
Neuronal Deletion of Tumor Susceptibility Gene 101 (Tsg101) Causes Rapid Apoptotic Loss of Hippocampal CA3 Neurons



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Mice
2.2. Genotyping
2.3. Activation and Verification of Cre Recombinase Activity
2.4. Histology
2.5. Immunoblotting
2.6. Antibodies
2.7. Statistics
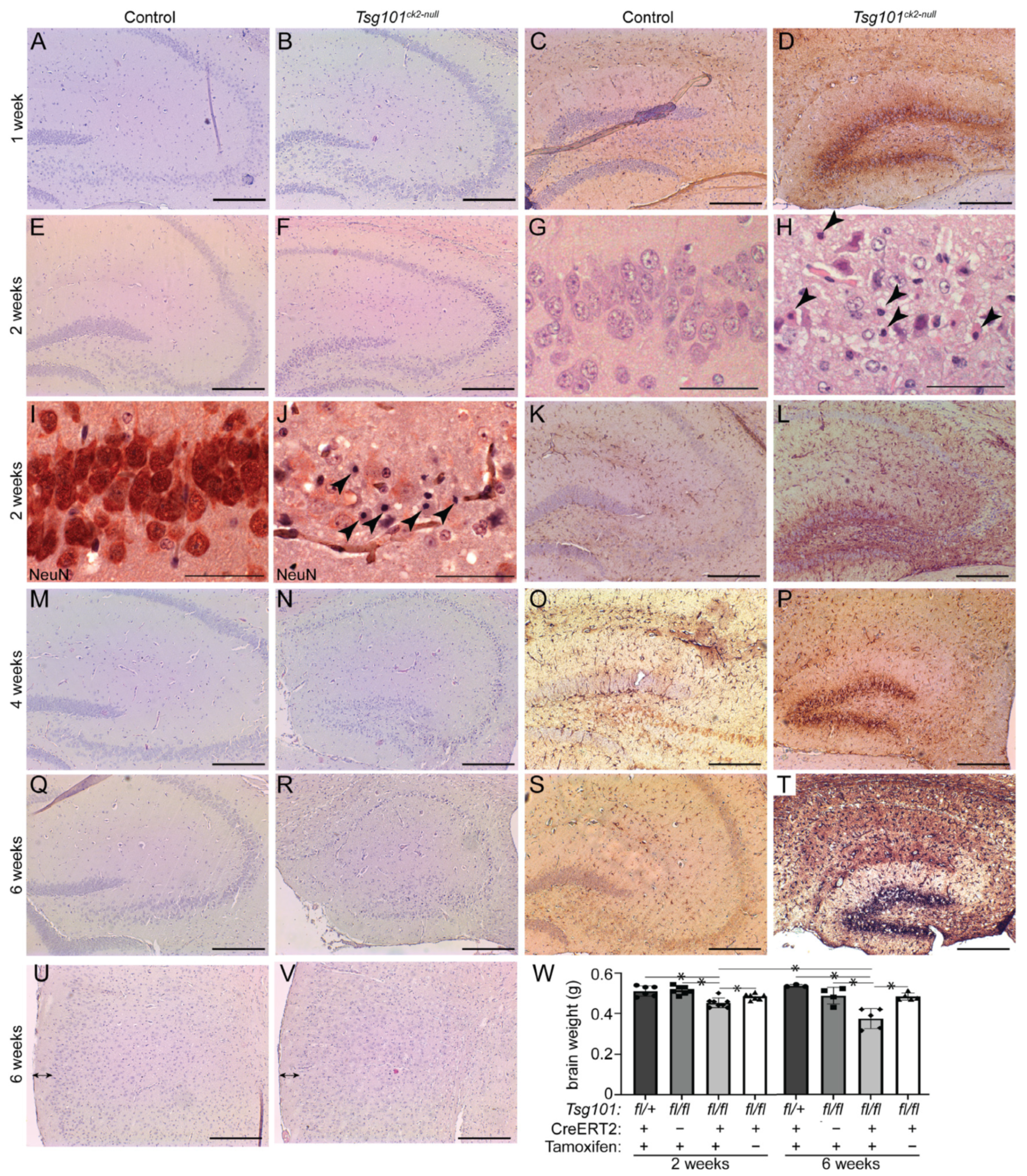
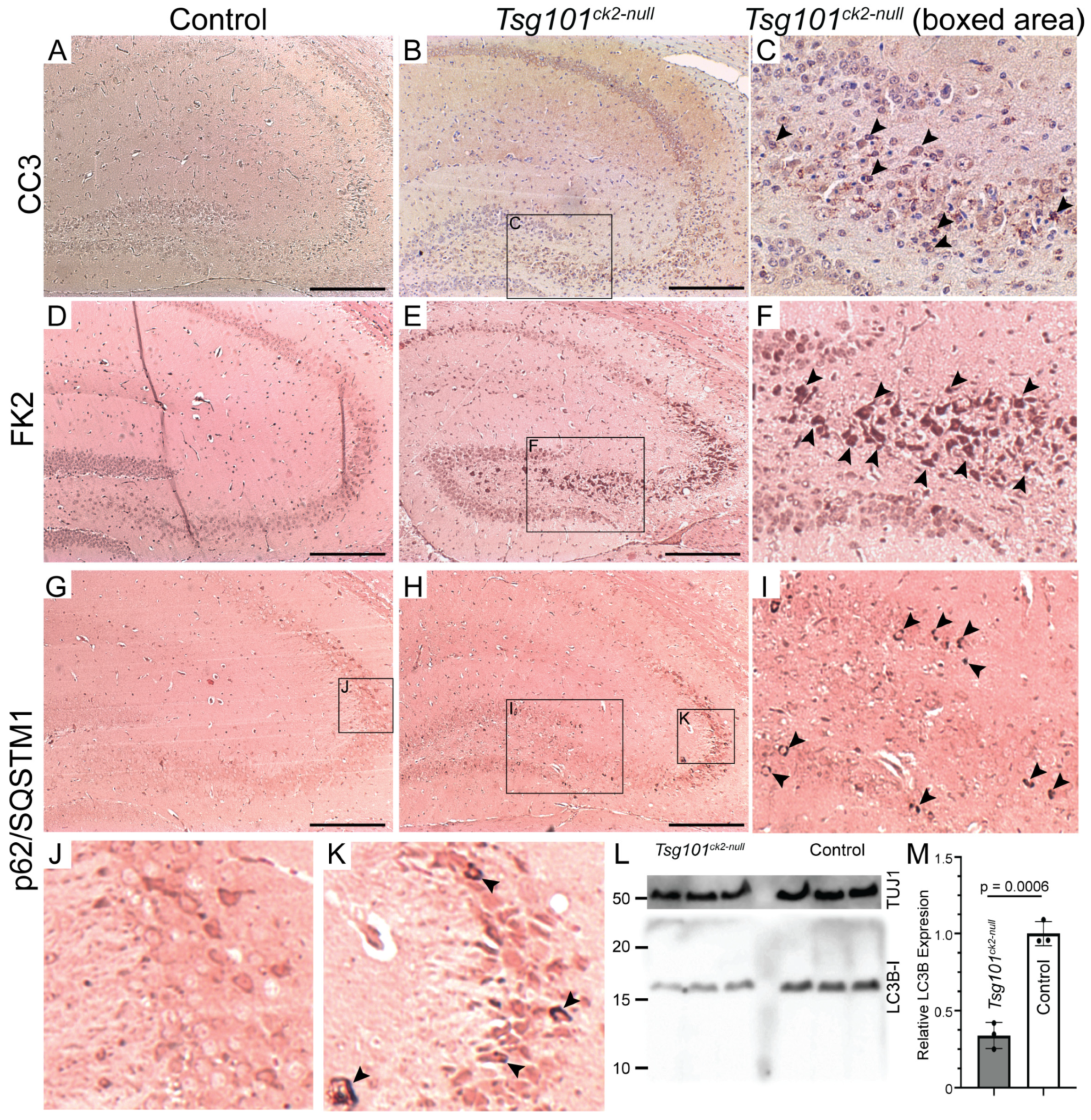
3. Results
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| TSG101 | Tumor susceptibility gene 101 |
| ESCRT | Endosomal sorting complex required for transport |
| p62/SQTM1 | Sequestosome-1 |
| RAB5 | Ras-related protein 5 |
| RAB7 | Ras-related protein 7 |
| LC3 | Microtubule-associated protein 1 light chain 3 beta (MAP1LC3B, ATG8) |
| CA3 | Cornu Ammonis subfield 3 of the hippocampus |
| AD | Alzheimer’s disease |
| Ab | A-beta or amyloid beta (proteolytic product of APP) |
| p-tau | Phosphorylated microtubule-associated protein tau (MAPT) |
| GTPase | Guanosine triphosphatase |
| APP | Amyloid precursor protein |
| APP-bCTF | APP β-cleavage C-terminal fragment (precursor to Aβ) |
| MVB | Multivesicular body |
| ILV | Intraluminal vesicle |
| EV | Extracellular vesicle |
| PCR | Polymerase chain reaction |
| Camk2a | Calcium/calmodulin-dependent protein kinase type II subunit alpha |
| Cre/ERT2 | Tamoxifen inducible Cre recombinase |
| IHC | Immunohistochemistry |
| HRP | Horseradish peroxidase |
| DAB | 3,3′-Diaminobenzidine |
| RIPA | Radioimmunoprecipitation assay buffer |
| TTBS | Tween Tris-buffered saline |
| CC3 | Cleaved caspase 3 |
| GFAP | Glial fibrillary acidic protein |
| NeuN | Neuronal nuclear antigen |
| 4G8 | Antibody reactive to amino acids 17–24 of APP and Aβ |
| CNS | Central nervous system |
| DNA | Deoxyribonucleic acid |
| H&E | Hematoxylin and eosin |
| TUJ1 | Tubulin, beta 3 class III (TUBB3) |
References
- Ginsberg, S.D.; Alldred, M.J.; Counts, S.E.; Cataldo, A.M.; Neve, R.L.; Jiang, Y.; Wuu, J.; Chao, M.V.; Mufson, E.J.; Nixon, R.A.; et al. Microarray analysis of hippocampal CA1 neurons implicates early endosomal dysfunction during Alzheimer’s disease progression. Biol. Psychiatry 2010, 68, 885–893. [Google Scholar] [CrossRef] [PubMed]
- Cataldo, A.; Rebeck, G.W.; Ghetri, B.; Hulette, C.; Lippa, C.; Van Broeckhoven, C.; van Duijn, C.; Cras, P.; Bogdanovic, N.; Bird, T.; et al. Endocytic disturbances distinguish among subtypes of Alzheimer’s disease and related disorders. Ann. Neurol. 2001, 50, 661–665. [Google Scholar] [CrossRef] [PubMed]
- Cataldo, A.M.; Hamilton, D.J.; Barnett, J.L.; Paskevich, P.A.; Nixon, R.A. Properties of the endosomal-lysosomal system in the human central nervous system: Disturbances mark most neurons in populations at risk to degenerate in Alzheimer’s disease. J. Neurosci. 1996, 16, 186–199. [Google Scholar] [CrossRef]
- Cataldo, A.M.; Peterhoff, C.M.; Troncoso, J.C.; Gomez-Isla, T.; Hyman, B.T.; Nixon, R.A. Endocytic pathway abnormalities precede amyloid beta deposition in sporadic Alzheimer’s disease and Down syndrome: Differential effects of APOE genotype and presenilin mutations. Am. J. Pathol. 2000, 157, 277–286. [Google Scholar] [CrossRef]
- Ginsberg, S.D.; Mufson, E.J.; Alldred, M.J.; Counts, S.E.; Wuu, J.; Nixon, R.A.; Che, S. Upregulation of select rab GTPases in cholinergic basal forebrain neurons in mild cognitive impairment and Alzheimer’s disease. J. Chem. Neuroanat. 2011, 42, 102–110. [Google Scholar] [CrossRef]
