
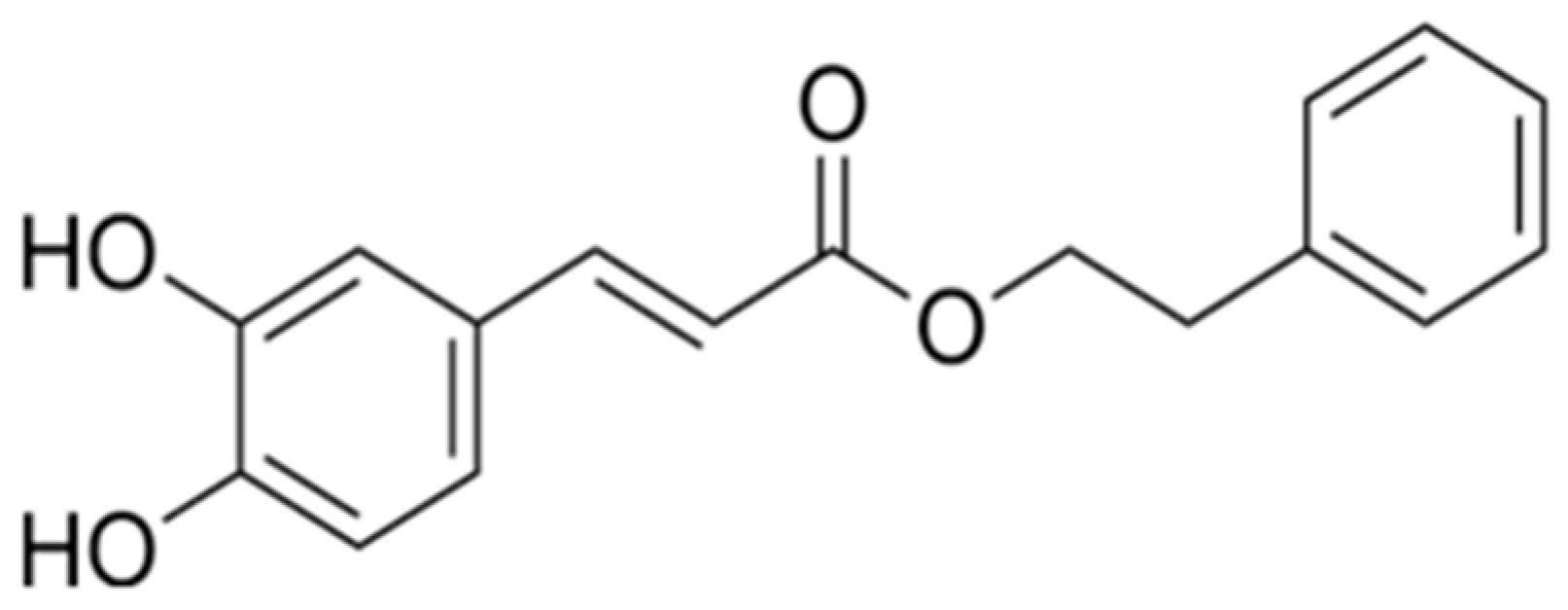
Caffeic Acid Phenethyl Ester Protects Against Doxorubicin-Induced Cardiotoxicity via Inhibiting the ROS-MLKL-Mediated Cross-Talk Between Oxidative Stress and Necroptosis
Abstract

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Reagents and Chemicals
2.2. Cell Lines and Culture
2.3. Animals and Treatments
2.4. Cell Viability Assay
2.5. Surface Electrocardiogram (ECG) Assessment of Cardiac Function
2.6. Histopathologic Assay
2.7. Measurement of Serum Levels of cTn-I, CK-MB and LDH
2.8. Transmission Electron Microscopy (TEM)
2.9. Oxidative Stress Detection
2.10. Mitochondrial Respiration Analysis
2.11. Western Blotting
2.12. Immunohistochemistry Assay (IHC)
2.13. Plasmid Transfection
2.14. Statistical Analysis
3. Results
3.1. CAPE Alleviated DOX-Induced Myocardial Injury In Vivo
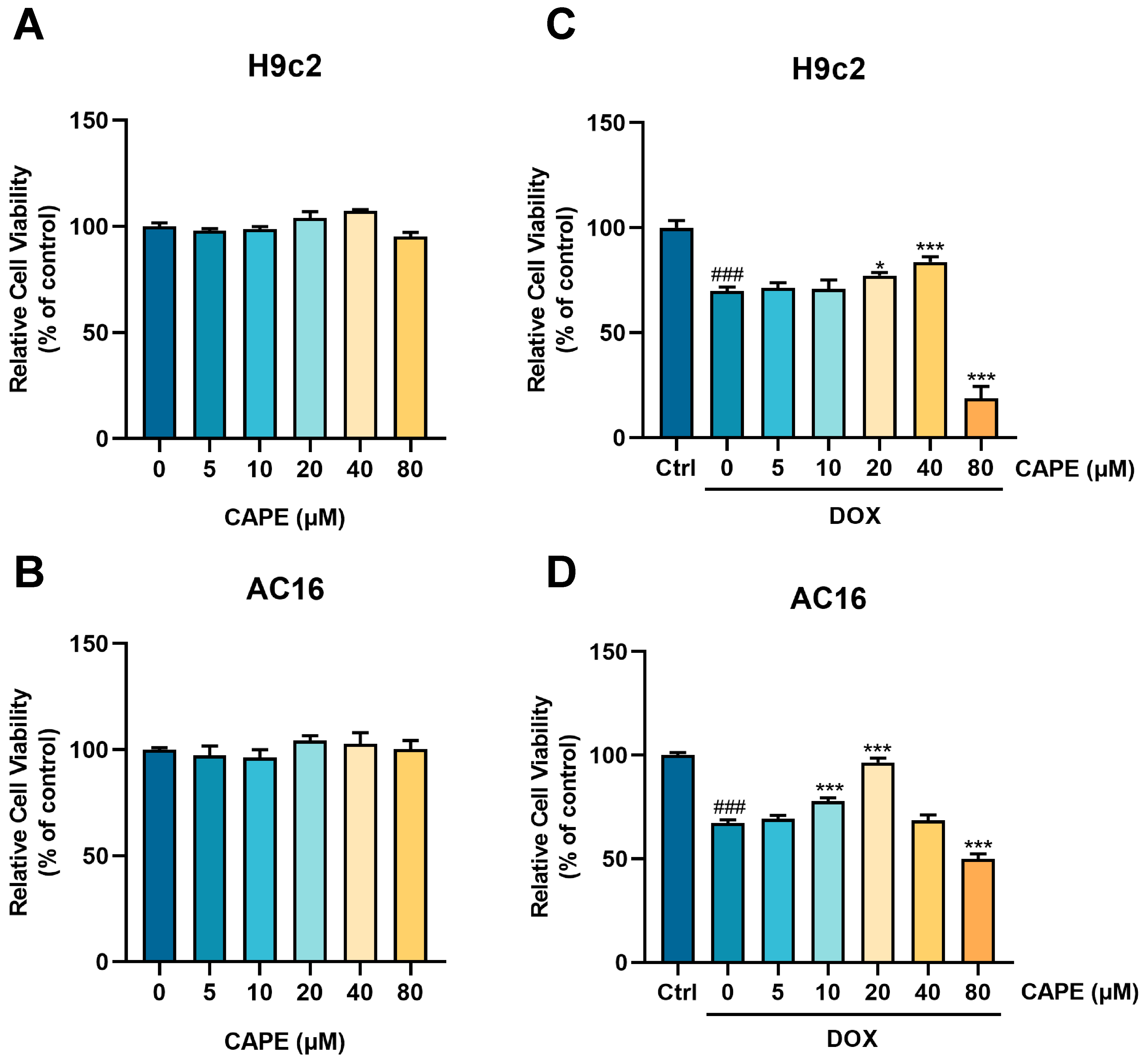
3.2. CAPE Alleviated DOX-Induced Myocardial Injury In Vitro
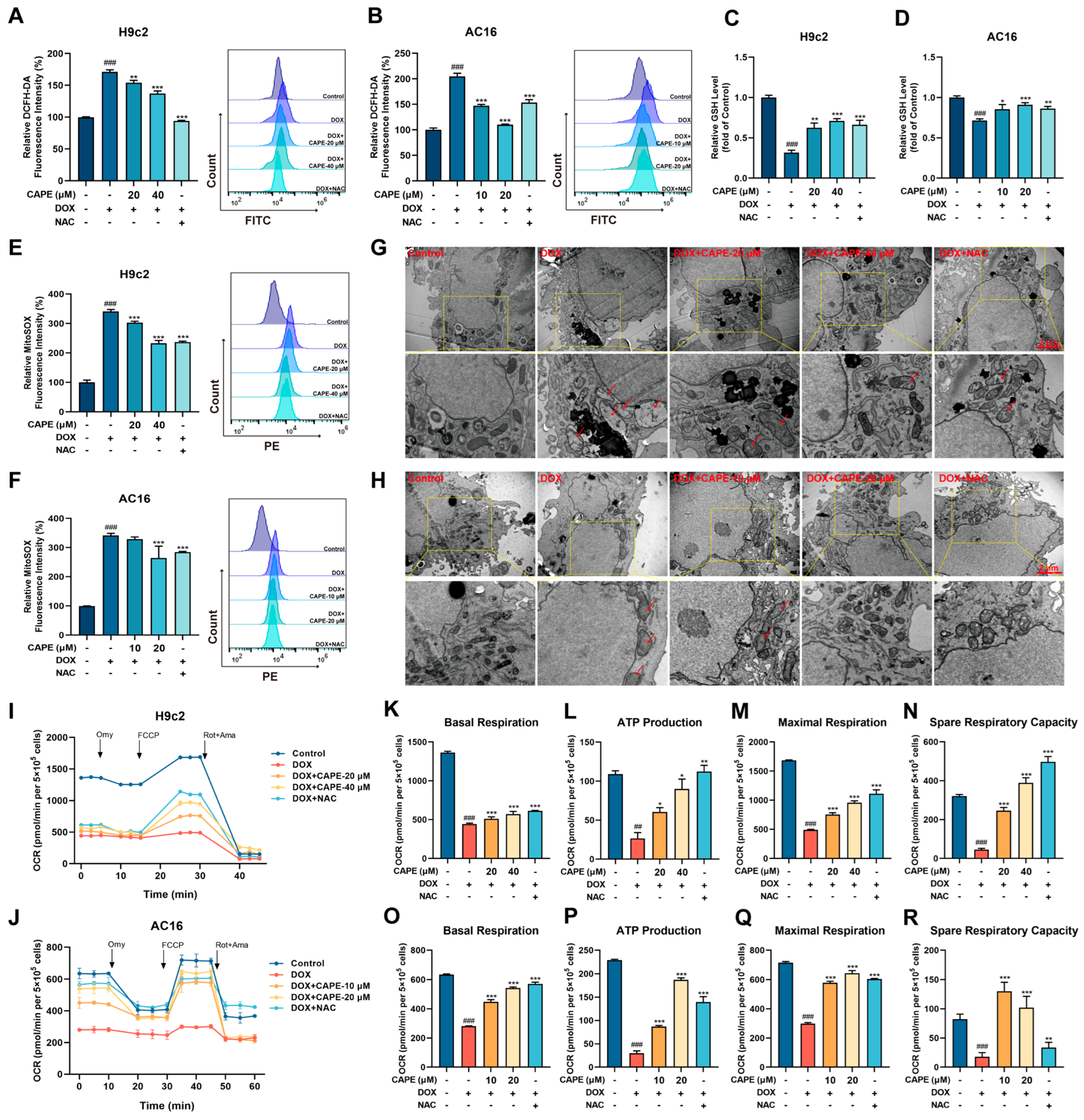
3.3. CAPE Attenuated DOX-Induced Oxidative Stress
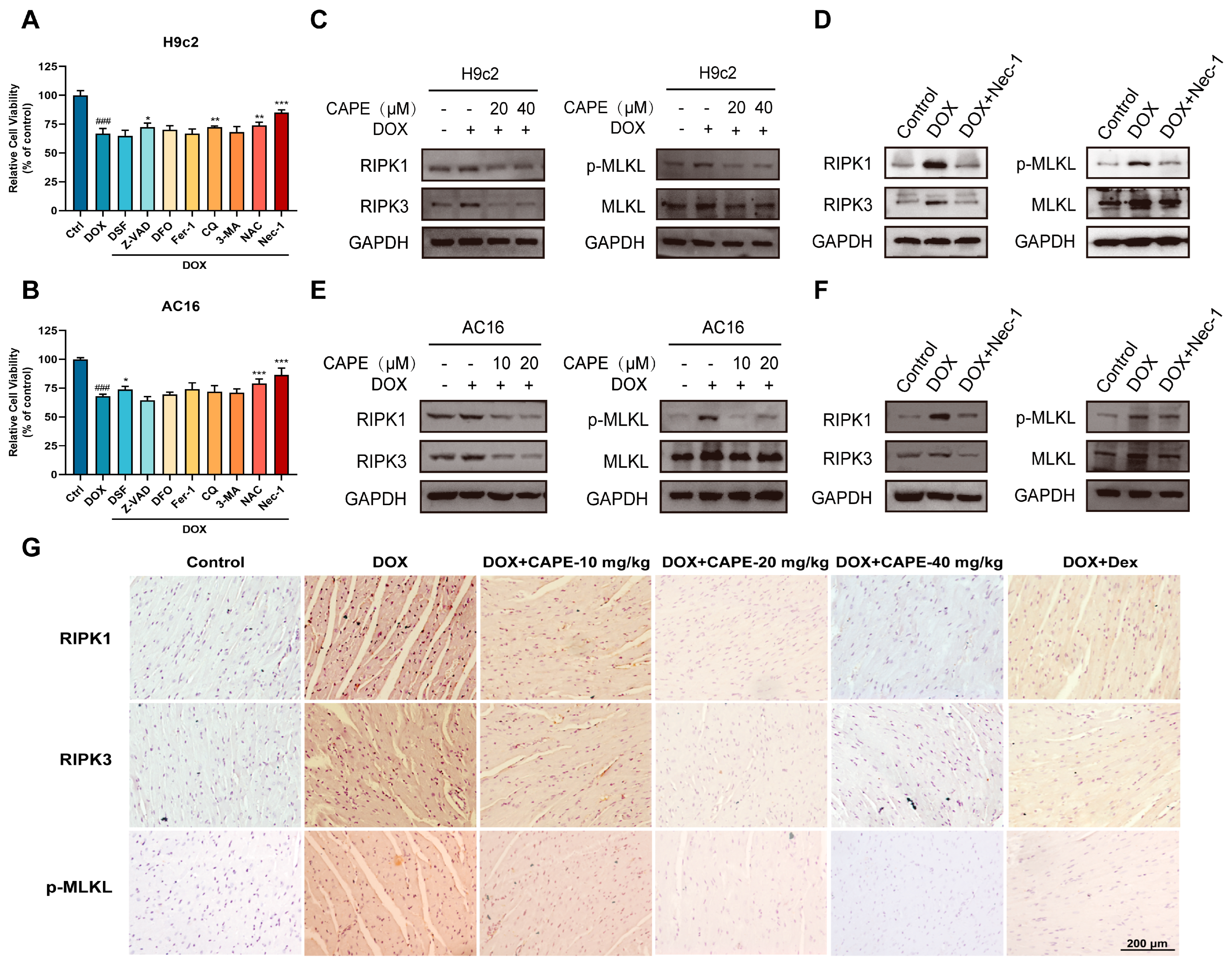
3.4. CAPE Inhibited DOX-Induced Necroptosis by Inhibiting the RIPK1/RIPK3/MLKL Pathway
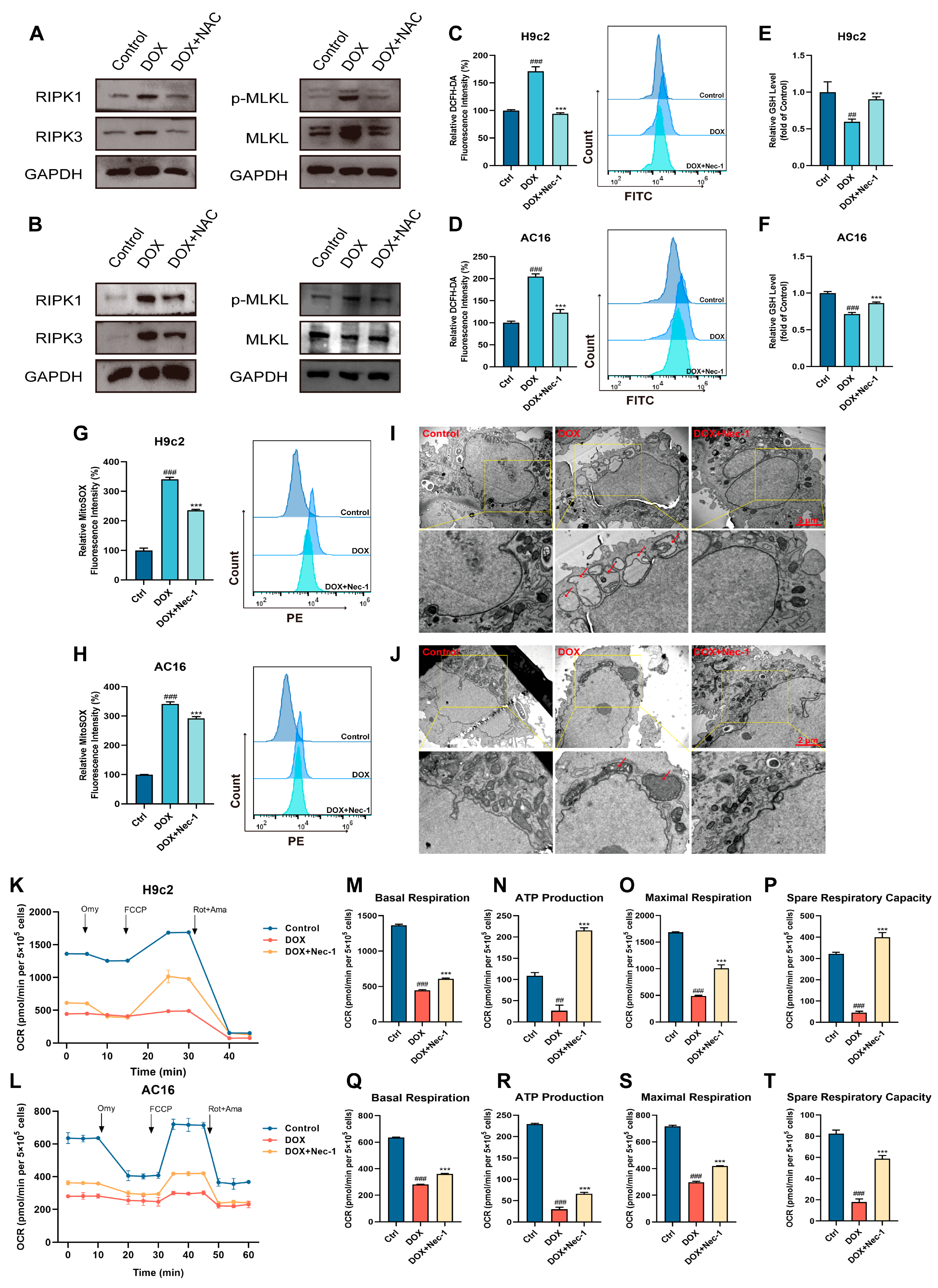
3.5. The Cross-Talk Between Oxidative Stress and Necroptosis Played a Vital Role in DOX-Induced Cardiotoxicity
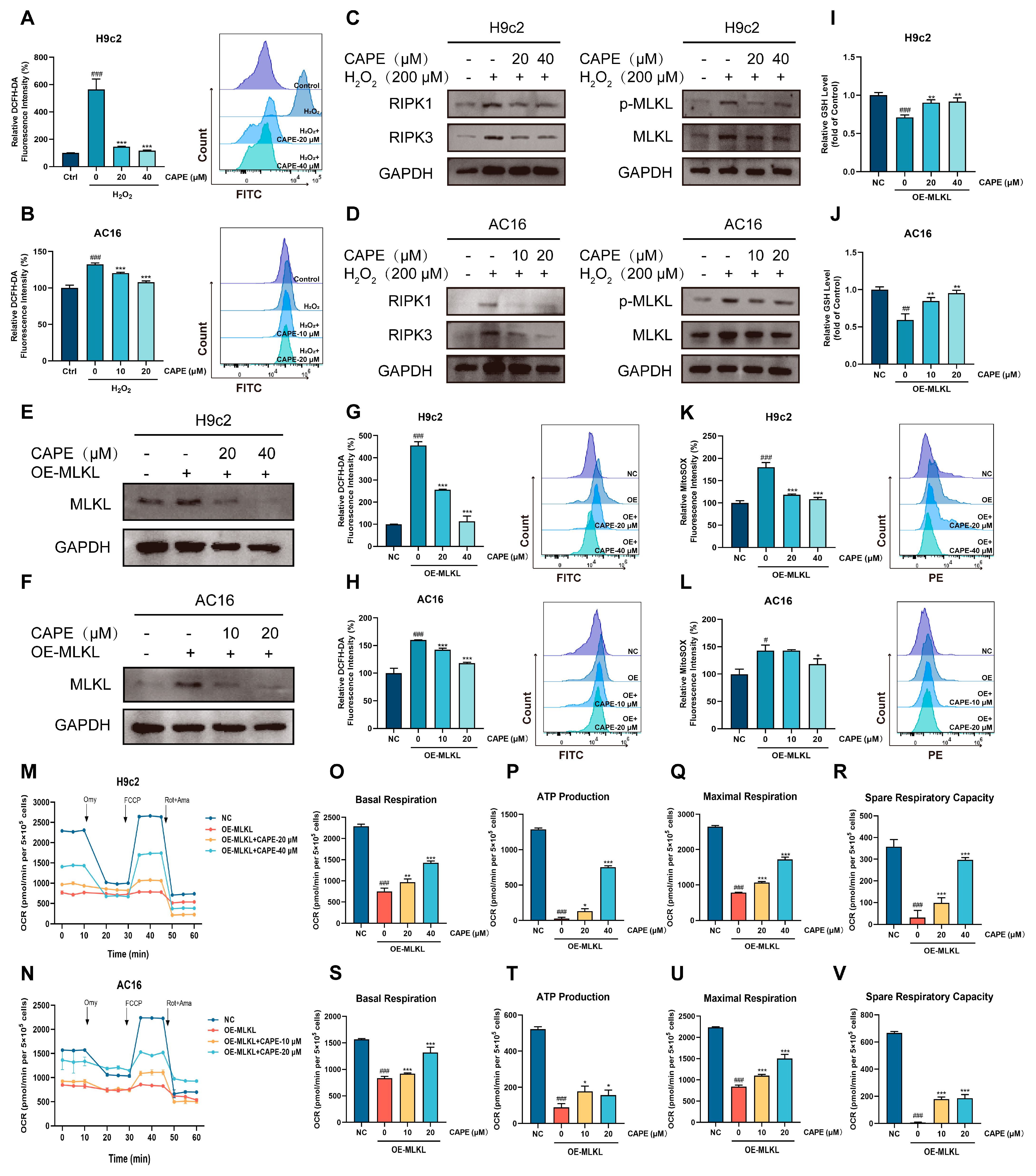
3.6. CAPE-Inhibited ROS-MLKL Signaling Mediated the Cross-Talk Between Oxidative Stress and Necroptosis
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Paridaens, R.; Biganzoli, L.; Bruning, P.; Klijn, J.G.; Gamucci, T.; Houston, S.; Coleman, R.; Schachter, J.; Van Vreckem, A.; Sylvester, R.; et al. Paclitaxel Versus Doxorubicin as First-Line Single-Agent Chemotherapy for Metastatic Breast Cancer: A European Organization for Research and Treatment of Cancer Randomized Study with Cross-Over. J. Clin. Oncol. 2000, 18, 724–733. [Google Scholar] [CrossRef] [PubMed]
- Seferović, P.M.; Polovina, M.; Bauersachs, J.; Arad, M.; Ben Gal, T.; Lund, L.H.; Felix, S.B.; Arbustini, E.; Caforio, A.L.P.; Farmakis, D.; et al. Heart Failure in Cardiomyopathies: A Position Paper from the Heart Failure Association of the European Society of Cardiology. Eur. J. Heart Fail. 2019, 21, 553–576. [Google Scholar] [CrossRef] [PubMed]
