

Discovery of Drugs Targeting Mutant p53 and Progress in Nano-Enabled Therapeutic Strategy for p53-Mutated Cancers

 , ,
, , 
Abstract
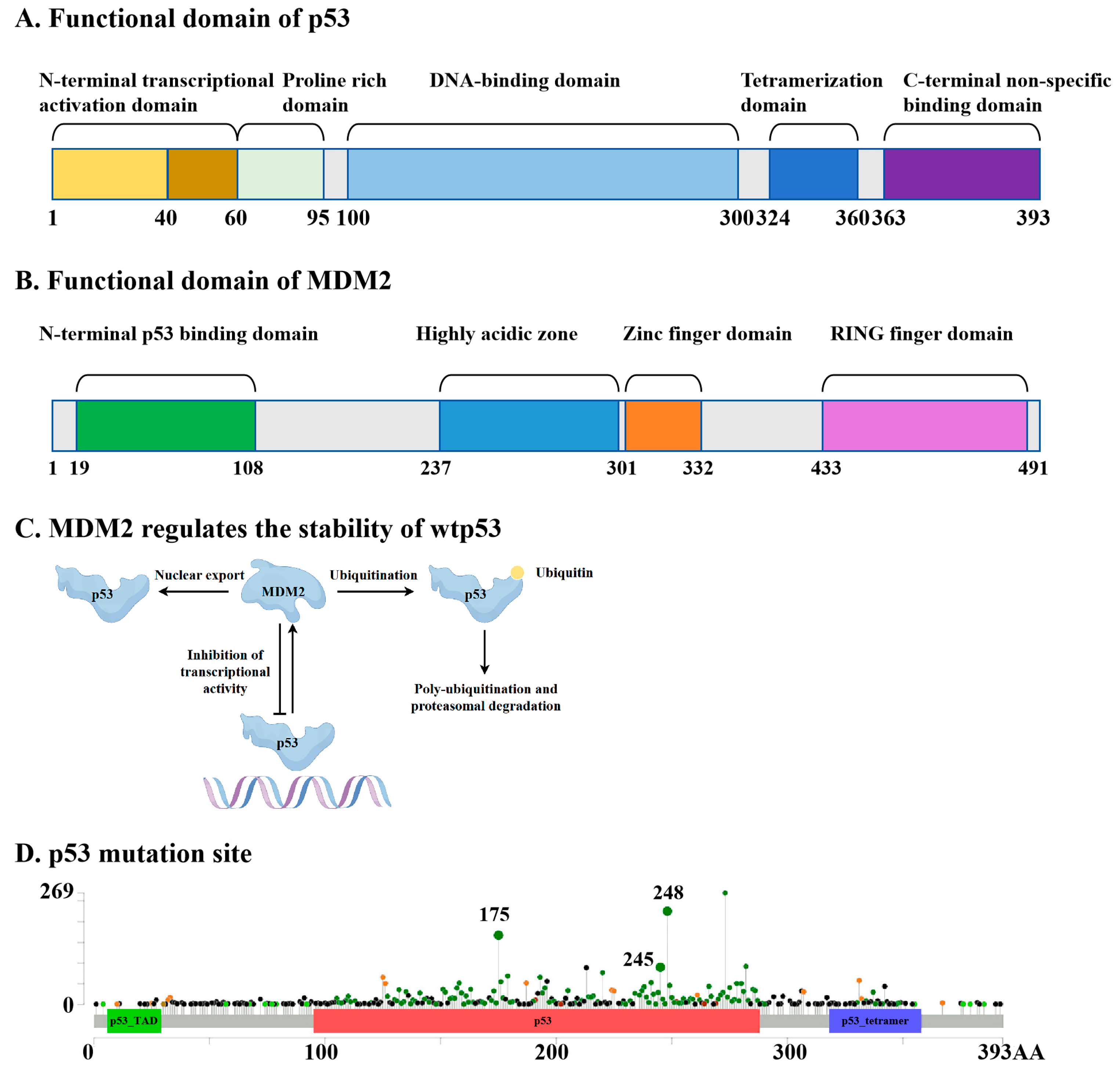
1. Introduction
2. Advances in mutp53-Targeting Drug Discovery
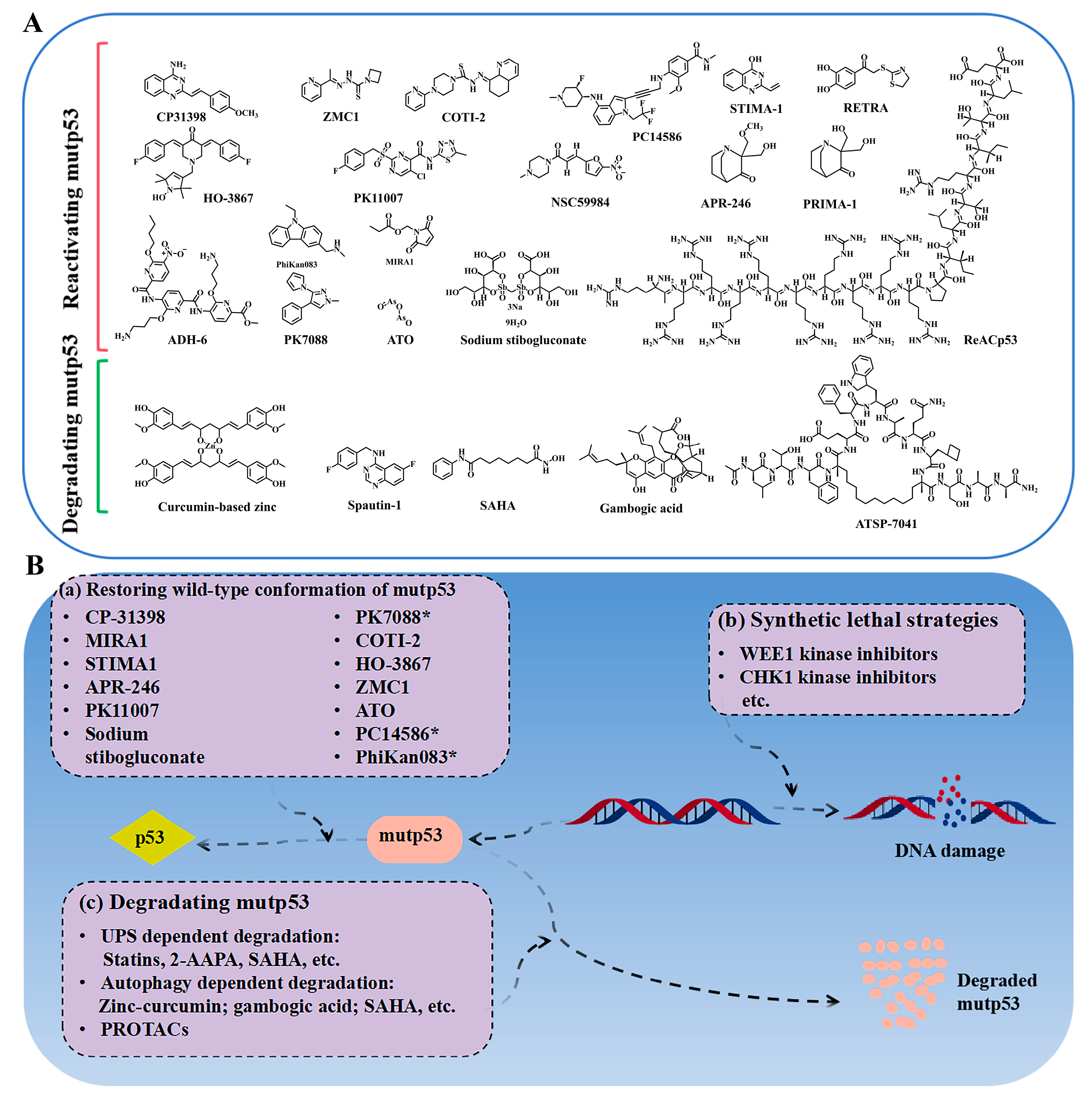
2.1. Compounds Reactivating mutp53

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compounds | Intervention | Cancer Type | Experimental or Clinical Results | Phase Status | Clinical Trial Identifier or Citations |
|---|---|---|---|---|---|
| APR-246 | APR-246 + azacitidine | P53-mutated AML or MDS following allogeneic stem cell transplant | One-year non-recurrence survival rate of 59.9% and a median overall survival of 20.6 months; well tolerated. | Phase II, completed | NCT03931291; [39] |
| APR-246 + azacitidine | P53-mutant myeloid neoplasms | The overall remission rate for 55 patients was 71%, with 44% achieving complete remission. The frequency of TP53 mutation alleles and p53 expression were significantly reduced. | Phase I b/II, completed | NCT03072043 [36] | |
| APR-246 + azacitidine | P53-mutant myeloid neoplasms | Compared to azacitidine monotherapy, it demonstrated a favorable safety profile and showed potentially superior ORR, complete response (CR) rate, and overall survival (OS). | Phase II, completed | NCT03588078 [37] | |
| APR-246 + azacitidine | P53-mutant MDS | Compared to azacitidine, the complete remission rate increased from 22.4% to 34.6%. | Phase III, completed | NCT03745716 [6] | |
| APR-246 + venetoclax + azacitidine | P53-mutant myeloid malignancies | Compared to the combination of APR-246 and venetoclax, an 8.91% reduction in serious adverse events and a 39.53% reduction in all-cause mortality. | Phase I, completed | NCT04214860 [40] | |
| APR-246 + carboplatin and pegylated liposomal doxorubicin hydrochloride | Platinum-sensitive recurrent high-grade serous ovarian cancer (HGSOC) with mutated p53 | CR: 9.5%. Partial responses (PRs): 40.0%. SD: 25.7%. | Phase I b/II, completed | NCT02098343 | |
| APR-246 + pegylated liposomal doxorubicin hydrochloride | Platinum-resistant recurrent high-grade serous ovarian cancer (HGSOC) with mutated TP53 | The ORR is 69.6%, and the incidence of severe adverse reactions is 39.29%. | Phase II, completed | NCT03268382 | |
| APR-246 + venetoclax | Relapsed refractory mantle cell lymphoma patients (with/without p53 aberrations) | Not disclosed. | Phase II, withdrawn | NCT04990778 | |
| APR-246, 5-FU, and cisplatin | Platinum-resistant advanced and metastatic oesophageal cancers | / | Phase I b/II, terminated | NCT02999893 | |
| ATO | ATO | Acute myeloid leukemia with p53 mutations | Not disclosed. | Phase II, unknown status | NCT03381781 |
| ATO | P53-mutated pediatric cancer | Not disclosed. | Phase II, recruiting | NCT06088030 | |
| ATO | Previously untreated or relapsed/refractory p53-mutated myeloid malignancies | Not disclosed. | Phase II, not yet recruiting | NCT06778187 | |
| ATO + decitabine, intravenously | AML/MDS-expressing mutant p53 | Not disclosed. | Phase I, unknown status | NCT03855371 | |
| PC14586 | PC14586 + pembrolizumab | Locally advanced or metastatic solid tumors harboring a p53 Y220C mutation | 21 efficacy-evaluable patients (out of 29 total); PRs were observed in 5 cases; treatment-related adverse events occurred in 79% of patients. | Phase I/II, recruiting | NCT04585750 [45] |
| PC14586 + azacitidine | TP53 Y220C-mutant AML/MDS | Not disclosed. | Phase I b, recruiting | NCT06616636; [51] | |
| COTI-2 | COTI-2 monotherapy and COTI-2/cisplatin combination therapy | Advanced or recurrent malignancies | Not disclosed. | Phase I, unknown status | NCT02433626 |
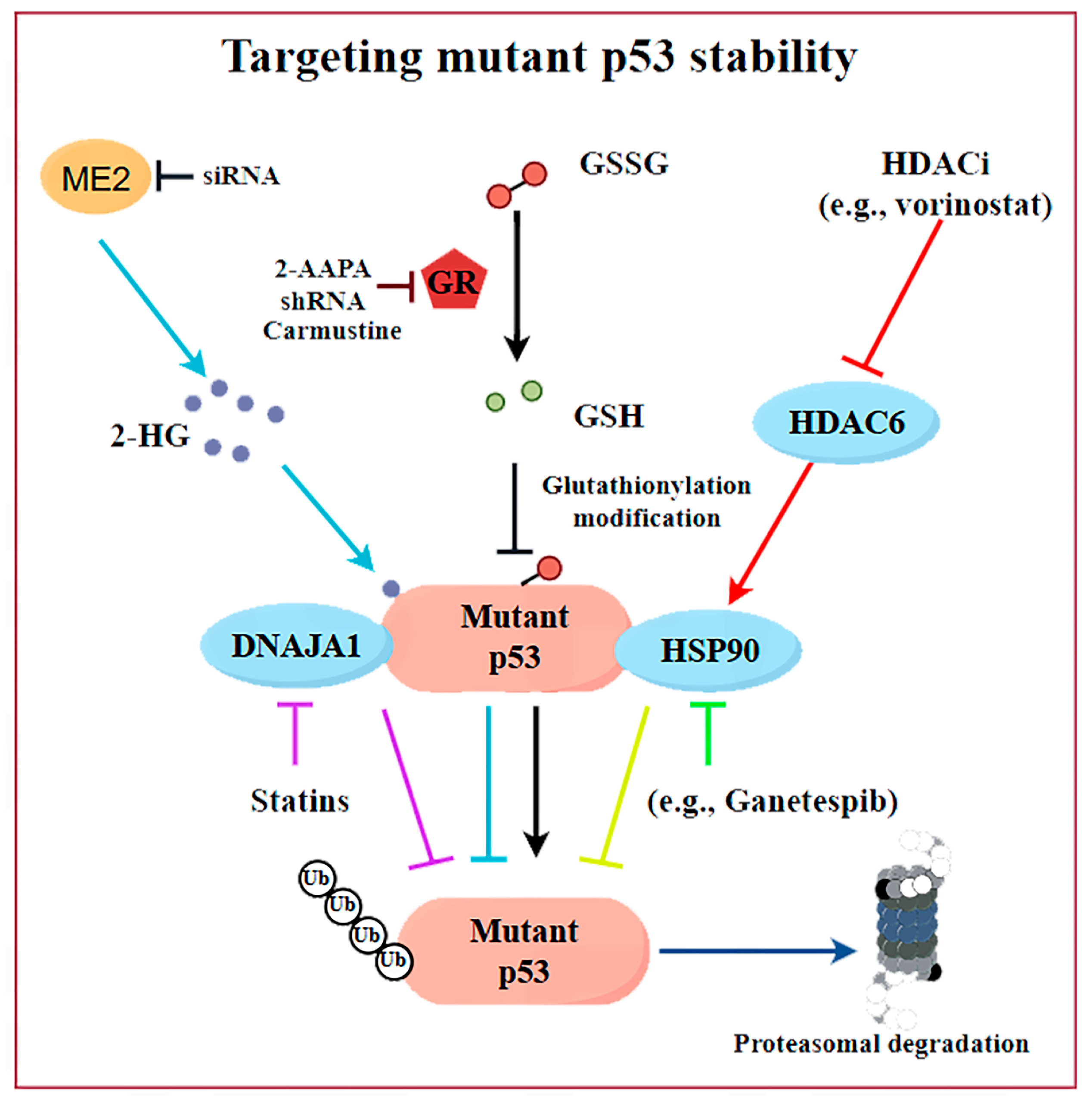
2.2. Targeting mutp53 Protein Stability
| Compounds | Mechanism of Action | Stage of Development (Up to Now) | Clinical Data | Citations |
|---|---|---|---|---|
| Hsp90 inhibitors (e.g., Ganetespib) | Promote the degradation of mutp53 via inhibiting HSP90 | Phase I/II clinical trial/NCT02012192/ terminated | p53-mutant platinum-resistant ovarian cancer: Ganetespib plus paclitaxel shows inferior safety profile compared to paclitaxel monotherapy. | [14] |
| HDACi (e.g., SAHA) | Promote the degradation of mutp53 through the inhibition of HDAC6, which acts as a positive regulator of HSP90 | Phase I clinical trial/NCT01339871/ terminated | Initial data warrant further investigation of this regimen in TP53-mutant cancers, especially metastatic sarcoma/colorectal cancer. | [56,60,71] |
| Disulfiram | Induces glutathionylation of p53 and degradation of both wild-type and mutant p53s | Phase I or II clinical trials for various cancers but not designed for p53-mutated tumors | / | [62] |
| GSH reductase inhibitors (e.g., 2-AAPA) | Selectively induce the proteasomal degradation of mutp53 via promoting glutathionylation and subsequent K48-linkage polyubiquitination of mutp53 proteins | Preclinical | / | [28] |
| Gambogic acid | Promotes the degradation of mutp53 (R280K and S241F) via inducing autophagy | Preclinical | / | [69] |
| Spautin-1 | Promotes the degradation of mutp53 via inducing chaperone- mediated autophagy | Preclinical | / | [70] |
| Curcumin-based zinc | Promotes the degradation of mutp53 (R175H) via inducing autophagy | Preclinical | / | [67] |
2.3. Synthetic Lethality
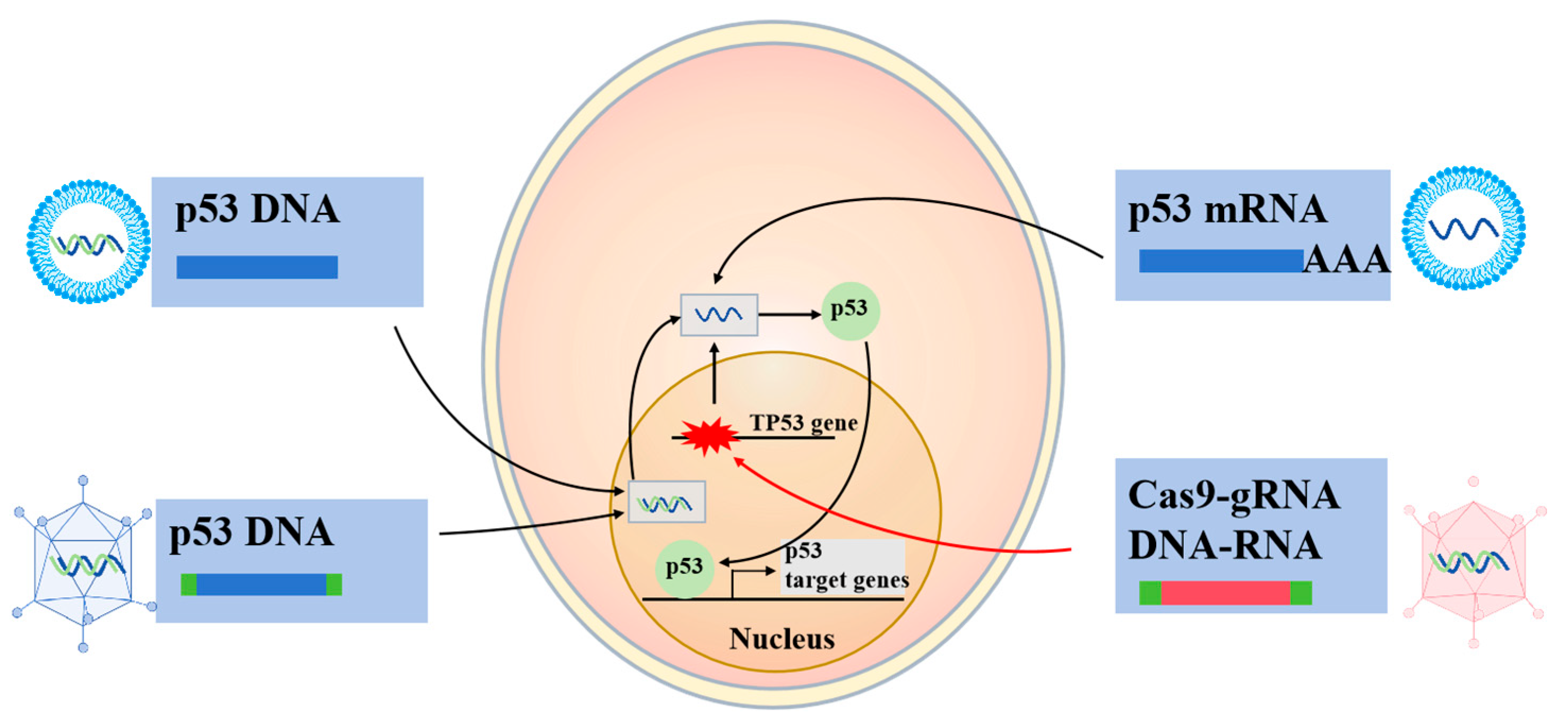
2.4. p53-Based Genetic Therapies
2.4.1. Wtp53-Based Gene Therapies
2.4.2. RNA Therapeutics
2.4.3. CRISPR-Cas9
2.5. PROTAC
3. Nanostrategies for Targeting mutp53 for Cancer Therapy
3.1. Delivering mutp53-Reactivating Agents
3.2. Triggering mutp53 Clearance
3.2.1. Nanodelivery of Zinc Ions
3.2.2. Delivering Compounds Capable of Clearing mutp53
3.2.3. Nanoformulations with Intrinsic Mutant p53 Clearance Ability
3.2.4. Biomimetic Nanoreceptor for a Specific Degradation of mutp53
| Biomaterial Types | Action on mutp53 | In Vitro/In Vivo | Mechanism | References |
|---|---|---|---|---|
| ZnFe-4 nanoparticles | Selective degradation of mutp53 | In vitro/in vivo | Induce degradation of mutp53 via UPS; reduce cell proliferation and cell migration | [123] |
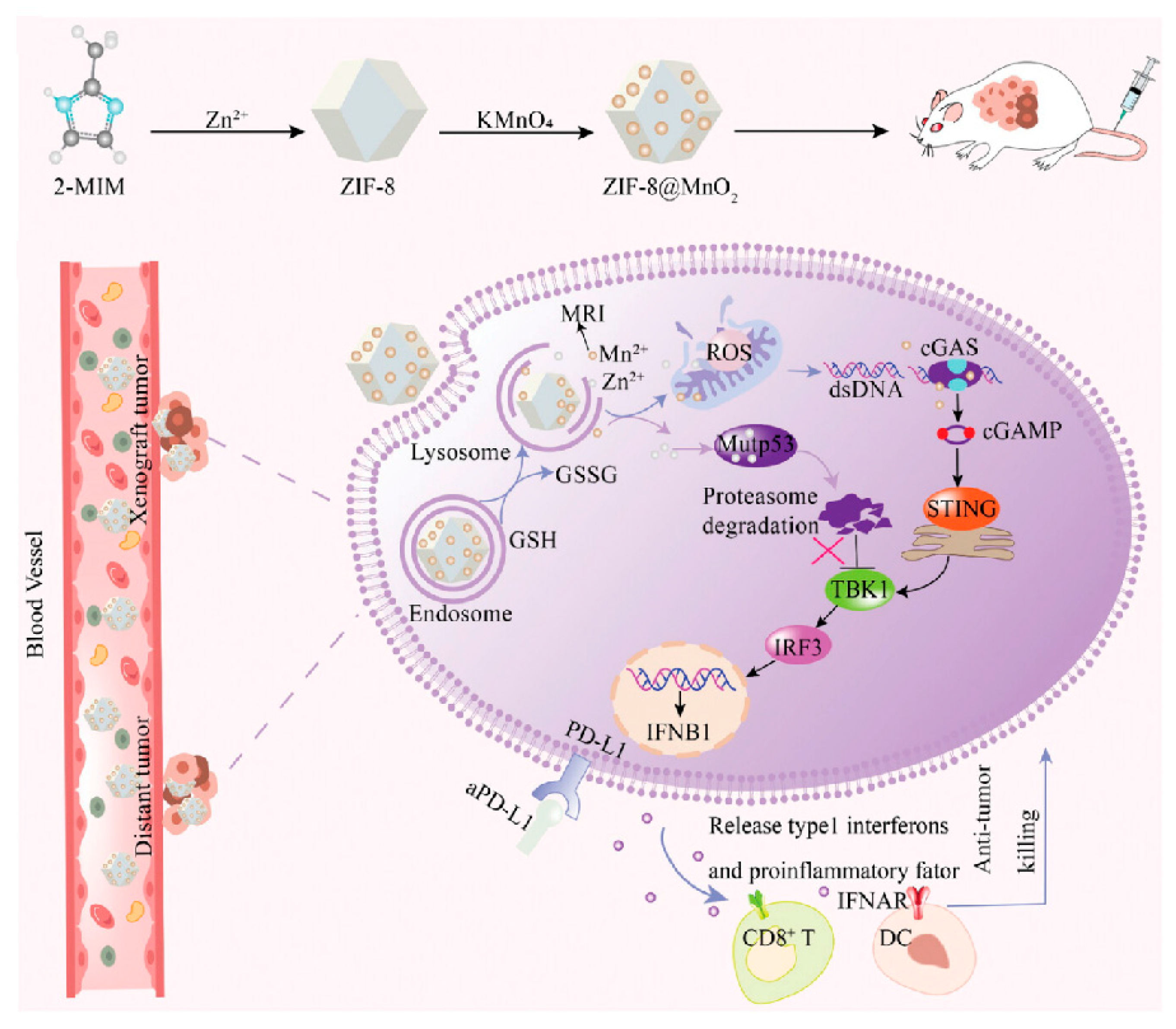
| ZIF-8; ZIF-8 modified with Z1-RGD peptides | Selective degradation of mutp53 | In vitro/in vivo | Induce degradation of mutp53 by UPS and glutathionylation-dependent proteasome; reduce the GSH: GSSG ratio | [124] |
| Mn-ZnO2 nanoparticles | Clearing mutp53 and enhancing wtp53 expression | In vitro/in vivo | Induce degradation of mutp53 by UPS, increase ROS level, and activate the ATM-p53-Bax pathway to elevate the wtp53 level | [125] |
| Zn-LDH@GOX | Degradation of mutp53 | In vitro/in vivo | Elevate intracellular Zn2+ concentration, promote the transformation of part of mutp53 conformation into wtp53 conformation, reactivate the function of wtp53, and promote the degradation of mutp53 via the autophagy pathway | [126] |
| PEGylated CeO2 NPs | Degradation of mutp53 | In vitro/in vivo | Increase the production of ROS, promote the degradation of mutp53, and reduce cell proliferation and migration | [135] |
| Black phosphorus nanosheets | Degradation of mutp53 | In vitro/in vivo | Reduce resistance of tumor cells to chemotherapy drugs and degrade mutp53 protein | [138] |
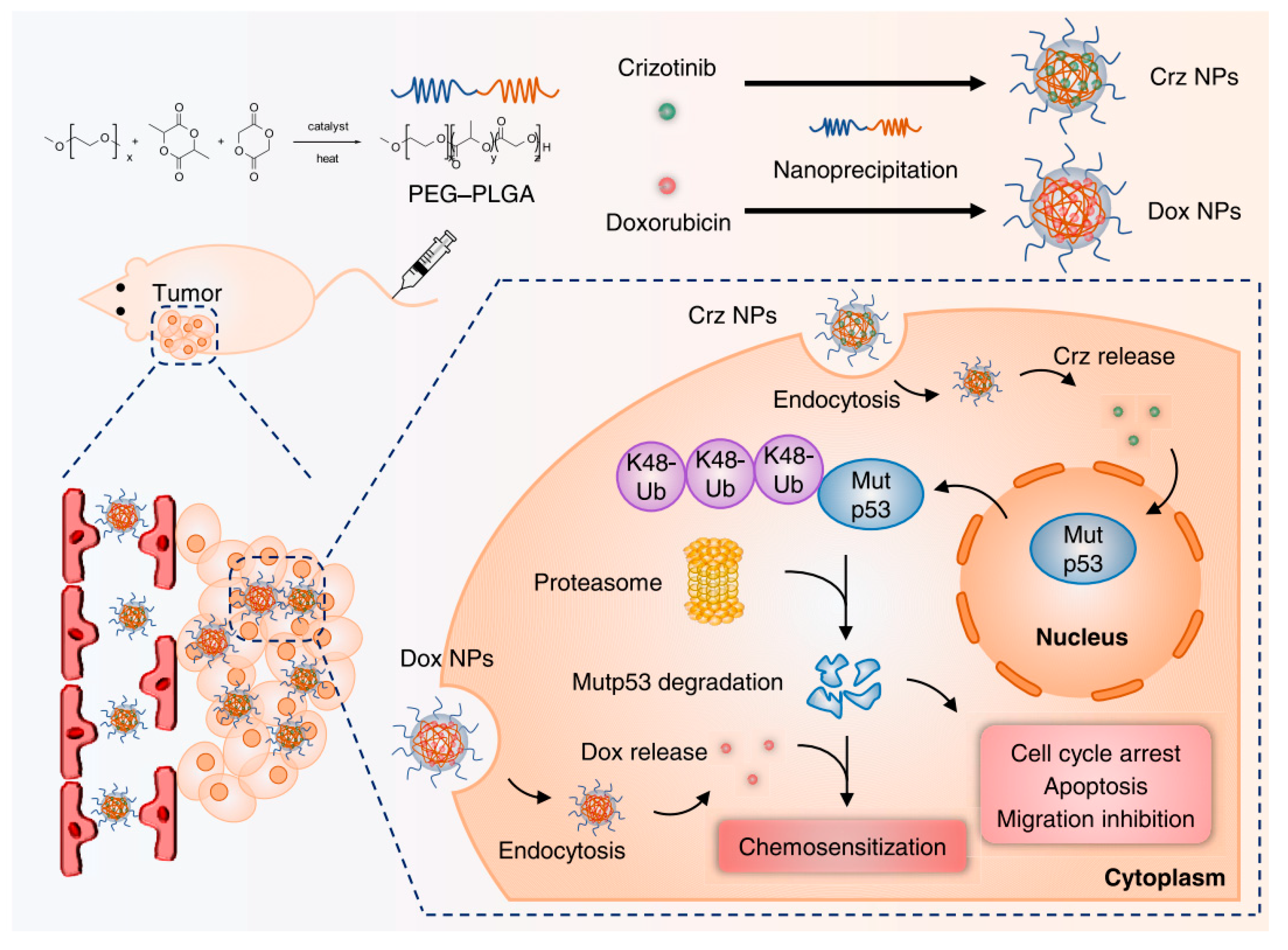
| Crizotinib nanomicelles | Degradation of mutp53 | In vitro/in vivo | Induce degradation of mutp53 via UPS, abrogate mutp53-manifested GOF, and reduce cell proliferation, migration, and cell cycle arrest | [130] |