- Cataldo, A.M.; Petanceska, S.; Peterhoff, C.M.; Terio, N.B.; Epstein, C.J.; Villar, A.; Carlson, E.J.; Staufenbiel, M.; Nixon, R.A. App gene dosage modulates endosomal abnormalities of Alzheimer’s disease in a segmental trisomy 16 mouse model of down syndrome. J. Neurosci. 2003, 23, 6788–6792. [Google Scholar] [CrossRef]
- Chen, X.Q.; Zuo, X.; Becker, A.; Mante, M.; Florio, J.B.; Jadhav, S.G.; Albay, R.; Johnstone, A.; Karachentsev, D.; Rissman, R.; et al. Antisense oligonucleotides directed against App and Rab5 normalized endosomal Rab activity and reversed DS-AD-linked degenerative phenotypes in the Dp16 mouse model of Down syndrome. Alzheimer’s Dement. J. Alzheimer’s Assoc. 2025, 21, e70022. [Google Scholar] [CrossRef]
- Xu, W.; Weissmiller, A.M.; White, J.A., 2nd; Fang, F.; Wang, X.; Wu, Y.; Pearn, M.L.; Zhao, X.; Sawa, M.; Chen, S.; et al. Amyloid precursor protein-mediated endocytic pathway disruption induces axonal dysfunction and neurodegeneration. J. Clin. Investig. 2016, 126, 1815–1833. [Google Scholar] [CrossRef]
- Cataldo, A.M.; Mathews, P.M.; Boiteau, A.B.; Hassinger, L.C.; Peterhoff, C.M.; Jiang, Y.; Mullaney, K.; Neve, R.L.; Gruenberg, J.; Nixon, R.A. Down syndrome fibroblast model of Alzheimer-related endosome pathology: Accelerated endocytosis promotes late endocytic defects. Am. J. Pathol. 2008, 173, 370–384. [Google Scholar] [CrossRef]
- Pensalfini, A.; Kim, S.; Subbanna, S.; Bleiwas, C.; Goulbourne, C.N.; Stavrides, P.H.; Jiang, Y.; Lee, J.H.; Darji, S.; Pawlik, M.; et al. Endosomal Dysfunction Induced by Directly Overactivating Rab5 Recapitulates Prodromal and Neurodegenerative Features of Alzheimer’s Disease. Cell Rep. 2020, 33, 108420. [Google Scholar] [CrossRef]
- Grbovic, O.M.; Mathews, P.M.; Jiang, Y.; Schmidt, S.D.; Dinakar, R.; Summers-Terio, N.B.; Ceresa, B.P.; Nixon, R.A.; Cataldo, A.M. Rab5-stimulated up-regulation of the endocytic pathway increases intracellular beta-cleaved amyloid precursor protein carboxyl-terminal fragment levels and Abeta production. J. Biol. Chem. 2003, 278, 31261–31268. [Google Scholar] [CrossRef] [PubMed]
- Xie, W.; Li, L.; Cohen, S.N. Cell cycle-dependent subcellular localization of the TSG101 protein and mitotic and nuclear abnormalities associated with TSG101 deficiency. Proc. Natl. Acad. Sci. USA 1998, 95, 1595–1600. [Google Scholar] [CrossRef] [PubMed]
- Babst, M.; Odorizzi, G.; Estepa, E.J.; Emr, S.D. Mammalian tumor susceptibility gene 101 (TSG101) and the yeast homologue, Vps23p, both function in late endosomal trafficking. Traffic 2000, 1, 248–258. [Google Scholar] [CrossRef]
- Bishop, N.; Woodman, P. TSG101/mammalian VPS23 and mammalian VPS28 interact directly and are recruited to VPS4-induced endosomes. J. Biol. Chem. 2001, 276, 11735–11742. [Google Scholar] [CrossRef]
- Garrus, J.E.; von Schwedler, U.K.; Pornillos, O.W.; Morham, S.G.; Zavitz, K.H.; Wang, H.E.; Wettstein, D.A.; Stray, K.M.; Cote, M.; Rich, R.L.; et al. Tsg101 and the vacuolar protein sorting pathway are essential for HIV-1 budding. Cell 2001, 107, 55–65. [Google Scholar] [CrossRef]