- Swain, S.M.; Whaley, F.S.; Ewer, M.S. Congestive Heart Failure in Patients Treated with Doxorubicin: A Retrospective Analysis of Three Trials. Cancer 2003, 97, 2869–2879. [Google Scholar] [CrossRef] [PubMed]
- De Baat, E.C.; Mulder, R.L.; Armenian, S.; Feijen, E.A.; Grotenhuis, H.; Hudson, M.M.; Mavinkurve-Groothuis, A.M.; Kremer, L.C.; van Dalen, E.C. Dexrazoxane for Preventing or Reducing Cardiotoxicity in Adults and Children with Cancer Receiving Anthracyclines. Cochrane Database Syst. Rev. 2022, 9, Cd014638. [Google Scholar] [CrossRef]
- Langer, S.W. Dexrazoxane for the Treatment of Chemotherapy-Related Side Effects. Cancer Manag. Res. 2014, 6, 357–363. [Google Scholar] [CrossRef]
- Van Dalen, E.C.; Caron, H.N.; Dickinson, H.O.; Kremer, L.C. Cardioprotective Interventions for Cancer Patients Receiving Anthracyclines. Cochrane Database Syst. Rev. 2011, 2011, Cd003917. [Google Scholar] [CrossRef]
- Bhargava, P.; Mahanta, D.; Kaul, A.; Ishida, Y.; Terao, K.; Wadhwa, R.; Kaul, S.C. Experimental Evidence for Therapeutic Potentials of Propolis. Nutrients 2021, 13, 2528. [Google Scholar] [CrossRef]
- Taysi, S.; Algburi, F.S.; Taysi, M.E.; Caglayan, C. Caffeic Acid Phenethyl Ester: A Review on Its Pharmacological Importance, and Its Association with Free Radicals, Covid-19, and Radiotherapy. Phytother. Res. PTR 2023, 37, 1115–1135. [Google Scholar] [CrossRef]
- Olgierd, B.; Kamila, Ż.; Anna, B.; Emilia, M. The Pluripotent Activities of Caffeic Acid Phenethyl Ester. Molecules 2021, 26, 1335. [Google Scholar] [CrossRef]
- Chung, L.C.; Chiang, K.C.; Feng, T.H.; Chang, K.S.; Chuang, S.T.; Chen, Y.J.; Tsui, K.H.; Lee, J.C.; Juang, H.H. Caffeic Acid Phenethyl Ester Upregulates N-Myc Downstream Regulated Gene 1 Via Erk Pathway to Inhibit Human Oral Cancer Cell Growth in Vitro and in Vivo. Mol. Nutr. Food Res. 2017, 61, 1600842. [Google Scholar] [CrossRef]
- Natella, F.; Nardini, M.; Di Felice, M.; Scaccini, C. Benzoic and Cinnamic Acid Derivatives as Antioxidants: Structure-Activity Relation. J. Agric. Food Chem. 1999, 47, 1453–1459. [Google Scholar] [CrossRef] [PubMed]
