
Bio-Inspired Mamba for Antibody–Antigen Interaction Prediction



Abstract
:1. Introduction
2. Materials and Methods
2.1. Data Preparation
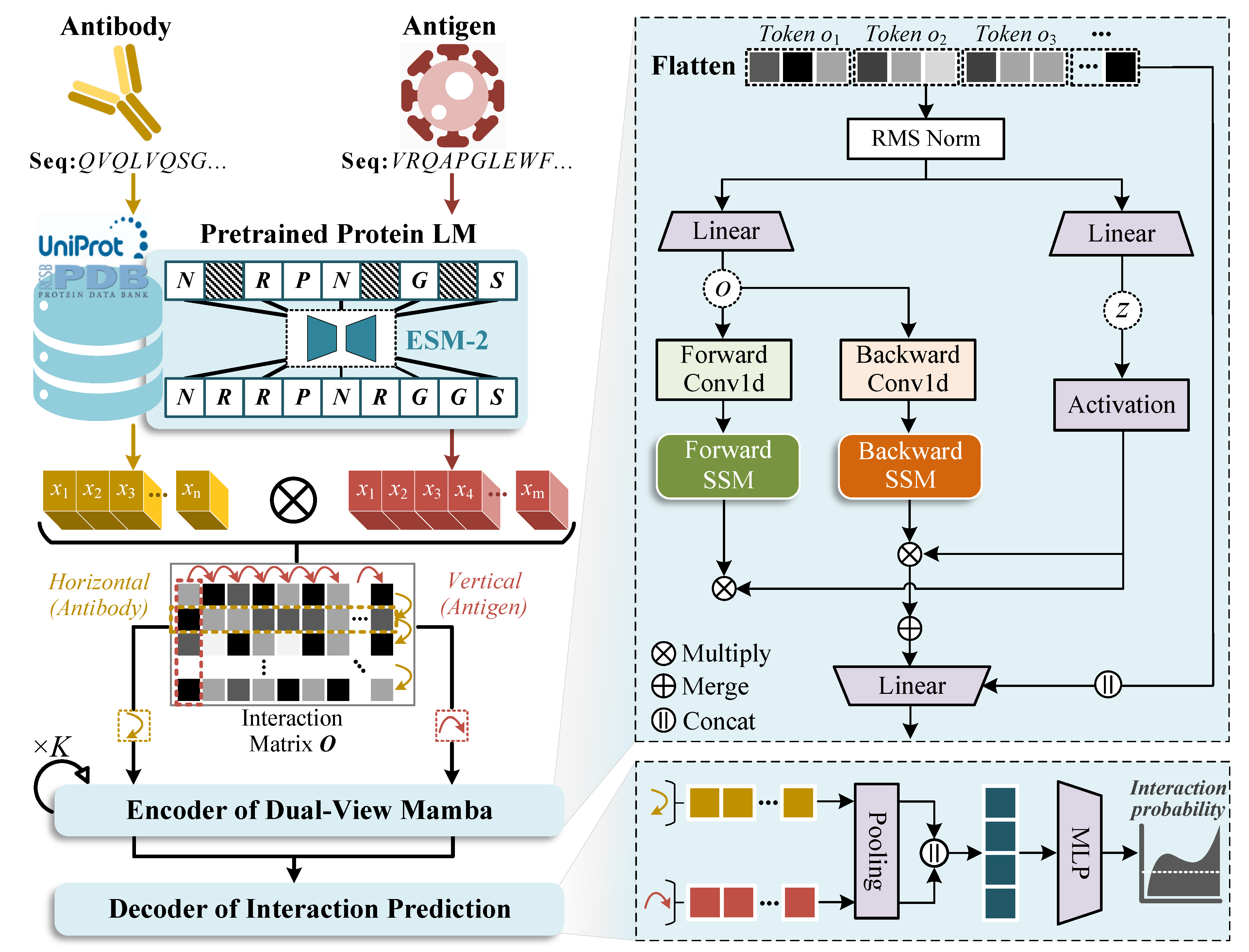
2.2. Model Architecture
2.2.1. Representation Extraction with PPLMs
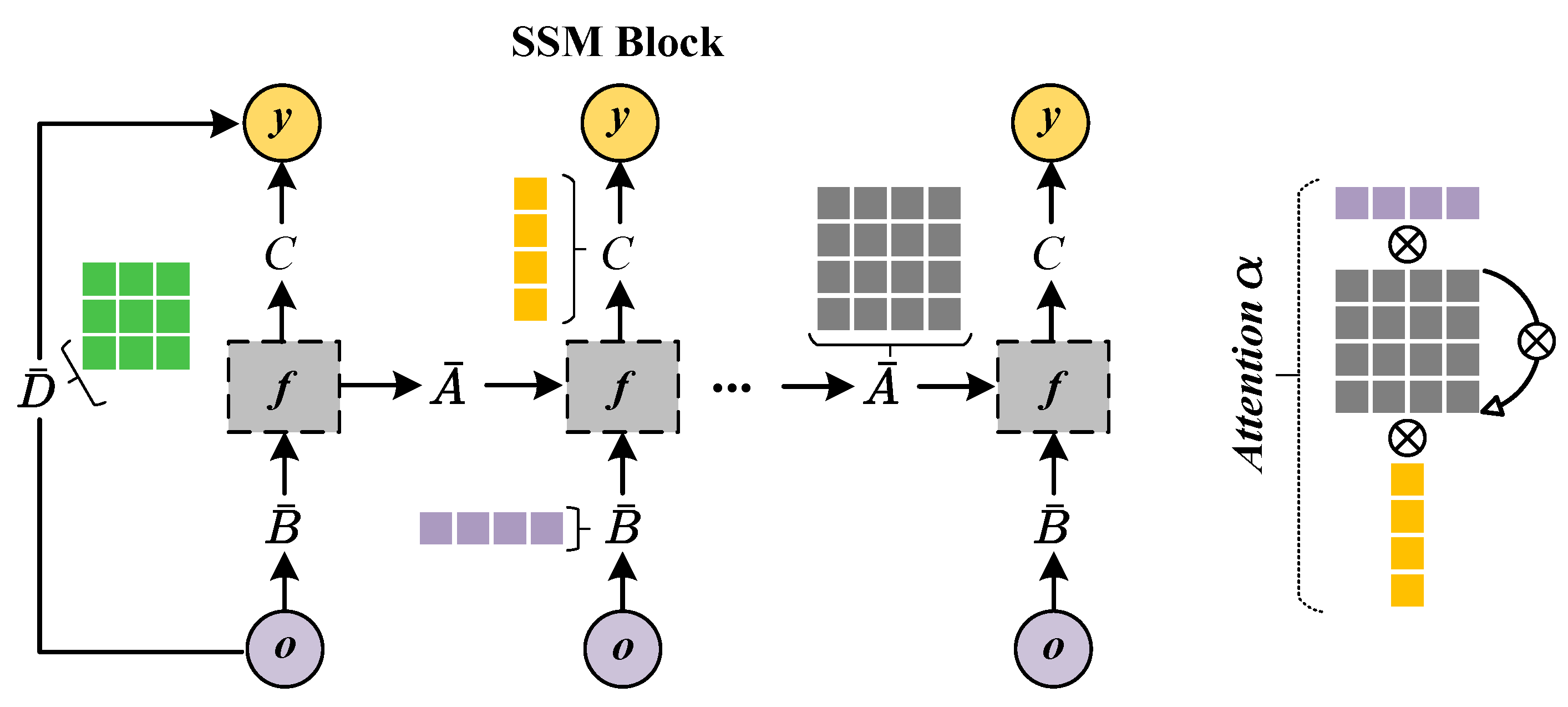
2.2.2. Encoder of Dual-View Mamba
2.2.3. Decoder of Interaction Prediction
3. Results
3.1. Evaluation Protocol
- •
- Cross-validation for seen antibodies and antigens. The regular 5-fold cross-validation (5-CV) is implemented by randomly dividing all samples of cross-validation set into five equal parts, iteratively using four parts for training and one part for validating across 5 times. This scenario is to rediscover known/seen AAIs.
- •
- Independent testing for unseen antibodies and antigens. The model is trained on the whole cross-validation set, and then makes predictions over the independent test set for objective evaluation. Since the independent test set is derived through partitioning at both the antibody or antigen levels, it guarantees only unseen antibodies and antigens are included in the testing stage.
3.2. Baselines
- •
- PIPR [41] introduced a deep residual recurrent CNN model for protein–protein interaction (e.g., AAI) prediction, which extracts both local features and contextualized information hidden in protein sequences.
- •
- DeepAAI [20] captured representations of unseen antibodies and seen antigens by constructing two adaptive relational graph neural networks, and leveraged laplacian smoothing to refine them for AAI predictions.
- •
- AttABseq [19] utilized CNNs to encode one-hot and PSSM features of antibodies and antigens, and then devised a multi-head mutual-attention mechanism to predict antigen–antibody binding affinity changes.
- •
- DeepInterAware [26] combined the pre-trained embeddings via the ESM-2 and AbLang, and incorporated both antigen–antibody specificity and sequence-derived contextual features for modeling dynamic interaction interface of AAIs.
- •
- PECAN [13] presented a unified deep learning framework that consists of a novel combination of graph convolution networks, attention mechanisms, and transfer learning, so as to enhance the representation learning in the AAI prediction.
- •
- AbAgIPA [42] constructed a hybrid neural network for AAI prediction, which extracts structural features of antibodies/antigens through physicochemical-based vectors and invariant point attention mechanisms.