| Fluplatin@PEG-PE nanoparticles | Degradation of mutp53 | In vitro/in vivo | Degrade mutp53, trigger endoplasmic reticulum stress (ERS), and mitigate cisplatin resistance caused by mutp53 | [131] |
| MBP-NPs-DOTAP | Clearing mutp53 | In vitro/in vivo | Elevate the levels of autophagosome formation; increase the degradation of mutp53 | [137] |
| HA-TPP/A | Degradation of mutp53 | In vitro/in vivo | Inhibit the signaling pathways of mutant KRAS and mutp53; degrade mutp53 proteins | [132] |
| P6@siKRAS | Degradation of mutp53 | In vitro/in vivo | Trigger UPS-mediated degradation of mutp53, inhibit KRAS signaling pathways, eliminate mutp53’s GOF effects, and suppress tumor growth | [134] |
3.3. Targeting Delivery of Synthetic Lethality Compounds
3.4. Nano-Enabling p53-Based Genetic Therapies
3.4.1. Nano-Enabling p53-Based Gene Therapies
| Biomaterial (Carrier) | Cell | Animal Models | Mechanism | References |
|---|---|---|---|---|
| MB-NSi–p53-CS ternary complexes | A549 cells | Male BALB/c nude mice of 5 weeks | Elevate expression level of p53 mRNA in tumor cells harboring mutp53; enhance antitumor efficacy | [143] |
| f-SWCNTs-p53 complexes | MCF-7 cells | / | Increase p53 mRNA levels in the cells and induce apoptosis | [147] |
| DOX/p53 mRNA complexes | HepG2 cells, Hela cells, and C6 cells | / | Activate the p53 pathway; increase the sensitivity of tumor cells to chemotherapeutic drugs | [144] |
| HMSNs-PEI-BTZ-p53 nanoparticles | CRL-5872 cells | / | HMSNs-PEI-BTZ-p53 nanoparticles | [145] |
| GOAS-pEFGP-p53 complexes | MCF-7 cells; BT-20 cells | / | Activate the p53 pathway by transferring therapeutic agents into tumor cells; induce cell apoptosis | [148] |
| p53-EGFP-C3 fusion construct | HeLa cells, H1299 cells, and HEK 293T cells | Healthy nude mice of 5–6 weeks | Enhance the expression of the p53 protein; induce cell death | [141] |
| TK-PEI/HAP/DNA NCs | B16F10 cells | Male C57BL/6 mice | Restore p53 expression, increase the generation of ROS, and strengthen the delivery of p53 genes | [149] |
| SAHP/p53 | HEK 293 cells; HepG2 cells | BALB/C nude mice | Trigger photothermal treatment, achieve the synergistic effects of photothermal therapy and chemotherapy, and enhance the joint delivery of sorafenib and p53 | [146] |
| PEN-p53 | HeLa cells; PC-3 cells | / | Up-regulate the expression of the p53 gene, inhibit cell proliferation, and activate cell apoptosis and cell cycle arrest | [150] |
| Chol-g-PMSC-PPDL/p53 nanoparticles | PC-3 cells | / | Increase the cellular expression level of p53, inhibit cell proliferation, and activate mitochondria-dependent apoptotic pathways and cell cycle arrest | [151] |
| p53/C-rNC/L-FA | MCF-7 cells | Famale BALB/c-nu mice, male BALB/c mice, female Sprague Dawley rats | Delivery of CytoC to the cytoplasm and the p53 | [152] |
| AP-PAMAM/p53 | HeLa cells | / | Enhance the expression of the p53 gene, inhibit cell proliferation, and activate apoptosis and cell cycle arrest | [153] |
3.4.2. Restoring wtp53 Expression Through mRNA Nanodelivery
| Biomaterial (Carrier) | Cell | Animal Models | Mechanism | References |
|---|---|---|---|---|
| p53 mRNA–lipid nanoparticles (p53 mRNA@LNPs) | MDA-MB-231, SK-OV-3, MDA-MB-453, SK-BR-3 cells, and BT-474 cells | 7-week-old female BALB/c nu/nu mice | Improve the expression levels of p53; induce cell death in a dose-dependent manner | [161] |
| DNA nanoparticles | RAW264.7 cells; DC2.4 cells | 8-week-old male BALB/c JGpt mice | Improve mRNA vaccine delivery and efficacy, activate immune responses, and induce the production of antigen-specific antibody | [155] |
| Lipofectamine Messenger MAX Transfection Reagent | Human ovarian cancer cell lines (SKOV3, OVCAR-3, OVCAR-4, OVCAR-5, OVCAR-8) | 6–8-week-old female nude mice | Lipofectamine Messenger MAX Transfection Reagent | [156] |
| A ROS-responsive polymeric nanoparticle | H1299 cells | Athymic nude mice | Promote mRNA translation efficiency and p53 expression; induce generation of ROS | [159] |
| PLGA/lipid–PEG/lipid nanoparticles | HCC cells, RIL-175 cells, and HCA-1 cells | 5–6-week-old or 6–8-week-old immunocompetent male and female C57BL/6 mice | Increase p53 expression, restore p53 functional activity, reduce cancer cell viability, and inhibit tumor growth improved tumor sensitivity to immunotherapies | [162] |
| HA/DSPE-PEG/mannosenanoparticles | H1299 cells; HCT116 cells | 4–6-week-old female BALB/c mice; 6-week-old C57BL mice | Deliver the targeted p53 proteins into lung tissues, accumulate p53 mRNA in lung tumor cells and inflammatory macrophages, and enhance the expression of p53 proteins | [160] |
| Redox-responsive polymer PDSA/DSPE-PEG/DMPE-PEG nanoparticles | Hep3B cells; H1299 cells | 4–6-week-old female athymic nude mice, 6-week-old wild-type BALB/c mice, and 4-week-old female C57BL/6 mice | Restore p53 function, impede the proliferation of p53-deficient liver and lung cancer cells, induce cell cycle arrest and apoptosis, reverse the resistance of cancer cells to the mTOR inhibitor | [157] |
| Paclitaxel amino lipid (PAL) nanoparticles | MDAMB-231 cells | athymic nude female mice | Enhance encapsulation efficiency for both paclitaxel and mRNA | [158] |
| PBA-BADP/mRNA nanoparticles | HeLa cells, SiHa cells, DU145 cells, CCC-HPF-1 cells, HK-2 cells, and HEK293 cells | / | Selectively prohibit cancer cell growth | [163] |
| PRIZE, a p53-repair nanosystem | 4T1 cells, MC38 cells, Luc-4T1 cells, 4T1-OVA cells, and MC38-OVA cells | BALB/C mice; C57BL/6 mice | Restore intracellular p53 levels; trigger immunogenic cell death | [164] |
3.4.3. Ablation of p53 Expression by Genetic Approach
3.5. Nanodelivery of wtp53 Proteins
4. Concluding Remarks and Future Directions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Linzer, D.I.H.; Levine, A.J. Characterization of a 54K Dalton cellular SV40 tumor antigen present in SV40-transformed cells and uninfected embryonal carcinoma cells. Cell 1979, 17, 43–52. [Google Scholar] [CrossRef] [PubMed]
- Parada, L.F.; Land, H.; Weinberg, R.A.; Wolf, D.; Rotter, V. Cooperation between gene encoding p53 tumour antigen and ras in cellular transformation. Nature 1984, 312, 649–651. [Google Scholar] [CrossRef] [PubMed]
- Lane, D.P.; Benchimol, S. p53: Oncogene or anti-oncogene? Genes Dev. 1990, 4, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Lane, D.P.; Crawford, L.V. T antigen is bound to a host protein in SY40-transformed cells. Nature 1979, 278, 261–263. [Google Scholar] [CrossRef]
- Levine, A.J.; Oren, M. The first 30 years of p53: Growing ever more complex. Nat. Rev. Cancer 2009, 9, 749–758. [Google Scholar] [CrossRef]
- Hassin, O.; Oren, M. Drugging p53 in cancer: One protein, many targets. Nat. Rev. Drug Discov. 2023, 22, 127–144. [Google Scholar] [CrossRef]
- Liu, J.; Zhang, C.; Hu, W.; Feng, Z.; Verma, C.S. Tumor suppressor p53 and metabolism. J. Mol. Cell Biol. 2019, 11, 284–292. [Google Scholar] [CrossRef]
- Grossman, S.R.; Deato, M.E.; Brignone, C.; Chan, H.M.; Kung, A.L.; Tagami, H.; Nakatani, Y.; Livingston, D.M. Polyubiquitination of p53 by a Ubiquitin Ligase Activity of p300. Science 2003, 300, 342–344. [Google Scholar] [CrossRef]
- Juven-Gershon, T.; Oren, M. Mdm2: The ups and downs. Mol. Med. 1999, 5, 71–83. [Google Scholar] [CrossRef]
- Freedman, D.A.; Wu, L.; Levine, A.J. Functions of the MDM2 oncoprotein. Cell. Mol. Life Sci. 1999, 55, 96–107. [Google Scholar] [CrossRef]
- Marvalim, C.; Datta, A.; Lee, S.C. Role of p53 in breast cancer progression: An insight into p53 targeted therapy. Theranostics 2023, 13, 1421–1442. [Google Scholar] [CrossRef]
