
The Role of Cancer Organoids in Ferroptosis, Pyroptosis, and Necroptosis: Functions and Clinical Implications


Abstract
1. Introduction
2. Cancer Organoids and Non-Apoptotic RCDs
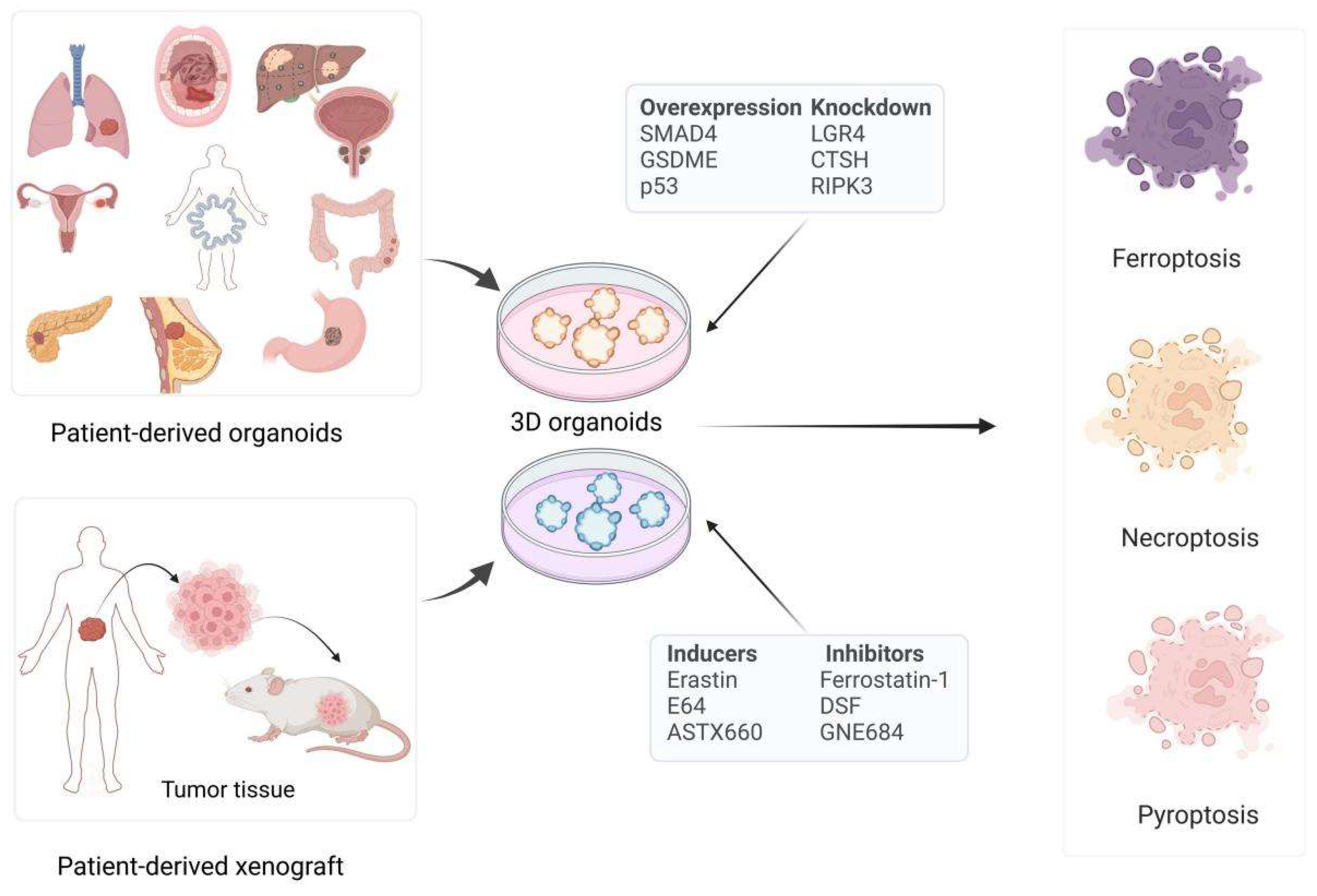
2.1. Cancer Organoids
2.2. Non-Apoptotic RCDs
2.2.1. Ferroptosis
2.2.2. Pyroptosis
2.2.3. Necroptosis
3. Various Cancer Organoid Models in Non-Apoptotic RCD
3.1. Ferroptosis in Cancer Organoid Models Research
3.1.1. Ferroptosis in Breast Cancer Models Research
3.1.2. Ferroptosis in Pancreatic Cancer Models Research
3.1.3. Ferroptosis in Liver Cancer Models Research
3.1.4. Ferroptosis in Gastric Cancer Models Research
3.1.5. Ferroptosis in Colorectal Cancer Models Research
3.1.6. Ferroptosis in Ovarian Cancer Models Research
3.1.7. Ferroptosis in Bladder Cancer Models Research
3.1.8. Ferroptosis in Other Cancer Models Research
3.2. Pyroptosis in Cancer Organoid Models Research
3.3. Necroptosis in Cancer Organoid Models Research
4. Therapeutic Implications of Non-Apoptotic RCD in Cancer Organoids
4.1. Functional Drug Screening
4.2. Enhancing Chemotherapy Efficacy
4.3. Exploring Combination Treatment Strategies
4.4. Facilitating Personalized Medicine
5. Discussion
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
References
- Yan, H.H.N.; Chan, A.S.; Lai, F.P.-L.; Leung, S.Y. Organoid cultures for cancer modeling. Cell Stem Cell 2023, 30, 917–937. [Google Scholar] [CrossRef] [PubMed]
- Drost, J.; Clevers, H. Organoids in cancer research. Nat. Rev. Cancer 2018, 18, 407–418. [Google Scholar] [CrossRef] [PubMed]
- Neal, J.T.; Li, X.; Zhu, J.; Giangarra, V.; Grzeskowiak, C.L.; Ju, J.; Liu, I.H.; Chiou, S.H.; Salahudeen, A.A.; Smith, A.R.; et al. Organoid Modeling of the Tumor Immune Microenvironment. Cell 2018, 175, 1972–1988.e16. [Google Scholar] [CrossRef]
- Strasser, A.; Vaux, D.L. Cell Death in the Origin and Treatment of Cancer. Mol. Cell 2020, 78, 1045–1054. [Google Scholar] [CrossRef]
- Galluzzi, L.; Vitale, I.; Aaronson, S.A.; Abrams, J.M.; Adam, D.; Agostinis, P.; Alnemri, E.S.; Altucci, L.; Amelio, I.; Andrews, D.W.; et al. Molecular mechanisms of cell death: Recommendations of the Nomenclature Committee on Cell Death 2018. Cell Death Differ. 2018, 25, 486–541. [Google Scholar] [CrossRef] [PubMed]
- Koren, E.; Fuchs, Y. Modes of Regulated Cell Death in Cancer. Cancer Discov. 2021, 11, 245–265. [Google Scholar] [CrossRef]
- Tang, D.; Kang, R.; Berghe, T.V.; Vandenabeele, P.; Kroemer, G. The molecular machinery of regulated cell death. Cell Res. 2019, 29, 347–364. [Google Scholar] [CrossRef]
- Tong, X.; Tang, R.; Xiao, M.; Xu, J.; Wang, W.; Zhang, B.; Liu, J.; Yu, X.; Shi, S. Targeting cell death pathways for cancer therapy: Recent developments in necroptosis, pyroptosis, ferroptosis, and cuproptosis research. J. Hematol. Oncol. 2022, 15, 174. [Google Scholar] [CrossRef]
- Tang, R.; Xu, J.; Zhang, B.; Liu, J.; Liang, C.; Hua, J.; Meng, Q.; Yu, X.; Shi, S. Ferroptosis, necroptosis, and pyroptosis in anticancer immunity. J. Hematol. Oncol. 2020, 13, 110. [Google Scholar] [CrossRef]
- Kim, N.; Andreas, S.; Nobuhiko, K.; Vishva, M. Cell death. Cell 2024, 187, 235–256. [Google Scholar] [CrossRef]
- Zheng, Y.; Huang, Y.; Xu, Y.; Sang, L.; Liu, X.; Li, Y. Ferroptosis, pyroptosis and necroptosis in acute respiratory distress syndrome. Cell Death Discov. 2023, 9, 91. [Google Scholar] [CrossRef] [PubMed]
- Dixon, S.J.; Lemberg, K.M.; Lamprecht, M.R.; Skouta, R.; Zaitsev, E.M.; Gleason, C.E.; Patel, D.N.; Bauer, A.J.; Cantley, A.M.; Yang, W.S.; et al. Ferroptosis: An iron-dependent form of nonapoptotic cell death. Cell 2012, 149, 1060–1072. [Google Scholar] [CrossRef] [PubMed]
- Roberts, J.Z.; Crawford, N.; Longley, D.B. The role of Ubiquitination in Apoptosis and Necroptosis. Cell Death Differ. 2022, 29, 272–284. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Liu, M.; Xie, N.; Wang, Z.; Yu, C.; Li, J.; Zhou, X. Cancer research revolutionized: Unveiling the power of organoids and their therapeutic potential in oncology. hLife 2024, in press. [Google Scholar] [CrossRef]
- LeSavage, B.L.; Suhar, R.A.; Broguiere, N.; Lutolf, M.P.; Heilshorn, S.C. Next-generation cancer organoids. Nat. Mater. 2022, 21, 143–159. [Google Scholar] [CrossRef]