- Martin-Serrano, J.; Zang, T.; Bieniasz, P.D. HIV-1 and Ebola virus encode small peptide motifs that recruit Tsg101 to sites of particle assembly to facilitate egress. Nat. Med. 2001, 7, 1313–1319. [Google Scholar] [CrossRef]
- Ruland, J.; Sirard, C.; Elia, A.; MacPherson, D.; Wakeham, A.; Li, L.; de la Pompa, J.L.; Cohen, S.N.; Mak, T.W. p53 accumulation, defective cell proliferation, and early embryonic lethality in mice lacking Tsg101. Proc. Natl. Acad. Sci. USA 2001, 98, 1859–1864. [Google Scholar] [CrossRef]
- Bishop, N.; Horman, A.; Woodman, P. Mammalian class E vps proteins recognize ubiquitin and act in the removal of endosomal protein-ubiquitin conjugates. J. Cell Biol. 2002, 157, 91–101. [Google Scholar] [CrossRef]
- Licata, J.M.; Simpson-Holley, M.; Wright, N.T.; Han, Z.; Paragas, J.; Harty, R.N. Overlapping motifs (PTAP and PPEY) within the Ebola virus VP40 protein function independently as late budding domains: Involvement of host proteins TSG101 and VPS-4. J. Virol. 2003, 77, 1812–1819. [Google Scholar] [CrossRef]
- Wagner, K.U.; Krempler, A.; Qi, Y.; Park, K.; Henry, M.D.; Triplett, A.A.; Riedlinger, G.; Rucker, I.E.; Hennighausen, L. Tsg101 is essential for cell growth, proliferation, and cell survival of embryonic and adult tissues. Mol. Cell. Biol. 2003, 23, 150–162. [Google Scholar] [CrossRef]
- Oh, K.B.; Stanton, M.J.; West, W.W.; Todd, G.L.; Wagner, K.U. Tsg101 is upregulated in a subset of invasive human breast cancers and its targeted overexpression in transgenic mice reveals weak oncogenic properties for mammary cancer initiation. Oncogene 2007, 26, 5950–5959. [Google Scholar] [CrossRef] [PubMed]
- Dennaoui, R.; Radler, P.D.; Wicker, M.N.; Vistisen, K.; Ferraiuolo, R.; Triplett, A.A.; Shrestha, H.; Liner, T.A.; Manthey, K.C.; Rui, H.; et al. Overexpression of TSG101 causes the development of adenosquamousmammary carcinoma. Breast Cancer Res. 2025, in press. [Google Scholar]
- Ferraiuolo, R.M.; Manthey, K.C.; Stanton, M.J.; Triplett, A.A.; Wagner, K.U. The Multifaceted Roles of the Tumor Susceptibility Gene 101 (TSG101) in Normal Development and Disease. Cancers 2020, 12, 450. [Google Scholar] [CrossRef]
- Colombo, M.; Raposo, G.; Théry, C. Biogenesis, secretion, and intercellular interactions of exosomes and other extracellular vesicles. Annu. Rev. Cell Dev. Biol. 2014, 30, 255–289. [Google Scholar] [CrossRef]
- Colombo, M.; Moita, C.; van Niel, G.; Kowal, J.; Vigneron, J.; Benaroch, P.; Manel, N.; Moita, L.F.; Théry, C.; Raposo, G. Analysis of ESCRT functions in exosome biogenesis, composition and secretion highlights the heterogeneity of extracellular vesicles. J. Cell Sci. 2013, 126, 5553–5565. [Google Scholar] [CrossRef]
- Morel, E.; Chamoun, Z.; Lasiecka, Z.M.; Chan, R.B.; Williamson, R.L.; Vetanovetz, C.; Dall’Armi, C.; Simoes, S.; Point Du Jour, K.S.; McCabe, B.D.; et al. Phosphatidylinositol-3-phosphate regulates sorting and processing of amyloid precursor protein through the endosomal system. Nat. Commun. 2013, 4, 2250. [Google Scholar] [CrossRef]
- Edgar, J.R.; Willen, K.; Gouras, G.K.; Futter, C.E. ESCRTs regulate amyloid precursor protein sorting in multivesicular bodies and intracellular amyloid-beta accumulation. J. Cell Sci. 2015, 128, 2520–2528. [Google Scholar] [CrossRef]
- Haass, C.; Koo, E.H.; Mellon, A.; Hung, A.Y.; Selkoe, D.J. Targeting of cell-surface beta-amyloid precursor protein to lysosomes: Alternative processing into amyloid-bearing fragments. Nature 1992, 357, 500–503. [Google Scholar] [CrossRef]