3.3. Hyperparameter Settings
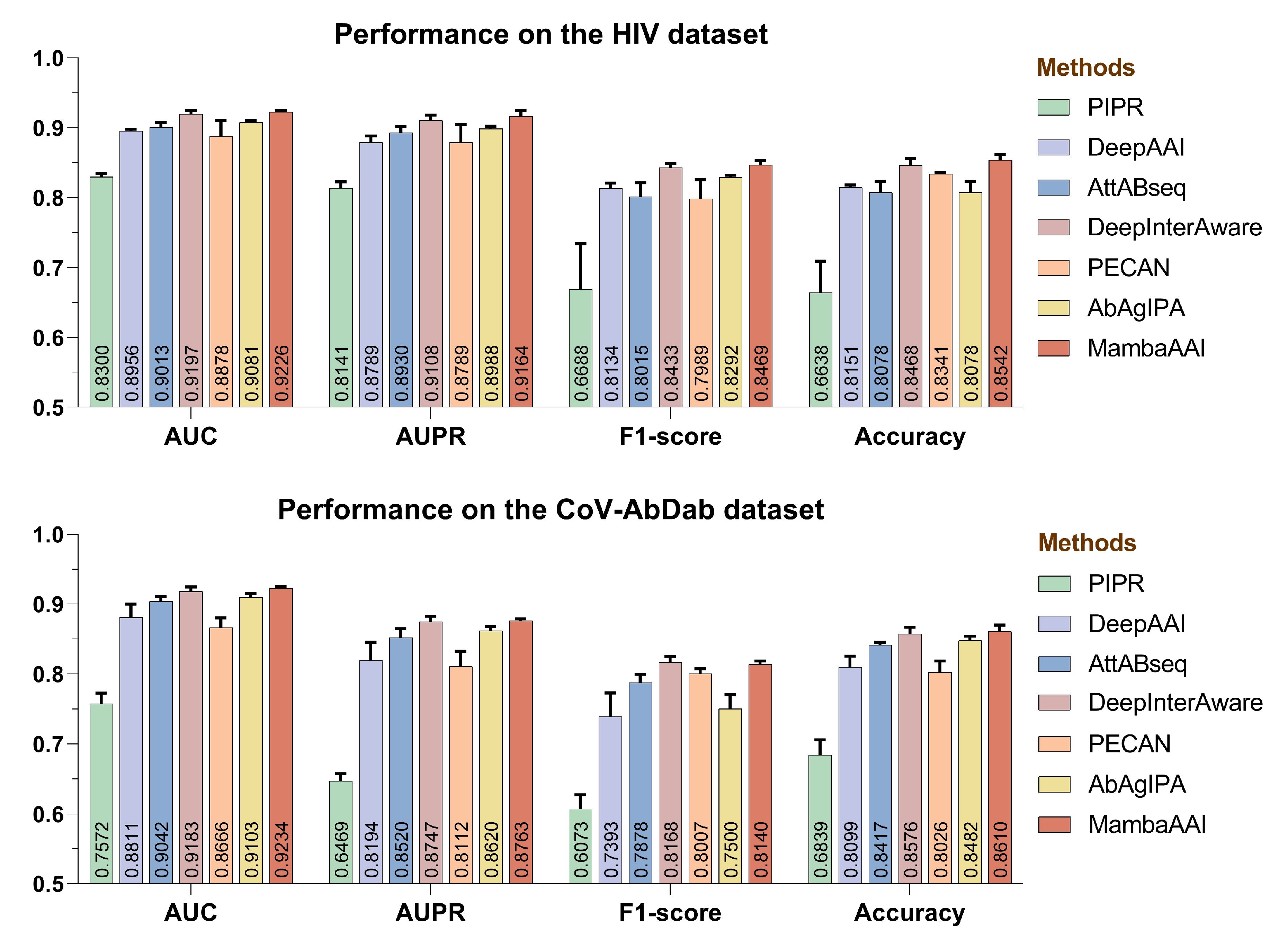
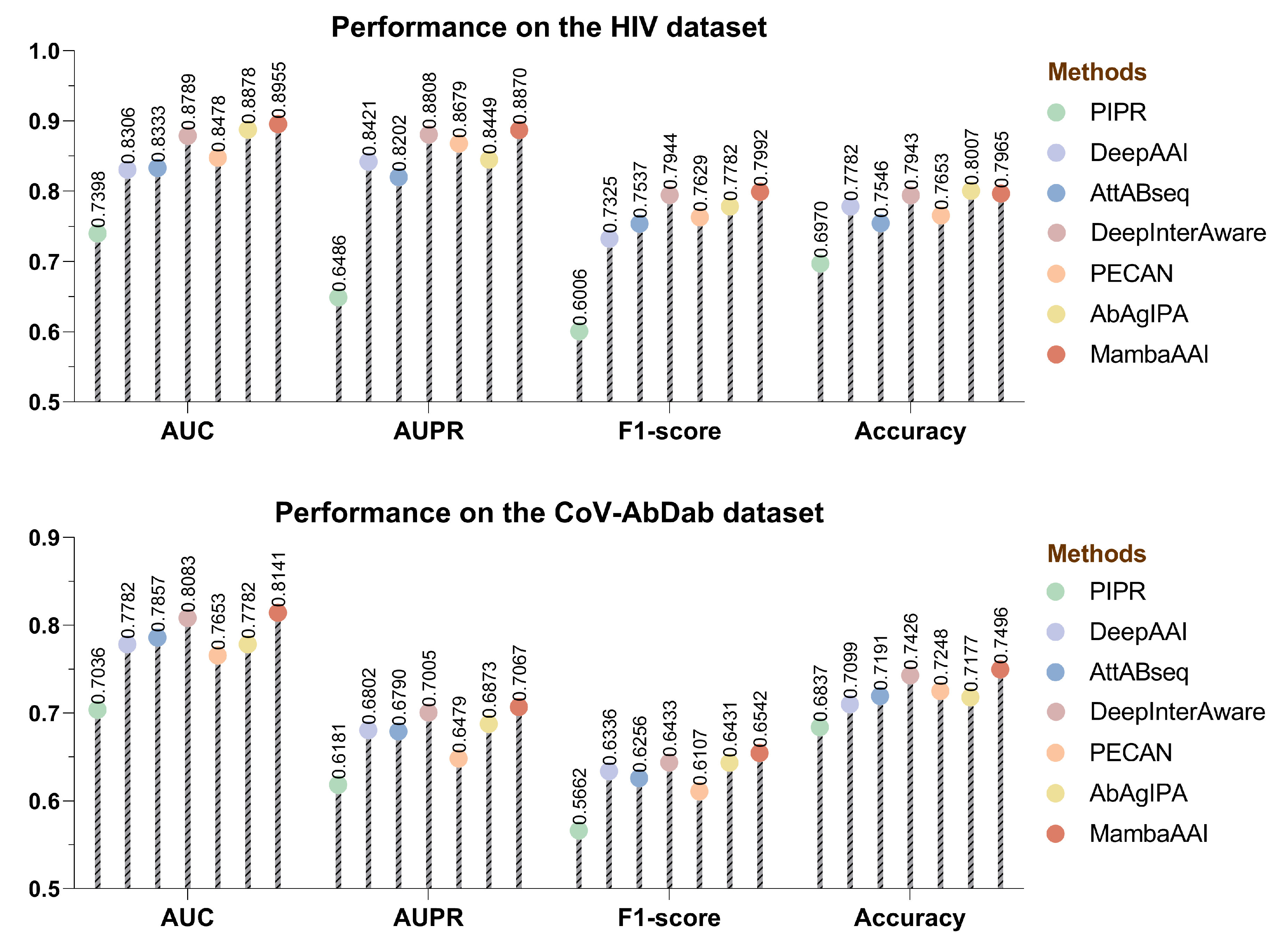
3.4. Performance Comparison
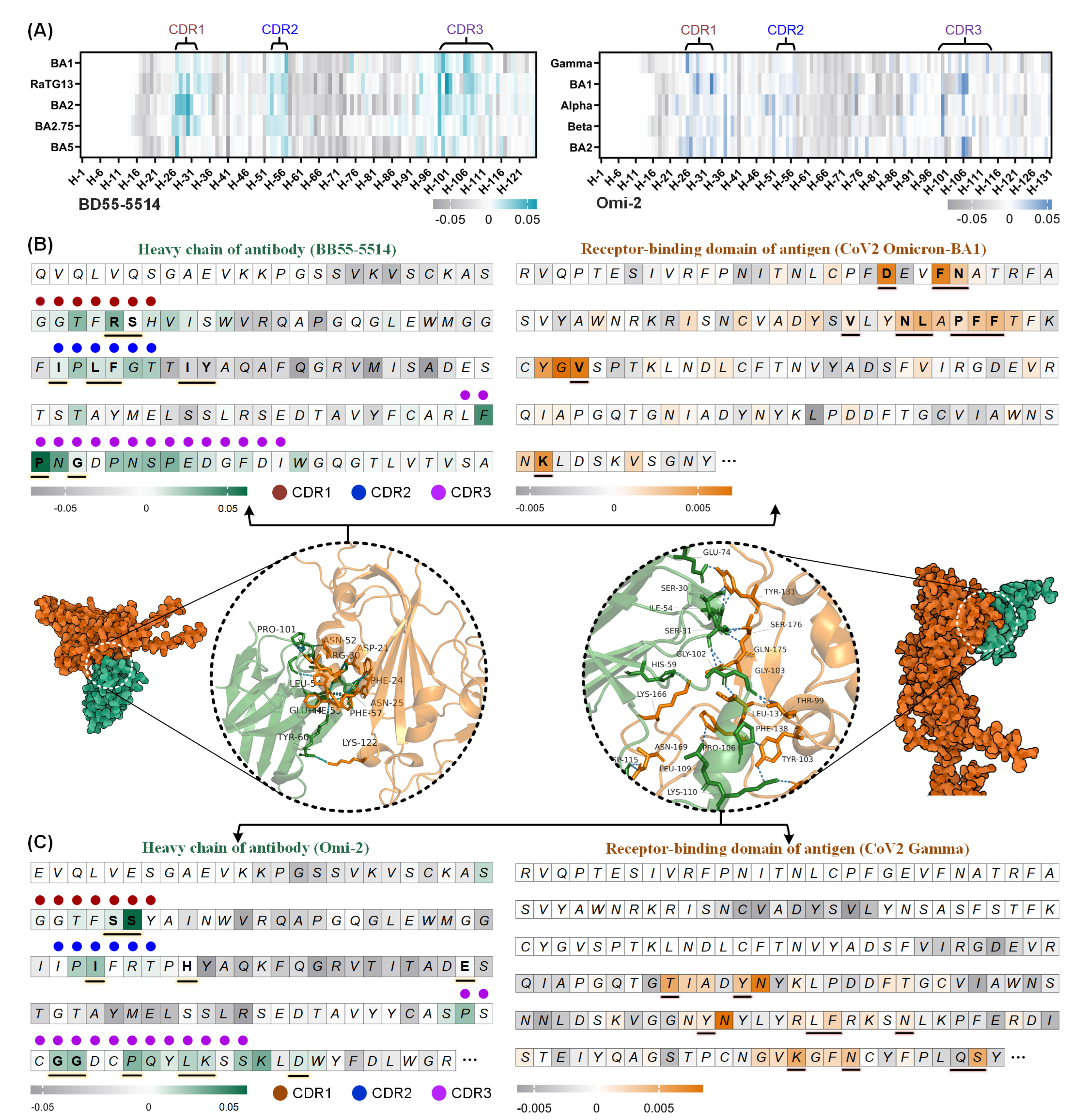
3.5. Interpretation Analysis of Binding Sites
3.6. Ablation Results
- •
- MambaAAI (-RE) eliminates the representation extraction using PPLMs, replacing it with the BLOSUM62 matrix to initialize antigen and antibody representations.