- Zheng, H.; Liu, J.; Cheng, Q.; Zhang, Q.; Zhang, Y.; Jiang, L.; Huang, Y.; Li, W.; Zhao, Y.; Chen, G.; et al. Targeted activation of ferroptosis in colorectal cancer via LGR4 targeting overcomes acquired drug resistance. Nat. Cancer 2024, 5, 572–589. [Google Scholar] [CrossRef]
- Hsu, S.K.; Li, C.Y.; Lin, I.L.; Syue, W.J.; Chen, Y.F.; Cheng, K.C.; Teng, Y.N.; Lin, Y.H.; Yen, C.H.; Chiu, C.C. Inflammation-related pyroptosis, a novel programmed cell death pathway, and its crosstalk with immune therapy in cancer treatment. Theranostics 2021, 11, 8813–8835. [Google Scholar] [CrossRef]
- Shi, S.; Verstegen, M.M.A.; Roest, H.P.; Ardisasmita, A.I.; Cao, W.; Roos, F.J.M.; de Ruiter, P.E.; Niemeijer, M.; Pan, Q.; JNM, I.J.; et al. Recapitulating Cholangiopathy-Associated Necroptotic Cell Death In Vitro Using Human Cholangiocyte Organoids. Cell. Mol. Gastroenterol. Hepatol. 2022, 13, 541–564. [Google Scholar] [CrossRef]
- Semertzidou, A.; Brosens, J.J.; McNeish, I.; Kyrgiou, M. Organoid models in gynaecological oncology research. Cancer Treat. Rev. 2020, 90, 102103. [Google Scholar] [CrossRef]
- Antoszczak, M.; Müller, S.; Cañeque, T.; Colombeau, L.; Dusetti, N.; Santofimia-Castaño, P.; Gaillet, C.; Puisieux, A.; Iovanna, J.L.; Rodriguez, R. Iron-Sensitive Prodrugs That Trigger Active Ferroptosis in Drug-Tolerant Pancreatic Cancer Cells. J. Am. Chem. Soc. 2022, 144, 11536–11545. [Google Scholar] [CrossRef]
- Polak, R.; Zhang, E.T.; Kuo, C.J. Cancer organoids 2.0: Modelling the complexity of the tumour immune microenvironment. Nat. reviews. Cancer 2024, 24, 523–539. [Google Scholar] [CrossRef] [PubMed]
- Clevers, H. Modeling Development and Disease with Organoids. Cell 2016, 165, 1586–1597. [Google Scholar] [CrossRef]
- Sato, T.; Vries, R.G.; Snippert, H.J.; van de Wetering, M.; Barker, N.; Stange, D.E.; van Es, J.H.; Abo, A.; Kujala, P.; Peters, P.J.; et al. Single Lgr5 stem cells build crypt-villus structures in vitro without a mesenchymal niche. Nature 2009, 459, 262–265. [Google Scholar] [CrossRef] [PubMed]
- Qiang, Y.; Yao, N.; Zuo, F.; Qiu, S.; Cao, X.; Zheng, W. Tumor Organoid Model and Its Pharmacological Applications in Tumorigenesis Prevention. Curr. Mol. Pharmacol. 2023, 16, 435–447. [Google Scholar] [CrossRef]
- Heydari, Z.; Moeinvaziri, F.; Agarwal, T.; Pooyan, P.; Shpichka, A.; Maiti, T.K.; Timashev, P.; Baharvand, H.; Vosough, M. Organoids: A novel modality in disease modeling. Bio-Des. Manuf. 2021, 4, 689–716. [Google Scholar] [CrossRef] [PubMed]
- Lv, J.; Du, X.; Wang, M.; Su, J.; Wei, Y.; Xu, C. Construction of tumor organoids and their application to cancer research and therapy. Theranostics 2024, 14, 1101–1125. [Google Scholar] [CrossRef]
- Zhenzhen, Z.; Yuan, P.; Jingyuan, J.; Jianyu, H.; Tiankun, L.; Liliang, O.; Wen, Z.; Xue-Li, Z.; Zhi-Gang, Z.; Kaitai, Z.; et al. Harnessing 3D in vitro systems to model immune responses to solid tumours: A step towards improving and creating personalized immunotherapies. Nat. Rev. Immunol. 2023, 24, 18–32. [Google Scholar] [CrossRef]
- Xu, H.; Jiao, D.; Liu, A.; Wu, K. Tumor organoids: Applications in cancer modeling and potentials in precision medicine. J. Hematol. Oncol. 2022, 15, 58. [Google Scholar] [CrossRef]
- Sachs, N.; de Ligt, J.; Kopper, O.; Gogola, E.; Bounova, G.; Weeber, F.; Balgobind, A.V.; Wind, K.; Gracanin, A.; Begthel, H.; et al. A Living Biobank of Breast Cancer Organoids Captures Disease Heterogeneity. Cell 2018, 172, 373–386.e10. [Google Scholar] [CrossRef]
- van de Wetering, M.; Francies, H.E.; Francis, J.M.; Bounova, G.; Iorio, F.; Pronk, A.; van Houdt, W.; van Gorp, J.; Taylor-Weiner, A.; Kester, L.; et al. Prospective derivation of a living organoid biobank of colorectal cancer patients. Cell 2015, 161, 933–945. [Google Scholar] [CrossRef]
- Melissa, C.S.; Dustin, A.D.; Jeremy, D.K. Technologies to Assess Drug Response and Heterogeneity in Patient-Derived Cancer Organoids. Annu. Rev. Biomed. Eng. 2022, 24, 157–177. [Google Scholar] [CrossRef]
- Lei, G.; Zhuang, L.; Gan, B. Targeting ferroptosis as a vulnerability in cancer. Nat. Rev. Cancer 2022, 22, 381–396. [Google Scholar] [CrossRef] [PubMed]
- Jiang, X.; Stockwell, B.R.; Conrad, M. Ferroptosis: Mechanisms, biology and role in disease. Nat. Rev. Mol. Cell Biol. 2021, 22, 266–282. [Google Scholar] [CrossRef] [PubMed]