- Setyawati, M.I.; Tay, C.Y.; Leong, D.T. Exploiting Cancer’s Antioxidative Weakness Through P53 with Nanotoxicology. Nanomedicine 2014, 9, 369–371. [Google Scholar] [CrossRef]
- Peuget, S.; Zhou, X.; Selivanova, G. Translating p53-based therapies for cancer into the clinic. Nat. Rev. Cancer 2024, 24, 192–215. [Google Scholar] [CrossRef] [PubMed]
- Alexandrova, E.M.; Yallowitz, A.R.; Li, D.; Xu, S.; Schulz, R.; Proia, D.A.; Lozano, G.; Dobbelstein, M.; Moll, U.M. Improving survival by exploiting tumour dependence on stabilized mutant p53 for treatment. Nature 2015, 523, 352–356. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.P.; Lozano, G. Mutant p53 partners in crime. Cell Death Differ. 2017, 25, 161–168. [Google Scholar] [CrossRef] [PubMed]
- Beyer, G.; Thayer, K. Total Mutagenesis and the Effects on the MDM2-p53 Interaction. FASEB J. 2015, 29, 712–719. [Google Scholar] [CrossRef]
- Shieh, S.Y.; Taya, Y.; Prives, C. DNA damage-inducible phosphorylation of p53 at N-terminal sites including a novel site, Ser20, requires tetramerization. EMBO J. 1999, 18, 1815–1823. [Google Scholar] [CrossRef]
- Cui, W.; Wu, R.; Cao, H.; Gao, J.; Wang, X.; Ren, Q. P53 gene mutation and expression of MDM2, P53, P16 protein and their relationship in human glioma. J. Huazhong Univ. Sci. Technol. 2005, 25, 622–624. [Google Scholar] [CrossRef]
- Liu, W.L.; Midgley, C.; Stephen, C.; Saville, M.; Lane, D.P. Biological significance of a small highly conserved region in the N terminus of the p53 tumour suppressor protein. J. Mol. Biol. 2001, 313, 711–731. [Google Scholar] [CrossRef]
- Hu, J.; Cao, J.; Topatana, W.; Juengpanich, S.; Li, S.; Zhang, B.; Shen, J.; Cai, L.; Cai, X.; Chen, M. Targeting mutant p53 for cancer therapy: Direct and indirect strategies. J. Hematol. Oncol. 2021, 14, 157. [Google Scholar] [CrossRef]
- Canon, J.; Rex, K.; Saiki, A.Y.; Mohr, C.; Cooke, K.; Bagal, D.; Gaida, K.; Holt, T.; Knutson, C.G.; Koppada, N.; et al. The clinical KRAS(G12C) inhibitor AMG 510 drives anti-tumour immunity. Nature 2019, 575, 217–223. [Google Scholar] [CrossRef] [PubMed]
- Zhu, C.X.; Guan, X.Q.; Zhang, X.N.; Luan, X.; Song, Z.B.; Cheng, X.D.; Zhang, W.D.; Qin, J.J. Targeting KRAS mutant cancers: From druggable therapy to drug resistance. Mol. Cancer 2022, 21, 159. [Google Scholar] [CrossRef]
- Wang, Y.-S.; Yin, J.-Z.; Shi, X.-Q.; Zhao, X.-W.; Li, B.; Yang, M.-H. MDM2-Mediated Inhibitory Effect of Arsenic Trioxide on Small Cell Lung Cancer Cell Line by Degrading Mutant p53. Anti-Cancer Agents Med. Chem. 2023, 23, 1829–1837. [Google Scholar] [CrossRef]
- Choudhary, D.; Kaur, A.; Singh, P.; Chaudhary, G.; Kaur, R.; Bayan, M.F.; Chandrasekaran, B.; Marji, S.M.; Ayman, R. Target protein degradation by protacs: A budding cancer treatment strategy. Pharmacol. Ther. 2023, 250, 108525. [Google Scholar] [CrossRef]
- Lu, Y.; Chen, Y.; Hou, G.; Lei, H.; Liu, L.; Huang, X.; Sun, S.; Liu, L.; Liu, X.; Na, J.; et al. Zinc–Iron Bimetallic Peroxides Modulate the Tumor Stromal Microenvironment and Enhance Cell Immunogenicity for Enhanced Breast Cancer Immunotherapy Therapy. ACS Nano 2024, 18, 10542–10556. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Zhang, C.; Xu, D.; Zhang, T.; Chang, C.-Y.; Wang, J.; Liu, J.; Zhang, L.; Haffty, B.G.; Zong, W.-X.; et al. The ubiquitin ligase TRIM21 regulates mutant p53 accumulation and gain of function in cancer. J. Clin. Investig. 2023, 133, e164354. [Google Scholar] [CrossRef] [PubMed]
- Zoltsman, G.; Dang, T.L.; Kuchersky, M.; Faust, O.; Silva, M.S.; Ilani, T.; Wentink, A.S.; Bukau, B.; Rosenzweig, R. A unique chaperoning mechanism in class A JDPs recognizes and stabilizes mutant p53. Mol. Cell 2024, 84, 1512–1526.e1519. [Google Scholar] [CrossRef]
- Wang, L.; Zhong, S.; Fan, X.; Xu, Y.; Wang, M.; Xu, Y.; Cai, Y.; Cao, Z.; Ye, Z.; Wen, L.; et al. Glutathione reductase underlies the stability of mutant p53 by antagonizing protein glutathionylation. Redox Biol. 2025, 81, 103522. [Google Scholar] [CrossRef]
- Selivanova, G.; Iotsova, V.; Okan, I.; Fritsche, M.; Ström, M.; Groner, B.; Grafström, R.C.; Wiman, K.G. Restoration of the growth suppression function of mutant p53 by a synthetic peptide derived from the p53 C-terminal domain. Nat. Med. 1997, 3, 632–638. [Google Scholar] [CrossRef]
- Wang, H.; Guo, M.; Wei, H.; Chen, Y. Targeting p53 pathways: Mechanisms, structures and advances in therapy. Signal Transduct. Target. Ther. 2023, 8, 92. [Google Scholar] [CrossRef]
- Lambert, J.M.R.; Gorzov, P.; Veprintsev, D.B.; Söderqvist, M.; Segerbäck, D.; Bergman, J.; Fersht, A.R.; Hainaut, P.; Wiman, K.G.; Bykov, V.J.N. PRIMA-1 Reactivates Mutant p53 by Covalent Binding to the Core Domain. Cancer Cell 2009, 15, 376–388. [Google Scholar] [CrossRef] [PubMed]
- Bykov, V.J.N.; Zhang, Q.; Zhang, M.; Ceder, S.; Abrahmsen, L.; Wiman, K.G. Targeting of Mutant p53 and the Cellular Redox Balance by APR-246 as a Strategy for Efficient Cancer Therapy. Front. Oncol. 2016, 6, 21. [Google Scholar] [CrossRef]
- Saha, M.N.; Chen, Y.; Chen, M.H.; Chen, G.; Chang, H. Small molecule MIRA-1 induces in vitro and in vivo anti-myeloma activity and synergizes with current anti-myeloma agents. Br. J. Cancer 2014, 110, 2224–2231. [Google Scholar] [CrossRef] [PubMed]
- Zache, N.; Lambert, J.M.R.; Rökaeus, N.; Shen, J.; Hainaut, P.; Bergman, J.; Wiman, K.G.; Bykov, V.J.N. Mutant p53 targeting by the low molecular weight compound STIMA-1. Mol. Oncol. 2008, 2, 70–80. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Wilcken, R.; Joerger, A.C.; Chuckowree, I.S.; Amin, J.; Spencer, J.; Fersht, A.R. Small molecule induced reactivation of mutant p53 in cancer cells. Nucleic Acids Res. 2013, 41, 6034–6044. [Google Scholar] [CrossRef]
- Sallman, D.A.; DeZern, A.E.; Garcia-Manero, G.; Steensma, D.P.; Roboz, G.J.; Sekeres, M.A.; Cluzeau, T.; Sweet, K.L.; McLemore, A.; McGraw, K.L.; et al. Eprenetapopt (APR-246) and Azacitidine in TP53-Mutant Myelodysplastic Syndromes. J. Clin. Oncol. 2021, 39, 1584–1594. [Google Scholar] [CrossRef]
- Cluzeau, T.; Sebert, M.; Rahmé, R.; Cuzzubbo, S.; Lehmann-Che, J.; Madelaine, I.; Peterlin, P.; Bève, B.; Attalah, H.; Chermat, F.; et al. Eprenetapopt Plus Azacitidine in TP53-Mutated Myelodysplastic Syndromes and Acute Myeloid Leukemia: A Phase II Study by the Groupe Francophone des Myélodysplasies (GFM). J. Clin. Oncol. 2021, 39, 1575–1583. [Google Scholar] [CrossRef]
- Pereira, M.P.; Herrity, E.; Kim, D.D.H. TP53-mutated acute myeloid leukemia and myelodysplastic syndrome: Biology, treatment challenges, and upcoming approaches. Ann. Hematol. 2023, 103, 1049–1067. [Google Scholar] [CrossRef]
- Mishra, A.; Tamari, R.; DeZern, A.E.; Byrne, M.T.; Gooptu, M.; Chen, Y.B.; Deeg, H.J.; Sallman, D.; Gallacher, P.; Wennborg, A.; et al. Eprenetapopt Plus Azacitidine After Allogeneic Hematopoietic Stem-Cell Transplantation for TP53-Mutant Acute Myeloid Leukemia and Myelodysplastic Syndromes. J. Clin. Oncol. 2022, 40, 3985–3993. [Google Scholar] [CrossRef]
- Garcia-Manero, G.; Goldberg, A.D.; Winer, E.S.; Altman, J.K.; Fathi, A.T.; Odenike, O.; Roboz, G.J.; Gallacher, P.; Wennborg, A.; Kaylor Hickman, D.; et al. Phase I and Expansion Study of Eprenetapopt (APR-246) in Combination with Venetoclax (VEN) and Azacitidine (AZA) in TP53-Mutant Acute Myeloid Leukemia (AML). Blood 2021, 138, 3409. [Google Scholar] [CrossRef]