- Tolba, M.F.; Omar, H.A.; Azab, S.S.; Khalifa, A.E.; Abdel-Naim, A.B.; Abdel-Rahman, S.Z. Caffeic Acid Phenethyl Ester: A Review of Its Antioxidant Activity, Protective Effects against Ischemia-Reperfusion Injury and Drug Adverse Reactions. Crit. Rev. Food Sci. Nutr. 2016, 56, 2183–2190. [Google Scholar] [CrossRef] [PubMed]
- Teke, Z.; Bostanci, E.B.; Yenisey, C.; Kelten, E.C.; Sacar, M.; Simsek, N.G.; Duzcan, S.E.; Akoglu, M. Caffeic Acid Phenethyl Ester Prevents Detrimental Effects of Remote Ischemia-Reperfusion Injury on Healing of Colonic Anastomoses. J. Investig. Surg. 2013, 26, 16–29. [Google Scholar] [CrossRef]
- Ozyurt, B.; Iraz, M.; Koca, K.; Ozyurt, H.; Sahin, S. Protective Effects of Caffeic Acid Phenethyl Ester on Skeletal Muscle Ischemia-Reperfusion Injury in Rats. Mol. Cell. Biochem. 2006, 292, 197–203. [Google Scholar] [CrossRef]
- Eşrefoğlu, M.; Gül, M.; Parlakpinar, H.; Acet, A. Effects of Melatonin and Caffeic Acid Phenethyl Ester on Testicular Injury Induced by Myocardial Ischemia/Reperfusion in Rats. Fundam. Clin. Pharmacol. 2005, 19, 365–372. [Google Scholar] [CrossRef] [PubMed]
- Calikoglu, M.; Tamer, L.; Sucu, N.; Coskun, B.; Ercan, B.; Gul, A.; Calikoglu, I.; Kanik, A. The Effects of Caffeic Acid Phenethyl Ester on Tissue Damage in Lung after Hindlimb Ischemia-Reperfusion. Pharmacol. Res. 2003, 48, 397–403. [Google Scholar] [CrossRef]
- Sonoki, H.; Tanimae, A.; Furuta, T.; Endo, S.; Matsunaga, T.; Ichihara, K.; Ikari, A. Caffeic Acid Phenethyl Ester Down-Regulates Claudin-2 Expression at the Transcriptional and Post-Translational Levels and Enhances Chemosensitivity to Doxorubicin in Lung Adenocarcinoma A549 Cells. J. Nutr. Biochem. 2018, 56, 205–214. [Google Scholar] [CrossRef]
- Takara, K.; Fujita, M.; Matsubara, M.; Minegaki, T.; Kitada, N.; Ohnishi, N.; Yokoyama, T. Effects of Propolis Extract on Sensitivity to Chemotherapeutic Agents in Hela and Resistant Sublines. Phytother. Res. PTR 2007, 21, 841–846. [Google Scholar] [CrossRef]
- Christidi, E.; Brunham, L.R. Regulated Cell Death Pathways in Doxorubicin-Induced Cardiotoxicity. Cell Death Dis. 2021, 12, 339. [Google Scholar] [CrossRef]
- Vitale, R.; Marzocco, S.; Popolo, A. Role of Oxidative Stress and Inflammation in Doxorubicin-Induced Cardiotoxicity: A Brief Account. Int. J. Mol. Sci. 2024, 25, 7477. [Google Scholar] [CrossRef]