- Brown, A.R.; Hirschhorn, T.; Stockwell, B.R. Ferroptosis-disease perils and therapeutic promise. Science 2024, 386, 848–849. [Google Scholar] [CrossRef] [PubMed]
- Koppula, P.; Zhuang, L.; Gan, B. Cystine transporter SLC7A11/xCT in cancer: Ferroptosis, nutrient dependency, and cancer therapy. Protein Cell 2021, 12, 599–620. [Google Scholar] [CrossRef]
- Chen, X.; Li, J.; Kang, R.; Klionsky, D.J.; Tang, D. Ferroptosis: Machinery and regulation. Autophagy 2021, 17, 2054–2081. [Google Scholar] [CrossRef]
- Chen, X.; Zeh, H.J.; Kang, R.; Kroemer, G.; Tang, D. Cell death in pancreatic cancer: From pathogenesis to therapy. Nat. Rev. Gastroenterol. Hepatol. 2021, 18, 804–823. [Google Scholar] [CrossRef]
- Mao, C.; Wang, M.; Zhuang, L.; Gan, B. Metabolic cell death in cancer: Ferroptosis, cuproptosis, disulfidptosis, and beyond. Protein Cell 2024, 15, 642–660. [Google Scholar] [CrossRef]
- Zheng, Y.; Li, L.; Chen, H.; Zheng, Y.; Tan, X.; Zhang, G.; Jiang, R.; Yu, H.; Lin, S.; Wei, Y.; et al. Luteolin exhibits synergistic therapeutic efficacy with erastin to induce ferroptosis in colon cancer cells through the HIC1-mediated inhibition of GPX4 expression. Free Radic. Biol. Med. 2023, 208, 530–544. [Google Scholar] [CrossRef]
- Müller, F.; Lim, J.K.M.; Bebber, C.M.; Seidel, E.; Tishina, S.; Dahlhaus, A.; Stroh, J.; Beck, J.; Yapici, F.I.; Nakayama, K.; et al. Elevated FSP1 protects KRAS-mutated cells from ferroptosis during tumor initiation. Cell Death Differ. 2023, 30, 442–456. [Google Scholar] [CrossRef]
- Yu, P.; Zhang, X.; Liu, N.; Tang, L.; Peng, C.; Chen, X. Pyroptosis: Mechanisms and diseases. Signal Transduct. Target. Ther. 2021, 6, 128. [Google Scholar] [CrossRef] [PubMed]
- Ai, Y.; Meng, Y.; Yan, B.; Zhou, Q.; Wang, X. The biochemical pathways of apoptotic, necroptotic, pyroptotic, and ferroptotic cell death. Mol. Cell 2024, 84, 170–179. [Google Scholar] [CrossRef]
- Hadian, K.; Stockwell, B.R. The therapeutic potential of targeting regulated non-apoptotic cell death. Nat. Rev. Drug Discov. 2023, 22, 723–742. [Google Scholar] [CrossRef] [PubMed]
- Su, L.; Chen, Y.; Huang, C.; Wu, S.; Wang, X.; Zhao, X.; Xu, Q.; Sun, R.; Kong, X.; Jiang, X.; et al. Targeting Src reactivates pyroptosis to reverse chemoresistance in lung and pancreatic cancer models. Sci. Transl. Med. 2023, 15, eabl7895. [Google Scholar] [CrossRef]
- Chen, T.; Zeineldin, M.; Johnson, B.A.; Dong, Y.; Narkar, A.; Li, T.; Zhu, J.; Li, R.; Larman, T.C. Colonic epithelial adaptation to EGFR-independent growth induces chromosomal instability and is accelerated by prior injury. Neoplasia 2021, 23, 488–501. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Wang, L.; Xu, J. The role of pyroptosis in modulating the tumor immune microenvironment. Biomark. Res. 2022, 10, 45. [Google Scholar] [CrossRef]
- Niu, X.; Chen, L.; Li, Y.; Hu, Z.; He, F. Ferroptosis, necroptosis, and pyroptosis in the tumor microenvironment: Perspectives for immunotherapy of SCLC. Semin. Cancer Biol. 2022, 86 Pt 3, 273–285. [Google Scholar] [CrossRef]
- Degterev, A.; Hitomi, J.; Germscheid, M.; Ch’en, I.L.; Korkina, O.; Teng, X.; Abbott, D.; Cuny, G.D.; Yuan, C.; Wagner, G.; et al. Identification of RIP1 kinase as a specific cellular target of necrostatins. Nat. Chem. Biol. 2008, 4, 313–321. [Google Scholar] [CrossRef]
- Di Grazia, A.; Marafini, I.; Pedini, G.; Di Fusco, D.; Laudisi, F.; Dinallo, V.; Rosina, E.; Stolfi, C.; Franzè, E.; Sileri, P.; et al. The Fragile X Mental Retardation Protein Regulates RIPK1 and Colorectal Cancer Resistance to Necroptosis. Cell. Mol. Gastroenterol. Hepatol. 2021, 11, 639–658. [Google Scholar] [CrossRef]
- Prado-Acosta, M.; Jeong, S.; Utrero-Rico, A.; Goncharov, T.; Webster, J.D.; Holler, E.; Morales, G.; Dellepiane, S.; Levine, J.E.; Rothenberg, M.E.; et al. Inhibition of RIP1 improves immune reconstitution and reduces GVHD mortality while preserving graft-versus-leukemia effects. Sci. Transl. Med. 2023, 15, eadf8366. [Google Scholar] [CrossRef]
- Tan, X.; Kong, D.; Tao, Z.; Cheng, F.; Zhang, B.; Wang, Z.; Mei, Q.; Chen, C.; Wu, K. Simultaneous inhibition of FAK and ROS1 synergistically repressed triple-negative breast cancer by upregulating p53 signalling. Biomark. Res. 2024, 12, 13. [Google Scholar] [CrossRef] [PubMed]