- •
- MambaAAI (-BP) eliminates the backward process in the bidirectional SSM block, retaining only the forward process for downstream prediction.
- •
- MambaAAI (-HV) eliminates the horizontal view, preserving only the encoded representations from the vertical view.
- •
- MambaAAI (-VV) eliminates the vertical view, retaining only the encoded representations from the horizontal view.
- •
- MambaAAI (-SSM) replaces the state space model (SSM) of Mamba with a conventional Transformer’s self-attention.
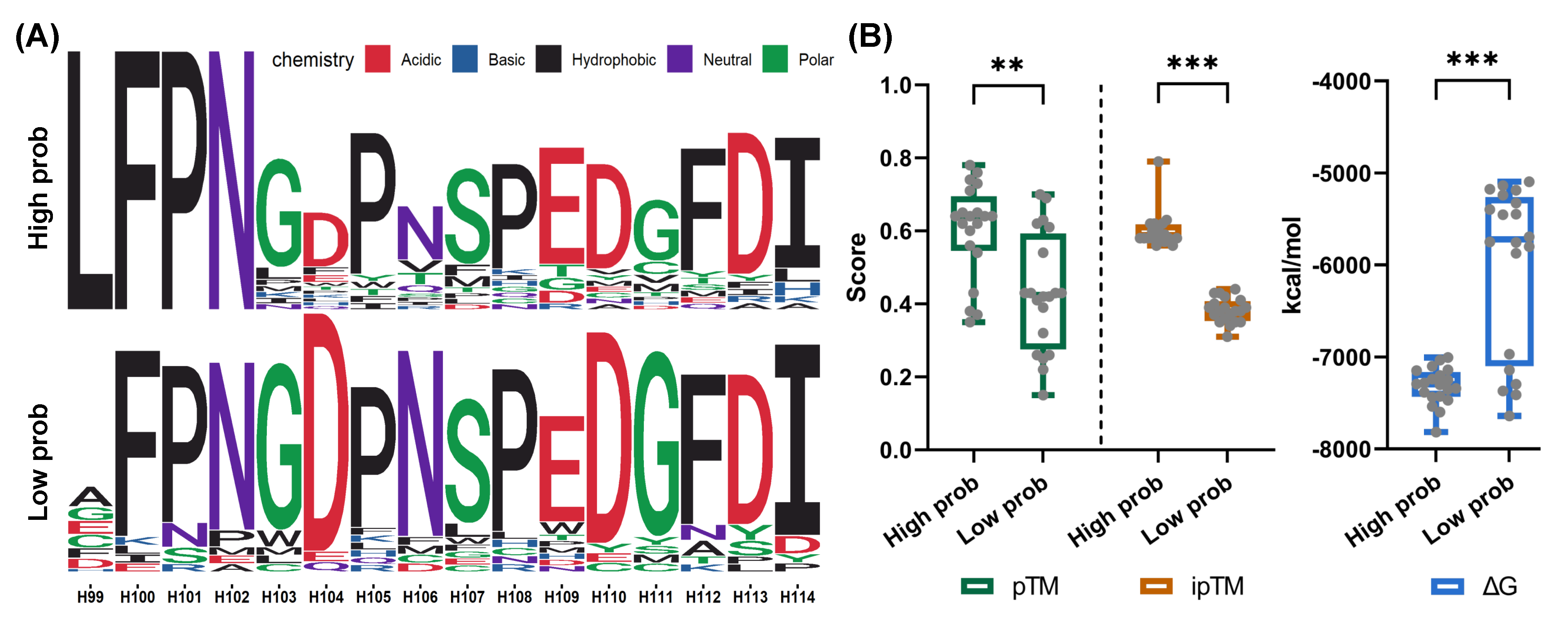
3.7. Screening Novel Antibodies from Mutants
4. Discussions
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Tiller, K.E.; Tessier, P.M. Advances in antibody design. Annu. Rev. Biomed. Eng. 2015, 17, 191–216. [Google Scholar] [CrossRef] [PubMed]
- Jarasch, A.; Koll, H.; Regula, J.T.; Bader, M.; Papadimitriou, A.; Kettenberger, H. Developability assessment during the selection of novel therapeutic antibodies. J. Pharm. Sci. 2015, 104, 1885–1898. [Google Scholar] [CrossRef]
- Kaplon, H.; Crescioli, S.; Chenoweth, A.; Visweswaraiah, J.; Reichert, J.M. Antibodies to watch in 2023. mAbs 2023, 15, 2153410. [Google Scholar] [CrossRef]
- Abbott, W.M.; Damschroder, M.M.; Lowe, D.C. Current approaches to fine mapping of antigen–antibody interactions. Immunology 2014, 142, 526–535. [Google Scholar] [CrossRef] [PubMed]
- Pucca, M.B.; Cerni, F.A.; Janke, R.; Bermúdez-Méndez, E.; Ledsgaard, L.; Barbosa, J.E.; Laustsen, A.H. History of envenoming therapy and current perspectives. Front. Immunol. 2019, 10, 1598. [Google Scholar] [CrossRef]
- Boder, E.T.; Raeeszadeh-Sarmazdeh, M.; Price, J.V. Engineering antibodies by yeast display. Arch. Biochem. Biophys. 2012, 526, 99–106. [Google Scholar] [CrossRef]
- Cheng, J.; Liang, T.; Xie, X.Q.; Feng, Z.; Meng, L. A new era of antibody discovery: An in-depth review of AI-driven approaches. Drug Discov. Today 2024, 6, 103984. [Google Scholar] [CrossRef]
- Dai, B.; Bailey-Kellogg, C. Protein interaction interface region prediction by geometric deep learning. Bioinformatics 2021, 37, 2580–2588. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Luo, Y.; Li, P.; Song, S.; Peng, J. Deep geometric representations for modeling effects of mutations on protein-protein binding affinity. PLoS Comput. Biol. 2021, 17, e1009284. [Google Scholar] [CrossRef]
- Davila, A.; Xu, Z.; Li, S.; Rozewicki, J.; Wilamowski, J.; Kotelnikov, S.; Kozakov, D.; Teraguchi, S.; Standley, D.M. AbAdapt: An adaptive approach to predicting antibody–antigen complex structures from sequence. Bioinform. Adv. 2022, 2, vbac015. [Google Scholar] [CrossRef]
- Xu, J.L.; Davis, M.M. Diversity in the CDR3 region of VH is sufficient for most antibody specificities. Immunity 2000, 13, 37–45. [Google Scholar] [CrossRef]
- Schneider, C.; Buchanan, A.; Taddese, B.; Deane, C.M. DLAB: Deep learning methods for structure-based virtual screening of antibodies. Bioinformatics 2022, 38, 377–383. [Google Scholar] [CrossRef]
- Pittala, S.; Bailey-Kellogg, C. Learning context-aware structural representations to predict antigen and antibody binding interfaces. Bioinformatics 2020, 36, 3996–4003. [Google Scholar] [CrossRef]
- Gao, M.; Skolnick, J. Improved deep learning prediction of antigen–antibody interactions. Proc. Natl. Acad. Sci. USA 2024, 121, e2410529121. [Google Scholar] [CrossRef]