- Soignet, S.L.; Maslak, P.; Wang, Z.-G.; Jhanwar, S.; Calleja, E.; Dardashti, L.J.; Corso, D.; DeBlasio, A.; Gabrilove, J.; Scheinberg, D.A.; et al. Complete Remission After Treatment of Acute Promyelocytic Leukemia with Arsenic Trioxide. N. Engl. J. Med. 1998, 339, 1341–1348. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.-W.; Yan, X.-J.; Zhou, Z.-R.; Yang, F.-F.; Wu, Z.-Y.; Sun, H.-B.; Liang, W.-X.; Song, A.-X.; Lallemand-Breitenbach, V.; Jeanne, M.; et al. Arsenic Trioxide Controls the Fate of the PML-RARα Oncoprotein by Directly Binding PML. Science 2010, 328, 240–243. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Wu, J.-L.; Liang, Y.; Tang, Y.-G.; Song, H.-X.; Wu, L.-L.; Xing, Y.-F.; Yan, N.; Li, Y.-T.; Wang, Z.-Y.; et al. Arsenic Trioxide Rescues Structural p53 Mutations Through a Cryptic Allosteric Site. Cancer Cell 2021, 39, 225–239.e228. [Google Scholar] [CrossRef]
- Song, H.; Wu, J.; Tang, Y.; Dai, Y.; Xiang, X.; Li, Y.; Wu, L.; Wu, J.; Liang, Y.; Xing, Y.; et al. Diverse rescue potencies of p53 mutations to ATO are predetermined by intrinsic mutational properties. Sci. Transl. Med. 2023, 15, eabn9155. [Google Scholar] [CrossRef]
- Dumbrava, E.E.; Johnson, M.L.; Tolcher, A.W.; Shapiro, G.; Thompson, J.A.; El-Khoueiry, A.B.; Vandross, A.L.; Kummar, S.; Parikh, A.R.; Munster, P.N.; et al. First-in-human study of PC14586, a small molecule structural corrector of Y220C mutant p53, in patients with advanced solid tumors harboring a TP53 Y220C mutation. J. Clin. Oncol. 2022, 40, 3003. [Google Scholar] [CrossRef]
- Garufi, A.; Ubertini, V.; Mancini, F.; D’Orazi, V.; Baldari, S.; Moretti, F.; Bossi, G.; D’Orazi, G. The beneficial effect of Zinc(II) on low-dose chemotherapeutic sensitivity involves p53 activation in wild-type p53-carrying colorectal cancer cells. J. Exp. Clin. Cancer Res. 2015, 34, 87. [Google Scholar] [CrossRef]
- Yu, X.; Vazquez, A.; Levine, A.J.; Carpizo, D.R. Allele-Specific p53 Mutant Reactivation. Cancer Cell 2012, 21, 614–625. [Google Scholar] [CrossRef]
- Yu, X.; Blanden, A.; Tsang, A.T.; Zaman, S.; Liu, Y.; Gilleran, J.; Bencivenga, A.F.; Kimball, S.D.; Loh, S.N.; Carpizo, D.R. Thiosemicarbazones Functioning as Zinc Metallochaperones to Reactivate Mutant p53. Mol. Pharmacol. 2017, 91, 567–575. [Google Scholar] [CrossRef]
- Salim, K.Y.; Maleki Vareki, S.; Danter, W.R.; San-Marina, S.; Koropatnick, J. COTI-2, a novel small molecule that is active against multiple human cancer cell lines in vitro and in vivo. Oncotarget 2016, 7, 41363–41379. [Google Scholar] [CrossRef]
- Lindemann, A.; Patel, A.A.; Silver, N.L.; Tang, L.; Liu, Z.Y.; Wang, L.; Tanaka, N.; Rao, X.Y.; Takahashi, H.; Maduka, N.K.; et al. COTI-2, A Novel Thiosemicarbazone Derivative, Exhibits Antitumor Activity in HNSCC Through p53-Dependent and -Independent Mechanisms. Clin. Cancer Res. 2019, 25, 5650–5662. [Google Scholar] [CrossRef]
- Ma, Z.; Shen, Q.; Zhou, J. Rezatapopt (PC14586): A First-in-Class Small Molecule p53 Y220C Mutant Protein Stabilizer in Clinical Trials. J. Med. Chem. 2025, 68, 6847–6849. [Google Scholar] [CrossRef] [PubMed]
- Bártek, J.; Bártková, J.; Vojtĕsek, B.; Stasková, Z.; Lukás, J.; Rejthar, A.; Kovarík, J.; Midgley, C.A.; Gannon, J.V.; Lane, D.P. Aberrant expression of the p53 oncoprotein is a common feature of a wide spectrum of human malignancies. Oncogene 1991, 6, 1699–1703. [Google Scholar] [PubMed]
- Suh, Y.A.; Post, S.M.; Elizondo-Fraire, A.C.; Maccio, D.R.; Jackson, J.G.; El-Naggar, A.K.; Van Pelt, C.; Terzian, T.; Lozano, G. Multiple Stress Signals Activate Mutant p53. Cancer Res. 2011, 71, 7168–7175. [Google Scholar] [CrossRef]
- Terzian, T.; Suh, Y.A.; Iwakuma, T.; Post, S.M.; Neumann, M.; Lang, G.A.; Van Pelt, C.S.; Lozano, G. The inherent instability of mutant p53 is alleviated by or loss. Genes Dev. 2008, 22, 1337–1344. [Google Scholar] [CrossRef]
- Muller, P.A.J.; Vousden, K.H. p53 mutations in cancer. Nat. Cell Biol. 2013, 15, 2–8. [Google Scholar] [CrossRef]
- Li, D.; Marchenko, N.D.; Moll, U.M. SAHA shows preferential cytotoxicity in mutant p53 cancer cells by destabilizing mutant p53 through inhibition of the HDAC6-Hsp90 chaperone axis. Cell Death Differ. 2011, 18, 1904–1913. [Google Scholar] [CrossRef]
- Parrales, A.; Ranjan, A.; Iyer, S.V.; Padhye, S.; Weir, S.J.; Roy, A.; Iwakuma, T. DNAJA1 controls the fate of misfolded mutant p53 through the mevalonate pathway. Nat. Cell Biol. 2016, 18, 1233–1243. [Google Scholar] [CrossRef] [PubMed]
- Tutuska, K.; Parrilla-Monge, L.; Di Cesare, E.; Nemajerova, A.; Moll, U.M. Statin as anti-cancer therapy in autochthonous T-lymphomas expressing stabilized gain-of-function mutant p53 proteins. Cell Death Dis. 2020, 11, 274. [Google Scholar] [CrossRef]
- Ingallina, E.; Sorrentino, G.; Bertolio, R.; Lisek, K.; Zannini, A.; Azzolin, L.; Severino, L.U.; Scaini, D.; Mano, M.; Mantovani, F.; et al. Mechanical cues control mutant p53 stability through a mevalonate–RhoA axis. Nat. Cell Biol. 2017, 20, 28–35. [Google Scholar] [CrossRef]
- Fu, S.; Hou, M.M.; Naing, A.; Janku, F.; Hess, K.; Zinner, R.; Subbiah, V.; Hong, D.; Wheler, J.; Piha-Paul, S.; et al. Phase I study of pazopanib and vorinostat: A therapeutic approach for inhibiting mutant p53-mediated angiogenesis and facilitating mutant p53 degradation. Ann. Oncol. 2015, 26, 1012–1018. [Google Scholar] [CrossRef]
- Zhao, M.; Yao, P.; Mao, Y.; Wu, J.; Wang, W.; Geng, C.; Cheng, J.; Du, W.; Jiang, P. Malic enzyme 2 maintains protein stability of mutant p53 through 2-hydroxyglutarate. Nat. Metab. 2022, 4, 225–238. [Google Scholar] [CrossRef] [PubMed]
- Paranjpe, A.; Srivenugopal, K.S. Degradation of NF-κB, p53 and other regulatory redox-sensitive proteins by thiol-conjugating and -nitrosylating drugs in human tumor cells. Carcinogenesis 2013, 34, 990–1000. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.B.; Wei, P.F.; Liu, L.; Ding, T.; Yang, Y.Y.; Jin, P.P.; Zhang, L.; Zhao, Z.B.; Wang, M.M.; Hu, B.C.; et al. AIE-enabled transfection-free identification and isolation of viable cell subpopulations differing in the level of autophagy. Autophagy 2023, 19, 3062–3078. [Google Scholar] [CrossRef]
- Cordani, M.; Butera, G.; Pacchiana, R.; Donadelli, M. Molecular interplay between mutant p53 proteins and autophagy in cancer cells. Biochim. Biophys. Acta-Rev. Cancer 2017, 1867, 19–28. [Google Scholar] [CrossRef]
- Rodriguez, O.C.; Choudhury, S.; Kolukula, V.; Vietsch, E.E.; Catania, J.; Preet, A.; Reynoso, K.; Bargonetti, J.; Wellstein, A.; Albanese, C.; et al. Dietary downregulation of mutant p53 levels via glucose restriction. Cell Cycle 2014, 11, 4436–4446. [Google Scholar] [CrossRef] [PubMed]
- Maan, M.; Pati, U. CHIP promotes autophagy-mediated degradation of aggregating mutant p53 in hypoxic conditions. FEBS J. 2018, 285, 3197–3214. [Google Scholar] [CrossRef]
- Garufi, A.; Pucci, D.; D’Orazi, V.; Cirone, M.; Bossi, G.; Avantaggiati, M.L.; D’Orazi, G. Degradation of mutant p53H175 protein by Zn(II) through autophagy. Cell Death Dis. 2014, 5, e1271. [Google Scholar] [CrossRef]
- Prasad, S.; DuBourdieu, D.; Srivastava, A.; Kumar, P.; Lall, R. Metal–Curcumin Complexes in Therapeutics: An Approach to Enhance Pharmacological Effects of Curcumin. Int. J. Mol. Sci. 2021, 22, 7094. [Google Scholar] [CrossRef]
- Foggetti, G.; Ottaggio, L.; Russo, D.; Monti, P.; Degan, P.; Fronza, G.; Menichini, P. Gambogic acid counteracts mutant p53 stability by inducing autophagy. Biochim. Biophys. Acta (BBA)-Mol. Cell Res. 2017, 1864, 382–392. [Google Scholar] [CrossRef]
- Vakifahmetoglu-Norberg, H.; Kim, M.; Xia, H.-g.; Iwanicki, M.P.; Ofengeim, D.; Coloff, J.L.; Pan, L.; Ince, T.A.; Kroemer, G.; Brugge, J.S.; et al. Chaperone-mediated autophagy degrades mutant p53. Genes Dev. 2013, 27, 1718–1730. [Google Scholar] [CrossRef]