- Zou, Y.; Zheng, S.; Xie, X.; Ye, F.; Hu, X.; Tian, Z.; Yan, S.M.; Yang, L.; Kong, Y.; Tang, Y.; et al. N6-methyladenosine regulated FGFR4 attenuates ferroptotic cell death in recalcitrant HER2-positive breast cancer. Nat. Commun. 2022, 13, 2672. [Google Scholar] [CrossRef] [PubMed]
- Ye, L.; Zhong, F.; Sun, S.; Ou, X.; Yuan, J.; Zhu, J.; Zeng, Z. Tamoxifen induces ferroptosis in MCF-7 organoid. J. Cancer Res. Ther. 2023, 19, 1627–1635. [Google Scholar] [CrossRef]
- Wang, X.; Li, Y.; Li, Z.; Lin, S.; Wang, H.; Sun, J.; Lan, C.; Wu, L.; Sun, D.; Huang, C.; et al. Mitochondrial Calcium Uniporter Drives Metastasis and Confers a Targetable Cystine Dependency in Pancreatic Cancer. Cancer Res. 2022, 82, 2254–2268. [Google Scholar] [CrossRef]
- Li, G.; Liao, C.; Chen, J.; Wang, Z.; Zhu, S.; Lai, J.; Li, Q.; Chen, Y.; Wu, D.; Li, J.; et al. Targeting the MCP-GPX4/HMGB1 Axis for Effectively Triggering Immunogenic Ferroptosis in Pancreatic Ductal Adenocarcinoma. Adv. Sci. 2024, 11, e2308208. [Google Scholar] [CrossRef]
- Chen, H.D.; Ye, Z.; Hu, H.F.; Fan, G.X.; Hu, Y.H.; Li, Z.; Li, B.R.; Ji, S.R.; Zhou, C.J.; Xu, X.W.; et al. SMAD4 endows TGF-β1-induced highly invasive tumor cells with ferroptosis vulnerability in pancreatic cancer. Acta Pharmacol. Sin. 2024, 45, 844–856. [Google Scholar] [CrossRef] [PubMed]
- Zheng, C.; Zhang, B.; Li, Y.; Liu, K.; Wei, W.; Liang, S.; Guo, H.; Ma, K.; Liu, Y.; Wang, J.; et al. Donafenib and GSK-J4 Synergistically Induce Ferroptosis in Liver Cancer by Upregulating HMOX1 Expression. Adv. Sci. 2023, 10, e2206798. [Google Scholar] [CrossRef]
- Zhang, Q.; Xiong, L.; Wei, T.; Liu, Q.; Yan, L.; Chen, J.; Dai, L.; Shi, L.; Zhang, W.; Yang, J.; et al. Hypoxia-responsive PPARGC1A/BAMBI/ACSL5 axis promotes progression and resistance to lenvatinib in hepatocellular carcinoma. Oncogene 2023, 42, 1509–1523. [Google Scholar] [CrossRef]
- Ding, Z.; Pan, Y.; Shang, T.; Jiang, T.; Lin, Y.; Yang, C.; Pang, S.; Cui, X.; Wang, Y.; Feng, X.F.; et al. URI alleviates tyrosine kinase inhibitors-induced ferroptosis by reprogramming lipid metabolism in p53 wild-type liver cancers. Nat. Commun. 2023, 14, 6269. [Google Scholar] [CrossRef]
- Conche, C.; Finkelmeier, F.; Pešić, M.; Nicolas, A.M.; Böttger, T.W.; Kennel, K.B.; Denk, D.; Ceteci, F.; Mohs, K.; Engel, E.; et al. Combining ferroptosis induction with MDSC blockade renders primary tumours and metastases in liver sensitive to immune checkpoint blockade. Gut 2023, 72, 1774–1782. [Google Scholar] [CrossRef]
- Yao, L.; Hou, J.; Wu, X.; Lu, Y.; Jin, Z.; Yu, Z.; Yu, B.; Li, J.; Yang, Z.; Li, C.; et al. Cancer-associated fibroblasts impair the cytotoxic function of NK cells in gastric cancer by inducing ferroptosis via iron regulation. Redox Biol. 2023, 67, 102923. [Google Scholar] [CrossRef]
- Ouyang, S.; Li, H.; Lou, L.; Huang, Q.; Zhang, Z.; Mo, J.; Li, M.; Lu, J.; Zhu, K.; Chu, Y.; et al. Inhibition of STAT3-ferroptosis negative regulatory axis suppresses tumor growth and alleviates chemoresistance in gastric cancer. Redox Biol. 2022, 52, 102317. [Google Scholar] [CrossRef]
- Callahan, R.C.; Bhagavatula, G.; Curry, J.; Staley, A.W.; Schaefer, R.E.M.; Minhajuddin, F.; Zhou, L.; Neuhart, R.; Atif, S.M.; Orlicky, D.J.; et al. Epithelial heme oxygenase-1 enhances colonic tumorigenesis by inhibiting ferroptosis. bioRxiv 2024. [Google Scholar] [CrossRef]
- Lv, X.; He, F.L.; Dai, Y.; Dai, X. IFNγ synergies with cold atmospheric plasma in triggering colorectal cancer cell ferroptosis via the IFNγ/IFNR2/APC/TCF4/GPX4 axis. Aging 2023, 15, 8692–8711. [Google Scholar] [CrossRef] [PubMed]
- Miyazaki, K.; Xu, C.; Shimada, M.; Goel, A. Curcumin and Andrographis Exhibit Anti-Tumor Effects in Colorectal Cancer via Activation of Ferroptosis and Dual Suppression of Glutathione Peroxidase-4 and Ferroptosis Suppressor Protein-1. Pharmaceuticals 2023, 16, 383. [Google Scholar] [CrossRef] [PubMed]
- Shen, L.; Zhang, J.; Zheng, Z.; Yang, F.; Liu, S.; Wu, Y.; Chen, Y.; Xu, T.; Mao, S.; Yan, Y.; et al. PHGDH Inhibits Ferroptosis and Promotes Malignant Progression by Upregulating SLC7A11 in Bladder Cancer. Int. J. Biol. Sci. 2022, 18, 5459–5474. [Google Scholar] [CrossRef] [PubMed]