- Jumper, J.; Evans, R.; Pritzel, A.; Green, T.; Figurnov, M.; Ronneberger, O.; Tunyasuvunakool, K.; Bates, R.; Žídek, A.; Potapenko, A.; et al. Highly accurate protein structure prediction with AlphaFold. Nature 2021, 596, 583–589. [Google Scholar] [CrossRef] [PubMed]
- Asti, L.; Uguzzoni, G.; Marcatili, P.; Pagnani, A. Maximum-entropy models of sequenced immune repertoires predict antigen-antibody affinity. PLoS Comput. Biol. 2016, 12, e1004870. [Google Scholar] [CrossRef] [PubMed]
- Rube, H.T.; Rastogi, C.; Feng, S.; Kribelbauer, J.F.; Li, A.; Becerra, B.; Melo, L.A.; Do, B.V.; Li, X.; Adam, H.H.; et al. Prediction of protein–ligand binding affinity from sequencing data with interpretable machine learning. Nat. Biotechnol. 2022, 40, 1520–1527. [Google Scholar] [CrossRef] [PubMed]
- Zhang, K.; Tao, Y.; Wang, F. AntiBinder: Utilizing bidirectional attention and hybrid encoding for precise antibody–antigen interaction prediction. Briefings Bioinform. 2025, 26, bbaf008. [Google Scholar] [CrossRef]
- Jin, R.; Ye, Q.; Wang, J.; Cao, Z.; Jiang, D.; Wang, T.; Kang, Y.; Xu, W.; Hsieh, C.Y.; Hou, T. AttABseq: An attention-based deep learning prediction method for antigen–antibody binding affinity changes based on protein sequences. Briefings Bioinform. 2024, 25, bbae304. [Google Scholar] [CrossRef]
- Zhang, J.; Du, Y.; Zhou, P.; Ding, J.; Xia, S.; Wang, Q.; Chen, F.; Zhou, M.; Zhang, X.; Wang, W.; et al. Predicting unseen antibodies’ neutralizability via adaptive graph neural networks. Nat. Mach. Intell. 2022, 4, 964–976. [Google Scholar] [CrossRef]
- Lin, Z.; Akin, H.; Rao, R.; Hie, B.; Zhu, Z.; Lu, W.; Smetanin, N.; dos Santos Costa, A.; Fazel-Zarandi, M.; Sercu, T.; et al. Language models of protein sequences at the scale of evolution enable accurate structure prediction. bioRxiv 2022. [Google Scholar] [CrossRef]
- Olsen, T.H.; Moal, I.H.; Deane, C.M. AbLang: An antibody language model for completing antibody sequences. Bioinform. Adv. 2022, 2, vbac046. [Google Scholar] [CrossRef]
- Ruffolo, J.A.; Gray, J.J.; Sulam, J. Deciphering antibody affinity maturation with language models and weakly supervised learning. arXiv 2021, arXiv:2112.07782. [Google Scholar]
- Yuan, Y.; Chen, Q.; Mao, J.; Li, G.; Pan, X. DG-Affinity: Predicting antigen–antibody affinity with language models from sequences. BMC Bioinform. 2023, 24, 430. [Google Scholar] [CrossRef]
- Rao, R.; Bhattacharya, N.; Thomas, N.; Duan, Y.; Chen, P.; Canny, J.; Abbeel, P.; Song, Y. Evaluating protein transfer learning with TAPE. Adv. Neural Inf. Process. Syst. 2019, 32, 9689–9701. [Google Scholar]
- Xia, Y.; Wang, Z.; Huang, F.; Xiong, M.; Zhang, W. DeepInterAware: Deep Interaction Interface-Aware Network for Improving Antigen-Antibody Interaction Prediction from Sequence Data. Adv. Sci. 2025, 12, 2412533. [Google Scholar] [CrossRef] [PubMed]
- Deac, A.; VeliČković, P.; Sormanni, P. Attentive cross-modal paratope prediction. J. Comput. Biol. 2019, 26, 536–545. [Google Scholar] [CrossRef]
- Parvizpour, S.; Pourseif, M.M.; Razmara, J.; Rafi, M.A.; Omidi, Y. Epitope-based vaccine design: A comprehensive overview of bioinformatics approaches. Drug Discovery Today 2020, 25, 1034–1042. [Google Scholar]
- Yang, Z.; Yang, D.; Dyer, C.; He, X.; Smola, A.; Hovy, E. Hierarchical attention networks for document classification. In Proceedings of the 2016 Conference of the North American Chapter of the Association for Computational Linguistics: Human Language Technologies, San Diego, CA, USA, 12–17 June 2016; pp. 1480–1489. [Google Scholar]