- Foggetti, G.; Ottaggio, L.; Russo, D.; Mazzitelli, C.; Monti, P.; Degan, P.; Miele, M.; Fronza, G.; Menichini, P. Autophagy induced by SAHA affects mutant P53 degradation and cancer cell survival. Biosci. Rep. 2019, 39, BSR20181345. [Google Scholar] [CrossRef] [PubMed]
- Nishikawa, S.; Iwakuma, T. Drugs Targeting p53 Mutations with FDA Approval and in Clinical Trials. Cancers 2023, 15, 429. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.E.; Pan, Y.R.; Yeh, C.N.; Lunec, J. Targeting P53 as a Future Strategy to Overcome Gemcitabine Resistance in Biliary Tract Cancers. Biomolecules 2020, 10, 1474. [Google Scholar] [CrossRef] [PubMed]
- Bukhari, A.B.; Lewis, C.W.; Pearce, J.J.; Luong, D.; Chan, G.K.; Gamper, A.M. Inhibiting Wee1 and ATR kinases produces tumor-selective synthetic lethality and suppresses metastasis. J. Clin. Investig. 2019, 129, 1329–1344. [Google Scholar] [CrossRef]
- Hu, W.W.; Feng, Z.H. Hypothermia Is a Potential New Therapy for a Subset of Tumors with Mutant p53. Cancer Res. 2021, 81, 3762–3763. [Google Scholar] [CrossRef]
- Li, S.J.; Topatana, W.; Juengpanich, S.; Cao, J.S.; Hu, J.H.; Zhang, B.; Ma, D.N.; Cai, X.J.; Chen, M.Y. Development of synthetic lethality in cancer: Molecular and cellular classification. Signal Transduct. Target. Ther. 2020, 5, 241. [Google Scholar] [CrossRef]
- Oza, A.M.; Estevez-Diz, M.; Grischke, E.-M.; Hall, M.; Marmé, F.; Provencher, D.; Uyar, D.; Weberpals, J.I.; Wenham, R.M.; Laing, N.; et al. A Biomarker-enriched, Randomized Phase II Trial of Adavosertib (AZD1775) Plus Paclitaxel and Carboplatin for Women with Platinum-sensitive TP53-mutant Ovarian Cancer. Clin. Cancer Res. 2020, 26, 4767–4776. [Google Scholar] [CrossRef]
- Qi, L.; Li, G.; Li, P.; Wang, H.; Fang, X.; He, T.; Li, J. Twenty years of Gendicine® rAd-p53 cancer gene therapy: The first-in-class human cancer gene therapy in the era of personalized oncology. Genes Dis. 2024, 11, 101155. [Google Scholar] [CrossRef]
- Li, Y.; Li, L.-J.; Wang, L.-J.; Zhang, Z.; Gao, N.; Liang, C.-Y.; Huang, Y.-D.; Han, B. Selective intra-arterial infusion of rAd-p53 with chemotherapy for advanced oral cancer: A randomized clinical trial. BMC Med. 2014, 12, 16. [Google Scholar] [CrossRef]
- Liu, S.; Chen, P.; Hu, M.; Tao, Y.; Chen, L.; Liu, H.; Wang, J.; Luo, J.; Gao, G. Randomized, controlled phase II study of post-surgery radiotherapy combined with recombinant adenoviral human p53 gene therapy in treatment of oral cancer. Cancer Gene Ther. 2013, 20, 375–378. [Google Scholar] [CrossRef]
- Zhang, W.W.; Li, L.J.; Li, D.G.; Liu, J.L.; Li, X.Q.; Li, W.; Xu, X.L.; Zhang, M.J.; Chandler, L.A.; Lin, H.; et al. The First Approved Gene Therapy Product for Cancer Ad-p53 (Gendicine): 12 Years in the Clinic. Hum. Gene Ther. 2018, 29, 160–179. [Google Scholar] [CrossRef]
- Paunovska, K.; Loughrey, D.; Dahlman, J.E. Drug delivery systems for RNA therapeutics. Nat. Rev. Genet. 2022, 23, 265–280. [Google Scholar] [CrossRef] [PubMed]
- Sasso, J.M.; Ambrose, B.J.B.; Tenchov, R.; Datta, R.S.; Basel, M.T.; DeLong, R.K.; Zhou, Q.A. The Progress and Promise of RNA Medicine-an Arsenal of Targeted Treatments. J. Med. Chem. 2022, 65, 6975–7015. [Google Scholar] [CrossRef]
- Kim, Y.K. RNA therapy: Rich history, various applications and unlimited future prospects. Exp. Mol. Med. 2022, 54, 455–465. [Google Scholar] [CrossRef] [PubMed]
- Roberts, T.C.; Langer, R.; Wood, M.J.A. Advances in oligonucleotide drug delivery. Nat. Rev. Drug Discov. 2020, 19, 673–694. [Google Scholar] [CrossRef] [PubMed]
- Yu, A.-M.; Choi, Y.H.; Tu, M.-J.; Touyz, R.M. RNA Drugs and RNA Targets for Small Molecules: Principles, Progress, and Challenges. Pharmacol. Rev. 2020, 72, 862–898. [Google Scholar] [CrossRef]
- Martinez, L.A.; Naguibneva, I.; Lehrmann, H.; Vervisch, A.; Tchénio, T.; Lozano, G.; Harel-Bellan, A. Synthetic small inhibiting RNAs: Efficient tools to inactivate oncogenic mutations and restore p53 pathways. Proc. Natl. Acad. Sci. USA 2002, 99, 14849–14854. [Google Scholar] [CrossRef]
- Ubby, I.; Krueger, C.; Rosato, R.; Qian, W.; Chang, J.; Sabapathy, K. Cancer therapeutic targeting using mutant–p53-specific siRNAs. Oncogene 2019, 38, 3415–3427. [Google Scholar] [CrossRef]
- Komor, A.C.; Kim, Y.B.; Packer, M.S.; Zuris, J.A.; Liu, D.R. Programmable editing of a target base in genomic DNA without double-stranded DNA cleavage. Nature 2016, 533, 420–424. [Google Scholar] [CrossRef]
- Enache, O.M.; Rendo, V.; Abdusamad, M.; Lam, D.; Davison, D.; Pal, S.; Currimjee, N.; Hess, J.; Pantel, S.; Nag, A.; et al. Cas9 activates the p53 pathway and selects for p53-inactivating mutations. Nat. Genet. 2020, 52, 662–668. [Google Scholar] [CrossRef]
- Haapaniemi, E.; Botla, S.; Persson, J.; Schmierer, B.; Taipale, J. CRISPR–Cas9 genome editing induces a p53-mediated DNA damage response. Nat. Med. 2018, 24, 927–930. [Google Scholar] [CrossRef] [PubMed]
- Ihry, R.J.; Worringer, K.A.; Salick, M.R.; Frias, E.; Ho, D.; Theriault, K.; Kommineni, S.; Chen, J.; Sondey, M.; Ye, C.; et al. p53 inhibits CRISPR–Cas9 engineering in human pluripotent stem cells. Nat. Med. 2018, 24, 939–946. [Google Scholar] [CrossRef] [PubMed]
- Békés, M.; Langley, D.R.; Crews, C.M. PROTAC targeted protein degraders: The past is prologue. Nat. Rev. Drug Discov. 2022, 21, 181–200. [Google Scholar] [CrossRef] [PubMed]
- Nalawansha, D.A.; Crews, C.M. PROTACs: An Emerging Therapeutic Modality in Precision Medicine. Cell Chem. Biol. 2020, 27, 998–1014. [Google Scholar] [CrossRef] [PubMed]
- Adams, C.M.; Mitra, R.; Xiao, Y.; Michener, P.; Palazzo, J.; Chao, A.; Gour, J.; Cassel, J.; Salvino, J.M.; Eischen, C.M. Targeted MDM2 Degradation Reveals a New Vulnerability for p53-Inactivated Triple-Negative Breast Cancer. Cancer Discov. 2023, 13, 1210–1229. [Google Scholar] [CrossRef]
- He, S.; Ma, J.; Fang, Y.; Liu, Y.; Wu, S.; Dong, G.; Wang, W.; Sheng, C. Homo-PROTAC mediated suicide of MDM2 to treat non-small cell lung cancer. Acta Pharm. Sin. B 2021, 11, 1617–1628. [Google Scholar] [CrossRef]
- Li, Y.; Yang, J.; Aguilar, A.; McEachern, D.; Przybranowski, S.; Liu, L.; Yang, C.-Y.; Wang, M.; Han, X.; Wang, S. Discovery of MD-224 as a First-in-Class, Highly Potent, and Efficacious Proteolysis Targeting Chimera Murine Double Minute 2 Degrader Capable of Achieving Complete and Durable Tumor Regression. J. Med. Chem. 2018, 62, 448–466. [Google Scholar] [CrossRef]
- Wang, B.; Liu, J.; Tandon, I.; Wu, S.; Teng, P.; Liao, J.; Tang, W. Development of MDM2 degraders based on ligands derived from Ugi reactions: Lessons and discoveries. Eur. J. Med. Chem. 2021, 219, 113425. [Google Scholar] [CrossRef]
- Kong, L.; Meng, F.; Wu, S.; Zhou, P.; Ge, R.; Liu, M.; Zhang, L.; Zhou, J.; Zhong, D.; Xie, S. Selective degradation of the p53-R175H oncogenic hotspot mutant by an RNA aptamer-based PROTAC. Clin. Transl. Med. 2023, 13, e1191. [Google Scholar] [CrossRef]
- Kong, L.; Meng, F.; Zhou, P.; Ge, R.; Geng, X.; Yang, Z.; Li, G.; Zhang, L.; Wang, J.; Ma, J.; et al. An engineered DNA aptamer-based PROTAC for precise therapy of p53-R175H hotspot mutant-driven cancer. Sci. Bull. 2024, 69, 2122–2135. [Google Scholar] [CrossRef]
- Peer, D.; Karp, J.M.; Hong, S.; Farokhzad, O.C.; Margalit, R.; Langer, R. Nanocarriers as an emerging platform for cancer therapy. Nat. Nanotechnol. 2007, 2, 751–760. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Ni, F.; Sun, D.; Peng, Y.; Zhao, Y.; Wu, X.; Li, S.; Qi, X.; He, X.; Li, M.; et al. Glucagon Enhances Chemotherapy Efficacy By Inhibition of Tumor Vessels in Colorectal Cancer. Adv. Sci. 2023, 11, e2307271. [Google Scholar] [CrossRef]