- Toh, W.H.; Tan, J.Z.; Zulkefli, K.L.; Houghton, F.J.; Gleeson, P.A. Amyloid precursor protein traffics from the Golgi directly to early endosomes in an Arl5b- and AP4-dependent pathway. Traffic 2017, 18, 159–175. [Google Scholar] [CrossRef]
- Mamde, S.; Rose, S.E.; Prater, K.E.; Cochoit, A.; Lin, Y.F.; Smith, I.; Johnson, C.S.; Reid, A.N.; Qiu, W.; Strohbehn, S.; et al. Genetic risk in endolysosomal network genes correlates with endolysosomal dysfunction across neural cell types in Alzheimer’s disease. bioRxiv 2025. [Google Scholar] [CrossRef]
- Szabo, M.P.; Mishra, S.; Knupp, A.; Young, J.E. The role of Alzheimer’s disease risk genes in endolysosomal pathways. Neurobiol. Dis. 2022, 162, 105576. [Google Scholar] [CrossRef] [PubMed]
- Gao, S.; Casey, A.E.; Sargeant, T.J.; Mäkinen, V.P. Genetic variation within endolysosomal system is associated with late-onset Alzheimer’s disease. Brain 2018, 141, 2711–2720. [Google Scholar] [CrossRef] [PubMed]
- Hung, C.; Tuck, E.; Stubbs, V.; van der Lee, S.J.; Aalfs, C.; van Spaendonk, R.; Scheltens, P.; Hardy, J.; Holstege, H.; Livesey, F.J. SORL1 deficiency in human excitatory neurons causes APP-dependent defects in the endolysosome-autophagy network. Cell Rep. 2021, 35, 109259. [Google Scholar] [CrossRef] [PubMed]
- Maninger, J.K.; Nowak, K.; Goberdhan, S.; O’Donoghue, R.; Connor-Robson, N. Cell type-specific functions of Alzheimer’s disease endocytic risk genes. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2024, 379, 20220378. [Google Scholar] [CrossRef]
- Li, J.; Kanekiyo, T.; Shinohara, M.; Zhang, Y.; LaDu, M.J.; Xu, H.; Bu, G. Differential regulation of amyloid-beta endocytic trafficking and lysosomal degradation by apolipoprotein E isoforms. J. Biol. Chem. 2012, 287, 44593–44601. [Google Scholar] [CrossRef]
- Yamazaki, T.; Koo, E.H.; Selkoe, D.J. Trafficking of cell-surface amyloid beta-protein precursor. II. Endocytosis, recycling and lysosomal targeting detected by immunolocalization. J. Cell Sci. 1996, 109 Pt 5, 999–1008. [Google Scholar] [CrossRef]
- Yu, Y.; Gao, Y.; Winblad, B.; Tjernberg, L.O.; Schedin-Weiss, S. A Super-Resolved View of the Alzheimer’s Disease-Related Amyloidogenic Pathway in Hippocampal Neurons. J. Alzheimers Dis 2021, 83, 833–852. [Google Scholar] [CrossRef]
- Koo, E.H.; Squazzo, S.L. Evidence that production and release of amyloid beta-protein involves the endocytic pathway. J. Biol. Chem. 1994, 269, 17386–17389. [Google Scholar] [CrossRef]
- Hu, X.; Crick, S.L.; Bu, G.; Frieden, C.; Pappu, R.V.; Lee, J.M. Amyloid seeds formed by cellular uptake, concentration, and aggregation of the amyloid-beta peptide. Proc. Natl. Acad. Sci. USA 2009, 106, 20324–20329. [Google Scholar] [CrossRef]
- Friedrich, R.P.; Tepper, K.; Rönicke, R.; Soom, M.; Westermann, M.; Reymann, K.; Kaether, C.; Fändrich, M. Mechanism of amyloid plaque formation suggests an intracellular basis of Abeta pathogenicity. Proc. Natl. Acad. Sci. USA 2010, 107, 1942–1947. [Google Scholar] [CrossRef]
- Willén, K.; Edgar, J.R.; Hasegawa, T.; Tanaka, N.; Futter, C.E.; Gouras, G.K. Aβ accumulation causes MVB enlargement and is modelled by dominant negative VPS4A. Mol. Neurodegener. 2017, 12, 61. [Google Scholar] [CrossRef] [PubMed]