- Villas-Boas, G.R.; Rescia, V.C.; Paes, M.M.; Lavorato, S.N.; Magalhães-Filho, M.F.D.; Cunha, M.S.; Oesterreich, S.A. The new coronavirus (SARS-CoV-2): A comprehensive review on immunity and the application of bioinformatics and molecular modeling to the discovery of potential anti-SARS-CoV-2 agents. Molecules 2020, 25, 4086. [Google Scholar]
- Gu, A.; Goel, K.; Ré, C. Efficiently modeling long sequences with structured state spaces. arXiv 2021, arXiv:2111.00396. [Google Scholar]
- Gupta, A.; Gu, A.; Berant, J. Diagonal state spaces are as effective as structured state spaces. Adv. Neural Inf. Process. Syst. 2022, 35, 22982–22994. [Google Scholar]
- Smith, J.; De Mello, S.; Kautz, J.; Linderman, S.; Byeon, W. Convolutional state space models for long-range spatiotemporal modeling. Adv. Neural Inf. Process. Syst. 2023, 36, 80690–80729. [Google Scholar]
- Gu, A.; Dao, T. Mamba: Linear-time sequence modeling with selective state spaces. arXiv 2023, arXiv:2312.00752. [Google Scholar]
- Zhu, L.; Liao, B.; Zhang, Q.; Wang, X.; Liu, W.; Wang, X. Vision Mamba: Efficient Visual Representation Learning with Bidirectional State Space Model. In Proceedings of the Forty-first International Conference on Machine Learning, Vancouver, BC, Canada, 13–19 July 2025. [Google Scholar]
- Ali, A.; Zimerman, I.; Wolf, L. The hidden attention of mamba models. arXiv 2024, arXiv:2403.01590. [Google Scholar]
- Yoon, H.; Macke, J.; West, A.P., Jr.; Foley, B.; Bjorkman, P.J.; Korber, B.; Yusim, K. CATNAP: A tool to compile, analyze and tally neutralizing antibody panels. Nucleic Acids Res. 2015, 43, W213–W219. [Google Scholar] [CrossRef]
- Raybould, M.I.J.; Kovaltsuk, A.; Marks, C.; Deane, C.M. CoV-AbDab: The coronavirus antibody database. Bioinformatics 2020, 37, 734–735. [Google Scholar] [CrossRef]
- Pitorro, H.; Treviso, M. LaTIM: Measuring Latent Token-to-Token Interactions in Mamba Models. arXiv 2025, arXiv:2502.15612. [Google Scholar]
- Vig, J.; Belinkov, Y. Analyzing the Structure of Attention in a Transformer Language Model. In Proceedings of the 2019 ACL Workshop, Florence, Italy, 28 July–2 August 2019; pp. 63–76. [Google Scholar]
- Chen, M.; Ju, C.J.T.; Zhou, G.; Chen, X.; Zhang, T.; Chang, K.W.; Zaniolo, C.; Wang, W. Multifaceted protein–protein interaction prediction based on Siamese residual RCNN. Bioinformatics 2019, 35, i305–i314. [Google Scholar] [CrossRef]
- Gu, M.; Yang, W.; Liu, M. Prediction of antibody-antigen interaction based on backbone aware with invariant point attention. BMC Bioinform. 2024, 25, 348. [Google Scholar] [CrossRef]
- Zuo, F.; Cao, Y.; Sun, R.; Wang, Q.; Simonelli, L.; Du, L.; Bertoglio, F.; Schubert, M.; Guerra, C.; Cavalli, A.; et al. Ultrapotent IgA dimeric antibodies neutralize emerging Omicron variants. J. Virol. 2025, 99, e01740-24. [Google Scholar] [CrossRef]
- Yan, Q.; Hou, R.; Huang, X.; Zhang, Y.; He, P.; Zhang, Y.; Liu, B.; Wang, Q.; Rao, H.; Chen, X.; et al. Shared IGHV1-69-encoded neutralizing antibodies contribute to the emergence of L452R substitution in SARS-CoV-2 variants. Emerg. Microbes Infect. 2022, 11, 2749–2761. [Google Scholar] [CrossRef] [PubMed]
- Lefranc, M.P.; Pommié, C.; Ruiz, M.; Giudicelli, V.; Foulquier, E.; Truong, L.; Thouvenin-Contet, V.; Lefranc, G. IMGT unique numbering for immunoglobulin and T cell receptor variable domains and Ig superfamily V-like domains. Dev. Comp. Immunol. 2003, 27, 55–77. [Google Scholar] [CrossRef] [PubMed]