- Danhier, F.; Feron, O.; Préat, V. To exploit the tumor microenvironment: Passive and active tumor targeting of nanocarriers for anti-cancer drug delivery. J. Control. Release 2010, 148, 135–146. [Google Scholar] [CrossRef]
- Chen, Q.; Feng, L.; Liu, J.; Zhu, W.; Dong, Z.; Wu, Y.; Liu, Z. Intelligent Albumin–MnO2 Nanoparticles as pH-/H2O2-Responsive Dissociable Nanocarriers to Modulate Tumor Hypoxia for Effective Combination Therapy. Adv. Mater. 2016, 28, 7129–7136. [Google Scholar] [CrossRef] [PubMed]
- Janib, S.M.; Moses, A.S.; MacKay, J.A. Imaging and drug delivery using theranostic nanoparticles. Adv. Drug Deliv. Rev. 2010, 62, 1052–1063. [Google Scholar] [CrossRef]
- Sun, D.; Yu, L.; Wang, G.; Xu, Y.; Wang, P.; Wang, N.; Wu, Z.; Zhang, G.; Zhang, J.; Zhang, Y.; et al. Rationally designed catalytic nanoplatform for enhanced chemoimmunotherapy via deploying endogenous plus exogenous copper and remodeling tumor microenvironment. J. Nanobiotechnol. 2024, 22, 551. [Google Scholar] [CrossRef]
- Davis, M.E.; Chen, Z.; Shin, D.M. Nanoparticle therapeutics: An emerging treatment modality for cancer. Nat. Rev. Drug Discov. 2008, 7, 771–782. [Google Scholar] [CrossRef] [PubMed]
- Shi, J.; Kantoff, P.W.; Wooster, R.; Farokhzad, O.C. Cancer nanomedicine: Progress, challenges and opportunities. Nat. Rev. Cancer 2016, 17, 20–37. [Google Scholar] [CrossRef]
- Kirk, R. The crystal ball clears for breast cancer therapy? Nat. Rev. Clin. Oncol. 2011, 8, 383. [Google Scholar] [CrossRef]
- Li, Q.; Gao, W.; Zhang, C.; Wang, P.; Wang, X.; Yan, M.; Jiang, W.; Wu, Z.; Wei, P.; Tian, G.; et al. A Biodegradable High-Efficiency Magnetic Nanoliposome Promotes Tumor Microenvironment-Responsive Multimodal Tumor Therapy Along with Switchable T2 Magnetic Resonance Imaging. ACS Appl. Mater. Interfaces 2022, 14, 24160–24173. [Google Scholar] [CrossRef]
- Xu, M.; Hu, Y.; Ding, W.; Li, F.; Lin, J.; Wu, M.; Wu, J.; Wen, L.-P.; Qiu, B.; Wei, P.-F.; et al. Rationally designed rapamycin-encapsulated ZIF-8 nanosystem for overcoming chemotherapy resistance. Biomaterials 2020, 258, 120308. [Google Scholar] [CrossRef] [PubMed]
- Fekry, B.; Jeffries, K.A.; Esmaeilniakooshkghazi, A.; Szulc, Z.M.; Knagge, K.J.; Kirchner, D.R.; Horita, D.A.; Krupenko, S.A.; Krupenko, N.I. C16-ceramide is a natural regulatory ligand of p53 in cellular stress response. Nat. Commun. 2018, 9, 4149. [Google Scholar] [CrossRef] [PubMed]
- Khiste, S.K.; Liu, Z.; Roy, K.R.; Uddin, M.B.; Hosain, S.B.; Gu, X.; Nazzal, S.; Hill, R.A.; Liu, Y.-Y. Ceramide–Rubusoside Nanomicelles, a Potential Therapeutic Approach to Target Cancers Carrying p53 Missense Mutations. Mol. Cancer Ther. 2020, 19, 564–574. [Google Scholar] [CrossRef]
- Bernhard, E.J.; Patwardhan, G.A.; Zhang, Q.-J.; Yin, D.; Gupta, V.; Bao, J.; Senkal, C.E.; Ogretmen, B.; Cabot, M.C.; Shah, G.V.; et al. A New Mixed-Backbone Oligonucleotide Against Glucosylceramide Synthase Sensitizes Multidrug-Resistant Tumors to Apoptosis. PLoS ONE 2009, 4, e6938. [Google Scholar] [CrossRef]
- Liu, Y.Y. Resuscitating Wild-Type p53 Expression by Disrupting Ceramide Glycosylation: A Novel Approach to Target Mutant p53 Tumors. Cancer Res. 2011, 71, 6295–6299. [Google Scholar] [CrossRef]
- Liu, Y.Y.; Patwardhan, G.A.; Bhinge, K.; Gupta, V.; Gu, X.; Jazwinski, S.M. Suppression of Glucosylceramide Synthase Restores p53-Dependent Apoptosis in Mutant p53 Cancer Cells. Cancer Res. 2011, 71, 2276–2285. [Google Scholar] [CrossRef]
- Li, F.Z.; Chen, D.L.; Sun, Q.Q.; Wu, J.L.; Gan, Y.L.; Leong, K.W.; Liang, X.J. MDM2-Targeting Reassembly Peptide (TRAP) Nanoparticles for p53-Based Cancer Therapy. Adv. Mater. 2023, 35, 2305164. [Google Scholar] [CrossRef]
- Schnorenberg, M.R.; Hawley, K.M.; Thomas-Toth, A.T.; Watkins, E.A.; Tian, Y.; Ting, J.M.; Leak, L.B.; Kucera, I.M.; Raczy, M.M.; Kung, A.L.; et al. Targeted Polymersome Delivery of a Stapled Peptide for Drugging the Tumor Protein p53:BCL-2-Family Axis in Diffuse Large B-Cell Lymphoma. ACS Nano 2023, 17, 23374–23390. [Google Scholar] [CrossRef]
- P, S.S.; Naresh, P.; Antony, J.; Wadhwani, A.; M, S.K.; Jubie, S. Dual Modulators of p53 and Cyclin D in ER Alpha Signaling by Albumin Nanovectors Bearing Zinc Chaperones for ER-Positive Breast Cancer Therapy. Mini-Rev. Med. Chem. 2021, 21, 792–802. [Google Scholar] [CrossRef]
- Gong, K.; Lin, J.; Chen, X.; Duan, Y.; Zhang, J.; Yu, J.; Wang, J.; Sun, R.; Li, J.; Duan, Y. Thermosensitive gel-nano system against esophageal cancer via restoring p53 activity and boosting T-cell immunity. J. Control. Release 2024, 371, 111–125. [Google Scholar] [CrossRef]
- Xhafa, S.; Di Nicola, C.; Tombesi, A.; Pettinari, R.; Pettinari, C.; Scarpelli, F.; Crispini, A.; La Deda, M.; Candreva, A.; Garufi, A.; et al. Pyrazolone-Based Zn(II) Complexes Display Antitumor Effects in Mutant p53-Carrying Cancer Cells. J. Med. Chem. 2024, 67, 15676–15690. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Z.; Qian, J.; Liu, K.; Zhang, Y.; Gao, M.; Tang, B.Z. A Water-Stable and Red-Emissive Radical Cation for Mutp53 Cancer Therapy. Angew. Chem. Int. Ed. 2022, 61, e202212671. [Google Scholar] [CrossRef]
- Qian, J.; Zhang, W.; Wei, P.; Yao, G.; Yi, T.; Zhang, H.; Ding, H.; Huang, X.; Wang, M.; Song, Y.; et al. Enhancing Chemotherapy of p53-Mutated Cancer Through Ubiquitination-Dependent Proteasomal Degradation of Mutant p53 Proteins by Engineered ZnFe-4 Nanoparticles. Adv. Funct. Mater. 2020, 30, 2001994. [Google Scholar] [CrossRef]
- Zhang, Y.J.; Huang, X.W.; Wang, L.S.; Cao, C.; Zhang, H.; Wei, P.F.; Ding, H.; Song, Y.; Chen, Z.Y.; Qian, J.Y.; et al. Glutathionylation-dependent proteasomal degradation of wide-spectrum mutant p53 proteins by engineered zeolitic imidazolate framework-8. Biomaterials 2021, 271, 120720. [Google Scholar] [CrossRef]
- Wang, J.; Qu, C.; Shao, X.; Song, G.; Sun, J.; Shi, D.; Jia, R.; An, H.; Wang, H. Carrier-free nanoprodrug for p53-mutated tumor therapy via concurrent delivery of zinc-manganese dual ions and ROS. Bioact. Mater. 2023, 20, 404–417. [Google Scholar] [CrossRef]
- Qiao, G.; Pan, X.; He, M.; Peng, R.; Huang, X.; Nie, C.; Jiang, J.; Chu, X. An ultrathin Zn-based layered double hydroxides augment degradation of mutant p53 to improve tumor therapy. Chem. Eng. J. 2023, 475, 146449. [Google Scholar] [CrossRef]
- Jin, Q.; Zuo, W.; Lin, Q.; Wu, T.; Liu, C.; Liu, N.; Liu, J.; Zhu, X. Zinc-doped Prussian blue nanoparticles for mutp53-carrying tumor ion interference and photothermal therapy. Asian J. Pharm. Sci. 2022, 17, 767–777. [Google Scholar] [CrossRef] [PubMed]
- Sun, L.; Gao, H.; Wang, H.; Zhou, J.; Ji, X.; Jiao, Y.; Qin, X.; Ni, D.; Zheng, X. Nanoscale Metal–Organic Frameworks-Mediated Degradation of Mutant p53 Proteins and Activation of cGAS-STING Pathway for Enhanced Cancer Immunotherapy. Adv. Sci. 2024, 11, e2307278. [Google Scholar] [CrossRef]
- Wu, F.; Zhang, M.; Chu, X.; Zhang, Q.; Su, Y.; Sun, B.; Lu, T.; Zhou, N.; Zhang, J.; Wang, J.; et al. Black phosphorus nanosheets-based nanocarriers for enhancing chemotherapy drug sensitiveness via depleting mutant p53 and resistant cancer multimodal therapy. Chem. Eng. J. 2019, 370, 387–399. [Google Scholar] [CrossRef]
- Yi, T.; Qian, J.; Ye, Y.; Zhang, H.; Jin, X.; Wang, M.; Yang, Z.; Zhang, W.; Wen, L.; Zhang, Y. Crizotinib Nanomicelles Synergize with Chemotherapy Through Inducing Proteasomal Degradation of Mutp53 Proteins. ACS Appl. Mater. Interfaces 2022, 15, 511–523. [Google Scholar] [CrossRef]
- Bi, Y.-Y.; Chen, Q.; Yang, M.-Y.; Xing, L.; Jiang, H.-L. Nanoparticles targeting mutant p53 overcome chemoresistance and tumor recurrence in non-small cell lung cancer. Nat. Commun. 2024, 15, 2759. [Google Scholar] [CrossRef] [PubMed]