- Kong, C.Y.; Guo, Z.; Song, P.; Zhang, X.; Yuan, Y.P.; Teng, T.; Yan, L.; Tang, Q.Z. Underlying the Mechanisms of Doxorubicin-Induced Acute Cardiotoxicity: Oxidative Stress and Cell Death. Int. J. Biol. Sci. 2022, 18, 760–770. [Google Scholar] [CrossRef] [PubMed]
- Yao, T.; Fujimura, T.; Murayama, K.; Okumura, K.; Seko, Y. Oxidative Stress-Responsive Apoptosis Inducing Protein (Oraip) Plays a Critical Role in Doxorubicin-Induced Apoptosis in Rat Cardiac Myocytes. Int. J. Cardiol. 2022, 348, 119–124. [Google Scholar] [CrossRef]
- Wallace, K.B.; Sardão, V.A.; Oliveira, P.J. Mitochondrial Determinants of Doxorubicin-Induced Cardiomyopathy. Circ. Res. 2020, 126, 926–941. [Google Scholar] [CrossRef]
- Ide, T.; Tsutsui, H.; Kinugawa, S.; Suematsu, N.; Hayashidani, S.; Ichikawa, K.; Utsumi, H.; Machida, Y.; Egashira, K.; Takeshita, A. Direct Evidence for Increased Hydroxyl Radicals Originating from Superoxide in the Failing Myocardium. Circ. Res. 2000, 86, 152–157. [Google Scholar] [CrossRef] [PubMed]
- Wu, B.B.; Leung, K.T.; Poon, E.N. Mitochondrial-Targeted Therapy for Doxorubicin-Induced Cardiotoxicity. Int. J. Mol. Sci. 2022, 23, 1912. [Google Scholar] [CrossRef]
- Gielecińska, A.; Kciuk, M.; Yahya, E.B.; Ainane, T.; Mujwar, S.; Kontek, R. Apoptosis, Necroptosis, and Pyroptosis as Alternative Cell Death Pathways Induced by Chemotherapeutic Agents? Biochim. Biophys. Acta Rev. Cancer 2023, 1878, 189024. [Google Scholar] [CrossRef]
- Reis-Mendes, A.; Ferreira, M.; Padrão, A.I.; Duarte, J.A.; Duarte-Araújo, M.; Remião, F.; Carvalho, F.; Sousa, E.; Bastos, M.L.; Costa, V.M. The Role of Nrf2 and Inflammation on the Dissimilar Cardiotoxicity of Doxorubicin in Two-Time Points: A Cardio-Oncology in Vivo Study through Time. Inflammation 2024, 47, 264–284. [Google Scholar] [CrossRef]
- Zhao, L.; Zhang, B. Doxorubicin Induces Cardiotoxicity through Upregulation of Death Receptors Mediated Apoptosis in Cardiomyocytes. Sci. Rep. 2017, 7, 44735. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Yang, Y.; He, W.; Sun, L. Necrosome Core Machinery: Mlkl. Cell Mol. Life Sci. 2016, 73, 2153–2163. [Google Scholar] [CrossRef]