- Sharma, P.; Shimura, T.; Banwait, J.K.; Goel, A. Andrographis-mediated chemosensitization through activation of ferroptosis and suppression of β-catenin/Wnt-signaling pathways in colorectal cancer. Carcinogenesis 2020, 41, 1385–1394. [Google Scholar] [CrossRef]
- Lorenzato, A.; Magrì, A.; Matafora, V.; Audrito, V.; Arcella, P.; Lazzari, L.; Montone, M.; Lamba, S.; Deaglio, S.; Siena, S.; et al. Vitamin C Restricts the Emergence of Acquired Resistance to EGFR-Targeted Therapies in Colorectal Cancer. Cancers 2020, 12, 685. [Google Scholar] [CrossRef]
- Asif, K.; Adeel, M.; Rahman, M.M.; Caligiuri, I.; Perin, T.; Cemazar, M.; Canzonieri, V.; Rizzolio, F. Iron nitroprusside as a chemodynamic agent and inducer of ferroptosis for ovarian cancer therapy. J. Mater. Chem. B 2023, 11, 3124–3135. [Google Scholar] [CrossRef]
- Xuan, Y.; Wang, H.; Yung, M.M.; Chen, F.; Chan, W.S.; Chan, Y.S.; Tsui, S.K.; Ngan, H.Y.; Chan, K.K.; Chan, D.W. SCD1/FADS2 fatty acid desaturases equipoise lipid metabolic activity and redox-driven ferroptosis in ascites-derived ovarian cancer cells. Theranostics 2022, 12, 3534–3552. [Google Scholar] [CrossRef]
- Hodara, E.; Mades, A.; Swartz, L.; Iqbal, M.; Xu, T.; Bsteh, D.; Farnham, P.J.; Rhie, S.K.; Goldkorn, A. m(6)A epitranscriptome analysis reveals differentially methylated transcripts that drive early chemoresistance in bladder cancer. NAR Cancer 2023, 5, zcad054. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.P.; Wang, Y.X.; Zhou, H.; Liu, Z.T.; Zhang, Z.J.; Xiong, L.; Zou, H.; Wen, Y. Surufatinib combined with photodynamic therapy induces ferroptosis to inhibit cholangiocarcinoma in vitro and in tumor models. Front. Pharmacol. 2024, 15, 1288255. [Google Scholar] [CrossRef] [PubMed]
- Qu, S.; Qi, S.; Zhang, H.; Li, Z.; Wang, K.; Zhu, T.; Ye, R.; Zhang, W.; Huang, G.; Yi, G.Z. Albumin-bound paclitaxel augment temozolomide treatment sensitivity of glioblastoma cells by disrupting DNA damage repair and promoting ferroptosis. J. Exp. Clin. Cancer Res. CR 2023, 42, 285. [Google Scholar] [CrossRef]
- Ni, Y.; Liu, J.; Zeng, L.; Yang, Y.; Liu, L.; Yao, M.; Chai, L.; Zhang, L.; Li, Y.; Zhang, L.; et al. Natural product manoalide promotes EGFR-TKI sensitivity of lung cancer cells by KRAS-ERK pathway and mitochondrial Ca(2+) overload-induced ferroptosis. Front. Pharmacol. 2022, 13, 1109822. [Google Scholar] [CrossRef]
- Sun, S.; Qi, G.; Chen, H.; He, D.; Ma, D.; Bie, Y.; Xu, L.; Feng, B.; Pang, Q.; Guo, H.; et al. Ferroptosis sensitization in glioma: Exploring the regulatory mechanism of SOAT1 and its therapeutic implications. Cell Death Dis. 2023, 14, 754. [Google Scholar] [CrossRef]
- Wang, Z.; Zong, H.; Liu, W.; Lin, W.; Sun, A.; Ding, Z.; Chen, X.; Wan, X.; Liu, Y.; Hu, Z.; et al. Augmented ERO1α upon mTORC1 activation induces ferroptosis resistance and tumor progression via upregulation of SLC7A11. J. Exp. Clin. Cancer Res. CR 2024, 43, 112. [Google Scholar] [CrossRef]
- Hsieh, M.S.; Ling, H.H.; Setiawan, S.A.; Hardianti, M.S.; Fong, I.H.; Yeh, C.T.; Chen, J.H. Therapeutic targeting of thioredoxin reductase 1 causes ferroptosis while potentiating anti-PD-1 efficacy in head and neck cancer. Chem.-Biol. Interact. 2024, 395, 111004. [Google Scholar] [CrossRef]
- Zhang, Y.; Xu, Z.; Feng, W.; Gao, H.; Xu, Z.; Miao, Y.; Li, W.; Chen, F.; Lv, Z.; Huo, J.; et al. Small molecule inhibitors from organoid-based drug screen induce concurrent apoptosis and gasdermin E-dependent pyroptosis in colorectal cancer. Clin. Transl. Med. 2022, 12, e812. [Google Scholar] [CrossRef]
- Wang, M.; Chen, X.; Tan, P.; Wang, Y.; Pan, X.; Lin, T.; Jiang, Y.; Wang, B.; Xu, H.; Wang, Y.; et al. Acquired semi-squamatization during chemotherapy suggests differentiation as a therapeutic strategy for bladder cancer. Cancer Cell 2022, 40, 1044–1059.e8. [Google Scholar] [CrossRef]
- Su, P.; Mao, X.; Ma, J.; Huang, L.; Yu, L.; Tang, S.; Zhuang, M.; Lu, Z.; Osafo, K.S.; Ren, Y.; et al. ERRα promotes glycolytic metabolism and targets the NLRP3/caspase-1/GSDMD pathway to regulate pyroptosis in endometrial cancer. J. Exp. Clin. Cancer Res. CR 2023, 42, 274. [Google Scholar] [CrossRef]
- Barber, G.; Anand, A.; Katarzyna, O.; Phelan, J.J.; Heeran, A.B.; Flis, E.; Clarke, N.E.; Watson, J.A.; Strangmann, J.; Flood, B.; et al. Characterizing caspase-1 involvement during esophageal disease progression. Cancer Immunol. Immunother. 2020, 69, 2635–2649. [Google Scholar] [CrossRef] [PubMed]