- Mei, Y.; Qin, X.; Yang, Z.; Song, S.; Liu, X.; Wu, C.; Qian, J.; Huang, X.; Zhang, Y.; He, W. Engineered a dual-targeting HA-TPP/A nanoparticle for combination therapy against KRAS-TP53 co-mutation in gastrointestinal cancers. Bioact. Mater. 2024, 32, 277–291. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Yang, Z.; Song, Y.; Wei, P.; Ishiwme, N.; Wang, L.; Zhang, H.; Jing, M.; Gao, M.; Wen, L.; et al. Proteasomal and autophagy-mediated degradation of mutp53 proteins through mitochondria-targeting aggregation-induced-emission materials. Acta Biomater. 2022, 150, 402–412. [Google Scholar] [CrossRef]
- Qian, J.; Yang, Z.; Zhang, W.; Ye, Y.; Song, Y.; Ye, Y.; Zhou, Y.; Zheng, Q.; Huang, X.; Du, S.; et al. NIR Emissive Functional Nanoparticles Promote Precise Pancreatic Cancer Therapy by Co-Targeting Mutant p53 and Oncogenic KRAS. Adv. Funct. Mater. 2024, 34, 2312610. [Google Scholar] [CrossRef]
- Zhang, H.; Zhang, W.; Hu, B.; Qin, X.; Yi, T.; Ye, Y.; Huang, X.; Song, Y.; Yang, Z.; Qian, J.; et al. Precise pancreatic cancer therapy through targeted degradation of mutant p53 protein by cerium oxide nanoparticles. J. Nanobiotechnol. 2023, 21, 117. [Google Scholar] [CrossRef]
- Vargas, J.N.S.; Hamasaki, M.; Kawabata, T.; Youle, R.J.; Yoshimori, T. The mechanisms and roles of selective autophagy in mammals. Nat. Rev. Mol. Cell Biol. 2022, 24, 167–185. [Google Scholar] [CrossRef] [PubMed]
- Huang, X.; Cao, Z.; Qian, J.; Ding, T.; Wu, Y.; Zhang, H.; Zhong, S.; Wang, X.; Ren, X.; Zhang, W.; et al. Nanoreceptors promote mutant p53 protein degradation by mimicking selective autophagy receptors. Nat. Nanotechnol. 2024, 19, 545–553. [Google Scholar] [CrossRef]
- Wu, T.; Lang, T.; Zheng, C.; Yan, W.; Li, Y.; Zhu, R.; Huang, X.; Xu, H.; Li, Y.; Yin, Q. Promote Intratumoral Drug Release and Penetration to Counteract Docetaxel-Induced Metastasis by Photosensitizer-Modified Red Blood Cell Membrane-Coated Nanoparticle. Adv. Funct. Mater. 2022, 33, 2212109. [Google Scholar] [CrossRef]
- Li, S.; Juengpanich, S.; Topatana, W.; Xie, T.; Hou, L.; Zhu, Y.; Chen, J.; Shan, Y.; Han, Y.; Lu, Z.; et al. Adavosertib-encapsulated metal-organic frameworks for p53-mutated gallbladder cancer treatment via synthetic lethality. Sci. Bull. 2024, 69, 1286–1301. [Google Scholar] [CrossRef]
- Chen, S.; Rong, L.; Lei, Q.; Cao, P.-X.; Qin, S.-Y.; Zheng, D.-W.; Jia, H.-Z.; Zhu, J.-Y.; Cheng, S.-X.; Zhuo, R.-X.; et al. A surface charge-switchable and folate modified system for co-delivery of proapoptosis peptide and p53 plasmid in cancer therapy. Biomaterials 2016, 77, 149–163. [Google Scholar] [CrossRef]
- Misra, S.K.; Naz, S.; Kondaiah, P.; Bhattacharya, S. A cationic cholesterol based nanocarrier for the delivery of p53-EGFP-C3 plasmid to cancer cells. Biomaterials 2014, 35, 1334–1346. [Google Scholar] [CrossRef] [PubMed]
- Rejeeth, C.; Kannan, S. p53 gene therapy of human breast carcinoma: Using a transferrin-modified silica nanoparticles. Breast Cancer 2014, 23, 101–110. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.; Zhao, Y.; Mu, X.; Wu, H.; Chen, L.; Liu, W.; Mu, Y.; Liu, J.; Wei, X. A silica–polymer composite nano system for tumor-targeted imaging and p53 gene therapy of lung cancer. J. Biomater. Sci. Polym. Ed. 2015, 26, 384–400. [Google Scholar] [CrossRef]
- Davoodi, P.; Srinivasan, M.P.; Wang, C.-H. Synthesis of intracellular reduction-sensitive amphiphilic polyethyleneimine and poly(ε-caprolactone) graft copolymer for on-demand release of doxorubicin and p53 plasmid DNA. Acta Biomater. 2016, 39, 79–93. [Google Scholar] [CrossRef]
- Li, C.; Hu, J.; Li, W.; Song, G.; Shen, J. Combined bortezomib-based chemotherapy and p53 gene therapy using hollow mesoporous silica nanospheres for p53 mutant non-small cell lung cancer treatment. Biomater. Sci. 2017, 5, 77–88. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Zhang, Q.; Li, J.; Yang, M.; Zhao, N.; Xu, F.-J. Rattle-Structured Rough Nanocapsules with In-Situ-Formed Gold Nanorod Cores for Complementary Gene/Chemo/Photothermal Therapy. ACS Nano 2018, 12, 5646–5656. [Google Scholar] [CrossRef]
- Karmakar, A.; Bratton, S.M.; Dervishi, E.; Ghosh, A.; Mahmood, M.; Xu, Y.; Saeed, L.M.; Mustafa, T.; Casciano, D.; Radominska-Pandya, A.; et al. Ethylenediamine functionalized-single-walled nanotube (f-SWNT)-assisted in vitro delivery of the oncogene suppressor p53 gene to breast cancer MCF-7 cells. Int. J. Nanomed. 2011, 2011, 1045–1055. [Google Scholar] [CrossRef]
- Mirzaie, V.; Ansari, M.; Nematollahi-Mahania, S.N.; Moballegh Nasery, M.; Karimi, B.; Eslaminejad, T.; Pourshojaei, Y. Nano-Graphene Oxide-Supported APTES-Spermine, as Gene Delivery System, for Transfection of pEGFP-p53 Into Breast Cancer Cell Lines. Drug Des. Dev. Ther. 2020, 14, 3087–3097. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; He, H.; Xu, X.; Wang, X.; Chen, Y.; Yin, L. Far-red light-mediated programmable anti-cancer gene delivery in cooperation with photodynamic therapy. Biomaterials 2018, 171, 72–82. [Google Scholar] [CrossRef]
- Zhang, J.; Wu, D.; Xing, Z.; Liang, S.; Han, H.; Shi, H.; Zhang, Y.; Yang, Y.; Li, Q. N-Isopropylacrylamide-modified polyethylenimine-mediated p53 gene delivery to prevent the proliferation of cancer cells. Colloids Surf. B Biointerfaces 2015, 129, 54–62. [Google Scholar] [CrossRef]
- Dong, M.M.; Chen, J.W.; Zhang, J.Y.; Liang, X.; Yang, J.B.; Li, D.; Li, Q.S. A chemoenzymatically synthesized cholesterol-g-poly(amine-co-ester)-mediated p53 gene delivery for achieving antitumor efficacy in prostate cancer. Int. J. Nanomed. 2019, 14, 1149–1161. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Zhu, Q.; Xu, X.; Shen, S.; Zhang, Y.; Mo, R. Sequentially Site-Specific Delivery of Apoptotic Protein and Tumor-Suppressor Gene for Combination Cancer Therapy. Small 2019, 15, 1902998. [Google Scholar] [CrossRef]
- Han, H.; Chen, W.; Yang, J.; Liang, X.; Wang, Y.; Li, Q.; Yang, Y.; Li, K. Inhibition of cell proliferation and migration through nucleobase-modified polyamidoamine-mediated p53 delivery. Int. J. Nanomed. 2018, 13, 1297–1311. [Google Scholar] [CrossRef]
- Huang, X.G.; Kong, N.; Zhang, X.C.; Cao, Y.H.; Langer, R.; Tao, W. The landscape of mRNA nanomedicine. Nat. Med. 2022, 28, 2273–2287. [Google Scholar] [CrossRef]
- Liu, H.L.; Li, X.X.; Yan, R.K.; Yang, J.; Lu, Q.; Wang, L.L. DNA tetrahedron nanoparticles service as a help carrier and adjvant of mRNA vaccine. J. Transl. Med. 2024, 22, 1024. [Google Scholar] [CrossRef]
- Raab, M.; Kostova, I.; Peña-Llopis, S.; Fietz, D.; Kressin, M.; Aberoumandi, S.M.; Ullrich, E.; Becker, S.; Sanhaji, M.; Strebhardt, K. Rescue of p53 functions by in vitro-transcribed mRNA impedes the growth of high-grade serous ovarian cancer. Cancer Commun. 2023, 44, 101–126. [Google Scholar] [CrossRef] [PubMed]
- Kong, N.; Tao, W.; Ling, X.; Wang, J.Q.; Xiao, Y.L.; Shi, S.J.; Ji, X.Y.; Shajii, A.; Gan, S.T.; Kim, N.Y.; et al. Synthetic mRNA nanoparticle-mediated restoration of p53 tumor suppressor sensitizes p53-deficient cancers to mTOR inhibition. Sci. Transl. Med. 2019, 11, eaaw1565. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.X.; Zhang, X.F.; Zhao, W.Y.; Zeng, C.X.; Li, W.Q.; Li, B.; Luo, X.; Li, J.N.; Jiang, J.; Deng, B.B.; et al. Chemotherapy drugs derived nanoparticles encapsulating mRNA encoding tumor suppressor proteins to treat triple-negative breast cancer. Nano Res. 2019, 12, 855–861. [Google Scholar] [CrossRef]
- Zhou, H.; Liao, Y.Q.; Han, X.F.; Chen, D.S.; Hong, X.C.; Zhou, K.; Jiang, X.Y.; Xiao, Y.L.; Shi, J.J. ROS-Responsive Nanoparticle Delivery of mRNA and Photosensitizer for Combinatorial Cancer Therapy. Nano Lett. 2023, 23, 3661–3668. [Google Scholar] [CrossRef]