- Poltorak, A. Cell Death: All Roads Lead to Mitochondria. Curr. Biol. 2022, 32, R891–R894. [Google Scholar] [CrossRef]
- Maeda, A.; Fadeel, B. Mitochondria Released by Cells Undergoing Tnf-A-Induced Necroptosis Act as Danger Signals. Cell Death Dis. 2014, 5, e1312. [Google Scholar] [CrossRef] [PubMed]
- Irrinki, K.M.; Mallilankaraman, K.; Thapa, R.J.; Chandramoorthy, H.C.; Smith, F.J.; Jog, N.R.; Gandhirajan, R.K.; Kelsen, S.G.; Houser, S.R.; May, M.J.; et al. Requirement of Fadd, Nemo, and Bax/Bak for Aberrant Mitochondrial Function in Tumor Necrosis Factor Alpha-Induced Necrosis. Mol. Cell Biol. 2011, 31, 3745–3758. [Google Scholar] [CrossRef] [PubMed]
- Weindel, C.G.; Martinez, E.L.; Zhao, X.; Mabry, C.J.; Bell, S.L.; Vail, K.J.; Coleman, A.K.; VanPortfliet, J.J.; Zhao, B.; Wagner, A.R.; et al. Mitochondrial Ros Promotes Susceptibility to Infection Via Gasdermin D-Mediated Necroptosis. Cell 2022, 185, 3214–3231.e3223. [Google Scholar] [CrossRef]
- Chen, H.; Li, Y.; Wu, J.; Li, G.; Tao, X.; Lai, K.; Yuan, Y.; Zhang, X.; Zou, Z.; Xu, Y. Ripk3 Collaborates with Gsdmd to Drive Tissue Injury in Lethal Polymicrobial Sepsis. Cell Death Differ. 2020, 27, 2568–2585. [Google Scholar] [CrossRef]
- Horvath, C.; Young, M.; Jarabicova, I.; Kindernay, L.; Ferenczyova, K.; Ravingerova, T.; Lewis, M.; Suleiman, M.S.; Adameova, A. Inhibition of Cardiac Rip3 Mitigates Early Reperfusion Injury and Calcium-Induced Mitochondrial Swelling without Altering Necroptotic Signalling. Int. J. Mol. Sci. 2021, 22, 7983. [Google Scholar] [CrossRef] [PubMed]
- Shen, C.; Yang, B.; Huang, L.; Chen, Y.; Zhao, H.; Zhu, Z. Cardioprotective Effect of Crude Polysaccharide Fermented by Trametes Sanguinea Lyoyd on Doxorubicin-Induced Myocardial Injury Mice. BMC Pharmacol. Toxicol. 2023, 24, 1. [Google Scholar]
- Martinez-Osorio, V.; Abdelwahab, Y.; Ros, U. The Many Faces of Mlkl, the Executor of Necroptosis. Int. J. Mol. Sci. 2023, 24, 10108. [Google Scholar] [CrossRef]
- Ren, J.; Zhang, N.; Liao, H.; Chen, S.; Xu, L.; Li, J.; Yang, Z.; Deng, W.; Tang, Q. Caffeic Acid Phenethyl Ester Attenuates Pathological Cardiac Hypertrophy by Regulation of Mek/Erk Signaling Pathway in Vivo and Vitro. Life Sci. 2017, 181, 53–61. [Google Scholar] [CrossRef]