- Lin, W.; Zhang, Y.; Yang, Y.; Lin, B.; Zhu, M.; Xu, J.; Chen, Y.; Wu, W.; Chen, B.; Chen, X.; et al. Anti-PD-1/Her2 Bispecific Antibody IBI315 Enhances the Treatment Effect of Her2-Positive Gastric Cancer through Gasdermin B-Cleavage Induced Pyroptosis. Adv. Sci. 2023, 10, e2303908. [Google Scholar] [CrossRef]
- Zou, C.; Shen, J.; Xu, F.; Ye, Y.; Wu, Y.; Xu, S. Immunoreactive Microenvironment Modulator GBP5 Suppresses Ovarian Cancer Progression by Inducing Canonical Pyroptosis. J. Cancer 2024, 15, 3510–3530. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.N.; Liang, Y.L.; Tsai, H.F.; Wu, P.Y.; Huang, L.Y.; Lin, Y.H.; Kang, C.Y.; Yao, C.L.; Shen, M.R.; Hsu, K.F. Adipocyte pyroptosis occurs in omental tumor microenvironment and is associated with chemoresistance of ovarian cancer. J. Biomed. Sci. 2024, 31, 62. [Google Scholar] [CrossRef]
- Yu, C.; Kang, R.; Tang, D. Organoids Models of Pancreatic Duct Adenocarcinoma. Methods Mol. Biol. 2023, 2712, 45–60. [Google Scholar] [CrossRef]
- Crawford, N.; Stott, K.J.; Sessler, T.; McCann, C.; McDaid, W.; Lees, A.; Latimer, C.; Fox, J.P.; Munck, J.M.; Smyth, T.; et al. Clinical Positioning of the IAP Antagonist Tolinapant (ASTX660) in Colorectal Cancer. Mol. Cancer Ther. 2021, 20, 1627–1639. [Google Scholar] [CrossRef]
- Watanabe, H.; Ishibashi, K.; Mano, H.; Kitamoto, S.; Sato, N.; Hoshiba, K.; Kato, M.; Matsuzawa, F.; Takeuchi, Y.; Shirai, T.; et al. Mutant p53-Expressing Cells Undergo Necroptosis via Cell Competition with the Neighboring Normal Epithelial Cells. Cell Rep. 2018, 23, 3721–3729. [Google Scholar] [CrossRef] [PubMed]
- Bockerstett, K.A.; Osaki, L.H.; Petersen, C.P.; Cai, C.W.; Wong, C.F.; Nguyen, T.M.; Ford, E.L.; Hoft, D.F.; Mills, J.C.; Goldenring, J.R.; et al. Interleukin-17A Promotes Parietal Cell Atrophy by Inducing Apoptosis. Cell. Mol. Gastroenterol. Hepatol. 2018, 5, 678–690.e1. [Google Scholar] [CrossRef]
- Bian, Y.; Shan, G.; Bi, G.; Liang, J.; Hu, Z.; Sui, Q.; Shi, H.; Zheng, Z.; Yao, G.; Wang, Q.; et al. Targeting ALDH1A1 to enhance the efficacy of KRAS-targeted therapy through ferroptosis. Redox Biol. 2024, 77, 103361. [Google Scholar] [CrossRef]
- Yao, Y.; Xu, X.; Yang, L.; Zhu, J.; Wan, J.; Shen, L.; Xia, F.; Fu, G.; Deng, Y.; Pan, M.; et al. Patient-Derived Organoids Predict Chemoradiation Responses of Locally Advanced Rectal Cancer. Cell Stem Cell 2020, 26, 17–26.e6. [Google Scholar] [CrossRef]
- Li, Y.; Guo, M.; Qiu, Y.; Li, M.; Wu, Y.; Shen, M.; Wang, Y.; Zhang, F.; Shao, J.; Xu, X.; et al. Autophagy activation is required for N6-methyladenosine modification to regulate ferroptosis in hepatocellular carcinoma. Redox Biol. 2024, 69, 102971. [Google Scholar] [CrossRef]
- Ballard, D.H.; Boyer, C.J.; Alexander, J.S. Organoids—Preclinical Models of Human Disease. N. Engl. J. Med. 2019, 380, 1981–1982. [Google Scholar] [CrossRef] [PubMed]
- Guo, L.; Li, C.; Gong, W. Toward reproducible tumor organoid culture: Focusing on primary liver cancer. Front. Immunol. 2024, 15, 1290504. [Google Scholar] [CrossRef]
- Gjorevski, N.; Nikolaev, M.; Brown, T.E.; Mitrofanova, O.; Brandenberg, N.; DelRio, F.W.; Yavitt, F.M.; Liberali, P.; Anseth, K.S.; Lutolf, M.P. Tissue geometry drives deterministic organoid patterning. Science 2022, 375, eaaw9021. [Google Scholar] [CrossRef]
- Brassard, J.A.; Nikolaev, M.; Hübscher, T.; Hofer, M.; Lutolf, M.P. Recapitulating macro-scale tissue self-organization through organoid bioprinting. Nat. Mater. 2021, 20, 22–29. [Google Scholar] [CrossRef] [PubMed]
- Paramanantham, A.; Asfiya, R.; Manjunath, Y.; Xu, L.; McCully, G.; Das, S.; Yang, H.; Kaifi, J.T.; Srivastava, A. Induction of Ferroptosis by an Amalgam of Extracellular Vesicles and Iron Oxide Nanoparticles Overcomes Cisplatin Resistance in Lung Cancer. bioRxiv 2024. [Google Scholar] [CrossRef]
- Sandbhor, P.; Palkar, P.; Bhat, S.; John, G.; Goda, J.S. Nanomedicine as a multimodal therapeutic paradigm against cancer: On the way forward in advancing precision therapy. Nanoscale 2024, 16, 6330–6364. [Google Scholar] [CrossRef]