- Perez-Gonzalez, R.; Gauthier, S.A.; Kumar, A.; Levy, E. The exosome secretory pathway transports amyloid precursor protein carboxyl-terminal fragments from the cell into the brain extracellular space. J. Biol. Chem. 2012, 287, 43108–43115. [Google Scholar] [CrossRef] [PubMed]
- Rajendran, L.; Honsho, M.; Zahn, T.R.; Keller, P.; Geiger, K.D.; Verkade, P.; Simons, K. Alzheimer’s disease beta-amyloid peptides are released in association with exosomes. Proc. Natl. Acad. Sci. USA 2006, 103, 11172–11177. [Google Scholar] [CrossRef]
- Sardar Sinha, M.; Ansell-Schultz, A.; Civitelli, L.; Hildesjö, C.; Larsson, M.; Lannfelt, L.; Ingelsson, M.; Hallbeck, M. Alzheimer’s disease pathology propagation by exosomes containing toxic amyloid-beta oligomers. Acta Neuropathol. 2018, 136, 41–56. [Google Scholar] [CrossRef]
- Winston, C.N.; Goetzl, E.J.; Akers, J.C.; Carter, B.S.; Rockenstein, E.M.; Galasko, D.; Masliah, E.; Rissman, R.A. Prediction of conversion from mild cognitive impairment to dementia with neuronally derived blood exosome protein profile. Alzheimer’s Dement. 2016, 3, 63–72. [Google Scholar] [CrossRef]
- Laulagnier, K.; Javalet, C.; Hemming, F.J.; Chivet, M.; Lachenal, G.; Blot, B.; Chatellard, C.; Sadoul, R. Amyloid precursor protein products concentrate in a subset of exosomes specifically endocytosed by neurons. Cell. Mol. Life Sci. 2018, 75, 757–773. [Google Scholar] [CrossRef]
- Takahashi, R.H.; Milner, T.A.; Li, F.; Nam, E.E.; Edgar, M.A.; Yamaguchi, H.; Beal, M.F.; Xu, H.; Greengard, P.; Gouras, G.K. Intraneuronal Alzheimer abeta42 accumulates in multivesicular bodies and is associated with synaptic pathology. Am. J. Pathol. 2002, 161, 1869–1879. [Google Scholar] [CrossRef]
- Goetzl, E.J.; Boxer, A.; Schwartz, J.B.; Abner, E.L.; Petersen, R.C.; Miller, B.L.; Kapogiannis, D. Altered lysosomal proteins in neural-derived plasma exosomes in preclinical Alzheimer disease. Neurology 2015, 85, 40–47. [Google Scholar] [CrossRef]
- Chen, J.J.; Nathaniel, D.L.; Raghavan, P.; Nelson, M.; Tian, R.; Tse, E.; Hong, J.Y.; See, S.K.; Mok, S.A.; Hein, M.Y.; et al. Compromised function of the ESCRT pathway promotes endolysosomal escape of tau seeds and propagation of tau aggregation. J. Biol. Chem. 2019, 294, 18952–18966. [Google Scholar] [CrossRef]
- Polanco, J.C.; Scicluna, B.J.; Hill, A.F.; Götz, J. Extracellular Vesicles Isolated from the Brains of rTg4510 Mice Seed Tau Protein Aggregation in a Threshold-dependent Manner. J. Biol. Chem. 2016, 291, 12445–12466. [Google Scholar] [CrossRef]
- Baker, S.; Polanco, J.C.; Götz, J. Extracellular Vesicles Containing P301L Mutant Tau Accelerate Pathological Tau Phosphorylation and Oligomer Formation but Do Not Seed Mature Neurofibrillary Tangles in ALZ17 Mice. J. Alzheimers Dis. 2016, 54, 1207–1217. [Google Scholar] [CrossRef] [PubMed]
- Jackson, N.A.; Guerrero-Muñoz, M.J.; Castillo-Carranza, D.L. The prion-like transmission of tau oligomers via exosomes. Front. Aging Neurosci. 2022, 14, 974414. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Balaji, V.; Kaniyappan, S.; Krüger, L.; Irsen, S.; Tepper, K.; Chandupatla, R.; Maetzler, W.; Schneider, A.; Mandelkow, E.; et al. The release and trans-synaptic transmission of Tau via exosomes. Mol. Neurodegener. 2017, 12, 5. [Google Scholar] [CrossRef]