- Ho, Y.J.; Lee, A.S.; Chen, W.P.; Chang, W.L.; Tsai, Y.K.; Chiu, H.L.; Kuo, Y.H.; Su, M.J. Caffeic Acid Phenethyl Amide Ameliorates Ischemia/Reperfusion Injury and Cardiac Dysfunction in Streptozotocin-Induced Diabetic Rats. Cardiovasc. Diabetol. 2014, 13, 98. [Google Scholar] [CrossRef]
- Park, S.H.; Min, T.S. Caffeic Acid Phenethyl Ester Ameliorates Changes in Igfs Secretion and Gene Expression in Streptozotocin-Induced Diabetic Rats. Life Sci. 2006, 78, 1741–1747. [Google Scholar] [CrossRef]
- Ozer, M.K.; Parlakpinar, H.; Cigremis, Y.; Ucar, M.; Vardi, N.; Acet, A. Ischemia-Reperfusion Leads to Depletion of Glutathione Content and Augmentation of Malondialdehyde Production in the Rat Heart from Overproduction of Oxidants: Can Caffeic Acid Phenethyl Ester (Cape) Protect the Heart? Mol. Cell Biochem. 2005, 273, 169–175. [Google Scholar] [CrossRef] [PubMed]
- Tan, J.; Ma, Z.; Han, L.; Du, R.; Zhao, L.; Wei, X.; Hou, D.; Johnstone, B.H.; Farlow, M.R.; Du, Y. Caffeic Acid Phenethyl Ester Possesses Potent Cardioprotective Effects in a Rabbit Model of Acute Myocardial Ischemia-Reperfusion Injury. Am. J. Physiol. Heart Circ. Physiol. 2005, 289, H2265–H2271. [Google Scholar] [CrossRef] [PubMed]
- Sun, X.; Du, J.; Meng, H.; Liu, F.; Yang, N.; Deng, S.; Wan, H.; Ye, D.; Song, E.; Zeng, H. Targeting Autophagy with Sar405 Alleviates Doxorubicin-Induced Cardiotoxicity. Cell Biol. Toxicol. 2023, 39, 3255–3267. [Google Scholar] [CrossRef]
- Shu, G.; Chen, K.; Li, J.; Liu, B.; Chen, X.; Wang, J.; Hu, X.; Lu, W.; Huang, H.; Zhang, S. Galangin Alleviated Doxorubicin-Induced Cardiotoxicity by Inhibiting Ferroptosis through Gstp1/Jnk Pathway. Phytomedicine 2024, 134, 155989. [Google Scholar] [CrossRef]
- Lin, Z.; Wu, C.; Song, D.; Zhu, C.; Wu, B.; Wang, J.; Xue, Y. Sarmentosin Alleviates Doxorubicin-Induced Cardiotoxicity and Ferroptosis Via the P62-Keap1-Nrf2 Pathway. Redox Rep. Commun. Free Radic. Res. 2024, 29, 2392329. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Ji, Y.; Jin, D.; Qi, J.; Hou, X.; Zhao, W.; Zhou, S.; Zhang, C.; Luo, Y.; An, P.; et al. Natural Polysaccharide Β-Glucan Protects against Doxorubicin-Induced Cardiotoxicity by Suppressing Oxidative Stress. Nutrients 2022, 14, 906. [Google Scholar] [CrossRef]