- Wang, X.; Wu, T. An update on the biological effects of quantum dots: From environmental fate to risk assessment based on multiple biological models. Sci. Total Environ. 2023, 879, 163166. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
| Cancer Types | Organoid Models | Functions | Reference |
|---|---|---|---|
| Ferroptosis | |||
| Breast cancer | Human TNBC organoid models | Simultaneous inhibition of FAK and ROS1 synergistically repress tumor growth by upregulating p53 signaling and inducing ferroptosis | [51] |
| Breast cancer | Breast cancer organoids | Combining anti-FGFR4 and anti-HER2 therapies induces ferroptosis in HER2-positive breast cancer | [52] |
| Breast cancer | MCF-7 organoid models | Tamoxifen can induce ferroptosis in MCF-7 organoid models | [53] |
| Pancreatic cancer | PDAC organoids | Iron-sensitive prodrugs that trigger active ferroptosis in drug-tolerant pancreatic cancer cells | [20] |
| Pancreatic cancer | Patient-derived organoids | Imidazole ketone erastin induces tumor regression and abrogates MCU-driven metastasis | [54] |
| Pancreatic cancer | Pancreatic organoids derived from a mouse model | Elevated FSP1 protects KRAS-mutated cells from ferroptosis during tumor initiation | [40] |
| Pancreatic cancer | Patient-derived organoids | Triggers immunogenic ferroptosis by targeting the MCP-GPX4/HMGB1 axis | [55] |
| Pancreatic cancer | SMAD family member 4 (SMAD4)-positive organoids | Enhances cytotoxic effects by combining gemcitabine with ferroptosis inducers | [56] |
| Liver cancer | Patient-derived organoids | Donafenib and GSK-J4 synergistically induced ferroptosis in liver cancer by upregulating HMOX1 expression | [57] |
| Liver cancer | Organoid models derived from HCC patients | Metformin restores PPARGC1A expression and enhances ferroptosis | [58] |
| Liver cancer | HCC organoids | URI mediates resistance to TKI-induced ferroptosis | [59] |
| Liver cancer | CRC organoids | Combining ferroptosis induction with MDSC blockade rendered primary tumors and metastases | [60] |
| Gastric cancer | Patient-derived organoid model | CAFs impair the cytotoxic function of NK cells in gastric cancer by inducing ferroptosis via iron regulation | [61] |
| Gastric cancer | Patient-derived organoids | Inhibition of the STAT3–ferroptosis negative regulatory axis suppresses tumor growth and alleviates chemoresistance | [62] |
| Colorectal cancer | Colonic epithelial organoids | Role in colitis-associated cancer, modulation of Hmox1 and ferroptosis | [63] |
| Colorectal cancer | Intestinal stem cell organoids | IFNγ synergies with cold atmospheric plasma to trigger ferroptosis | [64] |
| Colorectal cancer | Patient-derived organoids | Curcumin and andrographis exhibit anti-tumor effects via the activation of ferroptosis | [65] |
| Colorectal cancer | CRC organoids | Adipocyte-derived exosomal MTTP suppresses ferroptosis and promotes chemoresistance | [66] |
| Colorectal cancer | CRC-derived organoid | Overcome drug resistance through LGR4 targeting, enhance sensitivity to drug-induced ferroptosis | [16] |
| Colorectal cancer | Patient-derived organoids | Andrographis-mediated chemosensitization in CRC via the activation of ferroptosis | [67] |
| Colorectal cancer | CRC organoids | VitC disrupted iron homeostasis and increased ROS levels, ultimately leading to ferroptosis | [68] |
| Ovarian cancer | Ovarian cancer organoids derived from HGSOC | FeNP inhibits GPX4 activity, leading to the induction of ferroptosis | [69] |
| Ovarian cancer | Ovarian cancer organoids | SCD1/FADS2 fatty acid desaturases equipoise lipid metabolic activity and redox-driven ferroptosis in ascite-derived ovarian cancer cells | [70] |
| Bladder cancer | BCa cell organoid models | PHGDH inhibitor NCT-502 induces ferroptosis in BCa cell organoid models | [66] |
| Bladder cancer | Patient-derived organoids | m6A modifications affect chemoresistance by controlling SLC7A11 protein levels, influencing ferroptosis sensitivity | [71] |
| Cholangiocarcinoma | Patient-derived organoids | Combining SUR with PDT induces ferroptosis and inhibits tumor growth | [72] |
| Glioblastoma | GBM patient-derived organoid models | Combination treatment can enhance ferroptosis by regulating heme oxygenase 1 (HOXM1) and GPX4 expression | [73] |