- Erdmann, G.; Schutz, G.; Berger, S. Inducible gene inactivation in neurons of the adult mouse forebrain. BMC Neurosci. 2007, 8, 63. [Google Scholar] [CrossRef]
- Wells, G.A.; Wells, M. Neuropil vacuolation in brain: A reproducible histological processing artefact. J. Comp. Pathol. 1989, 101, 355–362. [Google Scholar] [CrossRef]
- Allen Reference Atlas—Mouse Brain [Brain Atlas]. 2004. Available online: https://atlas.brain-map.org (accessed on 30 November 2022).
- Cheng, J.T.; Liu, P.F.; Yang, H.C.; Huang, S.J.; Griffith, M.; Morgan, P.; Shu, C.W. Tumor Susceptibility Gene 101 facilitates rapamycin-induced autophagic flux in neuron cells. Biomed. Pharmacother. 2021, 134, 111106. [Google Scholar] [CrossRef]
- Chung, Y.; Lee, J.; Jung, S.; Lee, Y.; Cho, J.W.; Oh, Y.J. Dysregulated autophagy contributes to caspase-dependent neuronal apoptosis. Cell Death Dis. 2018, 9, 1189. [Google Scholar] [CrossRef]
- Lai, Y.; Hickey, R.W.; Chen, Y.; Bayir, H.; Sullivan, M.L.; Chu, C.T.; Kochanek, P.M.; Dixon, C.E.; Jenkins, L.W.; Graham, S.H.; et al. Autophagy is increased after traumatic brain injury in mice and is partially inhibited by the antioxidant gamma-glutamylcysteinyl ethyl ester. J. Cereb. Blood Flow Metab. 2008, 28, 540–550. [Google Scholar] [CrossRef]
- Amaral, D.G.; Scharfman, H.E.; Lavenex, P. The dentate gyrus: Fundamental neuroanatomical organization (dentate gyrus for dummies). Prog. Brain Res. 2007, 163, 3–22. [Google Scholar] [CrossRef]
- Hsu, D. The dentate gyrus as a filter or gate: A look back and a look ahead. Prog. Brain Res. 2007, 163, 601–613. [Google Scholar] [CrossRef]
- Price, B.R.; Johnson, L.A.; Norris, C.M. Reactive astrocytes: The nexus of pathological and clinical hallmarks of Alzheimer’s disease. Ageing Res. Rev. 2021, 68, 101335. [Google Scholar] [CrossRef] [PubMed]
- Mahmoud, S.; Gharagozloo, M.; Simard, C.; Gris, D. Astrocytes Maintain Glutamate Homeostasis in the CNS by Controlling the Balance between Glutamate Uptake and Release. Cells 2019, 8, 184. [Google Scholar] [CrossRef] [PubMed]
- Mira, R.G.; Cerpa, W. Building a Bridge Between NMDAR-Mediated Excitotoxicity and Mitochondrial Dysfunction in Chronic and Acute Diseases. Cell. Mol. Neurobiol. 2021, 41, 1413–1430. [Google Scholar] [CrossRef]
- Morris, C.R.; Stanton, M.J.; Manthey, K.C.; Oh, K.B.; Wagner, K.U. A Knockout of the Tsg101 Gene Leads to Decreased Expression of ErbB Receptor Tyrosine Kinases and Induction of Autophagy Prior to Cell Death. PLoS ONE 2012, 7, e34308. [Google Scholar] [CrossRef]
- Igarashi, K.M. Entorhinal cortex dysfunction in Alzheimer’s disease. Trends Neurosci. 2023, 46, 124–136. [Google Scholar] [CrossRef]
- Peric, A.; Annaert, W. Early etiology of Alzheimer’s disease: Tipping the balance toward autophagy or endosomal dysfunction? Acta Neuropathol. 2015, 129, 363–381. [Google Scholar] [CrossRef]
- Peng, K.Y.; Pérez-González, R.; Alldred, M.J.; Goulbourne, C.N.; Morales-Corraliza, J.; Saito, M.; Saito, M.; Ginsberg, S.D.; Mathews, P.M.; Levy, E. Apolipoprotein E4 genotype compromises brain exosome production. Brain 2019, 142, 163–175. [Google Scholar] [CrossRef]
- Hsu, C.; Morohashi, Y.; Yoshimura, S.; Manrique-Hoyos, N.; Jung, S.; Lauterbach, M.A.; Bakhti, M.; Grønborg, M.; Möbius, W.; Rhee, J.; et al. Regulation of exosome secretion by Rab35 and its GTPase-activating proteins TBC1D10A-C. J. Cell Biol. 2010, 189, 223–232. [Google Scholar] [CrossRef]