- Tang, Z.; You, X.; Xiao, Y.; Chen, W.; Li, Y.; Huang, X.; Liu, H.; Xiao, F.; Liu, C.; Koo, S.; et al. Inhaled mRNA nanoparticles dual-targeting cancer cells and macrophages in the lung for effective transfection. Proc. Natl. Acad. Sci. USA 2023, 120, e2304966120. [Google Scholar] [CrossRef]
- Park, W.; Choi, J.; Hwang, J.; Kim, S.; Kim, Y.; Shim, M.K.; Park, W.; Yu, S.; Jung, S.; Yang, Y.; et al. Apolipoprotein Fusion Enables Spontaneous Functionalization of mRNA Lipid Nanoparticles with Antibody for Targeted Cancer Therapy. ACS Nano 2025, 19, 6412–6425. [Google Scholar] [CrossRef]
- Xiao, Y.L.; Chen, J.; Zhou, H.; Zeng, X.D.; Ruan, Z.P.; Pu, Z.Y.; Jiang, X.Y.; Matsui, A.; Zhu, L.L.; Amoozgar, Z.; et al. Combining p53 mRNA nanotherapy with immune checkpoint blockade reprograms the immune microenvironment for effective cancer therapy. Nat. Commun. 2022, 13, 758. [Google Scholar] [CrossRef]
- Tang, Q.; Liu, J.; Jiang, Y.; Zhang, M.N.; Mao, L.Q.; Wang, M. Cell-Selective Messenger RNA Delivery and CRISPR/Cas9 Genome Editing by Modulating the Interface of Phenylboronic Acid-Derived Lipid Nanoparticles and Cellular Surface Sialic Acid. ACS Appl. Mater. Interfaces 2019, 11, 46585–46590. [Google Scholar] [CrossRef]
- Liang, Y.; Zhang, J.; Wang, J.; Yang, Y.; Tan, X.; Li, S.; Guo, Z.; Zhang, Z.; Liu, J.; Shi, J.; et al. Restoring Tumor Cell Immunogenicity Through Ion-Assisted p53 mRNA Domestication for Enhanced In Situ Cancer Vaccination Effect. Adv. Sci. 2025, 12, 2500825. [Google Scholar] [CrossRef] [PubMed]
- Ceña, V.; Kundu, A.K.; Iyer, S.V.; Chandra, S.; Adhikari, A.S.; Iwakuma, T.; Mandal, T.K. Novel siRNA formulation to effectively knockdown mutant p53 in osteosarcoma. PLoS ONE 2017, 12, e0179168. [Google Scholar] [CrossRef]
- García-Garrido, E.; Cordani, M.; Somoza, Á. Modified Gold Nanoparticles to Overcome the Chemoresistance to Gemcitabine in Mutant p53 Cancer Cells. Pharmaceutics 2021, 13, 2067. [Google Scholar] [CrossRef] [PubMed]
- Yoon, A.R.; Lee, S.; Kim, J.H.; Park, Y.; Koo, T.; Yun, C.O. CRISPR-mediated ablation of TP53 and EGFR mutations enhances gefitinib sensitivity and anti-tumor efficacy in lung cancer. Mol. Ther. 2024, 32, 3618–3628. [Google Scholar] [CrossRef] [PubMed]
- Zhao, M.X.; Liu, Y.R.; Hsieh, R.S.; Wang, N.; Tai, W.Y.; Joo, K.I.; Wang, P.; Gu, Z.; Tang, Y. Clickable Protein Nanocapsules for Targeted Delivery of Recombinant p53 Protein. J. Am. Chem. Soc. 2014, 136, 15319–15325. [Google Scholar] [CrossRef]
- Jiao, Y.X.; Tang, Y.Z.; Li, Y.; Liu, C.; He, J.C.; Zhang, L.K.; Guan, Y.Q. Tumor cell-derived extracellular vesicles for breast cancer specific delivery of therapeutic P53. J. Control. Release 2022, 349, 606–616. [Google Scholar] [CrossRef]
- Yang, Z.; Sun, J.K.-L.; Lee, M.M.; Chan, M.K. Restoration of p53 activity via intracellular protein delivery sensitizes triple negative breast cancer to anti-PD-1 immunotherapy. J. Immunother. Cancer 2022, 10, e005068. [Google Scholar] [CrossRef]
- Park, I.S.; Kim, S.; Yim, Y.; Park, G.; Choi, J.; Won, C.; Min, D.H. Multifunctional synthetic nano-chaperone for peptide folding and intracellular delivery. Nat. Commun. 2022, 13, 4568. [Google Scholar] [CrossRef] [PubMed]
- Kitamatsu, M.; Yuasa, H.; Ohtsuki, T.; Michiue, H. Complementary leucine zippering system for effective intracellular delivery of proteins by cell-penetrating peptides. Bioorg. Med. Chem. 2021, 33, 116036. [Google Scholar] [CrossRef] [PubMed]
- Funk, J.S.; Klimovich, M.; Drangenstein, D.; Pielhoop, O.; Hunold, P.; Borowek, A.; Noeparast, M.; Pavlakis, E.; Neumann, M.; Balourdas, D.-I.; et al. Deep CRISPR mutagenesis characterizes the functional diversity of TP53 mutations. Nat. Genet. 2025, 57, 140–153. [Google Scholar] [CrossRef]
- Sobol, R.E.; Menander, K.B.; Chada, S.; Wiederhold, D.; Sellman, B.; Talbott, M.; Nemunaitis, J.J. Meta-Analysis of Adenoviral p53 Gene Therapy Clinical Trials in Recurrent Head and Neck Squamous Cell Carcinoma. Front. Oncol. 2021, 11, 645745. [Google Scholar] [CrossRef] [PubMed]

| Biomaterial Types | p53 Reactivating Agents | In Vitro/In Vivo | Mechanism | References |
|---|---|---|---|---|
| Cer-RUB nanomicelles | C16-ceramides | In vitro/in vivo | Restore the expression of wtp53 in cancer cells or transgenic mice harboring mutp53 | [113] |
| MtrapNPs | ATO | In vitro/in vivo | Rescue p53 mutations and inhibit MDM2 | [117] |
| CD19-targeted polymersome | ATSP-7041 | In vitro/in vivo | Reactivate p53 and inhibit the function of the BCL-2 protein family | [118] |
| ANVs | ZMC | / | Restore the structure and function of wtp53 and restore DNA damage repair | [119] |
| Thermosensitive gel–nano system | PRIMA-1 | In vitro/in vivo | Restore p53 activity and boost T-cell immunity | [120] |
| Biomaterial Types | Cell | Animal Models | Mechanism | References |
|---|---|---|---|---|
| PLGA hybrid lipid | Mouse osteosarcoma cell line carries p53R172H mutant alone | / | Knocked down mutp53 efficiency | [165] |
| Adenovirus | H1975(EGFR-T790M; TP53-R273H) | 6-week-old male athymic nude mice | Increased drug sensitivity; suppressed tumor growth | [167] |
| Gold nanoparticles (AuNPs) modified with bPEI | PANC-1 (mutp53-R273H); MDA-MB-231 (mutp53-R280K) | / | Enhanced the delivery of gapmers targeting mutp53 protein | [166] |
| Biomaterial Types | Cell | Animal Models | Mechanism | References |
|---|---|---|---|---|
| TPP/P53@EVs | SK-BR-3 cells, MCF-7 cells, and 4T1 cells | 6-week-old BALB/c female mice | Induce tumor cell death with no obvious toxicity or side effects in vivo | [169] |
| Clickable p53 nanocapsules | MDA-MB-231 cells, SK-OV-3 cells, and HFF cells | / | Deliver recombinant human protein p53 to the targeted tumor cells, leading to the reactivation of apoptosis in these cells | [168] |
| Pos3Aa-p53 protein crystals | 4T1 cells | BALB/c mice | Restored p53 activity by delivering the p53 protein to tumor cells; induced anti-PD-1 immunotherapy | [170] |
| SNCPs@st-p53 peptide | HepG2 cells | 5-week-old Balb/c male nude mice | Delivered p53 peptide to tumor tissues and inhibited tumor growth and induced p53-mediated cell apoptosis | [171] |
| p53-Lzk/LzE-CPP | U251MG cells, T98G cells, LNZ308 cells, and U87ΔEGFR cells | C57BL6 pregnant female mice | Delivered the p53 protein into tumor cells by CPP; inhibited cell-specific proliferation | [172] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, N.; Jing, Z.; Song, J.; Liang, Q.; Xu, Y.; Xu, Z.; Wen, L.; Wei, P. Discovery of Drugs Targeting Mutant p53 and Progress in Nano-Enabled Therapeutic Strategy for p53-Mutated Cancers. Biomolecules 2025, 15, 763. https://doi.org/10.3390/biom15060763
Zhang N, Jing Z, Song J, Liang Q, Xu Y, Xu Z, Wen L, Wei P. Discovery of Drugs Targeting Mutant p53 and Progress in Nano-Enabled Therapeutic Strategy for p53-Mutated Cancers. Biomolecules. 2025; 15(6):763. https://doi.org/10.3390/biom15060763
Chicago/Turabian StyleZhang, Na, Zhiyuan Jing, Jie Song, Qiyue Liang, Yuxue Xu, Zhaowei Xu, Longping Wen, and Pengfei Wei. 2025. "Discovery of Drugs Targeting Mutant p53 and Progress in Nano-Enabled Therapeutic Strategy for p53-Mutated Cancers" Biomolecules 15, no. 6: 763. https://doi.org/10.3390/biom15060763
APA StyleZhang, N., Jing, Z., Song, J., Liang, Q., Xu, Y., Xu, Z., Wen, L., & Wei, P. (2025). Discovery of Drugs Targeting Mutant p53 and Progress in Nano-Enabled Therapeutic Strategy for p53-Mutated Cancers. Biomolecules, 15(6), 763. https://doi.org/10.3390/biom15060763