| Oral squamous cell carcinoma | Patient-derived organoid models | DRP1 inhibition-mediated mitochondrial elongation drives ferroptosis and abolishes cancer stemness | [42] |
| Lung cancer | Lung cancer organoids | MA promotes EGFR-TKI sensitivity by the KRAS-ERK pathway and mitochondrial Ca2+ overload | [74] |
| Prostate cancer | Organoid cultures derived from prostate cancer cells | TQB3720 promotes ferroptosis through the inhibition of the AR signaling pathway | [75] |
| Laryngeal squamous cell carcinoma | Organoid models derived from LSCC patients | Augmented ERO1α upon mTORC1 activation induces ferroptosis resistance and tumor progression via the upregulation of SLC7A11 | [76] |
| Head and neck cancer | HNSCC organoid models | TrxR1 induces ferroptosis and potentiates the efficacy of anti-PD-1 therapy | [77] |
| Pyroptosis | |||
| Pancreatic and lung cancers | Patient-derived tumor organoids | Src or ceramidase inhibitor reactivates pyroptosis to reverse chemoresistance | [44] |
| Colorectal cancer | Colorectal cancer patient-derived organoids | BI and CPT induce GSDME-mediated pyroptosis and apoptosis, with anti-tumoural activity | [78] |
| Colorectal cancer | Adult stem cell-derived murine colonic epithelial organoids | EGFR-signaling-independent colonoids show chromosomal instability and altered pyroptosis-related genes | [45] |
| Bladder cancer | MIBC mouse model with gene-edited organoids | Cathepsin inhibition induces pyroptosis, restraining chemoresistant MIBCs | [79] |
| Endometrial cancer | EC-derived organoids | ERRα regulates pyroptosis through the NLRP3/caspase-1/GSDMD pathway | [80] |
| Esophageal adenocarcinoma | Murine BE organoids | Caspase-1 inhibition reduces IL-1β and CXCL1 secretion, linked to BE to EAC progression | [81] |
| Her2-positive gastric cancer | Patient-derived organoids | IBI315 induces GSDMB-mediated pyroptosis in tumor cells, enhancing T cell activation and tumor killing | [82] |
| Ovarian cancer | Patient-derived ovarian cancer organoids | GBP5 induces canonical pyroptosis through the JAK2/STAT1 pathway, inhibiting cancer progression | [83] |
| Ovarian carcinoma | Adipocyte organoid model | Adipocyte pyroptosis in an omental tumor microenvironment is associated with chemoresistance | [84] |
| Necroptosis | |||
| Pancreatic duct adenocarcinoma | Human or mice PDAC organoids | Organoids used to study impaired cell death pathways, including necroptosis in PDAC | [85] |
| Colorectal cancer | Human colorectal cancer and murine organoid models | Tolinapant induces apoptosis, and its effect is augmented by FOLFOX treatment in the presence of HDAC inhibitors | [86] |
| Colorectal cancer | Patient-derived colon cancer organoids | FMRP regulates RIPK1 and colorectal cancer resistance to necroptosis | [49] |
| GVL | Mouse gastrointestinal organoids | Inhibition of RIP1 improves immune reconstitution and reduces GVHD mortality while preserving graft-versus-leukemia effects | [50] |
| Mutant p53-expressing cells | Intestinal organoids | Mutant p53-expressing cells undergo necroptosis via cell competition with the neighboring normal epithelial cells | [87] |
| Gastric cancer | Organoids derived from corpus glands | IL-17A is a cytokine that promotes parietal cell apoptosis during atrophic gastritis, a precursor lesion for gastric cancer | [88] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lu, D.; Xia, B.; Feng, T.; Qi, G.; Ma, Z. The Role of Cancer Organoids in Ferroptosis, Pyroptosis, and Necroptosis: Functions and Clinical Implications. Biomolecules 2025, 15, 659. https://doi.org/10.3390/biom15050659
Lu D, Xia B, Feng T, Qi G, Ma Z. The Role of Cancer Organoids in Ferroptosis, Pyroptosis, and Necroptosis: Functions and Clinical Implications. Biomolecules. 2025; 15(5):659. https://doi.org/10.3390/biom15050659
Chicago/Turabian StyleLu, Dingci, Bingqian Xia, Tianquan Feng, Gui Qi, and Zhaowu Ma. 2025. "The Role of Cancer Organoids in Ferroptosis, Pyroptosis, and Necroptosis: Functions and Clinical Implications" Biomolecules 15, no. 5: 659. https://doi.org/10.3390/biom15050659
APA StyleLu, D., Xia, B., Feng, T., Qi, G., & Ma, Z. (2025). The Role of Cancer Organoids in Ferroptosis, Pyroptosis, and Necroptosis: Functions and Clinical Implications. Biomolecules, 15(5), 659. https://doi.org/10.3390/biom15050659