- Zhang, Y.; Su, S.S.; Zhao, S.; Yang, Z.; Zhong, C.Q.; Chen, X.; Cai, Q.; Yang, Z.H.; Huang, D.; Wu, R.; et al. Rip1 Autophosphorylation Is Promoted by Mitochondrial Ros and Is Essential for Rip3 Recruitment into Necrosome. Nat. Commun. 2017, 8, 14329. [Google Scholar] [CrossRef]
- Liu, M.; Li, H.; Yang, R.; Ji, D.; Xia, X. Gsk872 and Necrostatin-1 Protect Retinal Ganglion Cells against Necroptosis through Inhibition of Rip1/Rip3/Mlkl Pathway in Glutamate-Induced Retinal Excitotoxic Model of Glaucoma. J. Neuroinflamm. 2022, 19, 262. [Google Scholar] [CrossRef]
- Vanlangenakker, N.; Vanden Berghe, T.; Bogaert, P.; Laukens, B.; Zobel, K.; Deshayes, K.; Vucic, D.; Fulda, S.; Vandenabeele, P.; Bertrand, M.J. Ciap1 and Tak1 Protect Cells from Tnf-Induced Necrosis by Preventing Rip1/Rip3-Dependent Reactive Oxygen Species Production. Cell Death Differ. 2011, 18, 656–665. [Google Scholar] [CrossRef]
- Xie, L.; Xue, F.; Cheng, C.; Sui, W.; Zhang, J.; Meng, L.; Lu, Y.; Xiong, W.; Bu, P.; Xu, F.; et al. Cardiomyocyte-Specific Knockout of Adam17 Alleviates Doxorubicin-Induced Cardiomyopathy Via Inhibiting Tnfα-Traf3-Tak1-Mapk Axis. Signal Transduct. Target. Ther. 2024, 9, 273. [Google Scholar] [CrossRef]
- Khames, A.; Khalaf, M.M.; Gad, A.M.; Abd El-Raouf, O.M.; Kandeil, M.A. Nicorandil Combats Doxorubicin-Induced Nephrotoxicity Via Amendment of Tlr4/P38 Mapk/Nfκ-B Signaling Pathway. Chem.-Biol. Interact. 2019, 311, 108777. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Kong, D.; Han, H.; Cao, Y.; Zhu, H.; Cui, G. Caffeic Acid Phenethyl Ester Protects against Doxorubicin-Induced Cardiotoxicity and Increases Chemotherapeutic Efficacy by Regulating the Unfolded Protein Response. Food Chem. Toxicol. 2022, 159, 112770. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Wu, F.; Tan, J.; Wang, K.; Zhang, C.; Zheng, H.; Hu, F. Caffeic Acid Phenethyl Ester Exhibiting Distinctive Binding Interaction with Human Serum Albumin Implies the Pharmacokinetic Basis of Propolis Bioactive Components. J. Pharm. Biomed. Anal. 2016, 122, 21–28. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Bowman, P.D.; Kerwin, S.M.; Stavchansky, S. Development and Validation of an Lcms Method to Determine the Pharmacokinetic Profiles of Caffeic Acid Phenethyl Amide and Caffeic Acid Phenethyl Ester in Male Sprague-Dawley Rats. Biomed. Chromatogr. BMC 2014, 28, 241–246. [Google Scholar] [CrossRef]