- Skibinski, G.; Parkinson, N.J.; Brown, J.M.; Chakrabarti, L.; Lloyd, S.L.; Hummerich, H.; Nielsen, J.E.; Hodges, J.R.; Spillantini, M.G.; Thusgaard, T.; et al. Mutations in the endosomal ESCRTIII-complex subunit CHMP2B in frontotemporal dementia. Nat. Genet. 2005, 37, 806–808. [Google Scholar] [CrossRef]
- Cox, L.E.; Ferraiuolo, L.; Goodall, E.F.; Heath, P.R.; Higginbottom, A.; Mortiboys, H.; Hollinger, H.C.; Hartley, J.A.; Brockington, A.; Burness, C.E.; et al. Mutations in CHMP2B in lower motor neuron predominant amyotrophic lateral sclerosis (ALS). PLoS ONE 2010, 5, e9872. [Google Scholar] [CrossRef]
- Isaacs, A.M.; Johannsen, P.; Holm, I.; Nielsen, J.E. Frontotemporal dementia caused by CHMP2B mutations. Curr. Alzheimer Res. 2011, 8, 246–251. [Google Scholar] [CrossRef] [PubMed]
- Ghazi-Noori, S.; Froud, K.E.; Mizielinska, S.; Powell, C.; Smidak, M.; Fernandez de Marco, M.; O’Malley, C.; Farmer, M.; Parkinson, N.; Fisher, E.M.; et al. Progressive neuronal inclusion formation and axonal degeneration in CHMP2B mutant transgenic mice. Brain 2012, 135, 819–832. [Google Scholar] [CrossRef] [PubMed]
- Vernay, A.; Therreau, L.; Blot, B.; Risson, V.; Dirrig-Grosch, S.; Waegaert, R.; Lequeu, T.; Sellal, F.; Schaeffer, L.; Sadoul, R.; et al. A transgenic mouse expressing CHMP2Bintron5 mutant in neurons develops histological and behavioural features of amyotrophic lateral sclerosis and frontotemporal dementia. Hum. Mol. Genet. 2016, 25, 3341–3360. [Google Scholar] [CrossRef] [PubMed]
- Tamai, K.; Toyoshima, M.; Tanaka, N.; Yamamoto, N.; Owada, Y.; Kiyonari, H.; Murata, K.; Ueno, Y.; Ono, M.; Shimosegawa, T.; et al. Loss of hrs in the central nervous system causes accumulation of ubiquitinated proteins and neurodegeneration. Am. J. Pathol. 2008, 173, 1806–1817. [Google Scholar] [CrossRef]
- Oshima, R.; Hasegawa, T.; Tamai, K.; Sugeno, N.; Yoshida, S.; Kobayashi, J.; Kikuchi, A.; Baba, T.; Futatsugi, A.; Sato, I.; et al. ESCRT-0 dysfunction compromises autophagic degradation of protein aggregates and facilitates ER stress-mediated neurodegeneration via apoptotic and necroptotic pathways. Sci. Rep. 2016, 6, 24997. [Google Scholar] [CrossRef]
- Silvius, D.; Hurley, E.; Poitelon, Y.; Wagner, K.U.; Feltri, M.L.; Gunn, T.M. Schwann cell deletion of Tumor Susceptibility Gene 101 (Tsg101) in mice results in severe peripheral neuropathy. Micropublication Biol. 2025, 2025, 10–17912. [Google Scholar] [CrossRef]
- McLean, J.W.; Wilson, J.A.; Tian, T.; Watson, J.A.; VanHart, M.; Bean, A.J.; Scherer, S.S.; Crossman, D.K.; Ubogu, E.; Wilson, S.M. Disruption of Endosomal Sorting in Schwann Cells Leads to Defective Myelination and Endosomal Abnormalities Observed in Charcot-Marie-Tooth Disease. J. Neurosci. 2022, 42, 5085–5101. [Google Scholar] [CrossRef]
- Walker, W.P.; Oehler, A.; Edinger, A.L.; Wagner, K.U.; Gunn, T.M. Oligodendroglial deletion of ESCRT-I component TSG101 causes spongiform encephalopathy. Biol. Cell 2016, 108, 324–337. [Google Scholar] [CrossRef]
- Bucci, C.; Thomsen, P.; Nicoziani, P.; McCarthy, J.; van Deurs, B. Rab7: A key to lysosome biogenesis. Mol. Biol. Cell 2000, 11, 467–480. [Google Scholar] [CrossRef]