- Abramson, J.; Adler, J.; Dunger, J.; Evans, R.; Green, T.; Pritzel, A.; Ronneberger, O.; Willmore, L.; Ballard, A.J.; Bambrick, J.; et al. Accurate structure prediction of biomolecular interactions with AlphaFold 3. Nature 2024, 630, 493–500. [Google Scholar] [CrossRef]
- Shrestha, L.B.; Foster, C.; Rawlinson, W.; Tedla, N.; Bull, R.A. Evolution of the SARS-CoV-2 omicron variants BA. 1 to BA. 5: Implications for immune escape and transmission. Rev. Med. Virol. 2022, 32, e2381. [Google Scholar] [CrossRef] [PubMed]
- Yuan, S.; Chan, H.S.; Hu, Z. Using PyMOL as a platform for computational drug design. Wiley Interdiscip. Rev. Comput. Mol. Sci. 2017, 7, e1298. [Google Scholar] [CrossRef]
- Gräf, T.; Martinez, A.A.; Bello, G.; Dellicour, S.; Lemey, P.; Colizza, V.; Mazzoli, M.; Poletto, C.; Cardoso, V.L.O.; da Silva, A.F.; et al. Dispersion patterns of SARS-CoV-2 variants gamma, lambda and mu in Latin America and the Caribbean. Nat. Commun. 2024, 15, 1837. [Google Scholar] [CrossRef]
- Zhang, Y.; Skolnick, J. Scoring function for automated assessment of protein structure template quality. Proteins Struct. Funct. Bioinform. 2004, 57, 702–710. [Google Scholar] [CrossRef]
- Xu, J.; Zhang, Y. How significant is a protein structure similarity with TM-score = 0.5? Bioinformatics 2010, 26, 889–895. [Google Scholar] [CrossRef]
- Genheden, S.; Ryde, U. The MM/PBSA and MM/GBSA methods to estimate ligand-binding affinities. Expert Opin. Drug Discov. 2015, 10, 449–461. [Google Scholar] [CrossRef]
- Wang, Z.; Wang, Y.; Zhang, W. Improving paratope and epitope prediction by multi-modal contrastive learning and interaction informativeness estimation. In Proceedings of the Thirty-Third International Joint Conference on Artificial Intelligence, Jeju, Republic of Korea, 3–9 August 2024; pp. 6053–6061. [Google Scholar]
- Li, W.; Zhou, H.; Yu, J.; Song, Z.; Yang, W. Coupled mamba: Enhanced multi-modal fusion with coupled state space model. arXiv 2024, arXiv:2405.18014. [Google Scholar]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Dataset | Methods | AUC | AUPR | F1-Score | Accurary |
|---|---|---|---|---|---|
| HIV | MambaAAI | 0.8955 | 0.8870 | 0.7992 | 0.7965 |
| MambaAAI (-RE) | 0.8507 | 0.8716 | 0.7365 | 0.7649 | |
| MambaAAI (-BP) | 0.8918 | 0.8652 | 0.7719 | 0.7810 | |
| MambaAAI (-HV) | 0.8699 | 0.8805 | 0.7835 | 0.7913 | |
| MambaAAI (-VV) | 0.8727 | 0.8836 | 0.7891 | 0.7872 | |
| MambaAAI (-SSM) | 0.8208 | 0.8344 | 0.7434 | 0.7587 | |
| CoV-AbDab | MambaAAI | 0.8141 | 0.7067 | 0.6542 | 0.7496 |
| MambaAAI (-RE) | 0.7813 | 0.6915 | 0.6263 | 0.7214 | |
| MambaAAI (-BP) | 0.8052 | 0.7009 | 0.6450 | 0.7435 | |
| MambaAAI (-HV) | 0.8063 | 0.7114 | 0.6393 | 0.7268 | |
| MambaAAI (-VV) | 0.8126 | 0.7044 | 0.6515 | 0.7330 | |
| MambaAAI (-SSM) | 0.7577 | 0.6673 | 0.6281 | 0.7117 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, X.; Fu, H.; Yang, Y.; Zhang, J. Bio-Inspired Mamba for Antibody–Antigen Interaction Prediction. Biomolecules 2025, 15, 764. https://doi.org/10.3390/biom15060764
Liu X, Fu H, Yang Y, Zhang J. Bio-Inspired Mamba for Antibody–Antigen Interaction Prediction. Biomolecules. 2025; 15(6):764. https://doi.org/10.3390/biom15060764
Chicago/Turabian StyleLiu, Xuan, Haitao Fu, Yuqing Yang, and Jian Zhang. 2025. "Bio-Inspired Mamba for Antibody–Antigen Interaction Prediction" Biomolecules 15, no. 6: 764. https://doi.org/10.3390/biom15060764
APA StyleLiu, X., Fu, H., Yang, Y., & Zhang, J. (2025). Bio-Inspired Mamba for Antibody–Antigen Interaction Prediction. Biomolecules, 15(6), 764. https://doi.org/10.3390/biom15060764