- Wang, X.; Pang, J.; Maffucci, J.A.; Pade, D.S.; Newman, R.A.; Kerwin, S.M.; Bowman, P.D.; Stavchansky, S. Pharmacokinetics of Caffeic Acid Phenethyl Ester and Its Catechol-Ring Fluorinated Derivative Following Intravenous Administration to Rats. Biopharm. Drug Dispos. 2009, 30, 221–228. [Google Scholar] [CrossRef]
- Liu, Y.; Li, J.; Zhu, H.J. Regulation of Carboxylesterases and Its Impact on Pharmacokinetics and Pharmacodynamics: An up-to-Date Review. Expert. Opin. Drug Metab. Toxicol. 2024, 20, 377–397. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jiang, C.; Zhang, T.; Gu, J.; Shen, C.; Gao, H.; An, H.; Wang, C.; Lu, J.; Lin, S.; Zhao, H.; et al. Caffeic Acid Phenethyl Ester Protects Against Doxorubicin-Induced Cardiotoxicity via Inhibiting the ROS-MLKL-Mediated Cross-Talk Between Oxidative Stress and Necroptosis. Biomolecules 2025, 15, 783. https://doi.org/10.3390/biom15060783
Jiang C, Zhang T, Gu J, Shen C, Gao H, An H, Wang C, Lu J, Lin S, Zhao H, et al. Caffeic Acid Phenethyl Ester Protects Against Doxorubicin-Induced Cardiotoxicity via Inhibiting the ROS-MLKL-Mediated Cross-Talk Between Oxidative Stress and Necroptosis. Biomolecules. 2025; 15(6):783. https://doi.org/10.3390/biom15060783
Chicago/Turabian StyleJiang, Chenying, Tinghuang Zhang, Jiawen Gu, Chenjun Shen, Hang Gao, Hai An, Chen Wang, Jiahui Lu, Shengzhang Lin, Huajun Zhao, and et al. 2025. "Caffeic Acid Phenethyl Ester Protects Against Doxorubicin-Induced Cardiotoxicity via Inhibiting the ROS-MLKL-Mediated Cross-Talk Between Oxidative Stress and Necroptosis" Biomolecules 15, no. 6: 783. https://doi.org/10.3390/biom15060783
APA StyleJiang, C., Zhang, T., Gu, J., Shen, C., Gao, H., An, H., Wang, C., Lu, J., Lin, S., Zhao, H., & Zhu, Z. (2025). Caffeic Acid Phenethyl Ester Protects Against Doxorubicin-Induced Cardiotoxicity via Inhibiting the ROS-MLKL-Mediated Cross-Talk Between Oxidative Stress and Necroptosis. Biomolecules, 15(6), 783. https://doi.org/10.3390/biom15060783