doi.org/10.3390/biom15060806