- Gutierrez, M.G.; Munafó, D.B.; Berón, W.; Colombo, M.I. Rab7 is required for the normal progression of the autophagic pathway in mammalian cells. J. Cell Sci. 2004, 117, 2687–2697. [Google Scholar] [CrossRef]
- Hyttinen, J.M.; Niittykoski, M.; Salminen, A.; Kaarniranta, K. Maturation of autophagosomes and endosomes: A key role for Rab7. Biochim. Biophys. Acta 2013, 1833, 503–510. [Google Scholar] [CrossRef] [PubMed]
- Bache, K.G.; Brech, A.; Mehlum, A.; Stenmark, H. Hrs regulates multivesicular body formation via ESCRT recruitment to endosomes. J. Cell Biol. 2003, 162, 435–442. [Google Scholar] [CrossRef] [PubMed]
- Lu, Q.; Hope, L.W.; Brasch, M.; Reinhard, C.; Cohen, S.N. TSG101 interaction with HRS mediates endosomal trafficking and receptor down-regulation. Proc. Natl. Acad. Sci. USA 2003, 100, 7626–7631. [Google Scholar] [CrossRef]
- Pornillos, O.; Higginson, D.S.; Stray, K.M.; Fisher, R.D.; Garrus, J.E.; Payne, M.; He, G.P.; Wang, H.E.; Morham, S.G.; Sundquist, W.I. HIV Gag mimics the Tsg101-recruiting activity of the human Hrs protein. J. Cell Biol. 2003, 162, 425–434. [Google Scholar] [CrossRef]
- Razi, M.; Futter, C.E. Distinct roles for Tsg101 and Hrs in multivesicular body formation and inward vesiculation. Mol. Biol. Cell 2006, 17, 3469–3483. [Google Scholar] [CrossRef]
- Raiborg, C.; Malerod, L.; Pedersen, N.M.; Stenmark, H. Differential functions of Hrs and ESCRT proteins in endocytic membrane trafficking. Exp. Cell Res. 2008, 314, 801–813. [Google Scholar] [CrossRef]
- Doyotte, A.; Russell, M.R.; Hopkins, C.R.; Woodman, P.G. Depletion of TSG101 forms a mammalian “Class E” compartment: A multicisternal early endosome with multiple sorting defects. J. Cell Sci. 2005, 118, 3003–3017. [Google Scholar] [CrossRef]
- Kaul, Z.; Mookherjee, D.; Das, S.; Chatterjee, D.; Chakrabarti, S.; Chakrabarti, O. Loss of tumor susceptibility gene 101 (TSG101) perturbs endoplasmic reticulum structure and function. Biochim. Biophys. Acta Mol. Cell Res. 2020, 1867, 118741. [Google Scholar] [CrossRef]
- Kilinc, S.; Paisner, R.; Camarda, R.; Gupta, S.; Momcilovic, O.; Kohnz, R.A.; Avsaroglu, B.; L’Etoile, N.D.; Perera, R.M.; Nomura, D.K.; et al. Oncogene-regulated release of extracellular vesicles. Dev. Cell 2021, 56, 1989–2006.e1986. [Google Scholar] [CrossRef]




Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Walker, W.P.; Ratz-Mitchem, M.L.; Wagner, K.-U.; Gunn, T.M. Neuronal Deletion of Tumor Susceptibility Gene 101 (Tsg101) Causes Rapid Apoptotic Loss of Hippocampal CA3 Neurons. Biomolecules 2025, 15, 786. https://doi.org/10.3390/biom15060786
Walker WP, Ratz-Mitchem ML, Wagner K-U, Gunn TM. Neuronal Deletion of Tumor Susceptibility Gene 101 (Tsg101) Causes Rapid Apoptotic Loss of Hippocampal CA3 Neurons. Biomolecules. 2025; 15(6):786. https://doi.org/10.3390/biom15060786
Chicago/Turabian StyleWalker, Will P., Megan Lea Ratz-Mitchem, Kay-Uwe Wagner, and Teresa M. Gunn. 2025. "Neuronal Deletion of Tumor Susceptibility Gene 101 (Tsg101) Causes Rapid Apoptotic Loss of Hippocampal CA3 Neurons" Biomolecules 15, no. 6: 786. https://doi.org/10.3390/biom15060786
APA StyleWalker, W. P., Ratz-Mitchem, M. L., Wagner, K.-U., & Gunn, T. M. (2025). Neuronal Deletion of Tumor Susceptibility Gene 101 (Tsg101) Causes Rapid Apoptotic Loss of Hippocampal CA3 Neurons. Biomolecules, 15(6), 786. https://doi.org/10.3390/biom15060786